Zusammensetzung
Wirkstoffe
Rilpivirin (als freie Rilpivirin-Base)
Hilfsstoffe
Zitronensäure-Monohydrat, Glucosemonohydrat, Poloxamer 338, Natriumdihydrogenphosphat-Monohydrat, Natriumhydroxid zum Einstellen des pH-Werts und zur Sicherstellung der Isotonizität, Wasser für Injektionszwecke.
Natrium-Gesamtgehalt bei Wirkstoffmenge 600 mg: 1,66 mg;
Natrium-Gesamtgehalt bei Wirkstoffmenge 900 mg: 2,49 mg.
Indikationen/Anwendungsmöglichkeiten
REKAMBYS ist in Kombination mit Cabotegravir zur Injektion für die Behandlung von Infektionen mit dem humanen Immunschwächevirus Typ 1 (HIV-1) bei Erwachsenen indiziert, die seit mindestens 6 Monaten vor Umstellung auf die Rilpivirin-Cabotegravir-Kombination unter einer stabilen antiretroviralen Therapie virologisch supprimiert (HIV-1 RNA < 50 Kopien /ml) sind und keine bekannten oder vermuteten Resistenzen gegen sowie in der Vorgeschichte kein virologisches Versagen mit Wirkstoffen aus der NNRTI- oder INI-Klasse aufweisen (siehe «Klinische Wirksamkeit»).
Dosierung/Anwendung
Übliche Dosierung
Erwachsene
Die Behandlung muss durch einen Arzt erfolgen, der Erfahrung in der Behandlung von HIV-Infektionen hat.
REKAMBYS-Injektionen müssen von einer medizinischen Fachperson verabreicht werden.
REKAMBYS sollte stets zusammen mit einer Cabotegravir-Injektion verabreicht werden. Daher sollte immer auch die Fachinformation für Cabotegravir-Injektionen beachtet werden.
REKAMBYS kann mit einer oralen Einleitungsbehandlung oder ohne (direkter Beginn mit Injektion) initiiert werden. Arzt und Patient können entscheiden, Rilpivirin-Tabletten als orale Einleitungsbehandlung vor Beginn der REKAMBYS-Injektionen zu verwenden, um die Verträglichkeit zu beurteilen (siehe Tabelle 1) oder direkt mit der REKAMBYS-Therapie zu beginnen. REKAMBYS-Injektionen können monatlich oder alle 2 Monate verabreicht werden (siehe Tabelle 2 für die monatliche und Tabelle 3 für die zweimonatliche Dosierungsempfehlung). Arzt und Patient sollten beide Dosierungsoptionen vor Beginn der REKAMBYS-Injektionen diskutieren und die für den Patienten geeignetste Dosierungsfrequenz wählen.
Vor dem Beginn der Behandlung mit REKAMBYS sollte der behandelnde Arzt sorgfältig Patienten auswählen, die mit dem erforderlichen Injektionsschema einverstanden sind, und die Patienten über die Bedeutung der Einhaltung der geplanten Termine zur Anwendung des Arzneimittels aufklären, um die Virussuppression aufrechtzuerhalten und das Risiko eines viralen Rebounds und einer möglichen Resistenzentwicklung im Zusammenhang mit verpassten Dosen zu verringern.
Nach Absetzen von REKAMBYS in Kombination mit einer Cabotegravir-Injektion ist es unerlässlich, ein alternatives, vollständig suppressives antiretrovirales Regime innerhalb eines Monats nach der letzten Injektion von REKAMBYS bei einem monatlichen Dosierungsschema und innerhalb von zwei Monaten nach der letzten Injektion von REKAMBYS bei einem zweimonatlichen Dosierungsschema einzuführen.
Wenn ein virologisches Versagen vermutet wird, sollte so schnell wie möglich ein Alternativregime initiiert werden.
Orale Einleitungsbehandlung (Lead-in-Phase)
Bei oraler Einleitungsbehandlung wird die orale Einnahme von Rilpivirin-Tabletten bei virologisch supprimierten Patienten für die Dauer von rund einem Monat (mindestens 28 Tage, maximal 2 Monate) vor Behandlungsbeginn mit REKAMBYS -Injektionen empfohlen, um die Verträglichkeit gegenüber Cabotegravir zu testen. Es sollte eine 25-mg-Tablette Rilpivirin einmal täglich zu einer Mahlzeit zusammen mit einer 30-mg-Tablette Cabotegravir (einmal tägliche Anwendung) eingenommen werden (siehe Fachinformation für Rilpivirin Tabletten).
|
Tabelle 1: Dosierungsplan der oralen Einleitungsbehandlung bei Erwachsenen | |
|
|
Orale Einleitungsbehandlung |
|
Arzneimittel |
1 Monat (für mindestens 28 Tage, maximal 2 Monate), gefolgt von der Erstinjektiona |
|
Rilpivirin |
25 mg einmal täglich zu einer Mahlzeit |
|
Cabotegravir |
30 mg einmal täglich |
|
a
siehe Tabelle 2 für den Dosierungsplan der monatlichen Injektionen und Tabelle 3 für den Dosierungsplan der Injektionen alle 2 Monate. | |
Monatliche Dosisgabe
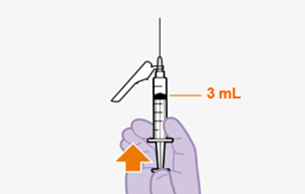
Erstinjektion (3-ml-Dosis)
Die empfohlene Anfangsdosis von REKAMBYS bei Erwachsenen besteht aus einer einzelnen intramuskulären Injektion von 3 ml (900 mg) am letzten Tag der derzeitigen antiretroviralen Therapie oder der oralen Einleitungsbehandlung. (Siehe «Art der Anwendung»).
Folgeinjektionen (2-ml-Dosis)
Nach der ersten Injektion besteht die empfohlene Dosis von REKAMBYS für weitere Injektionen bei Erwachsenen aus einer monatlichen intramuskulären Injektion von 2 ml (600 mg). (Siehe «Art der Anwendung»). Die Injektionen können den Patienten bis zu 7 Tage vor und nach dem geplanten Datum der monatlichen 2-ml-Injektion gegeben werden.
|
Tabelle 2: Dosierungsplan zur monatlichen intramuskulären Injektion bei Erwachsenen | ||
|
Arzneimittel |
Erstinjektion |
Folgeinjektionen |
|
Rilpivirine |
3 ml (900 mg) |
2 ml (600 mg) |
|
Cabotegravir |
3 ml (600 mg) |
2 ml (400 mg) |
Dosisgabe alle 2 Monate
Einleitungsinjektionen (3-ml Dosis)
Am letzten Tag der derzeitigen antriretroviralen Therapie oder der oralen Einleitungsbehandlung sollte die erste intramuskuläre REKAMBYS-Einleitungsinjektion von 3 ml (900 mg) bei Erwachsenen erfolgen. Einen Monat später sollte eine zweite intramuskuläre Einleitungsinjektion von 3 ml (900 mg) gegeben werden. (Siehe «Art der Anwendung»). Die zweite Einleitungsinjektion von 3 ml (900 mg) kann bei den Patienten bis zu 7 Tage vor oder nach der geplanten Dosisgabe erfolgen.
Folgeinjektionen (3-ml Dosis)
Nach der zweiten Einleitungsinjektion ist die empfohlene REKAMBYS-Dosis für weitere Injektionen bei Erwachsenen eine einzelne intramuskuläre Injektion von 3 ml (900 mg) alle 2 Monate. (Siehe «Art der Anwendung»). Die Injektionen können bei den Patienten bis zu 7 Tage vor oder der geplanten Gabe der 3-ml-Injektion alle 2 Monate erfolgen.
|
Tabelle 3: Dosierungsplan zur intramuskulären Injektion alle 2 Monate bei Erwachsenen | ||
|
Drug |
Einleitungsinjektionen |
Folgeinjektionen |
|
Einleitungsinjektionen in Monat 1 (am letzten Tag der laufenden ART oder der oralen Einleitungsphase, sofern angewendet) und Monat 2 |
Zwei Monate nach der letzten Einleitungsinjektion und danach alle 2 Monate | |
|
Rilpivirine |
3 ml (900 mg) |
3 ml (900 mg) |
|
Cabotegravir |
3 ml (600 mg) |
3 ml (600 mg) |
Anpassung der Dosierungsfrequenz
Dosierungsempfehlungen bei der Umstellung von monatlichen Injektionen auf Injektionen alle zwei Monate
Patienten, die von fortlaufenden monatlichen Injektionen auf fortlaufende Injektionen alle zwei Monate umgestellt werden, sollten einen Monat nach der letzten fortlaufenden Injektionsdosis von 2 ml (600 mg) eine intramuskuläre Injektion von 3 ml (900 mg) REKAMBYS erhalten. Die Folgeinjektionen von 3 ml (900 mg) erfolgen dann alle 2 Monate.
Dosierungsempfehlungen bei der Umstellung von zweimonatlichen Injektionen auf monatliche Injektionen
Patienten, die von fortlaufenden zweitmonatlichen Injektionen auf fortlaufende monatliche Injektionen in der Erhaltungsphase wechseln, sollten zwei Monate nach der letzten 3 ml (900 mg) REKAMBYS-Injektion eine einzige intramuskuläre Injektion von 2 ml (600 mg) REKAMBYS und danach monatlich 2 ml (600 mg) erhalten.
Versäumte Injektion
Die Einhaltung des Plans für die Injektionen wird dringend empfohlen. Bei Patienten, die einen Injektionstermin auslassen, sollte noch einmal klinisch abgeklärt werden, ob die Wiederaufnahme der Therapie angemessen ist. Siehe Tabellen 4 und 5 bezüglich Dosierungsempfehlungen nach einer versäumten Injektion.
Ausgelassene monatliche Injektionsdosis
Orale Überbrückungsbehandlung und Wiederaufnahme der monatlichen Injektionen:
Wenn eine Verzögerung um mehr als 7 Tage vom Datum einer geplanten Injektion unvermeidbar ist, kann eine orale Therapie (25 mg Rilpivirin und 30 mg Cabotegravir einmal täglich als Tabletten) für bis zu 2 aufeinanderfolgende Monate angewendet werden. Alternativ kann bis zur Wiederaufnahme der Injektionen eine andere vollständig suppressive orale antiretrovirale Therapie verwendet werden. Bei der Auswahl der Therapie sollen die aktuellen HIV-Behandlungsrichtlinien berücksichtigt werden. Daten zur oralen Überbrückungstherapie mit anderen vollständig suppressiven antiretroviralen Therapien siehe Abschnitt Klinische Wirksamkeit.
Die erste Dosis der oralen Therapie sollte 1 Monat (± 7 Tage) nach der letzten Injektionsdosis von REKAMBYS eingenommen werden. Die Injektionsdosierung sollte am Tag des Abschlusses der oralen Dosierung wieder aufgenommen werden (siehe Empfehlungen in Tabelle 4). Falls mehr als zwei aufeinanderfolgende monatliche Injektionen ausgelassen werden und ersetzt werden müssen, sollte einen Monat (± 7 Tage) nach der letzten Injektion von REKAMBYS eine alternative orale Behandlung eingeleitet werden.
|
Tabelle 4: Empfehlungen zur Wiederaufnahme der monatlichen Injektionen von REKAMBYS nach ausgelassenen Injektionen oder oraler Überbrückungsbehandlung | |
|
Zeitraum seit der letzten Injektion |
Empfehlung |
|
≤2 Monate: |
Frühestmögliche Fortsetzung der monatlichen Injektion von 2 ml (600 mg). |
|
> 2 Monate: |
Erneute Gabe der 3-ml-Dosis (900 mg), anschliessende Fortsetzung der monatlichen Injektion von 2 ml (600 mg). |
Ausgelassene 2-monatliche Injektionsdosis
Orale Überbrückungsbehandlung und Wiederaufnahme der zweimonatlichen Injektionen:
Wenn eine Verzögerung um mehr als 7 Tage vom Datum einer geplanten Injektion unvermeidbar ist, kann eine tägliche orale Therapie (25 mg Rilpivirin und 30 mg Cabotegravir als Tablette) für bis zu 2 aufeinanderfolgende Monate angewendet werden. Alternativ kann bis zur Wiederaufnahme der Injektionen eine andere vollständig suppressive orale antiretrovirale Therapie verwendet werden. Bei der Auswahl der Therapie sollen die aktuellen HIV-Behandlungsrichtlinien berücksichtigt werden. Daten zur oralen Überbrückungstherapie mit anderen vollständig suppressiven antiretroviralen Therapien siehe Abschnitt Klinische Wirksamkeit.
Die erste Dosis der oralen Therapie sollte zwei Monate (± 7 Tage) nach der letzten Injektion von REKAMBYS und Cabotegravir eingenommen werden. Die Dosierung per Injektion sollte an dem Tag fortgesetzt werden, an dem die orale Dosierung abgeschlossen ist (siehe die Empfehlungen in Tabelle 5). Falls mehr als zwei Monate ersetzt werden müssen, d.h. es wurde mehr als eine 2-monatliche Injektion ausgelassen, sollte zwei Monate (± 7 Tage) nach der letzten Injektion von REKAMBYS eine alternative orale Behandlung eingeleitet werden.
|
Tabelle 5: Empfehlungen zur Wiederaufnahme der zweimonatlichen Injektion von REKAMBYS nach ausgelassenen Injektionen oder nach der oralen Überbrückungstherapie | ||
|
Ausgelassener Besuch zur Injektion |
Vergangene Zeit seit der letzten Injektion |
Empfehlung (alle Injektionen sind 3 ml) |
|
Injektion 2 |
≤2 Monate |
Weiterbehandlung mit der Injektion von 3 ml (900 mg) so bald wie möglich und Fortsetzung des Dosierungsplans mit Injektionen alle 2 Monate. |
|
>2 Monate |
Wiedereinleitung der 3-ml-Dosis (900 mg) bei dem Patienten, gefolgt von einer zweiten 3-ml-Injektion (900 mg) als Anfangsdosis einen Monat später. Danach den Dosierungsplan zur Injektion alle 2 Monate befolgen. | |
|
Injektion 3 oder später |
≤3 Monate |
Weiterbehandlung mit der Injektion von 3 ml (900 mg) so bald wie möglich und Fortsetzung des Dosierungsplans mit Injektionen alle 2 Monate |
|
>3 Monate |
Wiedereinleitung der 3-ml-Dosis (900 mg) bei dem Patienten, gefolgt von einer zweiten 3-ml-Injektion (900 mg) als Anfangsdosis einen Monat später. Danach den Dosierungsplan zur Injektion alle 2 Monate befolgen | |
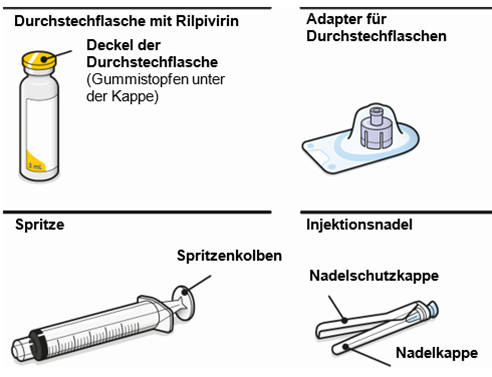
Art der Anwendung
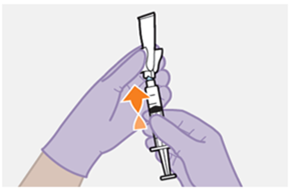
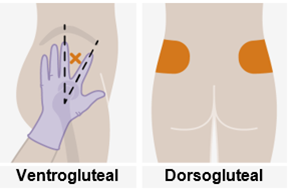
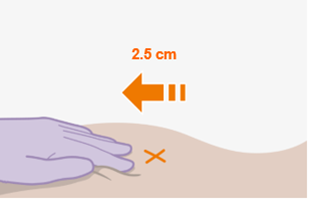
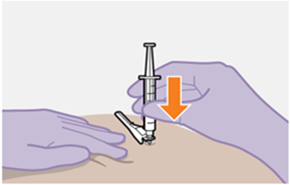
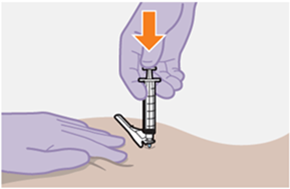
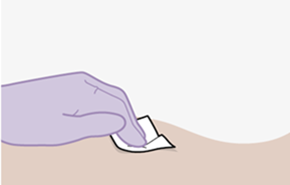
Nur zur intramuskulären (i. m.) Injektion in den Gesässmuskel (gluteal). Langsam injizieren (Siehe «Warnhinweise und Vorsichtsmassnahmen»). Nicht intravenös injizieren.
