Zusammensetzung
Wirkstoffe
Pegcetacoplan
Hilfsstoffe
Sorbit (E420), Eisessig (E260), Natriumacetat-Trihydrat (E262), Natriumhydroxid (zur pH-Einstellung, E524), Wasser für Injektionszwecke.
Enthält Sorbitol 41 mg/ml bzw. 820 mg/Durchstechflasche und Natrium max. 0,37 mg/ml bzw. 7,4 mg/Durchstechflasche.
Indikationen/Anwendungsmöglichkeiten
Aspaveli ist indiziert als Monotherapie für die Behandlung erwachsener Patienten mit paroxysmaler nächtlicher Hämoglobinurie (PNH), welche eine hämolytische Anämie aufweisen (siehe Dosierung/Anwendung und Klinische Wirksamkeit).
Dosierung/Anwendung
Aspaveli wird als subkutane Infusion angewendet.
Die Therapie muss unter der Aufsicht einer in der Behandlung von Patienten mit hämatologischen Erkrankungen erfahrener medizinischer Fachperson eingeleitet werden.
Aspaveli ist für die subkutane Verabreichung mit einer handelsüblichen Spritzeninfusionspumpe vorgesehen und kann vom Patienten selbst verabreicht werden. Aspaveli sollte in den Bauch, die Oberschenkel, die Hüften oder die Oberarme verabreicht werden.
Selbstmedikation und Infusion zu Hause sollten für Patienten in Betracht gezogen werden, die die Behandlung in erfahrenen Behandlungszentren gut vertragen haben. Die Entscheidung über eine mögliche Selbstmedikation und Infusion zu Hause sollte nach Bewertung und Empfehlung des behandelnden Arztes bzw. der behandelnden Ärztin getroffen werden.
Die paroxysmale nächtliche Hämoglobinurie (PNH) ist eine chronische Erkrankung, und es wird empfohlen, die Behandlung mit Aspaveli lebenslang fortzusetzen, es sei denn, das Absetzen von Aspaveli ist klinisch angezeigt (siehe Warnhinweise und Vorsichtsmassnahmen).
Übliche Dosierung
Aspaveli kann von medizinischem Fachpersonal verabreicht werden oder vom Patienten bzw. der Pflegeperson nach entsprechender Anleitung selbst verabreicht werden.
Aspaveli wird zweimal wöchentlich als subkutane Infusion von 1080 mg mit einer handelsüblichen Spritzeninfusionspumpe verabreicht, die Dosen von bis zu 20 ml abgeben kann. Die zweimal wöchentliche Dosis sollte an Tag 1 und Tag 4 jeder Behandlungswoche verabreicht werden (siehe Art der Anwendung).
Vor der Behandlung mit Aspaveli:
·Patienten, die in der Vergangenheit Impfungen erhalten haben: Es sollte sichergestellt werden, dass die Patienten innerhalb von 2 Jahren vor Beginn der Behandlung mit Aspaveli gegen bekapselte Bakterien wie Streptococcus pneumoniae, Neisseria meningitidis Typ A, C, W, Y und B sowie Haemophilus influenzae Typ B (Hib) geimpft wurden (siehe Warnhinweise und Vorsichtsmassnahmen).
·Patienten, die in der Vergangenheit keine Impfungen erhalten haben: Die erforderlichen Impfungen sollten mindestens 2 Wochen vor der ersten Dosis von Aspaveli verabreicht werden (siehe Warnhinweise und Vorsichtsmassnahmen).
oWenn eine sofortige Therapie mit Aspaveli angezeigt ist, sollte die erforderliche Impfung so bald wie möglich verabreicht werden und den Patienten eine zweiwöchige Prophylaxe mit antibakteriellen Medikamenten bereitgestellt werden (siehe Warnhinweise und Vorsichtsmassnahmen).
Patienten, die von einem C5- Hemmer auf Aspaveli umgestellt werden
·In den ersten 4 Wochen wird Aspaveli zweimal wöchentlichen in subkutanen Dosen von 1080 mg zusätzlich zur aktuellen C5-Hemmer Dosis des Patienten verabreicht, um das Risiko einer Hämolyse bei abruptem Absetzen der Behandlung zu minimieren.
·Nach 4 Wochen sollte der Patient den C5-Hemmer absetzen, und die Behandlung mit Aspaveli als Monotherapie fortsetzen.
·Die Umstellung von anderen Komplementinhibitoren als Eculizumab wurde nicht untersucht. Das Absetzen anderer Komplementinhibitoren vor Erreichen des Steady-State von Pegcetacoplan sollte mit Vorsicht erfolgen (siehe «Pharmakokinetik»).