Bei der Verabreichung von REKAMBYS sollte die medizinische Fachperson den Body-Mass-Index (BMI) des Patienten berücksichtigen, um sicherzustellen, dass die Nadellänge ausreicht, um den Gesässmuskel zu erreichen.
REKAMBYS und Cabotegravir sollten während desselben Besuchs an getrennten Injektionsstellen (auf gegenüberliegenden Seiten oder – falls dies nicht möglich ist - im Abstand von 2 cm) in den Gesässmuskel injiziert werden. Ein 2 cm Abstand sollte auch zu früheren Injektionsstellen oder eventuellen Reaktionen an früheren Injektionsstellen eingehalten werden.
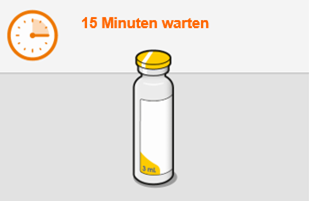
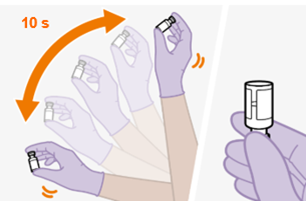
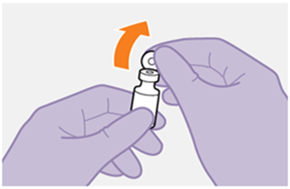
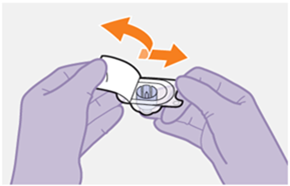
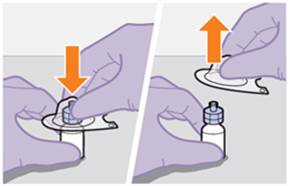
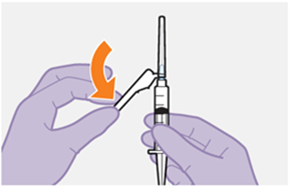
Die detaillierte Schritt-für-Schritt-Anleitung für die Verabreichung der Injektion in der Gebrauchsanweisung in der Packung ist zu beachten.
REKAMBYS sollte von einer medizinischen Fachperson verabreicht werden.
Spezielle Dosierungsanweisungen
Patienten mit eingeschränkter Leberfunktion
Es liegen nur begrenzte Informationen zur Anwendung von Rilpivirin bei Patienten mit leichter oder mittelschwerer Leberfunktionsstörung (Child-Pugh-Klasse A oder B) vor. Bei Patienten mit leichter bis mittelschwerer Leberfunktionsstörung (Child-Pugh-Klasse A oder B) ist keine Anpassung der REKAMBYS-Dosis erforderlich. Bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh-Klasse C) wurde REKAMBYS nicht untersucht (siehe «Pharmakokinetik»).
Patienten mit eingeschränkter Nierenfunktion
Bei Patienten mit leichter oder mittelschwerer Nierenfunktionsstörung ist keine Anpassung der Dosis von REKAMBYS erforderlich. Patienten mit einer geschätzten Kreatinin-Clearance < 50 ml/min/1,73 m2 wurden nicht in die Phase-III-Studien aufgenommen. REKAMBYS sollte bei Patienten mit schwerer Nierenfunktionsstörung (Kreatinin-Clearance 15-<30 ml/min) oder Nierenversagen im Endstadium (Kreatinin-Clearance <15 ml/min) mit Vorsicht (vermehrte Überwachung von unerwünschten Wirkungen) angewendet werden. Bei Patienten mit schwerer Nierenfunktionsstörung oder Niereninsuffizienz im Endstadium sollte die Kombination von REKAMBYS mit einem starken CYP3A-Inhibitor nur dann angewendet werden, wenn der Nutzen das Risiko überwiegt (siehe «Pharmakokinetik»).
Ältere Patienten (ab 65 Jahren)
Bei älteren Patienten ist keine Anpassung der REKAMBYS-Dosis erforderlich (siehe «Pharmakokinetik»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von REKAMBYS bei pädiatrischen Patienten wurden noch nicht nachgewiesen. Eine Behandlung mit REKAMBYS wird für diese Patientengruppe nicht empfohlen.
Kontraindikationen
Überempfindlichkeit gegenüber Rilpivirin oder einem der Hilfsstoffe.
Die gleichzeitige Gabe von REKAMBYS mit Arzneimitteln, die moderate bis starke Induktoren von CYP3A Enzymen sind, ist kontraindiziert, da erhebliche Verringerungen der Rilpivirin-Konzentrationen im Plasma auftreten können, was zum Verlust der therapeutischen Wirkung von REKAMBYS führen kann (siehe «Interaktionen»).
Beispiele für solche Arzneimitel sind:
·Die Antikonvulsiva Carbamazepin, Oxcarbazepin, Phenobarbital, Phenytoin
·Die antimykobakteriellen Wirkstoffe Rifabutin, Rifampicin, Rifapentin
·Das systemische Glukokortikoid Dexamethason, ausser zur Behandlung mit einer Einzeldosis
·Johanniskraut (Hypericum perforatum)
Bei der Anwendung von oralem Rilpivirin sind ebenfalls Protonenpumpenhemmer kontraindiziert.
Darüber hinaus ist auch die Fachinformation für Cabotegravir zu beachten.
Warnhinweise und Vorsichtsmassnahmen
Langwirkende Eigenschaften einer Rilpirivin-Injektion
Rilpivirin-Restkonzentrationen können für längere Zeit über den aktiven Dosierungszeitraum hinaus, bis zu 4 Jahre, im systemischen Kreislauf der Patienten verbleiben. Die lange Dauer der Freisetzung von Rilpivirin bei Anwendung von REKAMBYS sollte bei der individuellen Nutzen-Risiko-Abschätzung vor der Auswahl der Therapie, während der Therapie und nach Absetzen von REKAMBYS berücksichtigt werden (siehe «Interaktionen», «Unerwünschte Wirkungen», «Schwangerschaft, Stillzeit, Fertilität» und «Pharmakokinetik» sowie «Überdosierung»).
Aufgrund starker Proteinbindung gibt es keinen Mechanismus, durch den die Freisetzung von Rilpivirin gestoppt oder neutralisiert bzw. aus dem Muskel (z.B. durch Aspiration) oder aus dem Blut (z.B. durch Hämodialyse) entfernt werden kann.
Übertragung von HIV
Die Ergebnisse von Beobachtungstudien haben gezeigt, dass kein Risiko der sexuellen Übertragung von HIV besteht, wenn eine virale Suppression erreicht und aufrechterhalten wird. Allerdings kann das Risiko einer sexuellen Übertragung von HIV nicht ausgeschlossen werden, wenn die verordnete ART nicht regelmässig eingenommen wird und/oder die virale Suppression nicht erreicht und aufrechterhalten wird.
Patienten mit Hepatitis-B- und Hepatitis-C-Virus-Koinfektion
Patienten mit einer Hepatitis-B-Koinfektion waren von der Teilnahme an den Studien mit REKAMBYS ausgeschlossen. Die Einleitung von REKAMBYS, bei Patienten mit Hepatitis-B-Koinfektion wird nicht empfohlen. Bei Patienten mit Hepatitis-B-Koinfektion unter Behandlung mit oralem Rilpirivin war die Inzidenz von erhöhten Leberenzymen höher als bei Patienten unter Behandlung mit oralem Rilpirivin ohne Hepatitis-B-Koinfektion. Es sind die aktuellen Behandlungsleitlinien für die Behandlung einer HIV-Infektion bei Patienten mit Hepatitis-B-Virus-Koinfektion zu befolgen sowie der Abschnitt «Interaktionen mit Arzneimitteln» (siehe unten) zu beachten.
Es liegen begrenzte Daten bei Patienten mit Hepatitis-C-Koinfektion vor. Bei Patienten mit Hepatitis-C-Koinfektion unter Behandlung mit oralem Rilpirivin war die Inzidenz von erhöhten Leberenzymen höher als bei Patienten unter Behandlung mit oralem Rilpivirin ohne Hepatitis-C-Koinfektion. Die pharmakokinetische Exposition von oralem und injizierbarem Rilpivirin bei Hepatitis-C-koinfizierten Patienten war vergleichbar mit der bei Patienten ohne Hepatitis-C-Koinfektion. Bei Patienten mit Hepatitis-C-Koinfektion wird eine Überwachung der Leberfunktion empfohlen. Wenn während der Behandlung mit REKAMBYS eine Hepatitis-C-Infektion auftritt, sind die aktuellen Behandlungsrichtlinien für eine HIV-Infektion bei Patienten mit gleichzeitig bestehender Hepatitis-C-Virusinfektion zu befolgen sowie der Abschnitt «Interaktionen mit Arzneimitteln» (siehe unten) zu beachten.
Interaktionen mit Arzneimitteln
Wenn Rilpivirin zusammen mit Arzneimitteln verschrieben wird, welche die Rilpivirin-Exposition verringern können, ist Vorsicht geboten.
Bei gleichzeitiger Anwendung von REKAMBYS und einem Arzneimittel mit bekanntem Risiko für Torsade-de-Pointes-Arrhythmien ist Vorsicht geboten (siehe Abschnitt «Interaktionen»).
Für Hinweise zu Arzneimittelinteraktionen siehe «Interaktionen».
Opportunisische Infektionen
Bei Patienten, die Rilpivirin oder eine andere antiretrovirale Therapie erhalten, können weiterhin opportunistische Infektionen und andere Komplikationen der HIV-Infektion auftreten. Daher sollten die Patienten unter engmaschiger klinischer Beobachtung durch Ärzte bleiben, die Erfahrung mit der Behandlung solcher HIV-Begleiterkrankungen haben.
Immunrekonstitutions- und Inflammationssyndrom
Bei Patienten, die eine antiretrovirale Kombinationstherapie, beispielsweise mit oralem Rilpivirin, erhielten, wurde über ein Immunrekonstitutions- und Inflammationssyndrom berichtet. In der Anfangsphase einer antiretroviralen Kombinationstherapie können Patienten, deren Immunsystem auf die Behandlung anspricht, eine entzündliche Reaktion gegen indolente oder residuelle opportunistische Infektionen (z.B. mit Mycobacterium-avium-Komplex, Zytomegalievirus, Pneumocystis-jiroveci-Pneumonie und Tuberkulose) entwickeln, was eine weitere Abklärung und Behandlung erforderlich machen kann. Es liegen auch Berichte über Autoimmunerkrankungen (wie z.B. Morbus Basedow und Autoimmunhepatitis) vor, die im Rahmen eines Immunrekonstitutions- und Inflammationssyndrom auftraten. Allerdings ist der Zeitpunkt des Auftretens variabler, und diese Ereignisse können viele Monate nach Beginn der Behandlung auftreten (siehe «Unerwünschte Wirkungen»).
Hepatotoxizität
Bei einer begrentzen Anzahl von Patienten mit oder ohne bekannter Lebervorerkrankung wurde unter Rilpivirin und Cabotegravir eine Lebertoxizität festgestellt (siehe Abschnitt «Unerwünschte Wirkungen»).
Es wird eine Überwachung der Leberfunktionswerte empfohlen. Bei vermuteter Lebertoxizität muss Rilpivirin abgesetzt werden (siehe Abschnitt «Langwirkende Eigenschaften einer Rilpivirin-Injektion»).
Risiko einer Resistenzentwicklung nach Behandlungsabbruch
Um das Risiko einer Virusresistenz zu minimieren, muss spätestens einen Monat nach der letzten Injektion von REKAMBYS resp. spätestens 2 Monate nach der letzten Injektion von REKAMBYS bei Dosierung alle 2 Monate ein alternatives, vollständig supprimierendes antiretrovirales Regime angewendet werden. Wenn ein virologisches Versagen vermutet wird, sollte so schnell wie möglich ein Alternativregime initiiert werden.
Versehentliche intravenöse Verabreichung / Reaktionen nach der Injektion
Gelegentlich traten in klinischen Studien innerhalb von Minuten nach der Injektion von Rilpivirin schwerwiegende Reaktionen auf, mit Symptomen wie Dyspnoe, Bronchospasmus, Unruhe, Abdominalkrämpfe, Ausschlag/Urtikaria, Schwindel, Flushing, Schwitzen, Taubheitsgefühl im Mund, Veränderungen des Blutdrucks und Pulses und Schmerzen (z.B. Rücken und Brust). Diese Symptome traten bei <1% der Patienten auf und begannen innert Minuten nach der Injektion abzuklingen, wobei ihr vollständiges Verschwinden bis zum nächsten Tag dauern konnte. Einige Patienten erhielten eine unterstützende Behandlung. Sie können mit einer versehentlichen intravenösen Verabreichung während der intramuskulären Injektion im Zusammenhang stehen, die zu vorübergehend hohen Plasmakonzentrationen von Rilpivirin führt.
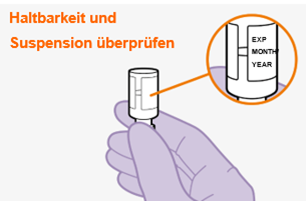
Bei der Vorbereitung und Verabreichung von REKAMBYS sind sorgfältig die Anwendungshinweise zu befolgen.Vor der Verabreichung sollte die Durchstechflasche von REKAMBYS auf Raumtemperatur gebracht werden. Die Suspension sollte langsam injiziert werden, und es sollte darauf geachtet werden, dass eine versehentliche intravenöse Verabreichung vermieden wird. Die Patienten sind über einen kurzen Zeitraum (ca. 10 Minuten) nach der Injektion unter Beobachtung zu halten. Wenn bei einem Patienten nach der Injektion eine Reaktion auftritt, ist diese zu überwachen und der klinischen Indikation entsprechend zu behandeln (siehe «Überdosierung»).
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) je Dosierungseinheit, d.h. es ist nahezu «natriumfrei».
Interaktionen
Es wurden keine Studien zur Untersuchung von Arzneimittelinteraktionen nach Verabreichung von REKAMBYS durchgeführt. Die folgenden Angaben und Empfehlungen beruhen auf Studien nach oraler Verabreichung von Rilpivirin oder auf theoretischen Überlegungen.
Auswirkungen anderer Arzneimittel auf Rilpivirin
CYP3A Induktoren und Inhibitoren
Rilpivirin wird hauptsächlich über Cytochrom-P450-3A (CYP3A) metabolisiert. Arzneimittel, die CYP3A induzieren oder hemmen, können daher die Clearance von Rilpivirin beeinflussen (siehe «Pharmakokinetik»). Bei gleichzeitiger Anwendung von REKAMBYS mit Arzneimitteln, die CYP3A induzieren, wurden reduzierte Plasmakonzentrationen von Rilpivirin festgestellt, wodurch sich die therapeutische Wirkung von REKAMBYS möglicherweise verringern kann. Die gleichzeitige Verabreichung von REKAMBYS mit moderaten und starken CYP3A Induktoren ist daher kontraindiziert.
Bei gleichzeitiger Anwendung von REKAMBYS mit Arzneimitteln, die CYP3A hemmen, wurden erhöhte Plasmakonzentrationen von Rilpivirin festgestellt.
Arzneimittel, die das QT-Intervall verlängern
Es liegen nur begrenzte Informationen zur Möglichkeit einer pharmakodynamischen Interaktion zwischen Rilpivirin und Arzneimitteln vor, die das QTc-Intervall im Elektrokardiogramm verlängern. In einer Studie mit gesunden Probanden verlängerten supratherapeutische Dosen von oralem Rilpivirin (75 mg einmal täglich und 300 mg einmal täglich) das QTc-Intervall im Elektrokardiogramm. Die Rilpivirin-Konzentrationen im Plasma nach REKAMBYS-Injektionen und nach Einnahme von Edurant in einer Dosis von 25 mg einmal täglich sind vergleichbar (siehe «Pharmakodynamik»). Bei gleichzeitiger Anwendung von REKAMBYS und einem Arzneimittel mit bekanntem Risiko für Torsade-de-Pointes-Arrhythmien ist Vorsicht geboten.