Dosisanpassung
·Das Dosierungsschema kann auf 1080 mg jeden dritten Tag (d.h. Tag 1, Tag 4, Tag 7, Tag 10, Tag 13 usw.) geändert werden, wenn ein Patient einen Laktatdehydrogenase (LDH)-Wert von mehr als dem 2fachen des oberen Grenzwerts (ULN, upper limit of normal) aufweist.
·Im Falle einer Dosissteigerung ist der LDH-Wert mindestens 4 Wochen lang zweimal wöchentlich zu überwachen.
Vergessene Dosisgabe
Wenn eine Dosis von Aspaveli versäumt wird, sollte sie so bald wie möglich nachgeholt werden, und dann die Behandlung mit dem regulären Dosierungsschema wieder aufgenommen werden.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Die Sicherheit und Wirksamkeit von Pegcetacoplan wurde bei Patienten mit Leberfunktionsstörungen nicht untersucht; es wird jedoch keine Dosisanpassung empfohlen, da bei Leberfunktionsstörungen keine Auswirkungen auf die Clearance von Pegcetacoplan zu erwarten sind (siehe Pharmakokinetik).
Patienten mit Nierenfunktionsstörungen
Eine schwere Niereninsuffizienz (Kreatinin-Clearance <30 ml/min) hatte keinen Einfluss auf die Pharmakokinetik (PK) von Pegcetacoplan; daher ist eine Dosisanpassung von Pegcetacoplan bei Patienten mit Niereninsuffizienz nicht erforderlich. Es liegen keine Daten für die Anwendung von Pegcetacoplan bei Patienten mit terminaler Niereninsuffizienz (ESRD) vor, die eine Hämodialyse benötigen (siehe Pharmakokinetik).
Ältere Patienten
Obwohl in klinischen Studien keine offensichtlichen altersbedingten Unterschiede beobachtet wurden und es keine Hinweise darauf gibt, dass bei der Behandlung älterer Menschen besondere Vorsichtsmassnahmen erforderlich sind, war die Zahl der Patienten im Alter von 65 Jahren und älter nicht ausreichend, um festzustellen, ob es altersbedingte Unterschiede gibt.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Pegcetacoplan bei Kindern mit PNH von der Geburt bis zu einem Alter von unter 18 Jahren wurde nicht nachgewiesen. Es liegen keine Daten vor.
Art der Anwendung
Aspaveli sollte nur über eine subkutane Verabreichung mit einer Spritzeninfusionspumpe verabreicht werden, die ein nominales Fördervolumen von 20 ml erreichen kann.
Wenn die Therapie mit Aspaveli eingeleitet wird, sollte der Patient von einer qualifizierten medizinischen Fachperson in Infusionstechniken, in der Verwendung einer Spritzeninfusionspumpe, dem Führen eines Behandlungsprotokolls, dem Erkennen möglicher unerwünschter Wirkungen und den zu ergreifenden Massnahmen bei deren Auftreten, unterrichtet werden.
Aspaveli ist durch eine subkutane Infusion in den Bauch, die Oberschenkel, die Hüfte oder die Oberarme zu verabreichen. Die Infusionsstellen sollten mindestens 7,5 cm voneinander entfernt sein. Die Infusionsstellen sollten zwischen den Verabreichungen gewechselt werden. Infusionen in Bereiche, in denen die Haut empfindlich, gequetscht, rot oder hart ist und Infusionen in Tattoos, Narben oder Dehnungsstreifen sollten vermieden werden.
Die typische Infusionsdauer beträgt etwa 30 Minuten (bei Infusion an zwei Stellen) bzw. etwa 60 Minuten (bei einer Infusionsstelle). Die Infusion sollte sofort nach dem Aufziehen von Aspaveli in die Spritze begonnen werden. Die Verabreichung sollte innerhalb von 2 Stunden nach der Vorbereitung der Spritze beendet werden.
Siehe Hinweise für die Handhabung und Gebrauchsanweisung in der Packungsbeilage für weitere Hinweise zur Vorbereitung und Verabreichung des Arzneimittels.
Kontraindikationen
Aspaveli ist kontraindiziert bei Patienten mit:
·Überempfindlichkeit gegen Pegcetacoplan oder gegen einen der Hilfsstoffe.
·nicht ausgeheilten Infektionen durch bekapselte Bakterien wie Streptococcus pneumoniae, Neisseria meningitidis und Haemophilus influenzae.
·ohne aktuellen Impfschutz gegen Neisseria meningitidis, Streptococcus pneumoniae und Haemophilus influenzae, es sei denn, sie erhalten eine geeignete Antibiotikaprophylaxe bis zwei Wochen nach der Impfung.
Warnhinweise und Vorsichtsmassnahmen
Durch bekapselte Bakterien verursachte schwere Infektionen
Die Anwendung von Pegcetacoplan kann zu schweren Infektionen führen, die durch bekapselte Bakterien wie Streptococcus pneumoniae, Neisseria meningitidis und Haemophilus influenzae verursacht werden. Um das Infektionsrisiko zu verringern, müssen alle Patienten mindestens 2 Wochen vor Beginn der Behandlung mit Pegcetacoplan gemäss den geltenden lokalen Richtlinien gegen diese Bakterien geimpft werden, es sei denn, das Risiko einer Verzögerung der Therapie mit Pegcetacoplan überwiegt das Risiko einer Infektion.
Patienten mit bekannter Impfanamnese
Vor der Behandlung mit Pegcetacoplan ist bei Patienten mit bekannter Impfanamnese sicherzustellen, dass die Patienten Impfstoffe gegen bekapselte Bakterien, darunter Streptococcus pneumoniae, Neisseria meningitidis Serotypen A, C, W, Y und B sowie Haemophilus influenzae Typ B in den letzten 2 Jahren vor Beginn der Therapie mit Pegcetacoplan erhalten haben.
Patienten ohne bekannte Impfanamnese
Bei Patienten ohne bekannte Impfanamnese müssen die erforderlichen Impfungen mindestens zwei Wochen vor Erhalt der ersten Dosis von Pegcetacoplan verabreicht werden. Falls eine sofortige Therapie angezeigt ist, müssen die erforderlichen Impfstoffe so bald wie möglich verabreicht werden, und der Patient muss bis zwei Wochen nach der Impfung mit geeigneten Antibiotika behandelt werden.
Eine Impfung reicht möglicherweise nicht aus, um eine schwere Infektion zu verhindern. Die offiziellen Leitlinien für den angemessenen Einsatz von Antibiotika sollten in Betracht gezogen werden. Alle Patienten sollten auf frühe Anzeichen einer Infektion mit bekapselten Bakterien wie Neisseria meningitidis, Streptococcus pneumoniae und Haemophilus influenzae überwacht werden, bei Verdacht auf eine Infektion sofort untersuchen werden und bei Bedarf mit geeigneten Antibiotika behandeln werden. Die Patienten sollten über diese Anzeichen und Symptome informiert werden und darüber, dass sie sofort einen Arzt aufsuchen sollten.
Überempfindlichkeit
Es wurde über Überempfindlichkeitsreaktionen berichtet. Wenn eine schwere Überempfindlichkeitsreaktion (einschliesslich Anaphylaxie) auftritt, ist die Infusion mit Pegcetacoplan sofort abzubrechen und eine angemessene Behandlung einzuleiten.
Überwachung von PNH-Manifestationen nach Absetzen von Pegcetacoplan
Wenn Patienten mit PNH die Behandlung mit Pegcetacoplan abbrechen, müssen sie engmaschig auf Anzeichen und Symptome einer schweren intravaskulären Hämolyse überwacht werden. Eine intravaskuläre Hämolyse zeigt sich durch erhöhte LDH-Werte in Verbindung mit einer plötzlichen Abnahme der PNH-Klongrösse oder des Hämoglobins oder durch das erneute Auftreten von Symptomen wie Müdigkeit, Hämoglobinurie, Bauchschmerzen, Dyspnoe, schwerwiegenden unerwünschten vaskulären Ereignissen (einschliesslich Thrombose), Dysphagie oder erektiler Dysfunktion. Wenn ein Absetzen von Pegcetacoplan erforderlich ist, sollte eine alternative Therapie in Betracht gezogen werden, da PNH unbehandelt lebensbedrohlich ist. Falls nach dem Absetzen eine schwerwiegende Hämolyse auftritt, sind folgende Verfahren/Therapien in Erwägung zu ziehen: Bluttransfusion (Erythrozytenkonzentrat), Austauschtransfusion, Antikoagulation oder Kortikosteroide. Patienten sind nach der letzten Dosis über mindestens 8 Wochen engmaschig auf eine schwerwiegende Hämolyse und andere Reaktionen zu überwachen. Darüber hinaus sollte ein langsames Ausschleichen in Betracht gezogen werden.
Verhütung bei Frauen im gebärfähigen Alter
Es wird empfohlen, dass Frauen im gebärfähigen Alter zuverlässige Verhütungsmethoden anwenden, um eine Schwangerschaft während der Behandlung mit Pegcetacoplan und eines Zeitraums von mindestens acht Wochen nach der letzten Dosis von Pegcetacoplan zu vermeiden (siehe Schwangerschaft, Stillzeit).
Akkumulierung von Polyethylenglykol (PEG)
Aspaveli ist ein PEGyliertes Arzneimittel. Die potenziellen Langzeitwirkungen der PEG-Akkumulation in den Nieren, im Plexus choroideus des Gehirns und in anderen Organen sind nicht bekannt (siehe Präklinische Daten). Regelmässige Laboruntersuchungen der Nierenfunktion werden empfohlen.