Auswirkung von Rilpivirin auf andere Arzneimittel
CYP Substrate
Es ist nicht davon auszugehen, dass Rilpivirin klinisch relevante Auswirkungen auf die Exposition gegenüber Arzneimitteln hat, die von CYP-Enzymen metabolisiert werden.
Andere antiretrovirale Arzneimittel
REKAMBYS ist in Kombination mit Cabotegravir zur Injektion als vollständiges Regime zur Behandlung von HIV-1-Infektionen indiziert und sollte nicht zusammen mit anderen antiretroviralen Arzneimitteln zur Behandlung von HIV-1 angewendet werden.
In Tabelle 7 sind Informationen zu Arzneimittel-Interaktionen mit anderen antiretroviralen HIV-Arzneimitteln aufgeführt, um die Wahl eines alternativen antiretroviralen Regimes nach Absetzen von REKAMBYS zu erleichtern (siehe «Risiko einer Resistenzentwicklung nach Behandlungsabbruch»). Was die Arzneimittel-Interaktionen anbelangt, können nach dem Absetzen von REKAMBYS alle in Tabelle 7 gelisteten antiretroviralen HIV-Arzneimittel angewendet werden. Aufgrund der langen Dauer der Freisetzung von Rilpivirin nach dem Absetzen von REKAMBYS-Injektionen sind einige Interaktionen mit anderen antiretroviralen HIV-Arzneimitteln möglich. Rilpivirin hat wahrscheinlich keinen klinisch relevanten Einfluss auf die Pharmakokinetik anderer antiretroviraler HIV-Arzneimittel. Geboosterte Integrase-Inhibitoren und geboosterte oder ungeboosterte Proteaseinhibitoren können zu erhöhten Restmengen von Rilpivirin im Plasma führen. Die nicht-nukleosidischen Reverse-Transkriptase-Inhibitoren Efavirenz, Etravirin und Nevirapin können dagegen zu einer Verringerung der Restmengen von Rilpivirin im Plasma führen. Diese Arzneimittel-Interaktionen nach dem Absetzen von REKAMBYS werden als klinisch nicht relevant erachtet und verändern die lange Dauer der Freisetzung von Rilpivirin von der Injektionsstelle nicht.
In Tabelle 6 und Tabelle 7 sind ausgewählte bekannte und theoretische Interaktionen zwischen Rilpivirin und gleichzeitig angewendeten Arzneimitteln und für antiretroviralen HIV-Arzneimittel die nach Absetzen von REKAMBYS angewendet werden sollen, aufgeführt. Daten aus Studien zur Arzneimittel-Interaktion zeigen die Quotienten der geometrischen Mittelwerte (Geometric Mean Ratios, GMR) für die pharmakokinetischen Parameter bei Gabe mit/ohne Begleitmedikation mit 90 %-Konfidenzintervallen (KI).
|
Tabelle 6: Interaktionen und Empfehlungen zur Dosierung von REKAMBYS mit anderen Arzneimitteln | ||
|
Wirkstoff nach Therapiefeld (Dosierungsregime) |
Auswirkungen auf die Wirkstoffkonzentration |
Empfehlungen für die gleichzeitige Anwendung |
|
ANTIVIRALE HIV-MITTEL (zur Anwendung mit REKAMBYS) | ||
|
Cabotegravir* Ω (30 mg q.d. für 12 Tage) |
Cabotegravir: |
Keine Dosisanpassung empfohlen. |
|
ANDERE ANTIVIRALE MITTEL | ||
|
Ribavirin |
Nicht untersucht. Es ist keine klinisch relevante Arzneimittel-Interaktion zu erwarten |
Keine Dosisanpassung empfohlen. |
|
Telbivudin, Entecavir, Lamivudin, Tenofoviralafenamid |
Nicht untersucht. Es ist keine klinisch relevante Arzneimittel-Interaktion zu erwarten. |
Keine Dosisanpassung empfohlen. |
|
Sofosbuvir/Velpatasvir, Sofosbuvir/Velpatasvir/Voxilaprevir, Ledipasvir, Pibrentasvir/Glecaprivir, Elbasvir/Grazoprevir |
Nicht untersucht. |
Keine Dosisanpassung empfohlen. |
|
ANTIKONVULSIVA | ||
|
Carbamazepin |
Nicht untersucht. Es sind erhebliche Verringerungen der Rilpivirin-Konzentrationen im Plasma zu erwarten. |
Die gleichzeitige Anwendung von REKAMBYS ist kontraindiziert. |
|
AZOL-ANTIMYKOTIKA | ||
|
Ketoconazol*#Ω (400 mg q.d. für 22 Tage) |
Ketoconazol: |
Keine Dosisanpassung empfohlen. |
|
Fluconazol |
Nicht untersucht. Die gleichzeitige Anwendung von REKAMBYS und Azol-Antimykotika kann eine Erhöhung der Rilpivirin-Konzentrationen im Plasma bewirken. |
Keine Dosisanpassung empfohlen. |
|
ANTIMYKOBAKTERIELLE MITTEL | ||
|
Rifabutin*#Ω |
Rifabutin: |
Die gleichzeitige Anwendung ist kontraindiziert. Eine gleichzeitige Anwendung kann zu einem Verlust der therapeutischen Wirkung von Rilpivirin führen (siehe «Kontraindikationen»). |
|
Rifabutin*#Ω |
Rilpivirin: | |
|
Rifabutin*#Ω |
Rilpivirin: | |
|
Rifampicin*#Ω |
Rifampicin: |
Die gleichzeitige Anwendung ist kontraindiziert. Die gleichzeitige Anwendung kann zu einem Verlust der therapeutischen Wirkung von Rilpivirin führen (siehe «Kontraindikationen»). |
|
Rifapentin |
Nicht untersucht. Es sind erhebliche Verringerungen der Rilpivirin-Konzentrationen im Plasma zu erwarten. |
Die gleichzeitige Anwendung ist kontraindiziert. Die gleichzeitige Anwendung kann zu einem Verlust der therapeutischen Wirkung von Rilpivirin führen |
|
MAKROLIDANTIBIOTIKA | ||
|
Clarithromycin |
Nicht untersucht. Es ist eine erhöhte Rilpivirin-Exposition zu erwarten. |
Wenn möglich, sollten Alternativen wie Azithromycin in Betracht gezogen werden. |
|
GLUKOKORTIKOIDE oder Kortikosteroide | ||
|
Dexamethason (systemisch, ausser bei Anwendung als Einzeldosis) |
Nicht untersucht. Bedeutende Verringerungen der Rilpivirin-Konzentrationen im Plasma sind zu erwarten. |
Die gleichzeitige Anwendung ist kontraindiziert (ausser als Einzeldosis). Die gleichzeitige Anwendung kann zu einem Verlust der therapeutischen Wirkung von Rilpivirin führen (siehe «Kontraindikationen»). Es sollten Alternativen in Betracht gezogen werden. |
|
NARKOTISCHE ANALGETIKA | ||
|
Methadon*Ω |
R(-)-Methadon: |
Zu Beginn der gleichzeitigen Anwendung von Methadon sind keine Dosisanpassungen erforderlich. Allerdings empfiehlt sich eine klinische Überwachung, da die Methadon-Erhaltungstherapie bei manchen Patienten möglicherweise angepasst werden muss. |
|
ANTIARRHYTHMIKA | ||
|
Digoxin*Ω (0,5 mg Einzeldosis) |
Digoxin: |
Keine Dosisanpassung empfohlen. |
|
ANTIDIABETIKA | ||
|
Metformin*Ω (850 mg als Einzeldosis) |
Metformin; |
Keine Dosisanpassung empfohlen. |
|
PFLANZLICHE PRÄPARATE | ||
|
Johanniskraut (Hypericum perforatum) |
Nicht untersucht. Bedeutende Verringerungen der Rilpivirin-Konzentrationen im Plasma sind zu erwarten. |
Die gleichzeitige Anwendung ist kontraindiziert. Die gleichzeitige Anwendung kann zu einem Verlust der therapeutischen Wirkung von Rilpivirin führen (siehe «Kontraindikationen»). |
|
ANALGETIKA | ||
|
Paracetamol*#Ω |
Paracetamol: |
Keine Dosisanpassung empfohlen. |
|
ORALE KONTRAZEPTIVA | ||
|
Ethinylestradiol*Ω |
Ethinylestradiol: |
Keine Dosisanpassung empfohlen. |
|
HMG-CO-A-REDUKTASEHEMMER | ||
|
Atorvastatin*#Ω |
Atorvastatin: |
Keine Dosisanpassung empfohlen. |
|
Fluvastatin |
Nicht untersucht. |
Keine Dosisanpassung empfohlen. |
|
PHOSPHODIESTERASE-TYP-5-HEMMER (PDE-5-HEMMER) | ||
|
Sildenafil*#Ω |
Sildenafil: |
Keine Dosisanpassung empfohlen. |
|
Vardenafil |
Nicht untersucht. |
Keine Dosisanpassung empfohlen. |
|
Ω
GMR und 90 % KI auf der Basis von Studien zu Arzneimittelinteraktionen mit oralem Rilpivirin | ||
|
Tabelle 7: Interaktionen und Dosisempfehlungen für ANTIVIRALE HIV-ARZNEIMITTEL (zur Anwendung bei Absetzen von REKAMBYS) | ||
|
Wirkstoff nach Therapiefeld (Dosierungsregime) |
Auswirkungen auf die Wirkstoffkonzentration |
Empfehlung zur Anwendung nach dem Absetzen von REKAMBYS |
|
Dolutegravir*Ω (50 mg q.d. für 5 Tage) |
Dolutegravir: |
Keine Dosisanpassung empfohlen. |
|
Raltegravir*Ω (400 mg b.i.d. für 15 Tage) |
Raltegravir: |
Keine Dosisanpassung empfohlen. |
|
Bictegravir |
Nicht untersucht. Es ist keine klinisch relevante Arzneimittel-Interaktion zu erwarten. |
Keine Dosisanpassung empfohlen. |
|
Darunavir/Ritonavir*Ω (800 mg q.d. für 22 Tage) |
Darunavir: |
Keine Dosisanpassung empfohlen. |
|
Lopinavir/Ritonavir* Ω (400 mg b.i.d. für 20 Tage) |
Lopinavir: |
Keine Dosisanpassung empfohlen. |
|
Darunavir/Cobicistat |
Nicht untersucht. Es ist keine klinisch relevante Arzneimittel-Interaktion zu erwarten. |
Keine Dosisanpassung empfohlen. |
|
Tenofovirdisoproxilfumarat*Ω (300 mg q.d. für 16 Tage) |
Tenofovir (Plasma): |
Keine Dosisanpassung empfohlen. |
|
Didanosin*Ω (400 mg q.d. für 14 Tage) |
Didanosin: |
Keine Dosisanpassung empfohlen. |
|
Abacavir, Emtricitabin, Lamivudin, Stavudin, Tenofoviralafenamid, Zidovudin |
Nicht untersucht. Es ist keine klinisch relevante Arzneimittel-Interaktion zu erwarten. |
Keine Dosisanpassung empfohlen. |
|
Efavirenz, Doravirin, Etravirin, Nevirapin |
Nicht untersucht. Bei Anwendung nach dem Absetzen von REKAMBYS ist keine klinisch relevante Arzneimittel-Interaktion zu erwarten. |
Keine Dosisanpassung empfohlen. |
|
Maraviroc |
Nicht untersucht. Es ist keine klinisch relevante Arzneimittel-Interaktion zu erwarten. |
Keine Dosisanpassung empfohlen. |
|
Enfuvirtid |
Nicht untersucht. Es ist keine klinisch relevante Arzneimittel-Interaktion zu erwarten |
Keine Dosisanpassung empfohlen. |
|
Fostemsavir |
Nicht untersucht. Es ist keine klinisch relevante Arzneimittel-Interaktion zu erwarten. |
Keine Dosisanpassung empfohlen. |
|
* Die Interaktion zwischen Rilpivirin und dem Wirkstoff wurde in einer klinischen Studie untersucht. Bei allen anderen gezeigten Arzneimittel-Interaktionen handelt es sich um Prognosen. | ||
Schwangerschaft, Stillzeit
Schwangerschaft
Die Wirkung von REKAMBYS auf Schwangerschaften beim Menschen ist nicht bekannt. Es liegen keine klinischen Studien zur Anwendung von REKAMBYS bei Schwangeren vor.
Tierexperimentelle Studien zeigten keine direkte oder indirekte Toxizität mit Rilpivirin und keine Auswirkung auf Schwangerschaft, Embryonalentwicklung, Entwicklung des Föten und postnatale Entwicklung (siehe «Präklinische Daten»).
REKAMBYS sollte bei Frauen im gebärfähigen Alter, die eine Schwangerschaft planen oder keine zuverlässige kontrazeptive Methode anwenden nicht eingesetzt werden, ausser der erwartete Nutzen rechtfertigt die möglichen Risiken für das Ungeborene.
Die Patientinnen sollten hinsichtlich der Anwendung wirksamer Verhütungsmittel beraten werden. REKAMBYS und Kontrazeptiva auf Östrogen- und/oder Progesteronbasis können gleichzeitig ohne Dosisanpassung angewendet werden (siehe «Interaktionen»).
REKAMBYS sollte nicht während der Schwangerschaft angewendet werden, es sei denn, der erwartete Nutzen rechtfertigt das mögliche Risiko für das Ungeborene. Während der Schwangerschaft wurde eine geringere Verfügbarkeit von oralem Rilpivirin beobachtet, daher sollte die Viruslast engmaschig überwacht werden.
Nach dem Absetzen von REKAMBYS könnte Rilpivirin bei einigen Patienten bis zu 4 Jahre lang im systemischen Kreislauf vorkommen. Daher sollte das Potenzial einer Exposition des ungeborenen Kindes während der Schwangerschaft berücksichtigt werden.
Zur Überwachung des Schwangerschaftsausgangs in Bezug auf die Mutter und den Fötus wurde eine Registerstudie für Schwangerschaften bei antiretroviral behandelten Patientinnen (Antiretroviral Pregnancy Registry) gestartet (http://www.apregistry.com). Dabei handelt es sich um eine prospektive Beobachtungsstudie mit Expositionserfassung zur freiwilligen Teilnahme zur Erhebung und Bewertung von Daten zum Ausgang von Schwangerschaften, bei denen die Mütter während der Schwangerschaft antiretroviral behandelt wurden. Für Rilpivirin liegen ausreichende Daten zur Exposition im ersten Trimenon vor, um eine Erhöhung des Risikos von Geburtsfehlern um mindestens das Doppelte erkennen zu können. Bislang wurden keine solchen Erhöhungen festgestellt.
Stillzeit
Es ist nicht bekannt, ob Rilpivirin beim Menschen in die Muttermilch übergeht. In tierexperimentellen Studien wurde Rilpirivin in die Milch abgegeben. Unter Berücksichtigung, dass Rilpivirin nach dem Absetzen von REKAMBYS bei einigen Patientinnen bis zu 4 Jahre lang im systemischen Kreislauf vorkommen könnte, könnte es auch für diese Zeit in der Muttermilch vorhanden sein.
Sowohl wegen der Möglichkeit einer HIV-Übertragung als auch wegen der Möglichkeit unerwünschter Wirkungen bei gestillten Säuglingen sollten Mütter dazu angehalten werden, nicht zu stillen.
Fertilität
Es liegen keine Daten über die Auswirkung von Rilpivirin auf die Fertilität beim Menschen vor. In tierexperimentellen Studien wurde bei oraler Gabe von Rilpivirin keine Wirkung auf die Fertilität festgestellt (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Der Einfluss von REKAMBYS auf die Verkehrstüchtigkeit oder die Fähigkeit, Maschinen zu bedienen, wurde nicht untersucht.
Unerwünschte Wirkungen
Unerwünschte Wirkungen aus klinischen Studien
Nachstehend sind die unter Rilpivirin allein oder unter einer Kombinationstherapie von Rilpivirin plus Cabotegravir aufgetretenen unerwünschte Wirkungen (UW) aufgeführt (monatliche Dosierung oder Dosierung alle 2 Monate) einschliesslich solcher, die den oralen als auch den injizierbaren Formulierungen von Rilpivirin plus Cabotegravir zuzuschreiben sind. Wenn sich die Häufigkeiten zwischen Phase-III-Studien unterschieden haben, ist die Kategorie mit der höchsten Häufigkeit angegeben.
Die am häufigsten berichteten UW in den Studien mit monatlicher Dosisgabe waren Reaktionen an der Injektionsstelle (bis zu 84 % der Patienten), Kopfschmerzen (bis zu 12 % der Patienten) und Pyrexie (10 % der Patienten).
Die am häufigsten berichteten UW aus der Studie ATLAS-2M mit Dosisgabe alle 2 Monate waren Reaktionen an der Injektionsstelle (76 % der Patienten), Kopfschmerzen (7 % der Patienten) und Pyrexie (7 % der Patienten).
Die UW sind nach Systemorganklasse (SOC) und Häufigkeit aufgeführt1. Die Häufigkeitskategorien werden definiert als «sehr häufig» (≥1/10), «häufig» (≥1/100 bis < 1/10), «gelegentlich» (≥1/1'000 bis < 1/100), «selten» (≥1/10'000 bis < 1/1'000) und «sehr selten» (< 1/10'000).
Stoffwechsel- und Ernährungsstörungen
Häufig: Gewichtszunahme.
Psychiatrische Erkrankungen
Häufig: Depression, Angstzustände, abnormale Träume, Schlaflosigkeit.
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (12 %).
Häufig: Schwindel.
Gelegentlich: Schläfrigkeit, vasovagale Reaktionen (auf Injektionen).
Erkrankungen des Gastrointestinaltrakts
Häufig: Übelkeit, Erbrechen, Bauchschmerzen2, Flatulenz, Durchfall, Lipase erhöht (Grad 3-4).
Leber- und Gallenerkrankungen
Gelegentlich: Transaminase erhöht (AST/ALT), Hepatotoxizität (AST/ALT).
Erkrankungen der Haut und des Unterhautgewebes
Häufig: Ausschlag3.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Myalgie, Kreatinphosphokinase erhöht (Grad 3–4).
Allgemeine Erkrankungen
Sehr häufig: Pyrexie4 (10 %).
Häufig: Fatigue, Asthenie, Krankheitsgefühl.
Gelegentlich: Postinjektionssyndrom (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Beschwerden am Verabreichungsort
Reaktionen an der Injektionsstelle5 (84 %).
Sehr häufig: Schmerzen (79 %), Knötchen (17 %), Verhärtung (12 %).
Häufig: Beschwerden, Schwellung, Erythem, Pruritus, Blutergüsse, Wärmegefühl, Hämatom.
Gelegentlich: Cellulitis, Abszess, Anästhesie, Hämorrhagie, Verfärbung.
1 Die Häufigkeit der identifizierten unerwünschen Wirkungen basiert auf allen gemeldeten Ereignissen und ist nicht auf diejenigen beschränkt, die vom Prüfer zumindest als möglicherweise zusammenhängend angesehen werden.
2 Bauchschmerzen beinhaltet die folgenden gruppierten bevorzugten MedDRA-Begriffe: Bauchschmerzen, Oberbauchschmerzen.
3 Ausschlag beinhaltet die folgenden gruppierten bevorzugten MedDRA-Begriffe: Ausschlag, Ausschlag erythematös, Ausschlag generalisiert, Ausschlag makulös, Ausschlag makulo-papulös, Ausschlag morbilliform, Ausschlag papulös, Ausschlag pruritisch.
4 Pyrexie beinhaltet die folgenden gruppierten bevorzugten MedDRA-Begriffe: Wärmegefühl, Körpertemperatur erhöht, Pyrexie. Die Mehrzahl der Pyrexie-Ereignisse wurde innerhalb einer Woche nach der Injektion berichtet.
5 Die aufgeführten Reaktionen an der Injektionsstelle wurden jeweils bei mindestens 2 Teilnehmern berichtet.
Das allgemeine Sicherheitsprofil in Woche 96 und Woche 124 der FLAIR-Studie entsprach dem in Woche 48 beobachteten Profil, wobei keine neuen Sicherheitsbefunde festgestellt wurden. In der Verlängerungsphase der FLAIR-Studie in Woche 124 ergaben sich bei der direkten Einführung von Rilpivirin- plus CabotegravirInjektionen ohne orale Einleitungsbehandlung keine neuen Sicherheitsbedenken im Zusammenhang mit dem Weglassen der oralen Einleitungsbehandlung.
Das allgemeine Sicherheitsprofil in Woche 152 der ATLAS-2M-Studie entsprach dem in Woche 48 und in Woche 96 beobachteten Profil, wobei keine neuen Sicherheitsbefunde festgestellt wurden.
Für unerwünschte Wirkungen, die bei Anwendung von oralem Rilpivirin (Edurant) berichtet wurden, siehe Fachinformation von Rilpivirin Tabletten.
Beschreibung ausgewählter unerwünschter Wirkungen und Zusatzinformationen
Häufige Unerwünschter Arzneimittelwirkungen unter Vocabria/Rekambys einmal monatlich im Vergleich zur täglichen oralen Standardtherapie (CAR)
|
Tabelle 8: Systemische unerwünschte Wirkungen bei ≥1 % der virologisch supprimierten Teilnehmenden mit HIV-1-Infektion in den gepoolten Studien FLAIR und ATLAS (Woche 48) | ||
|
Unerwünschte Wirkung |
CAB+RPV |
CAR |
|
Kopfschmerz |
12% |
6% |
|
Pyrexie3 |
10% |
2% |
|
Durchfall |
9% |
7% |
|
Kreatinphosphokinase erhöht (Grad 3-4) |
8% |
4% |
|
Lipase erhöht (Grad 3-4) |
6% |
3% |
|
Übelkeit |
5% |
3% |
|
Fatigue |
5% |
2% |
|
Hautausschlag2 |
5% |
3% |
|
Schwindelgefühl |
4% |
1% |
|
Myalgie |
4% |
1% |
|
Bauchschmerzen1 |
4% |
2% |
|
Schlaflosigkeit |
4% |
1% |
|
Angstzustände |
4% |
2% |
|
Asthenie |
3% |
<1% |
|
Erbrechen |
2% |
1% |
|
Depression |
2% |
2% |
|
Malaise |
2% |
<1% |
|
Abnormale Träume |
1% |
<1% |
|
Flatulenz |
1% |
<1% |
1 Bauchschmerzen beinhaltet die folgenden gruppierten bevorzugten MedDRA-Begriffe: Bauchschmerzen, Oberbauchschmerzen.
2 Ausschlag beinhaltet die folgenden gruppierten bevorzugten MedDRA-Begriffe: Ausschlag, Ausschlag erythematös, Ausschlag generalisiert, Ausschlag makulös, Ausschlag makulo-papulös, Ausschlag morbilliform, Ausschlag papulös, Ausschlag pruritisch.
3 Pyrexie beinhaltet die folgenden gruppierten bevorzugten MedDRA-Begriffe: Pyrexie, Hitzegefühl, Körpertemperatur erhöht.
CAR = aktuelle antiretrovirale Behandlung (Current Antiretroviral Regimen)
Es ist zu beachten, dass es sich bei den Studien FLAIR und ATLAS um offene Studien mit Medikationsumstellung handelte (siehe die Details auch im Abschnitt «Klinische Wirksamkeit»). Im Behandlungsarm mit Cabotegravir und Rilpivirin wurde über ein häufigeres Auftreten von unerwünschten Wirkungen berichtet, was entweder auf das Behandlungsregime oder auf eine Verzerrung infolge des Designs der Studien zurückzuführen ist.
Lokale Reaktionen an der Injektionsstelle (Injection Site Reactions, ISR)
Bei monatlicher Dosisgabe
In den Phase-III-Studien und der Phase-IIIB-Studie (ATLAS, FLAIR und ATLAS-2M) brachen insgesamt 1 % der Patienten die Behandlung mit REKAMBYS- und Cabotegravir-Injektionen aufgrund von ISR ab.
Von 30'393 Injektionen wurden 6'815 ISR berichtet und die Reaktionen an der Injektionsstelle waren im Allgemeinen leicht (Grad 1, 75 % der Patienten) oder mittelstark (Grad 2, 36 % der Patienten). 4 % der Patienten entwickelten schwere (Grad 3) ISRs, und kein Studienteilnehmender entwickelte ISR vom Grad 4.
Die mediane Dauer der ISR-Ereignisse insgesamt betrug 3 Tage (1 Tag bis 341 Tage), wobei 11 % der Studienteilnehmer zum Zeitpunkt ihrer nächsten Injektion über nicht abgeklungene ISR berichteten.
Der prozentuale Anteil der Patienten, die über ISR berichteten, ging im Lauf der Zeit zurück von 70 % in Woche 4 auf 19 % in Woche 48.
Bei Dosisgabe alle 2 Monate
In der Studie ATLAS-2M brach weniger als 1 % der Studienteilnehmer die Behandlung mit REKAMBYS plus Vocabria aufgrund von ISR ab. Bei 8'470 Injektionen wurden 2'507 ISR berichtet, und die Reaktionen an der Injektionsstelle waren im Allgemeinen leicht (Grad 1, 71 % der Patienten) oder mittelstark (Grad 2, 27 % der Patienten). Bei 3 % der Patienten traten schwere (Grad 3) ISR auf, kein Patient entwickelte ISR vom Grad 4.
Die mediane Dauer der ISR-Ereignisse insgesamt betrug 3 Tage (1 Tag bis 424 Tage), wobei 5 % der Patienten zum Zeitpunkt ihrer nächsten Injektion über nicht abgeklungene ISR berichteten.
Der prozentuale Anteil der Patienten, die über ISR berichteten, ging im Lauf der Zeit von 70 % in Woche 4 auf 20 % in Woche 48 zurück.
Gewichtszunahme
Zum Analysezeitpunkt in Woche 48 hatten die Patienten in den Phase-III-Studien FLAIR und ATLAS, die i.m. Rilpivirin plus i.m. Cabotegravir erhielten, eine mediane Gewichtszunahme von 1,5 kg; die Patienten der Vergleichsgruppe, bei denen das jeweiliges antiretrovirale Standardtherapie-Regime (CAR) fortgeführt wurde, hatten eine mediane Gewichtszunahme von1,0 kg (kombinierte Analyse). In den individuellen Auswertungen der Phase-III-Studien FLAIR und ATLAS nahmen die Patienten in den mit i.m. Rilpivirin plus i.m. Cabotegravir behandelten Armen im Median 1,3 kg bzw. 1,8 kg zu, verglichen mit 1,5 kg bzw. 0,3 kg in den CAR-Armen. Nach 48 Wochen Behandlung betrug die mediane Gewichtszunahme in der ATLAS-2M-Studie sowohl im Arm mit monatlicher Dosierung von i.m. Rilpivirin plus i.m. Cabotegravir als auch im Arm mit Dosierung alle 2 Monate 1,0 kg.
Laborwertveränderungen
In den klinischen Studien mit REKAMBYS plus Cabotegravir wurden erhöhte Lipasewerte beobachtet; Lipaseerhöhungen der Grade 3 und 4 traten bei REKAMBYS plus Cabotegravir häufiger auf als bei der CAR-Gruppe. Diese Erhöhungen waren im Allgemeinen asymptomatisch und führten nicht zum Abbruch der Behandlung.
Unter Behandlung mit i.m. Rilpivirin plus i.m. Cabotegravir wurden ausserdem asymptomatische Erhöhungen der Kreatinphosphokinase (CPK) berichtet, hauptsächlich im Zusammenhang mit körperlicher Bewegung.
Hepatotoxizität
In den klinischen Studien wurden erhöhte Transaminasen (ALT/AST) bei Patienten beobachtet, die Rilpivirin plus Cabotegravir erhielten. Diese Erhöhungen wurden hauptsächlich einer akuten Virushepatitis (Hepatitis A, B, C) zugeschrieben. Einige Teilnehmende, die mit oralem Rilpivirin plus oralem Cabotegravir behandelt wurden, wiesen Transaminase-Erhöhungen auf, die einer vermuteten arzneimittelbedingten Hepatotoxizität zugeschrieben wurden. Diese Veränderungen waren nach Absetzen der Behandlung reversibel (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Unter Behandlung mit Rilpivirin plus Cabotegravir wurde ein geringer, nicht progressiver Anstieg des Gesamtbilirubins (ohne klinischen Ikterus) festgestellt. Diese Veränderungen werden als klinisch nicht relevant erachtet, da sie wahrscheinlich die Kompetition zwischen Cabotegravir und unkonjugiertem Bilirubin um einen gemeinsamen Clearance-Weg (UGT1A1) widerspiegeln.
Zusätzliche Informationen über spezielle Patientengruppen
Anwendung bei Kindern und Jugendlichen
Die Sicherheit und Wirksamkeit von REKAMBYS bei Kindern < 18 Jahren wurden noch nicht nachgewiesen.
Bezüglich unerwünschter Wirkungen im Zusammenhang mit Cabotegravir ist die massgebliche Arzneimittelinformation zu beachten.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Es gibt kein spezielles Antidot bei einer Überdosierung von REKAMBYS. Die Erfahrungen mit Überdosierungen von Rilpivirin beim Menschen sind begrenzt. Überdosierungen können u.a. durch versehentliche intravenöse Verabreichung auftreten (siehe «Warhnhinweise und Vorsichtsmassnahmen»). Die Behandlung im Falle einer Überdosierung mit REKAMBYS besteht aus allgemeinen unterstützenden Massnahmen, wie der Überwachung der Vitalzeichen und des EKGs (QT-Intervall, siehe «Interaktionen»/«Arzneimittel, die das QT-Intervall verlängern») sowie der Beobachtung des klinischen Status des Patienten. Es ist ratsam, hinsichtlich der neuesten Empfehlungen für die Behandlung einer Überdosierung eine Giftnotrufzentrale zu kontaktieren. Da Rilpivirin in hohem Umfang an Plasmaproteine gebunden ist, ist es unwahrscheinlich, dass eine Dialyse zu einer bedeutsamen Entfernung des Wirkstoffs führt.
Eigenschaften/Wirkungen
ATC-Code
J05AG05
Pharmakotherapeutische Gruppe: Virustatikum zur systemischen Anwendung, NNRTI (nichtnukleosidischer Reverse-Transkriptase-Inhibitor).
Wirkungsmechanismus
Rilpivirin ist ein Diarylpyrimidin-NNRTI von HIV-1. Die Aktivität von Rilpivirin wird durch nichtkompetitive Hemmung der reversen Transkriptase (RT) von HIV-1 vermittelt. Rilpivirin hat keine Hemmwirkung auf die zellulären DNA-Polymerasen α, β und γ beim Menschen.
Pharmakodynamik
Mikrobiologie
Antivirale In-vitro-Aktivität
Rilpivirin erwies sich als wirksam gegen Laborstämme vom HIV-1-Wildtyp bei akut infizierten T-Zell-Linien mit einem medianen EC50-Wert für HIV-1/IIIB von 0,73 nM (0,27 ng/ml). Wenngleich Rilpivirin gegen HIV-2 in vitro mit EC50-Werten im Bereich von 2'510 bis 10'830 nM (920 bis 3'970 ng/ml) begrenzte Wirkung aufwies, wird die Behandlung einer HIV-2-Infektion mit REKAMBYS aufgrund fehlender klinischer Daten nicht empfohlen.
Des Weiteren wurde eine antivirale Aktivität von Rilpivirin gegen ein breites Spektrum von primären Isolaten der HIV-1-Gruppe M (Subtypen A, B, C, D, F, G, H) mit EC50-Werten im Bereich von 0,07 bis 1,01 nM (0,03 bis 0,37 ng/ml) und der Gruppe O mit EC50-Werten im Bereich von 2,88 bis 8,45 nM (1,06 bis 3,10 ng/ml) festgestellt.
Resistenz
Unter Einbeziehung aller verfügbaren In-vitro- und In-vivo-Daten, die mit oralem Rilpivirin bei vorgängig unbehandelten Patienten erhalten wurden, können die folgenden Aminosäuresubstitutionen die Aktivität von Rilpivirin beeinflussen, wenn sie zu Behandlungsbeginn vorliegen: K101E, K101P, E138A, E138G, E138K, E138Q, E138R, V179L, Y181C, Y181I, Y181V, Y188L, H221Y, F227C, M230I und M230L sowie die Kombination von L100I und K103N.