Schulungsmaterial
Alle Ärzte, die beabsichtigen, Aspaveli zu verschreiben, müssen sicherstellen, dass sie das
Schulungsmaterial für Ärzte erhalten haben und mit ihm vertraut sind. Die Ärzte müssen den Nutzen und die Risiken von Aspaveli mit den Patienten besprechen und ihnen das Informationspaket für
Patienten sowie den Patientenausweis aushändigen. Die Patienten sind anzuweisen, sich umgehend in ärztliche Behandlung zu begeben, wenn Anzeichen oder Symptome einer schweren Infektion oder
Überempfindlichkeitsreaktion während der Therapie mit Aspaveli bei ihnen auftreten,insbesondere wenn diese auf eine Infektion mit bekapselten Bakterien hindeuten.
Auswirkungen auf Labortests
Es können Interferenzen zwischen Silica-Reagenzien in Gerinnungsprofilen und Pegcetacoplan auftreten, die zu künstlich verlängerter aktivierter partieller Thromboplastinzeit (aPTT) führen; daher ist die Verwendung von Silica-Reagenzien in Gerinnungstests zu vermeiden.
Sorbitol
Dieses Arzneimittel enthält 820 mg Sorbitol pro Durchstechflasche à 20 ml. Patienten mit hereditärer Fructoseintoleranz (HFI) dürfen dieses Arzneimittel nicht erhalten.
Natrium
Dieses Arzneimittel enthält 7,4 mg Natrium pro Durchstechflasche à 20 ml, d.h. es ist nahezu „natriumfrei“.
Interaktionen
Es wurden keine Interaktionsstudien durchgeführt. Auf der Grundlage von In-vitro Daten hat Pegcetacoplan ein geringes Potenzial für klinische Wechselwirkungen mit anderen Arzneimitteln.
Schwangerschaft, Stillzeit
Frauen im gebärfähigen Alter
Es wird empfohlen, dass Frauen im gebärfähigen Alter während der Behandlung mit Pegcetacoplan und für mindestens 8 Wochen nach der letzten Pegcetacoplan-Dosis wirksame Verhütungsmethoden anwenden, um eine Schwangerschaft zu verhindern.
Bei Frauen, die eine Schwangerschaft planen, sollte die Anwendung von Pegcetacoplan nur nach Abwägung der Risiken und des Nutzens erwogen werden (siehe Schwangerschaft).
Schwangerschaft
Es liegen keine oder begrenzte Daten über die Anwendung von Pegcetacoplan bei schwangeren Frauen vor. Tierexperimentelle Studien weisen auf Reproduktionstoxizität hin (siehe Präklinische Daten).
Pegcetacoplan darf während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, nicht angewendet werden, es sei denn, dass eine Behandlung mit Pegcetacoplan aufgrund des klinischen Zustandes der Frau erforderlich ist.
Stillzeit
Es ist nicht bekannt, ob Pegcetacoplan in die Muttermilch übergeht. Bei Affen wurde eine minimale (weniger als 1 %, pharmakologisch nicht signifikant) Ausscheidung von Pegcetacoplan in die Milch nachgewiesen. Es ist unwahrscheinlich, dass ein gestillter Säugling eine klinisch relevante Exposition erfährt (siehe Präklinische Daten).
Es wird empfohlen, während der Behandlung mit Pegcetacoplan nicht zu stillen.
Fertilität
Die Auswirkungen von Pegcetacoplan auf die Fruchtbarkeit wurden an Tieren nicht untersucht. In Toxizitätsstudien an Affen wurden keine mikroskopischen Anomalien der männlichen oder weiblichen Fortpflanzungsorgane festgestellt (siehe Präklinische Daten).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Aspaveli hat keinen oder einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
Die am häufigsten gemeldeten Nebenwirkungen bei Patienten, die mit Pegcetacoplan behandelt wurden, waren Reaktionen an der Injektionsstelle: Erythem an der Injektionsstelle, Juckreiz an der Injektionsstelle, Schwellung an der Injektionsstelle, Schmerzen an der Injektionsstelle und Hämatom an der Injektionsstelle. Andere Nebenwirkungen, die bei mehr als 10 % der Patienten während der klinischen Studien gemeldet wurden, waren Infektionen der oberen Atemwege, Diarrhoe, Hämolyse, Abdominalschmerz, Kopfschmerzen, Ermüdung (Fatigue), Fieber, Husten, Harnwegsinfektionen, Impfkomplikation, Schmerzen in den Extremitäten, Schwindelgefühl, Arthralgie, und Rückenschmerzen. Die am häufigsten gemeldeten schwerwiegenden unerwünschten Wirkungen waren Hämolyse und Sepsis.