Virologisch supprimierte Patienten
Die Anzahl der Patienten, welche die Kriterien (zweimal hintereinander HIV-1-RNA-Mengen im Plasma von ≥200 Kopien/ml nach vorhergehender Suppression auf < 200 Kopien/ml) für bestätigtes virologisches Versagen (CVF) erfüllten, war in den kombinierten Phase-III-Studien ATLAS und FLAIR (siehe auch Abschnitt »Klinische Wirksamkeit») niedrig. In der kombinierten Analyse gab es bis Woche 48 7 Fälle von CVF unter Rilpivirin plus Cabotegravir (7/591, 1,2 %) und 7 CVF unter CAR (7/591, 1,2 %). Ein Patient mit CVF (FLAIR) hatte nie eine Injektionsdosis erhalten und keine Resistenz entwickelt. In der Rilpivirin-plus-Cabotegravir-Gruppe in der kombinierten Analyse wurde bei 5/591 (0,8 %) Teilnehmenden Resistenzentwicklung festgestellt: 5/591 (0,8 %) bzw. 4/591 (0,7 %) mit Rilpivirin- und/oder Cabotegravirresistenz-assoziierten Mutationen (Tabelle 9).
|
Tabelle 9: Resistenzassoziierte Mutationen vor Beginn der Studienbehandlung und beim vermuteten virologischen Versagen bei Fällen mit bestätigtem virologischem Versagen in den Studien ATLAS und FLAIR | |||||
|
Studie |
HIV-1 Subtyp |
«Baseline»* |
Vermutetes virologisches Versagen** | ||
|
|
|
Reverse Transkriptase |
Integrase |
Reverse Transkriptase |
Integrase |
|
ATLAS |
A |
E138A/E |
Keine |
E138A |
Keine |
|
AG |
V108I/V+E138K |
Keine |
V108I+E138K |
Keine | |
|
A |
Keine |
Keine |
E138E/K |
N155H | |
|
FLAIR |
A1 |
Keine |
Keine |
E138A/E/K/T |
Q148R |
|
A1 |
Keine |
Keine |
K101E |
G140R | |
|
A1 |
Keine |
Keine |
E138K |
Q148R | |
|
*FLAIR: Post-hoc Tests auf INI-Resistenz und Screening-Tests auf NNRTI-Resistanz mit zur Baseline gewonnenem Plasma, ATLAS: Post-hoc -Tests auf INI- und NNRTI-Resistenz wurden mit mononukleären Zellen aus dem peripheren Blut durchgeführt, die in beiden Studien vor Beginn der Studienbehandlung zur Baseline gewonnen wurden. | |||||
In der Phase-3B-Studie ATLAS-2M (siehe auch Abschnitt »Klinische Wirksamkeit») erfüllten 10/1045 (1,0 %) Teilnehmenden die CVF-Kriterien bis Woche 48: 8/522 (1,5 %) im q8w-Arm (Dosisgabe alle 2 Monate) und 2/523 (0,4 %) im q4w-Arm (monatliche Dosierung). In der q8w-Gruppe wurde bei 5/522 (1,0 %) Resistenzentwicklung festgestellt: 4/522 (0,8 %) bzw. 5/522 (1,0 %) mit Rilpivirin- und/oder Cabotegravirresistenz-assoziierten Mutationen. In der q4w-Gruppe wurde bei 2/523 (0,4 %) Resistenzentwicklung festgestellt: 1/523 (0,2 %) bzw. 2/523 (0,4 %) mit Rilpivirin- und/oder Cabotegravirresistenz-assoziierten Mutationen (Tabelle 10).
|
Tabelle 10: Resistenzassoziierte Mutationen vor Beginn der Studienbehandlung und beim vermuteten virologischen Versagen bei Fällen mit bestätigtem virologischem Versagen in der Studie ATLAS-2M | |||||
|
Studie |
HIV-1 Subtyp |
«Baseline»* |
Vermutetes virologisches Versagen ** | ||
|
|
|
Reverse Transkriptase |
Integrase |
Reverse Transkriptase |
Integrase |
|
ATLAS-2M q8w Arm |
A |
E138A/E |
Keine |
K101E+E138A |
N155H |
|
|
A1 |
Keine |
Keine |
E138E/K |
Q148Q/R+N155N/H |
|
|
A1 |
Y188L+P225H |
Keine |
Y188L+P225H |
NA |
|
|
B |
Keine |
Keine |
Keine |
Keine |
|
|
B |
K103N+V108I/V+E138A |
Keine |
K103N+E138A |
N155H |
|
|
C |
V108I/V+H221H/Y+Y181C/Y |
Keine |
K103N |
Keine |
|
|
C |
Y188F/H/L/Y |
G140G/R |
Y188L |
Q148Q/R+N155H/N |
|
|
Complex |
Keine |
Keine |
K101E |
Q148R |
|
ATLAS-2M q4w Arm |
B |
Keine |
Keine |
Keine |
N155N/H |
|
|
B |
Keine |
Keine |
K101E+M230L |
E138E/K+Q148R |
|
* Post-hoc-Resistenztests wurden mit vor Beginn der Studienbehandlung zur Baseline gewonnenen mononukleären Zellen aus dem peripheren Blut durchgeführt. n.v.: nicht verfügbar. | |||||
Bis Woche 152 erfüllten 13 Patienten die CVF-Kriterien während der Erhaltungs- und Verlängerungsphase; 2 Patienten (Q8W-Arm) erfüllten die CVF-Kriterien seit der Analyse in Woche 96 (siehe Tabelle 11). Bei zehn Patienten trat CVF vor Woche 48 auf (8 Patienten im Q8W-Arm und 2 Patienten im Q4W-Arm) und 1 Patient (Q8W-Arm) erfüllte die CVF-Kriterien zwischen Woche 48 und Woche 96.
|
Tabelle 11: Kumulativer Anteil der Patienten, die die CVF erfüllen, nach Visite bis zu Woche 152 Erhaltungsphase + Verlängerungsphase (ITT-E-Population): Woche 152 Analyse | ||
|
SVF-Zeitpunkta |
Q8W |
Q4W |
|
Woche 8 |
1 (0.2) |
0 |
|
Woche 16 |
4 (0.8) |
1 (0.2) |
|
Woche 24 |
7 (1.3) |
1 (0.2) |
|
Woche 32 |
7 (1.3) |
2 (0.4) |
|
Woche 48 |
8 (1.5) |
2 (0.4) |
|
Woche 88 |
9 (1.7) |
2 (0.4) |
|
Woche 112 |
10 (1.9) |
2 (0.4) |
|
Woche 120 |
11 (2.1) |
2 (0.4) |
|
a
Erster der 2 aufeinanderfolgenden HIV-1-RNA-Werte ≥200 c/ml. | ||
Zusätzlich zu den 9 Patienten mit CVF in der Q8W-Gruppe erreichten 2 Patienten CVF zwischen den Zeitpunkten Woche 96 und Woche 152 (siehe Tabelle 11). Ein Patient, der aus der Studie ATLAS wechselte, nachdem er 1 bis 24 Wochen CAB + RPV LA erhalten hatte, erfüllte die CVF-Kriterien in Woche 112. Zu Studienbeginn wies dieser Patient den IN-Polymorphismus L74I auf. Zum SVF Zeitpunkt wurden 3 NNRTI-Mutationen beobachtet, K103N und RPV-Resistenz-assoziierte Mutationen, E138A und Y181Y/C. Die INI-Resistenz-assoziierte Mutation Q148R wurde zusammen mit dem L74I-Polymorphismus nachgewiesen. Es wurde eine verminderte phänotypische Anfälligkeit für RPV (FC=3,4) und CAB (FC=9,5) beobachtet. Der HIV-1-Virus-Subtyp war zum Zeitpunkt der SVF A. Der andere Patient erreichte die CVF-Kriterien in Woche 120 und wies zum Zeitpunkt der Baseline keine Resistenz-assoziierten Mutationen auf. Zum Zeitpunkt der SVF wurden die RPV-Resistenzmutationen E138A und M230M/L sowie die INI-Resistenzmutation Q148R festgestellt. Die phänotypische Analyse zeigte eine verminderte RPV- (FC= 16) und CAB-Empfindlichkeit (FC= 3,3). Der Patient trug zum Zeitpunkt der SVF Viren des HIV-1-Subtyps B/C in sich. In der Q4W-Gruppe gab es keine weiteren CVF-Patienten.
Kreuzresistenz
Viren mit zielgerichteten Mutationen an der NNRTI-Bindungsstelle
In einem Panel von 67 rekombinanten HIV-1-Laborstämmen mit einer Aminosäuresubstitution an RT-Positionen, die mit NNRTI-Resistenz assoziiert sind, einschliesslich den am häufigsten festgestellten Substitutionen K103N und Y181C, zeigte Rilpivirin antivirale Aktivität gegen 64 dieser Stämme (96 %). Die mit einem Verlust der Empfindlichkeit gegenüber Rilpivirin verbundenen Einzelaminosäuresubstitutionen waren: K101P, Y181I und Y181V. Während die K103N-Substitution nicht zu einer reduzierten Empfindlichkeit gegenüber Rilpivirin führte, ergab sich bei der Kombination von K103N und L100I eine 7-fach reduzierte Empfindlichkeit gegenüber Rilpivirin.
Rekombinante klinische Isolate
Rilpivirin behielt seine Empfindlichkeit (FC ≤ BCO) gegenüber 62 % der 4'786 rekombinanten klinischen HIV-1-Isolate mit Resistenz gegenüber Efavirenz und/oder Nevirapin bei.
Virologisch supprimierte Patienten
In den Phase-III-Studien FLAIR und ATLAS lag bei der Analyse in Woche 48 bei 5/7 Patienten mit CVF zum Zeitpunkt des Versagens ein Phänotyp mit Resistenz gegenüber Rilpivirin vor. Bei diesen 5 Patienten wurde ein Phänotyp mit Kreuzresistenz gegen Efavirenz (n = 4), Etravirin (n = 3) und Nevirapin (n = 4) festgestellt.
Wirkungen auf das QT/QTc-Intervall und die kardiale Elektrophysiologie
Die Rilpivirin-Konzentrationen im Plasma nach REKAMBYS-Injektionen und nach Einnahme von Rilpivirin in einer Dosis von 25 mg einmal täglich sind vergleichbar. REKAMBYS ist in der empfohlenen monatlichen Dosis von 600 mg oder 900 mg alle 2 Monate nicht mit einer klinisch relevanten Wirkung auf das QTc-Intervall verbunden. Die Wirkung von oralem Rilpivirin in der empfohlenen Dosis von 25 mg einmal täglich auf das QTcF-Intervall wurde in einer randomisierten, placebo- und aktiv (einmal täglich 400 mg Moxifloxacin) kontrollierten Crossover-Studie bei 60 gesunden Erwachsenen mit 13 Messungen über 24 Stunden im Steady-State untersucht. Orales Rilpivirin ist in der empfohlenen Dosis von 25 mg einmal täglich nicht mit klinisch relevanten Auswirkungen auf das QTc-Intervall verbunden.
Bei der Untersuchung supratherapeutischer Dosen von oralem Rilpivirin (75 mg einmal täglich und 300 mg einmal täglich) bei gesunden Erwachsenen betrugen die mittleren maximalen, zeitgematchten (obere Konfidenzgrenze 95 %) Unterschiede in Bezug auf das QTcF-Intervall gegenüber dem Placebo nach Baseline-Korrektur 10,7 (15,3) bzw. 23,3 (28,4) ms. Die Einnahme von Rilpivirin in den Dosierungen von einmal täglich 75 mg und einmal täglich 300 mg ergab eine mittlere Cmax, die um etwa das 4,4- bzw. 11,6-Fache höher lag als die mittlere Cmax im Steady-State bei der empfohlenen monatlichen Dosis von 600 mg REKAMBYS. Die orale Gabe von 75 mg oder 300 mg Rilpivirin einmal täglich im Steady-State führte zu einer mittleren Cmax, die ungefähr um das 4,1-Fache bzw. 10,7-Fache höher war als die mittlere Cmax im Steady-State, die mit der empfohlenen Dosis von 900 mg REKAMBYS alle 2 Monate gemessen wurde.
Klinische Wirksamkeit
Klinische Studien
Bei monatlicher Dosisgabe
Virologisch supprimierte HIV-1-infizierte Patienten
Die Wirksamkeit von REKAMBYS plus Cabotegravir-Injektionen wurde in zwei randomisierten, multizentrischen, aktiv kontrollierten, unverblindeten Nichtunterlegenheitsstudien der Phase-III mit parallelen Prüfarmen, FLAIR und ATLAS, untersucht. Die Primäranalyse wurde durchgeführt, nachdem alle Patienten ihren Besuch in Woche 48 abgeschlossen hatten oder vorzeitig aus der Studie ausgeschieden waren.
In der Studie FLAIR erhielten 629 HIV-1-infizierte, antiretroviral nicht vorbehandelte (ART-naive) Patienten 20 Wochen lang eine Behandlung mit einem Dolutegravir-Integrase-Strangtransferinhibitor (INI) (entweder Dolutegravir/Abacavir/Lamivudin oder Dolutegravir + 2 andere nukleosidische Reverse-Transkriptase-Inhibitoren, wenn die Patienten HLA-B*5701-positiv waren). Virologisch supprimierte Patienten (HIV-1-RNA < 50 Kopien je ml, n = 566) wurden dann randomisiert (1:1), um entweder eine Behandlung mit Rilpivirin plus Cabotegravir zu erhalten oder weiterhin ihre jeweilige aktuelle antiretrovirale (CAR) Therapie beizubehalten. Bei den Patienten, die randomisiert wurden, um die Behandlung mit Rilpivirin plus Cabotegravir zu erhalten, wurde die Therapie mit einer etwa 1-monatigen (mindestens 28-tägigen, maximal 2-monatigen) oralen Einleitungsbehandlung mit einer 30 mg Cabotegravir-Tablette plus einer 25 mg Rilpivirin-Tablette begonnen. Anschliessend erfolgte die Behandlung mit Cabotegravir als Injektion (Monat 1: Injektion von 600 mg, ab Monat 2: Injektion von 400 mg) plus Rilpivirin als Injektion (Monat 1: Injektion von 900 mg, ab Monat 2: Injektion von 600 mg) monatlich, für bis zu 96 Wochen.
In der Studie ATLAS wurden 616 HIV-1-infizierte, ART-erfahrene und virologisch seit mindestens 6 Monaten supprimierte Patienten (HIV-1-RNA < 50 Kopien je ml) randomisiert (1:1) und erhielten entweder eine Behandlung mit Rilpivirin plus Cabotegravir oder behielten ihre jeweilige aktuelle antiretrovirale (CAR) Therapie bei. Bei den Patienten, die randomisiert wurden, um die Behandlung mit Rilpivirin plus Cabotegravir zu erhalten, wurde die Therapie mit einer etwa 1-monatigen (mindestens 28-tägigen und maximal 2-monatigen) oralen Einleitungsbehandlung mit einer 30 mg Cabotegravir-Tablette plus einer 25 mg Rilpivirin-Tablette einmal täglich begonnen. Anschliessend erfolgte die Behandlung mit Cabotegravir als Injektion (Monat 1: Injektion von 600 mg, ab Monat 2: Injektion von 400 mg) plus Rilpivirin als Injektion (Monat 1: Injektion von 900 mg, ab Monat 2: Injektion von 600 mg) monatlich für weitere 44 Wochen. Zu Beginn der Studie ATLAS waren vor der Randomisierung 50 %, 17 % und 33 % der Patienten mit einem NNRTI, PI bzw. INI als dritte Wirkstoffklasse behandelt worden. Die Verteilung bleibt nach der Randomisierung im Kontrollarm (CAR) ähnlich.
Die kombinierte Analyse der Studien FLAIR und ATLAS ergab, dass im Behandlungsarm mit Rilpivirin plus Cabotegravir zu Studienbeginn das mediane Alter der Patienten 38 Jahre betrug, 27 % weiblich waren, 27 % nicht weiss waren und 7 % eine CD4+-Zellzahl von weniger als 350 Zellen je mm3 hatten; bezüglich dieser Merkmale unterschieden sich die Behandlungsarme nicht wesentlich voneinander.
In beiden Studien war der primäre Endpunkt der Anteil der Patienten mit Plasma-HIV-1-RNA ≥50 Kopien/ml in Woche 48 (Schnappschussalgorithmus für das ITT-E-Kollektiv).
In einer kombinierten Analyse der beiden Phase-III-Studien FLAIR und ATLAS war Rilpivirin plus Cabotegravir hinsichtlich des Anteils der Patienten mit Plasma-HIV-1-RNA ≥50 Kopien/ml (1,9 % bzw. 1,7 %) in Woche 48 gegenüber der CAR nicht unterlegen. Der adjustierte Behandlungsunterschied zwischen Rilpivirin plus Cabotegravir und CAR (0,2; 95-%-KI: –1,4; 1,7) erfüllte das Kriterium der Nichtunterlegenheit (obere Grenze des 95-%-KI unter 4 %).
Die Nichtunterlegenheitsergebnisse aus den Studien FLAIR und ATLAS zeigten, dass die Dauer der virologischen Suppression der HIV-1-RNA vor Einleitung der Behandlung mit Rilpivirin plus Cabotegravir (d.h. 5 Monate oder ≥6 Monate) keine Auswirkung auf die Gesamtansprechraten hatte.
In Tabelle 12 und Tabelle 13 sind der primäre Endpunkt und andere Ergebnisse in Woche 48, einschliesslich der Ergebnisse in Bezug auf zentrale Ausgangsmerkmale der Patienten («Baseline»-Faktoren), für die Studien FLAIR und ATLAS sowie deren kombinierte Daten gezeigt.
|
Tabelle 12: Virologische Ergebnisse der randomisierten Behandlung in den Studien FLAIR und ATLAS in Woche 48 (Schnappschussanalyse) | ||||||
|
|
FLAIR |
ATLAS |
Kombinierte Daten | |||
|
|
RPV + CAB |
CAR |
RPV + CAB |
CAR |
RPV + CAB |
CAR |
|
HIV-1-RNA ≥50 Kopien/ml, n (%)† |
6 (2,1) |
7 (2,5) |
5 (1,6) |
3 (1,0) |
11 (1,9) |
10 (1,7) |
|
Behandlungsunterschied % (95%-KI)* |
–0,4 (–2,8, 2,1) |
0,7 (–1,2, 2,5) |
0,2 (–1,4, 1,7) | |||
|
HIV-1-RNA < 50 Kopien/ml, n (%) |
265 |
264 (93,3) |
285 |
294 (95,5) |
550 |
558 (94,4) |
|
Keine virologischen Daten im Woche-48-Fenster, n (%) |
12 |
12 (4,2) |
18 |
11 (3,6) |
30 |
23 (3,9) |
|
Gründe | ||||||
|
Studie/Studienmedikament aufgrund von UE abgesetzt oder Tod, |
8 |
2 (0,7) |
11 |
5 (1,6) |
19 |
7 |
|
Studie/Studienmedikament aus anderen Gründen beendet/abgesetzt, n (%) |
4 (1,4) |
10 (3,5) |
7 (2,3) |
6 (1,9) |
11 (1,9) |
16 (2,7) |
|
Fehlende Daten in diesem Fenster, aber in der Studie |
0 |
0 |
0 |
0 |
0 |
0 |
|
* Adjustiert nach Stratifizierungsfaktoren zum Baseline-Zeitpunkt. | ||||||
|
Tabelle 13: Anteil der Patienten mit Plasma-HIV-1-RNA ≥50 Kopien/ml in der Subgruppenanalyse von Woche 48 in Bezug auf zentrale Ausgangsmerkmale der Patienten («Baseline»-Faktoren) (Schnappschuss-Outcomes) | |||
|
Ausgangsmerkmale der Patienten |
Kombinierte Daten aus FLAIR und ATLAS | ||
|
RPV + CAB |
CAR | ||
|
CD4+ zum «Baseline»-Zeitpunkt (Zellen/mm3) |
<350 |
0/42 |
2/54 (3,7) |
|
≥350 bis <500 |
5/120 (4,2) |
0/117 | |
|
≥500 |
6/429 (1,4) |
8/420 (1,9) | |
|
Geschlecht |
Männlich |
6/429 (1,4) |
9/423 (2,1) |
|
Weiblich |
5/162 (3,1) |
1/168 (0,6) | |
|
Ethnizität |
Weiss |
9/430 (2,1) |
7/408 (1,7) |
|
Schwarz/afroamerikanisch |
2/109 (1,8) |
3/133 (2,3) | |
|
Asiatisch/andere |
0/52 |
0/48 | |
|
BMI |
<30 kg/m2 |
6/491 (1,2) |
8/488 (1,6) |
|
≥30 kg/m2 |
5/100 (5,0) |
2/103 (1,9) | |
|
Alter (Jahre) |
<50 |
9/492 (1,8) |
8/466 (1,7) |
|
≥50 |
2/99 (2,0) |
2/125 (1,6) | |
|
Antivirale «Baseline»-Therapie bei der Randomisierung |
PI |
1/51 (2,0) |
0/54 |
|
INI |
6/385 (1,6) |
9/382 (2,4) | |
|
NNRTI |
4/155 (2,6) |
1/155 (0,6) | |
|
BMI = Body Mass Index, PI = Proteaseinhibitor, INI =Integraseinhibitor, NNRTI = nicht-nukleosidischer Reverse-Transkriptase-Inhibitor | |||
In den Studien FLAIR und ATLAS waren die Behandlungsunterschiede in Bezug auf die Ausgangsmerkmale der Patienten («Baseline»-Faktoren) (CD4+-Zahl, Geschlecht, Alter, Ethnizität, BMI, dritte «Baseline»-Wirkstoffklasse zur Behandlung) klinisch nicht bedeutend.
Sowohl in der FLAIR- als auch in der ATLAS-Studie waren die Patienten vor Tag 1 bzw. vor Randomisierung virologisch supprimiert, und es wurde keine klinisch relevante Veränderung der CD4+-Zellzahlen gegenüber dem Ausgangswert beobachtet.
Patienten, die i.m. Rilpivirin plus i.m. Cabotegravir erhielten, berichteten von einer erheblichen Verbesserung der Behandlungszufriedenheit im Vergleich zu Anwendung einer täglichen oralen antiretroviralen Therapie. Bei der Messung anhand des Fragebogens zur Erfassung der Zufriedenheit mit der HIV-Behandlung (HIV-Treatment Satisfaction Questionnaire, HIV-TSQ) betrug der Unterschied hinsichtlich der Behandlungszufriedenheit im HIV-TSQc (Veränderung) in Woche 48 in der FLAIR + 4,1 (95-%-KI: 2,8; 5,5; p < 0,001) und im HIV-TSQs (Status) in Woche 44 in der ATLAS + 5,7 (95-%-KI: 4,37; 7,0; p < 0,001).
Woche 96 FLAIR
In der FLAIR-Studie blieben die Wirksamkeits-Ergebnisse nach 96 Wochen mit den Ergebnissen nach 48 Wochen konsistent. Der Anteil der Patienten mit HIV-1-RNA ≥50 Kopien/ml Plasma unter i.m. Rilpivirin plus i.m. Cabotegravir (n = 283) und CAR (n = 283) betrug 3,2 % bzw. 3,2 % (bereinigter Behandlungsunterschied zwischen i.m. Rilpivirin plus i.m. Cabotegravir und CAR [0,0; 95-%-KI: –2,9; 2,9]).
Woche 124 FLAIR - direkt zur Injektion versus orale Einleitungsbehandlung
In der FLAIR-Studie wurde in Woche 124 eine Bewertung der Sicherheit und Wirksamkeit bei Patienten durchgeführt, die sich in Woche 100 für eine Umstellung von Abacavir/Dolutegravir/Lamivudin auf Rilpivirin plus Cabotegravir in der Verlängerungsphase mit oder ohne orale Einleitungsbehandlung entschieden hatten. Es wurde dabei eine Gruppe mit oraler Einleitungsbehandlung («Oral Lead-in» (OLI)- Gruppe) (n = 121) und eine mit direkter Injektion («Direct to Injecjtion» (DTI)-Gruppe) (n = 111) gebildet.
In Woche 124 betrug der Anteil der Patienten mit einer HIV-1-RNA ≥50 Kopien/ml (1/111 [0,9%]) bzw. (1/121 [0,8%]) in der Gruppe mit direkter Injektion bzw. der Gruppe mit oraler Einleitungsbehandlung. Die Raten der virologischen Suppression (HIV-1-RNA < 50 Kopien/ml) waren sowohl in der Gruppe mit direkter Injektion (110/111 [99,1%]) als auch in der Gruppe mit oraler Einleitungsbehandlung (113/121 [93,4%]) ähnlich.
Bei Dosierungen alle 2 Monate
Virologisch supprimierte HIV-1-infizierte Patienten
Die Wirksamkeit von alle 2 Monate verabreichten Rilpivirin-Injektionen wurden in einer randomisierten, multizentrischen, offenen Nichtunterlegenheitsstudie der Phase-IIIB mit Parallelgruppen, ATLAS-2M, bewertet. Die primäre Analyse wurde durchgeführt, nachdem alle Teilnehmenden ihren Besuch in Woche 48 abgeschlossen hatten oder vorzeitig aus der Studie ausgeschieden waren.
In der ATLAS-2M-Studie wurden 1'045 HIV-1-infizierte, mit ART vorbehandelte, virologisch supprimierte Teilnehmende randomisiert (1:1) und erhielten Rilpivirin-plus-Cabotegravir-Injektionen, die entweder alle 2 Monate oder monatlich verabreicht wurden. Patienten, die vor Studienbeginn nicht mit Cabotegravir/Rilpivirin behandelt worden waren, erhielten eine orale Einleitungs-Behandlung, die aus einer einmal täglichen 25 mg Rilpivirin Tablette plus einer 30 mg Cabotegravir Tablette bestand, für etwa 1 Monat (mindesten 28 Tage, maximal 2 Monate). Patienten, die nach der Randomisierung monatliche Rilpivirin-Injektionen (Monat 1: Injektion von 900 mg, ab Monat 2: Injektion von 600 mg) und Cabotegravir-Injektionen (Monat 1: Injektion von 600 mg, ab Monat 2: Injektion von 400 mg) erhielten, wurden über weitere 44 Wochen behandelt. Patienten, die nach der Randomisierung alle 2 Monate Rilpivirin-Injektionen (Injektion von 900 mg in den Monaten 1, 2 und 4 und danach alle 2 Monate) und Cabotegravir-Injektionen (Injektion von 600 mg in den Monaten 1, 2 und 4 und danach alle 2 Monate) erhielten, wurden über weitere 44 Wochen behandelt.
Vor der Randomisierung hatten 63 %, 13 % und 24 % der Teilnehmenden Rilpivirin + Cabotegravir über 0 Wochen, 1 bis 24 Wochen bzw. > 24 Wochen erhalten.
Zu Beginn der Studie betrug das mediane Alter der Teilnehmenden 42 Jahre, 27 % waren weiblich, 27 % waren nicht weiss und 6 % hatten eine CD4+-Zellzahl von unter 350 Zellen pro mm3. Diese Merkmale waren in den verschiedenen Behandlungsarmen ähnlich.
Der primäre Endpunkt in der Studie ATLAS-2M war der Anteil der Patienten mit HIV-1 RNA ≥50 Kopien/ml Plasma in Woche 48 (Schnappschussalgorithmus für das ITT-E-Kollektiv). In Tabelle 14 und Tabelle 15 sind der primäre Endpunkt und andere Ergebnisse von Woche 48, einschliesslich der Ergebnisse in Bezug auf zentrale Ausgangsmerkmale der Patienten («Baseline»-Charakteristika), für die ATLAS-2M Studie gezeigt.
In der Studie ATLAS-2M war die Behandlung alle 2 Monate mit i.m. Cabotegravir plus i.m. Rilpivirin gegenüber der monatlichen Gabe von i.m. Cabotegravir und i.m. Rilpivirin in Bezug auf den Anteil der Patienten mit HIV-1-RNA ≥50 Kopien/ml Plasma (1,7 % bzw. 1,0 %) in Woche 48 nicht unterlegen. Der bereinigte Behandlungsunterschied zwischen alle 2 Monate und monatlich verabreichtem i.m. Cabotegravir + i.m. Rilpivirin (0,8; 95-%-KI: –0,6; 2,2) erfüllte das Kriterium der Nichtunterlegenheit (Obergrenze des 95-%-KI unter 4 %). Die Ergebnisse zur Wirksamkeit in Woche 96 und Woche 152 stimmen mit den Ergebnissen für den primären Endpunkt in Woche 48 überein (siehe Tabelle 14).
Tabelle 14: Virologische Ergebnisse der randomisierten Behandlung in der ATLAS-2M-Studie nach 48, 96 und 152 Wochen (Schnappschussanalyse)
|
|
Dosierung alle 2 Monate (q8w) |
Monatliche Dosierung |
|
|
n=522 (%) |
n=523 (%) |
|
Woche 48 |
|
|
|
HIV-1 RNA ≥50 Kopien/ml, n (%)† |
9 (1.7) |
5 (1.0) |
|
Behandlungsunterschied % (95-%-KI)* |
0.8 (-0.6, 2.2) | |
|
HIV-1 RNA < 50 Kopien/ml, n (%) |
492 (94.3) |
489 (93.5) |
|
Keine virologischen Daten im Woche-48-Fenster, n (%) |
21 (4.0) |
29 (5.5) |
|
Gründe: | ||
|
Studienabbruch aufgrund eines UE oder Tod, n (%) |
9 (1.7) |
13 (2.5) |
|
Studienabbruch aus anderen Gründen, n (%)a |
12 (2.3) |
16 (3.1) |
|
In der Studie, aber fehlende Daten in dem Zeitfenster |
0 |
0 |
|
Woche 96 |
|
|
|
HIV-1 RNA ≥50 Kopien/ml, n (%)† |
11 (2.1) |
6 (1.1) |
|
HIV-1 RNA < 50 Kopien/ml, n (%) |
475 (91.0) |
472 (90.2) |
|
Keine virologischen Daten im Woche-48-Fenster, n (%) |
36 (6.9) |
45 (8.6) |
|
Gründe: | ||
|
Studienabbruch aufgrund eines UE oder Tod, n (%) |
17 (3.3) |
17 (3.3) |
|
Studienabbruch aus anderen Gründen, n (%)a |
16 (3.1) |
27 (5.2) |
|
In der Studie, aber fehlende Daten in dem Zeitfenster |
3 (0.6) |
1 (0.2) |
|
Woche 152 |
|
|
|
HIV-1 RNA ≥50 Kopien/ml, n (%)† |
14 (2.7) |
5 (1.0) |
|
HIV-1 RNA < 50 Kopien/ml, n (%) |
456 (87.4) |
449 (85.9) |
|
Keine virologischen Daten im Woche-48-Fenster, n (%) |
52 (10.0) |
69 (3.2) |
|
Gründe: | ||
|
Studienabbruch aufgrund eines UE oder Tod, n (%) |
23 (4.4) |
24 (4.6) |
|
Studienabbruch aus anderen Gründen, n (%)a |
28 (5.4) |
44 (8.4) |
|
In der Studie, aber fehlende Daten in dem Zeitfenster |
1 (0.2) |
1 (0.2) |
|
* Bereinigt in Bezug auf Baseline-Stratifizierungsfaktoren | ||
Tabelle 15: Anteil der Teilnehmenden mit HIV-1 RNA ≥50 Kopien/ml Plasma in der ATLAS-2M-Studie in der Subgruppenanalyse von Woche 48 nach wichtigen Ausgangsmerkmalen der Patienten («Baseline»-Faktoren) (Schnappschuss Ergebnisse).
|
Anzahl Patienten mit HIV-1 RNA ≥50 Kopien/ml/Insgesamt beurteilt (%) | |||
|
Ausgangsmerkmale der Patienten |
|
Dosierung alle 2 Monate (q8w) |
Monatliche Dosierung (q4w) |
|
|
n/N (%) |
n/N (%) | |
|
CD4+-Zellzahl zur Baseline (Zellen/mm3) |
<350 |
1/ 35 (2.9) |
1/ 27 (3.7) |
|
350 bis <500 |
1/ 96 (1.0) |
0/ 89 | |
|
≥500 |
7/391 (1.8) |
4/407 (1.0) | |
|
Geschlecht |
Männlich |
4/385 (1.0) |
5/380 (1.3) |
|
Weiblich |
5/137 (3.5) |
0/143 | |
|
Ethnie |
Weiss |
5/370 (1.4) |
5/393 (1.3) |
|
Nicht weiss |
4/152 (2.6) |
0/130 | |
|
schwarz /Afroamerikaner |
4/101 (4.0) |
0/ 90 | |
|
Nicht schwarz /Afroamerikaner |
5/421 (1.2) |
5/421 (1.2) | |
|
BMI |
<30 kg/m2 |
3/409 (0.7) |
3/425 (0.7) |
|
≥30 kg/m2 |
6/113 (5.3) |
2/98 (2.0) | |
|
Alter (Jahre) |
<35 |
4/137 (2.9) |
1/145 (0.7) |
|
35 bis <50 |
3/242 (1.2) |
2/239 (0.8) | |
|
>50 |
2/143 (1.4) |
2/139 (1.4) | |
|
Vorgängige Behandlung mit CAB/RPV |
Keine |
5/327 (1.5) |
5/327 (1.5) |
|
1-24 Wochen |
3/69 (4.3) |
0/68 | |
|
>24 Wochen |
1/126 (0.8) |
0/128 | |
|
BMI = Body Mass Index, CAB = Cabotegravir, RPV = Rilpivirin | |||
In der ATLAS-2M Studie gab es, was den primären Endpunkt anbelangte, keine klinisch bedeutsamen Behandlungsunterschiede in Korrelation mit den Ausgangsmerkmalen der Patienten («Baseline»-Merkmalen) (Anzahl der CD4+-Lymphozyten, Geschlecht, Ethnie, BMI, Alter und vorgängige Behandlung mit Cabotegravir/Rilpivirin).