Tabellarische Auflistung der unerwünschten Wirkungen
Tabelle 1 zeigt die in den klinischen Studien und nach der Markteinführung mit Pegcetacoplan beobachteten Nebenwirkungen bei Patienten mit PNH. Die unerwünschten Wirkungen sind nach MedDRA-Systemorganklassen und Häufigkeit gemäss folgender Konvention geordnet: sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1‘000, <1/100), selten (≥1/10‘000, <1/1‘000), sehr selten (<1/10‘000), nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen in der Reihenfolge ihrer abnehmenden Schwere aufgeführt.
Tabelle 1 Unerwünschte Wirkungen, die in den klinischen Studien1 und nach der Markteinführung beobachtet wurden
|
MedDRA-Systemorganklasse |
Häufigkeit |
Unerwünschte Wirkung |
|
Infektionen und parasitäre Erkrankungen |
Sehr häufig |
Infektion der oberen Atemwege |
|
Häufig |
Sepsis2 | |
|
Gelegentlich |
Zervizitis | |
|
Erkrankungen des Immunsystems |
Gelegentlich |
Anaphylaktische Reaktion3 |
|
Erkrankungen des Blutes und des Lymphsystems |
Sehr häufig |
Hämolyse |
|
Häufig |
Thrombozytopenie | |
|
Stoffwechsel- und Ernährungsstörungen |
Häufig |
Hypokalaemia |
|
Erkrankungen des Nervensystems |
Sehr häufig |
Kopfschmerzen |
|
Gefässerkrankungen |
Häufig |
Hypertension |
|
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Sehr häufig |
Husten |
|
Häufig |
Dyspnoe | |
|
Erkrankungen des Gastrointestinaltrakts |
Sehr häufig |
Abdominalschmerz |
|
Häufig |
Übelkeit | |
|
Erkrankungen der Haut und des Unterhautgewebes |
Häufig |
Erythema |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Sehr häufig |
Arthralgie |
|
Häufig |
Myalgie | |
|
Erkrankungen der Nieren und Harnwege |
Häufig |
Akute Nierenschäden |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Sehr häufig |
Erythem an der Injektionsstelle |
|
Häufig |
Reaktion an der Injektionsstelle | |
|
Leber- und Gallenerkrankungen |
Häufig |
Erhöhte Alanin-Aminotransferase |
|
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen |
Sehr häufig |
Impfkomplikation4 |
1 Studien APL2-302, APL2-308, APL2-202, APL2-CP-PNH-204 und APL2-CP0514 zu Patienten mit PNH. Sofern angemessen, sind medizinisch ähnliche Begriffe anhand eines ähnlichen medizinischen Konzepts gruppiert.
2 Sepsis beinhaltet einen Fall eines septischen Schocks.
3 Geschätzt anhand der Daten nach der Markteinführung.
4 Impfkomplikationen standen im Zusammenhang mit den Pflichtimpfungen.
Beschreibung spezifischer unerwünschter Wirkungen
Infektionen
Aufgrund seines Wirkmechanismus kann die Anwendung von Pegcetacoplan möglicherweise das Risiko von Infektionen erhöhen, insbesondere von Infektionen, die durch bekapselte Bakterien wie Streptococcus pneumoniae, Neisseria meningitidis Typ A, C, W, Y und B sowie Haemophilus influenzae verursacht werden (siehe Warnhinweise und Vorsichtsmassnahmen). Während der APL2-302 Studie wurde keine schwerwiegende Infektion durch bekapselte Bakterien gemeldet. Achtundvierzig Patienten bekamen während der Studie eine Infektion. Die häufigsten Infektionen bei Patienten, die während der APL2-302 Studie mit Pegcetacoplan behandelt wurden, waren Infektionen der oberen Atemwege (28 Fälle, 35 %). Die meisten Infektionen, die bei den im Rahmen der APL2-302 Studie mit Pegcetacoplan behandelten Patienten gemeldet wurden, waren nicht schwerwiegend und verliefen überwiegend mild. Zehn Patienten entwickelten Infektionen, die als schwerwiegend gemeldet wurden, darunter ein Patient, der an COVID-19 starb. Die häufigsten schwerwiegenden Infektionen waren Sepsis (3 Fälle) (was bei einem Patienten zum Absetzen von Pegcetacoplan führte) und Gastroenteritis (3 Fälle), die alle abheilten. 11 Patienten wiesen in der Studie APL2 308 eine Infektion auf. Mit einer Ausnahme wurden alle Infektionen als leicht oder mässig schwer berichtet. Ein Patient, der eine Infektion hatte, entwickelte einen septischen Schock und starb.