Posthoc-Analysen
Multivariable Analysen (MVA) von gepoolten Phase-III-Studien (ATLAS über 96 Wochen, FLAIR über 124 Wochen, ATLAS-2M über 152 Wochen), untersuchten den Einfluss verschiedener Faktoren auf das Risiko eines virologischen Versagens (CVF). Die Analyse der Ausgangsfaktoren (Baseline Factors Analysis, BFA) untersuchte die Ausgangs-(«Baseline»)-Merkmale des Virus und der Studienteilnehmerund das Dosierungsschema; die MVA umfasste die Faktoren bei Studienbeginn und beinhaltete die prognostizierten Plasmakonzentrationen des Wirkstoffs nach Studienbeginn bei Patienten mit bestätigtem virologischen Versagen (CVF) unter Verwendung einer Regressionsmodellierung mit einem kovariaten Auswahlverfahren. Nach insgesamt 4291 Personenjahren betrug die nicht bereinigte CVF-Inzidenzrate 0,54 pro 100 Personenjahre; es wurden 23 Fälle von CVF berichtet (1,4 % von 1651 Personen in diesen Studien).
Die BFA zeigte, dass Baseline-Rilpivirin-Resistenzmutationen (Inzidenzratenverhältnis IRR=21,65; p < 0,0001), HIV-1-Subtyp A6/A1 (IRR=12,87; p < 0,0001) und Body-Mass-Index (IRR=1,09 pro Einheitsanstieg; p=0,04; IRR=3,97 von ≥30 kg/m²; p=0,01) mit CVF assoziiert waren. Andere Variable einschliesslich Q4W- oder Q8W-Dosierung, weibliches Geschlecht oder CAB/INSTI-Resistenzmutationen wiesen keine signifikante Assoziation mit CVF auf. Eine Kombination von mindestens 2 der folgenden Schlüsselfaktoren bei Baseline war mit einem erhöhten Risiko für ein CVF verbunden: Rilpivirin-Resistenz assoziierte Mutationen, HIV-1-Subtyp A6/A1 oder BMI ≥30 kg/m2 (Tabelle 16).
|
Tabelle 16: Virologische Ergebnisse nach Vorhandensein der wichtigsten Ausgangsfaktoren von Rilpivirin assoziierten Mutationen, HIV-1 Subtyp A6/A11 und BMI ≥30 kg/m2 | ||
|
Ausgangsfaktor (Anzahl) |
Virologische Erfolge2 |
Bestätigtes virologisches Versagen (%)3 |
|
0 |
844/970 (87,0) |
4/970 (0,4) |
|
1 |
343/404 (84,9) |
8/404 (2,0)4 |
|
≥2 |
44/57 (77,2) |
11/57 (19,3)5 |
|
Insgesamt |
1231/1431 (86,0) |
23/1431 (1,6)6 |
|
1
HIV-1-Subtyp A1- oder A6-Klassifikation basierend auf der Los Alamos HIV-Sequenzdatenbank (Juni 2020) | ||
Der Anteil der Patienten mit CVF war bei den Patienten mit mindestens zwei dieser Risikofaktoren höher als bei Patienten mit keinem oder nur einem Risikofaktor; dabei trat ein CVF bei 6 von 24 Patienten [25,0 %, 95 %-KI (9,8 %; 46,7 %)] mit der zweimonatlichen Injektion und bei 5 von 33 Patienten mit der monatlichen Injektion auf [15,2 %, 95 %-KI (5,1 %; 31,9 %)]
Orale Überbrückungstherapie mit anderen ART
In einer retrospektiven Analyse gepoolter Daten aus 3 klinischen Studien (FLAIR, ATLAS-2M und LATTE-2/Studie 200056) waren 29 Patienten eingeschlossen, die eine orale Überbrückungstherapie für eine durchschnittliche Dauer von 59 Tagen (25. und 75. Perzentile 53-135) mit einer anderen ART als Vocabria plus Rilpivirin (alternative orale Überbrückungstherapie) während der Behandlung mit intramuskulären (i.m.) langwirksamen (LA) Vocabria- plus Rilpivirin-Injektionen erhielten. Das mediane Alter der Patienten betrug 32 Jahre, 14 % davon waren weiblich, 31 % waren nichtkaukasisch, 97 % erhielten ein Integrase-Inhibitor (INI)-basiertes Regime als alternative orale Überbrückungstherapie, 41 % erhielten einen NNRTI als Teil ihrer alternativen oralen Überbrückungstherapie (einschlisslich Rilpivirin bei 11 von 12 Fällen) und 62 % erhielten einen NRTI. Drei Patienten brachen die Behandlung während oder kurz nach der oralen Überbrückungsphase aufgrund von nicht-sicherheitsrelevanten Gründen ab. Bei der Mehrheit (≥96 %) der Patienten konnte die virale Suppression (Plasma HIV-1-RNA < 50 Kopien/ml) aufrechterhalten werden. Während der Überbrückung mit einer alternativen oralen Überbrückungstherapie und während der Phase nach der alternativen oralen Überbrückungstherapie (bis zu 2 Injektionen mit Vocabria plus Rilpivirin nach der oralen Überbrückung) wurden keine Fälle von CVF (Plasma HIV-1- RNA ≥200 Kopien/ml) beobachtet.
Pharmakokinetik
Die pharmakokinetischen Eigenschaften von REKAMBYS wurden bei gesunden und bei HIV-1-infizierten Erwachsenen untersucht.
Absorption
REKAMBYS weist eine durch die Absorption begrenzte (Flip-Flop-) Kinetik auf, die sich aus der langsamen Absorption aus dem Gesässmuskel in den systemischen Kreislauf ergibt und zu anhaltenden Rilpivirin-Konzentrationen im Plasma führt.
Nach einer intramuskulären Einzeldosis sind Rilpivirin-Konzentrationen im Plasma am ersten Tag nachweisbar und erreichen nach 3 bis 4 Tagen (Medianwert) einen Höchstwert. Rilpivirin wurde nach Gabe einer Einzeldosis REKAMBYS bis zu 52 Wochen lang oder länger im Plasma nachgewiesen. Nach monatlichen oder alle 2 Monate verabreichten Injektionen für die Dauer von 1 Jahr ist die Steady-State Exposition von Rilpivirin zu ungefähr 80 % erreicht.
Die Exposition von Rilpivirin im Plasma steigt nach einmaliger und wiederholter i. m. Injektion von Dosen zwischen 300 und 1200 mg proportional oder geringfügig unterproportional zur Dosis an.
Distribution
Rilpivirin ist in vitro zu ungefähr 99,7 % an Plasmaproteine gebunden, hauptsächlich an Albumin. Basierend auf einer populationspharmakokinetischen Analyse wurde das typische scheinbare Volumen des zentralen Kompartiments (Vc/F) für Rilpivirin nach i. m. Gabe auf 132 l geschätzt.
Rilpivirin ist in der Cerebrospinalflüssigkeit (CSF) vorhanden. Bei HIV-1-infizierten Patienten unter Behandlung mit i.m. Rilpivirin plus i.m. Cabotegravir betrug das mediane Verhältnis zwischen der CSF- und der Plasmakonzentration von Rilpivirin (n = 16) 1,07 bis 1,32 % (Bereich: nicht quantifizierbar bis 1,69 %).
Metabolismus
Biotransformation
In-vitro-Versuche weisen darauf hin, dass Rilpivirin hauptsächlich einer oxidativen Metabolisierung durch das Cytochrom-P450-3A-System (CYP3A-System) unterliegt.
Elimination
Die mittlere scheinbare Halbwertszeit von Rilpivirin nach Gabe von REKAMBYS ist durch die Absorptionsrate begrenzt und wurde auf 13 bis 28 Wochen geschätzt.
Die scheinbare Plasma-Clearance (CL/F) von Rilpivirin nach Gabe von REKAMBYS beträgt schätzungsweise 5,08 l/h.
Nach Gabe einer Einzeldosis von oralem 14C-Rilpivirin wurden im Durchschnitt 85 % der Radioaktivität in den Fäzes und 6,1 % im Urin detektiert. In den Fäzes lag der Anteil des unveränderten Rilpivirins bei durchschnittlich 25 % der verabreichten Dosis. Im Urin wurden nur Spuren von unverändertem Rilpivirin (< 1 % der Dosis) detektiert.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Rilpivirin wird hauptsächlich in der Leber metabolisiert und eliminiert. In einer Studie, in der 8 Patienten mit leichter Leberfunktionsstörung (Child-Pugh-Klasse A) und 8 Patienten mit mittelschwerer Leberfunktionsstörung (Child-Pugh-Klasse B) jeweils mit 8 gematchten Kontrollen verglichen wurden, war die Verfügbarkeit von oralem Rilpivirin bei mehrfacher oraler Gabe bei Patienten mit leichter Leberfunktionsstörung 47 % höher und bei Patienten mit mittelschwerer Leberfunktionsstörung 5 % höher. Bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh-Klasse C) wurde REKAMBYS nicht untersucht.
Nierenfunktionsstörungen
Die Pharmakokinetik von Rilpivirin wurde bei Patienten mit Nierenfunktionsstörung nicht untersucht. Die renale Elimination von Rilpivirin ist vernachlässigbar. Daher ist davon auszugehen, dass die Auswirkung einer Nierenfunktionsstörung auf die Elimination von Rilpivirin gering ist. Weil Rilpivirin in hohem Mass an Plasmaproteine gebunden sind, ist es unwahrscheinlich, dass es durch eine Hämodialyse oder Peritonealdialyse in wesentlichem Umfang entfernt wird.
Ältere Patienten
Es wurde kein klinisch relevanter Einfluss des Alters auf die Verfügbarkeit von Rilpivirin nach intramuskulärer Gabe festgestellt. Die pharmakokinetischen Daten für Rilpivirin bei Patienten über 65 Jahren sind begrenzt.
Kinder und Jugendliche
Die Pharmakokinetik von Rilpivirin nach Verabreichung von REKAMBYS wurde bei Kindern und Jugendlichen nicht untersucht.
Geschlecht
Es wurden zwischen Männern und Frauen keine klinisch relevanten Unterschiede bezüglich der Verfügbarkeit von Rilpivirin nach intramuskulärer Gabe festgestellt.
Ethnizität
Es wurde kein klinisch relevanter Einfluss der ethnischen Zugehörigkeit auf die Verfügbarkeit von Rilpivirin nach intramuskulärer Gabe festgestellt.
BMI
Es wurde kein klinisch relevanter Einfluss des BMI auf die Verfügbarkeit von Rilpivirin nach intramuskulärer Gabe festgestellt.
Patienten mit HBV/HCV-Koinfektion
Eine populationsspezifische pharmakokinetische Analyse ergab keinen Hinweis, dass eine Koinfektion mit dem Hepatitis-B- und/oder Hepatitis-C-Virus klinisch relevante Auswirkung auf die Verfügbarkeit von Rilpivirin nach Einnahme von oralem Rilpivirin hat.
Präklinische Daten
Toxizität bei wiederholter Verabreichung
Es wurden tierexperimentelle Toxizitätsstudien bei Mäusen, Ratten, Kaninchen, Hunden und Cynomolgusaffen mit oral verabreichtem Rilpivirin durchgeführt. Von Toxizität betroffene Zielorgane und -systeme waren die Nebennierenrinde und die dort stattfindende Steroidbiosynthese (Maus, Ratte, Hund, Cynomolgusaffe), die Fortpflanzungsorgane (weibliche Tiere bei der Maus, männliche und weibliche Tiere beim Hund), die Leber (Maus, Ratte, Hund), die Schilddrüse und Hypophyse (Ratte), die Niere (Maus, Hund), das hämatopoietische System (Maus, Ratte, Hund) und das Gerinnungssystem (Ratte).
Karzinogenität
Das karzinogene Potenzial von Rilpivirin wurde durch orale Verabreichung über Sonden an Mäuse und Ratten über einen Zeitraum von bis zu 104 Wochen bewertet. Den Mäusen wurden Dosen von 20, 60 und 160 mg/kg pro Tag verabreicht, die Ratten erhielten 40, 200, 500 und 1500 mg/kg pro Tag. Es wurde ein Anstieg der Inzidenz von hepatozellulären Adenomen und Karzinomen bei Mäusen und Ratten festgestellt. Bei Ratten ergab sich ein Anstieg der Inzidenzen von Follikelzelladenomen und/oder -karzinomen in der Schilddrüse. Die Verabreichung von Rilpivirin führte nicht zu einer statistisch signifikanten Erhöhung der Inzidenz von jeglichen anderen benignen oder malignen Neoplasien bei Mäusen oder Ratten. Die beobachteten hepatozellulären Befunde bei Mäusen und Ratten werden als nagerspezifisch und im Zusammenhang mit der Induktion von Leberenzymen stehend erachtet. Beim Menschen gibt es keinen vergleichbaren Mechanismus. Daher sind diese Tumoren für den Menschen nicht relevant. Die Follikelzellbefunde werden als spezifisch für Ratten und im Zusammenhang mit einer erhöhten Clearance von Thyroxin stehend erachtet, sodass sie für den Menschen keine Relevanz haben. Bei den niedrigsten getesteten Dosen betrug die systemische Verfügbarkeit (auf Basis des AUC-Werts) von Rilpivirin das ≥17-Fache (Mäuse) bzw. das 2-Fache (Ratten) der Verfügbarkeit beim Menschen in der empfohlenen Höchstdosis von 25 mg Rilpivirin einmal täglich bei HIV-1-infizierten Patienten bzw. 600 mg oder 900 mg Rilpivirin als Depot-Injektionssuspension.
Rilpivirin war im In-vitro-Ames-Umkehrmutationstest, im In-vitro-Chromosomenaberrationstest in menschlichen Lymphozyten und im In-vitro-Mauslymphom-Test auf Klastogenität in Abwesenheit und Gegenwart eines metabolischen Aktivierungssystems negativ. Rilpivirin induzierte im In-vivo-Mikronukleustest bei Mäusen nach oraler Gabe keine Chromosomenschädigung.
Reproduktionstoxizität
In einer bei Ratten durchgeführten Studie mit Rilpivirin in einer oralen Dosis von bis zu 400 mg/kg/Tag, einer Dosis, bei der bei Muttertieren Toxizität festgestellt wurde, wurden keine Auswirkungen auf das Paarungsverhalten oder die Fertilität festgestellt. Diese Dosis ist mit einer Exposition verbunden, die ungefähr ≥28-mal höher ist als die Exposition beim Menschen bei der maximal empfohlenen humanen Dosis (MRHD) von 25 mg einmal täglich oder bei Gabe einer intramuskulären Injektionsdosis von 600 mg oder 900 mg Rilpivirin als Depot-Injektionssuspension. Tierexperimentelle Studien ergaben keine Hinweise auf relevante embryonale oder fötale Toxizität oder Auswirkungen auf die Fortpflanzungsfunktion. Bei Ratten und Kaninchen wurde mit Rilpivirin keine Teratogenität festgestellt. Die verfügbaren Mengen bei den embryofötalen NOAEL-Werten bei Ratten und Kaninchen waren ≥12-mal bzw. ≥57-mal höher als die verfügbare Menge beim Menschen in der MRHD von 25 mg einmal täglich oder bei Gabe der intramuskulären Injektionsdosis von 600 mg oder 900 mg Rilpivirin Depot-Injektionssuspension. Bei der Bewertung der prä- und postnatalen Entwicklung bei Ratten hatte Rilpivirin keinen Einfluss auf die Entwicklung von Nachkommen während der Laktation oder nach dem Absetzen, wenn die Mütter eine Dosis von bis zu 400 mg/kg/Tag erhielten.
Lokale Toxizität
Nach i.m. Injektion von Rilpivirin bei Hunden und Minischweinen über längere Zeiträume wurde ein leichtes, kurz anhaltendes Erythem beobachtet und bei der Autopsie wurden an den Injektionsstellen weisse Einlagerungen festgestellt, begleitet von Schwellung und Verfärbung der drainierenden Lymphknoten. Die mikroskopische Untersuchung ergab eine Infiltration von Makrophagen und eosinophile Einlagerungen an den Injektionsstellen. Eine Makrophagen-Infiltrationsreaktion wurde auch in den drainierenden/regionalen Lymphknoten festgestellt. Diese Befunde wurden als Reaktion auf die Einlagerungen betrachtet und weniger als Manifestation einer lokalen Reizung.
Sonstige Hinweise
Inkompatibilitäten
Da keine Kompatibilitätsstudien vorliegen, darf dieses Arzneimittel nicht mit anderen Arzneimitteln oder einem Verdünnungsmittel gemischt werden
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2–8 °C) lagern.
REKAMBYS bis zur Anwendung in der Originalverpackung aufbewahren. Nicht einfrieren.
Vor der Anwendung sollte die Durchstechflasche auf Raumtemperatur gebracht werden (max. 25 °C). Die Durchstechflasche kann bis zu 6 Stunden bei Raumtemperatur im Umkarton stehen bleiben; nicht wieder in den Kühlschrank stellen. Wird sie innerhalb von 6 Stunden nicht verwendet, muss sie verworfen werden.
Sobald die Suspension in die Spritze aufgezogen worden ist, sollte die Injektion so bald wie möglich verabreicht werden, sie kann jedoch bis zu 2 Stunden in der Spritze verbleiben. Nach Ablauf von 2 Stunden müssen das Medikament, die Spritze und die Nadel entsorgt werden.
Aus mikrobiologischer Sicht ist die Suspension sofort zu verwenden. Wird die Suspension nicht sofort verwendet, so liegen die Lagerungsbedingungen und die Dauer der Lagerung in der Verantwortung des Anwenders.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer
67742 (Swissmedic)
Packungen
Rilpivirin Depot-Injektionssuspension liegt in einer Durchstechflasche aus Glas vor.
Packung mit einer 2-ml-Durchstechflasche (600 mg)
Jede Packung enthält 1 Durchstechflasche (600 mg), 1 Spritze, 1 Adapter für die Durchstechflasche und 1 Injektionsnadel. [A].
Packung mit einer 3-ml-Durchstechflasche (900 mg)
Jede Packung enthält 1 Durchstechflasche (900 mg), 1 Spritze, 1 Adapter für die Durchstechflasche und 1 Injektionsnadel. [A].
Hinweise für die Handhabung
Es sind die Hinweise für die Handhabung am Ende dieser Fachinformation für medizinisches Fachpersonal zu beachten.
Zulassungsinhaberin
Janssen-Cilag AG, Zug
Stand der Information
September 2023
Die folgenden Informationen sind nur für Ärzte oder medizinische Fachpersonen bestimmt und sollten nur vom Arzt oder medizinischen Fachpersonen in Verbindung mit der vollständigen Fachinformation gelesen werden
|
Überblick |
| ||
|
|
| ||
|
Im Kühlschrank (2–8 °C) lagern. |
| ||
|
|
| ||
|
Inhalt Ihrer Packung | |||
|
·1 Durchstechflasche mit Rilpivirin | |||
|
Zusätzlich benötigtes Material | |||
|
·Unsterile Handschuhe | |||
|
Vorbereitung | |||
|
1. Durchstechflasche inspizieren | |||
|
|
|
·Es ist zu überprüfen, ob das Haltbarkeitsdatum noch gültig ist. |
|
|
2. 15 Minuten warten | |||
|
|
|
·Vor der Injektion mindestens 15 Minuten warten, damit das Medikament Raumtemperatur annehmen kann. | |
|
3. Kräftig schütteln | |||
|
|
|
·Die Durchstechflasche festhalten und 10 Sekunden lang kräftig schütteln (siehe Abbildung). | |
|
4. Kappe von der Durchstechflasche entfernen | |||
|
|
|
·Die Kappe von der Durchstechflasche entfernen. |
|
|
5. Packung mit dem Adapter für Durchstechflaschen öffnen | |||
|
|
|
·Die Papierfolie von der Packung mit dem Adapter für Durchstechflaschen abziehen. |
|
|
6. Durchstechflaschenadapter aufsetzen | |||
|
|
|
·Den Adapter für die Durchstechflasche in seiner Verpackung in einer geraden Linie nach unten auf die Durchstechflasche drücken (siehe Abbildung). | |
|
7. Spritze vorbereiten | |||
|
|
|
·Die Spritze aus ihrer Verpackung nehmen. | |
|
8. Spritze anbringen | |||
|
|
|
·Den Durchstechflaschenadapter und die Durchstechflasche festhalten (siehe Abbildung). | |
|
9. Langsam die Dosis aufziehen | |||
|
|
|
·Die Spritze und die Durchstechflasche umdrehen und langsam so viel von der Flüssigkeit wie möglich in die Spritze aufziehen. Möglicherweise ist mehr Flüssigkeit vorhanden, als für die Dosismenge benötigt wird. | |
|
10. Die Spritze abnehmen | |||
|
|
|
·Die Spritze vom Durchstechflaschenadapter in einer Drehbewegung abnehmen, und den Durchstechflaschenadapter dabei festhalten (siehe Abbildung). |
|
|
11. Nadel aufsetzen | |||
|
|
|
·Die Verpackung mit der Nadel so weit aufziehen, dass man an die Nadelbasis gelangt. | |
|
Injektion | |||
|
12. Injektionsstelle vorbereiten | |||
|
|
|
Die Injektionen sind in den Gesässmuskel zu verabreichen. Verabreichen Sie die Injektionen an gegenüberliegenden Seiten oder 2 cm voneinander entfernt. Halten Sie einen Abstand von 2 cm von früheren Injektionsstellen ein. Für die Injektionen eine der folgenden Stellen wählen: |
Nicht intravenös injizieren. |
|
13. Schutzkappe entfernen | |||
|
|
|
·Die Nadelschutzkappe an der Nadel nach unten klappen. | |
|
14. Überschüssige Flüssigkeit entfernen | |||
|
|
|
·Die Spritze so halten, dass die Nadel nach oben zeigt. Den Kolben bis zur 2-ml-Dosis drücken, um überschüssige Flüssigkeit und etwaige Luftblasen zu entfernen. |
|
|
15. Haut straff ziehen | |||
|
|
|
Die «Zickzack»-Injektionstechnik anwenden, um das Austreten von Medikamentenflüssigkeit aus der Injektionsstelle zu minimieren. | |
|
16. Nadel einführen | |||
|
|
|
·Die Nadel bis zur vollen Tiefe bzw. bis zum Muskel einführen. | |
|
17. Dosis injizieren | |||
|
|
|
·Die Haut weiterhin straff halten und den Kolben langsam bis zum Anschlag nach unten drücken. | |
|
18. Injektionsstelle prüfen | |||
|
|
|
·Einen Gazetupfer auf die Injektionsstelle pressen. |
|
|
19. Nadel sichern | |||
|
|
|
·Die Nadelschutzkappe über die Nadel klappen. | |
|
Nach der Injektion | |||
|
20. Sicher entsorgen | |||
|
|
|
·Benutzte Nadeln, Spritzen, Durchstechflaschen und Durchstechflaschenadapter den geltenden Gesetzen für Sicherheit und Gesundheitsschutz entsorgen. | |
|
Die beschriebenen Schritte für das 2. Medikament wiederholen | |||
|
|
|
Wurden noch nicht beide Medikamente injiziert, sind die Schritte zur Zubereitung und Injektion von Cabotegravir zu befolgen, für das eigene spezielle Anwendungshinweise gelten. | |
|
Fragen und Antworten | |||
|
1. Wie lange kann das Medikament ausserhalb des Kühlschranks aufbewahrt werden? | |||

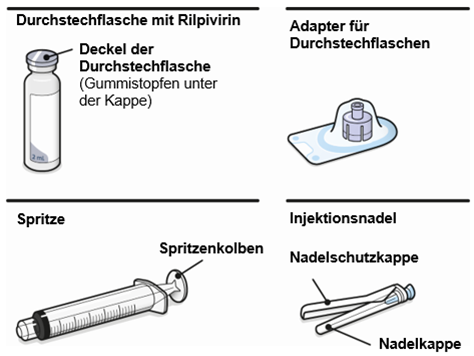
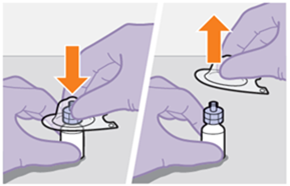
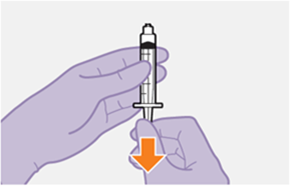
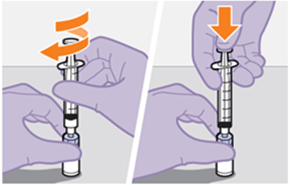
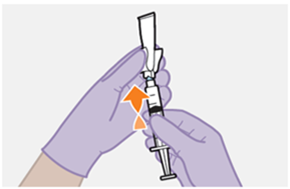
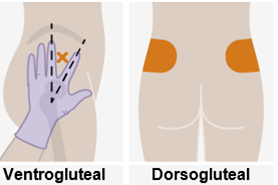
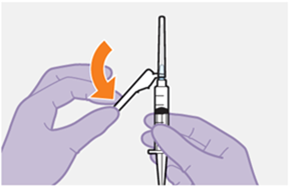
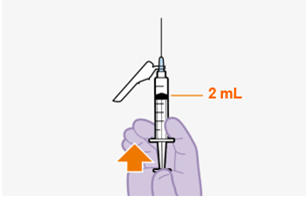
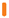
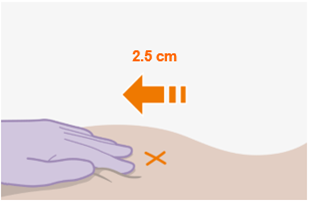
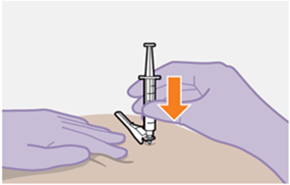

Die folgenden Informationen sind nur für Ärzte oder medizinische Fachpersonen bestimmt und sollten nur vom Arzt oder medizinischen Fachpersonen in Verbindung mit der vollständigen Fachinformation gelesen werden.
|
Überblick |
| ||
|
|
| ||
|
• Im Kühlschrank (2–8 °C) lagern. |
| ||
|
|
| ||
|
Inhalt Ihrer Packung | |||
|
·1 Durchstechflasche mit Rilpivirin | |||
|
Zusätzlich benötigtes Material | |||
|
·Unsterile Handschuhe | |||
|
Vorbereitung | |||
|
1. Durchstechflasche inspizieren | |||
|
|
|
·Es ist zu überprüfen, ob das Haltbarkeitsdatum noch gültig ist. |
|
|
2. 15 Minuten warten | |||
|
|
|
·Vor der Injektion mindestens 15 Minuten warten, damit das Medikament Raumtemperatur annehmen kann. | |
|
3. Kräftig schütteln | |||
|
|
|
·Die Durchstechflasche festhalten und 10 Sekunden lang kräftig schütteln (siehe Abbildung). | |
|
4. Kappe von der Durchstechflasche entfernen | |||
|
|
|
·Die Kappe von der Durchstechflasche entfernen. |
|
|
5. Packung mit dem Adapter für Durchstechflaschen öffnen | |||
|
|
|
·Die Papierfolie von der Packung mit dem Adapter für Durchstechflaschen abziehen. |
|
|
6. Durchstechflaschenadapter aufsetzen | |||
|
|
|
·Den Adapter für die Durchstechflasche in seiner Verpackung in einer geraden Linie nach unten auf die Durchstechflasche drücken (siehe Abbildung). | |
|
7. Spritze vorbereiten | |||
|
|
|
·Die Spritze aus ihrer Verpackung nehmen. | |
|
8. Spritze anbringen | |||
|
|
|
·Den Durchstechflaschenadapter und die Durchstechflasche festhalten (siehe Abbildung). | |
|
9. Langsam die Dosis aufziehen | |||
|
|
|
·Die Spritze und die Durchstechflasche umdrehen und langsam so viel von der Flüssigkeit wie möglich in die Spritze aufziehen. Möglicherweise ist mehr Flüssigkeit vorhanden, als für die Dosismenge benötigt wird. | |
|
10. Die Spritze abnehmen | |||
|
|
|
·Die Spritze vom Durchstechflaschenadapter in einer Drehbewegung abnehmen, und den Durchstechflaschenadapter dabei festhalten (siehe Abbildung). |
|
|
11. Nadel aufsetzen | |||
|
|
|
·Die Verpackung mit der Nadel so weit aufziehen, dass man an die Nadelbasis gelangt. | |
|
Injektion | |||
|
12. Injektionsstelle vorbereiten | |||
|
|
|
Die Injektionen sind in den Gesässmuskel zu verabreichen. Verabreichen Sie die Injektionen an gegenüberliegenden Seiten oder 2 cm voneinander entfernt. Halten Sie einen Abstand von 2 cm von früheren Injektionsstellen ein. Für die Injektionen eine der folgenden Stellen wählen: |
Nicht intravenös injizieren. |
|
13. Schutzkappe entfernen | |||
|
|
|
·Die Nadelschutzkappe an der Nadel nach unten klappen. | |
|
14. Überschüssige Flüssigkeit entfernen | |||
|
|
|
·Die Spritze so halten, dass die Nadel nach oben zeigt. Den Kolben bis zur 3-ml-Dosis drücken, um überschüssige Flüssigkeit und etwaige Luftblasen zu entfernen. |
|
|
15. Haut straff ziehen | |||
|
|
|
Die «Zickzack»-Injektionstechnik anwenden, um das Austreten von Medikamentenflüssigkeit aus der Injektionsstelle zu minimieren. | |
|
16. Nadel einführen | |||
|
|
|
·Die Nadel bis zur vollen Tiefe bzw. bis zum Muskel einführen. | |
|
17. Dosis injizieren | |||
|
|
|
·Die Haut weiterhin straff halten und den Kolben langsam bis zum Anschlag nach unten drücken. | |
|
18. Injektionsstelle prüfen | |||
|
|
|
·Einen Gazetupfer auf die Injektionsstelle pressen. |
|
|
19. Nadel sichern | |||
|
|
|
·Die Nadelschutzkappe über die Nadel klappen. | |
|
Nach der Injektion | |||
|
20. Sicher entsorgen | |||
|
|
|
·Benutzte Nadeln, Spritzen, Durchstechflaschen und Durchstechflaschenadapter den geltenden Gesetzen für Sicherheit und Gesundheitsschutz entsorgen. | |
|
Die beschriebenen Schritte für das 2. Medikament wiederholen | |||
|
|
|
Wurden noch nicht beide Medikamente injiziert, sind die Schritte zur Zubereitung und Injektion von Cabotegravir zu befolgen, für das eigene spezielle Anwendungshinweise gelten. | |
|
Fragen und Antworten | |||
|
1. Wie lange kann das Medikament ausserhalb des Kühlschranks aufbewahrt werden? | |||