Hämolyse
Neunzehn Patienten, die während der APL2-302 Studie mit Pegcetacoplan behandelt wurden, berichteten über Hämolyse. Sieben Fälle wurden als schwerwiegend eingestuft, 5 Fälle führten zum Absetzen von Pegcetacoplan und bei 10 Patienten wurde die Dosis von Pegcetacoplan erhöht. Während der Studie APL2-308 traten bei den mit Pegcetacoplan behandelten Patienten 3 Fälle von Hämolyse auf. Keiner dieser Fälle wurde als schwerwiegend berichtet oder führte zum Absetzen von Pegcetacoplan. Bei allen 3 Patienten wurde die Pegcetacoplan-Dosis erhöht.
Reaktionen an der Injektionsstelle
Während der Studie APL2-302 wurde über Reaktionen an der Injektionsstelle (z. B. Erythem, Schwellung, Juckreiz und Schmerzen) berichtet. Diese Reaktionen waren von leichter bis mittlerer Intensität und führten nicht zum Abbruch der Behandlung.
Diarrhöe
Während der Studie APL2-302 wurden Fälle von Durchfall gemeldet, von denen keiner schwerwiegend war oder zum Abbruch der Behandlung führte.
Immunogenität
Die Immunogenität von Aspaveli wurde mit Hilfe spezifischer Anti-Arzneimittel-Antikörper (ADA) untersucht, von denen einer spezifisch für den Nachweis von ADAs gegen die Peptidkomponente von Pegcetacoplan (Anti-Pegcetacoplan-Peptid) und ein zweiter spezifisch für ADAs gegen die Polyethylenglykol-Komponente (PEG) von Pegcetacoplan (Anti-PEG) ist.
Die Inzidenz von Anti-Arzneimittel-Antikörpern (serokonvertierte ADA oder erhöhte ADA-Werte) war gering und hatte, falls vorhanden, keine nennenswerten Auswirkungen auf die Pharmakokinetik/Pharmakodynamik (PK/PD), die Wirksamkeit oder das Sicherheitsprofil von Pegcetacoplan. Während der Studien APL2-302 und APL2-308 entwickelten 3 von 126 Patienten, die Pegcetacoplan erhielten, Anti-Pegcetacoplan-Peptid-Antikörper. Alle 3 Patienten wurden auch positiv auf neutralisierende Antikörper (NAb) getestet. Das Ansprechen auf NAb hatte keinen offensichtlichen Einfluss auf die PK oder die klinische Wirksamkeit. Bei 18 von 126 Patienten traten Anti-PEG-Antikörper auf; bei 9 Patienten handelte es sich um eine Serokonversion und bei 9 Patienten um eine behandlungsbedingte Verstärkung.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Es wurden keine Fälle einer Überdosierung berichtet.
Eigenschaften/Wirkungen
ATC-Code
L04AJ03
Pegcetacoplan ist ein symmetrisches Molekül, das aus zwei identischen Pentadecapeptiden besteht, die kovalent an die Enden eines linearen Polyethylenglykol (PEG)-Moleküls gebunden sind. Das Molekulargewicht von Pegcetacoplan beträgt etwa 43,5 Kilodalton (kDa). Die Peptideinheiten binden an Komplement C3 und üben eine breite Hemmung auf die Komplementkaskade aus. Der 40-kDa-PEG-Anteil sorgt für eine bessere Löslichkeit und eine längere Verweildauer im Körper nach Verabreichung des Arzneimittels.
Wirkungsmechanismus
Pegcetacoplan bindet mit hoher Affinität an das Komplementprotein C3 und sein Aktivierungsfragment C3b und reguliert dadurch die Spaltung von C3 und die Bildung von nachgeschalteten Effektoren der Komplementaktivierung. Bei PNH wird die extravaskuläre Hämolyse (EVH) durch die C3b-Opsonisierung gefördert, während die intravaskuläre Hämolyse (IVH) durch den nachgeschalteten Membranangriffskomplex (MAC) vermittelt wird. Pegcetacoplan übt eine umfassende Regulierung der Komplementkaskade aus, indem es proximal zur C3b- und MAC-Bildung wirkt und so die Mechanismen kontrolliert, die zu EVH und IVH führen.
Pharmakodynamik
In der Studie APL2 302 stieg die mittlere C3-Konzentration in der Pegcetacoplan-Gruppe von 0,94 g/l bei Studienbeginn auf 3,83 g/l in Woche 16 und verblieb auf diesem Niveau bis Woche 48. In der Studie APL2-308 stieg die mittlere C3-Konzentration von einem Ausgangswert von 0,95 g/l auf 3,56 g/l in Woche 26.
In der Studie APL2-302 stieg der mittlere prozentuale Anteil an PNH-Typ II + PNH-Typ III Erythrozyten von einem Ausgangswert von 66,80 % auf 93,85 % in Woche 16 und verblieb auf diesem Niveau bis Woche 48. In der Studie APL2 308 stieg der mittlere prozentuale Anteil an PNH-Typ II + PNH-Typ III Erythrozyten von einem Ausgangswert von 42,4 % auf 90,0 % in Woche 26.In der Studie APL2-302 sank der mittlere prozentuale Anteil an PNH-Typ-II- + PNH-Typ-III-Erythrozyten mit C3-Ablagerung von einem Ausgangswert von 17,73 % auf 0,20 % in Woche 16 und verblieb auf diesem Niveau bis Woche 48. In der Studie APL2-308 sank der mittlere prozentuale Anteil an PNH-Typ-II-+PNH-Typ-III-Erythrozyten mit C3-Ablagerung von einem Ausgangswert von 2,85 % auf 0,09 % in Woche 26.
Herz-Elektrophysiologie
Es wurden keine spezifischen Studien durchgeführt, um das Potenzial von Pegcetacoplan zur Verzögerung der kardialen Repolarisation zu bestimmen. Pegcetacoplan ist eine PEGylierte Peptidstruktur und zeigte keine Hemmung im humanen Ether-a-go-go-Gen (hERG)-Ionenkanaltest. Die Analyse der Konzentration-QTc bestätigte keine Auswirkungen auf die kardiale Repolarisation (QT-Intervall um die Herzfrequenz korrigiert).
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von Pegcetacoplan bei Patienten mit PNH wurde in zwei offenen, randomisierten, kontrollierten Phase-3-Studien untersucht: in Studie APL2-302 an Patienten, die bereits mit einem Komplementinhibitor vorbehandelt waren, und in Studie APL2-308 an Patienten, die zuvor mit keinem Komplementinhibitor behandelt wurden. In beiden Studien lag die Dosis von Pegcetacoplan bei zweimal wöchentlich 1080 mg. Bei Bedarf konnte die Dosis von Pegcetacoplan auf 1080 mg alle 3 Tage angepasst werden.
Studie an erwachsenen Patienten, die bereits mit einem Komplementinhibitor vorbehandelt waren (APL2-302)
Bei der Studie APL2-302 handelte es sich um eine offene, randomisierte Studie mit einem aktiven, Vergleichspräparat-kontrollierten Zeitraum von 16 Wochen, gefolgt von einem 32-wöchigen offenen Zeitraum (OLP). An dieser Studie nahmen Patienten mit PNH teil, die mindestens in den vorangegangenen drei Monaten mit einer stabilen Eculizumab-Dosis behandelt worden waren und einen Hb-Wert <10,5 g/dl aufwiesen.
Die in Frage kommenden Patienten nahmen an einer vierwöchigen Vorlaufphase teil, während der sie zusätzlich zu ihrer bestehenden Eculizumab-Dosis zweimal wöchentlich 1080 mg Pegcetacoplan subkutan erhielten. Anschliessend wurden die Patienten im Verhältnis 1:1 randomisiert und erhielten entweder zweimal wöchentlich 1080 mg Pegcetacoplan oder ihre bestehende Eculizumab-Dosis für die Dauer des 16-wöchigen randomisierten Kontrollzeitraums (RCP). Die Randomisierung wurde anhand der Anzahl der Transfusionen von Erythrozytenkonzentraten innerhalb der letzten 12 Monate vor Tag -28 (<4; ≥4) und der Thrombozytenzahl beim Screening (<100’000/μl; ≥100’000/μl) stratifiziert. Patienten, die die RCP abschlossen, kamen anschliessend in die OLP, in der alle Patienten Pegcetacoplan für bis zu 32 Wochen erhielten (Patienten, die während der RCP Eculizumab erhalten hatten, kamen zunächst in eine 4-wöchige Vorlaufphase, bevor sie zur Pegcetacoplan-Monotherapie wechselten).
Die Patienten wurden gegen Streptococcus pneumoniae, Neisseria meningitidis Typ A, C, W, Y und B sowie Haemophilus influenzae Typ B (Hib) geimpft, und zwar entweder innerhalb von zwei Jahren vor dem ersten Tag der Behandlung oder innerhalb von zwei Wochen nach Beginn der Behandlung mit Pegcetacoplan. Patienten, die nach Tag 1 geimpft wurden, erhielten bis 2 Wochen nach der Impfung eine prophylaktische Behandlung mit geeigneten Antibiotika. Darüber hinaus wurde eine prophylaktische Antibiotikatherapie nach Ermessen des Prüfarztes in Übereinstimmung mit den lokalen Behandlungsrichtlinien für Patienten mit PNH, die mit einem Komplementärinhibitor behandelt werden, verabreicht. Pegcetacoplan wurde als subkutane Infusion verabreicht; die Infusionszeit betrug etwa 20 bis 40 Minuten.
Der primäre und die sekundären Wirksamkeitsendpunkte wurden in Woche 16 bewertet. Der primäre Wirksamkeitsendpunkt war die Veränderung des Hämoglobinspiegels vom Ausgangswert bis zur Woche 16 (während der RCP). Der Ausgangswert wurde als der Durchschnitt der Messwerte definiert, die vor der Verabreichung der ersten Dosis von Pegcetacoplan aufgezeichnet wurden. Wichtige sekundäre Wirksamkeitsendpunkte waren die Vermeidung von Transfusionen, definiert als der Anteil der Patienten, die während der RCP keine Transfusion benötigten, sowie die Veränderung der absoluten Retikulozytenzahl (ARC), des LDH-Spiegels und des FACIT-Fatigue Scale Score vom Ausgangswert bis zur Woche 16. Insgesamt wurden 80 Patienten in die Vorlaufphase aufgenommen. Am Ende der Vorlaufphase wurden alle 80 Patienten randomisiert, 41 für Pegcetacoplan und 39 für Eculizumab. Die demografischen Daten und die Ausgangskrankheitscharakteristika waren im Allgemeinen zwischen den Behandlungsgruppen ausgewogen (siehe Tabelle 2). Insgesamt 38 Patienten in der mit Pegcetacoplan behandelten Gruppe und 39 Patienten in der Eculizumab-Gruppe schlossen die 16-wöchige RCP ab und setzten die 32-wöchige offene Behandlungsphase fort. Insgesamt brachen 12 von 80 (15 %) Patienten, die Pegcetacoplan erhielten, die Studie wegen unerwünschter Ereignisse ab. Gemäss Prüfplan wurde die Dosis bei 15 Patienten auf 1080 mg alle 3 Tage angepasst. Bei 12 Patienten wurde der Nutzen beurteilt und bei 8 von 12 Patienten erwies sich die Dosisanpassung als wirksam.
Tabelle 2: Demografische Grunddaten und Merkmale der Patienten in der Studie APL2-302
|
Parameter |
Statistik |
Pegcetacoplan (n=41) |
Eculizumab (n=39) |
|
Alter (Jahre) |
Mittelwert (SD) |
50,2 (16,3) |
47,3 (15,8) |
|
Baseline-Dosis von Eculizumab |
n (%) |
26 (63,4) |
29 (74,4) |
|
Frauen |
n (%) |
27 (65,9) |
22 (56,4) |
|
Zeit seit PNH Diagnose (Jahre) bis Tag -28 |
Mittelwert (SD) |
8,7 (7,4) |
11,4 (9,7) |
|
Hämoglobinspiegel (g/dl) |
Mittelwert (SD) |
8,7 (1,1) |
8,7 (0,9) |
|
Absolute Reticulozyten Anzahl (109 /l) |
Mittelwert (SD) |
218 (75,0) |
216 (69,1) |
|
LDH Spiegel (U/l) |
Mittelwert (SD) |
257,5 (97,6) |
308,6 (284,8) |
|
FACIT-Fatigue Gesamt-Score |
Mittelwert (SD) |
32,2 (11,4) |
31,6 (12,5) |
|
Anzahl Transfusionen in den letzten 12 Monaten vor Tag -28 |
Mittelwert (SD) |
6,1 (7,3) |
6,9 (7,7) |
|
<4 |
n (%) |
20 (48,8) |
16 (41,0) |
|
≥4 |
n (%) |
21 (51,2) |
23 (59,0) |
|
Thrombozytenzahl beim Screening (109 /l) |
Mittelwert (SD) |
167 (98,3) |
147 (68,8) |
|
Thrombozytenzahl beim Screening |
n (%) |
12 (29,3) |
9 (23,1) |
|
Thrombozytenzahl beim Screening |
n (%) |
29 (70,7) |
30 (76,9) |
|
Aplastische Anämie in der Anamnese |
n (%) |
11 (26,8) |
9 (23,1) |
|
Myelodysplastisches Syndrom in der Anamnese |
n (%) |
1 (2,4) |
2 (5,1) |
Pegcetacoplan war Eculizumab in Bezug auf den primären Endpunkt der Hämoglobinveränderung gegenüber dem Ausgangswert überlegen (p<0,0001). Die korrigierte mediane Veränderung des Hb-Wertes gegenüber dem Ausgangswert betrug 2,4 g/dl in der mit Pegcetacoplan behandelten Gruppe gegenüber -1,5 g/dl in der Eculizumab-Gruppe, was einen korrigierten medianen Anstieg von 3,8 g/dl unter Pegcetacoplan im Vergleich zu Eculizumab in Woche 16 entspricht (Abbildung 1).
Abbildung 1: Korrigierter Mittelwert (± SE) der Veränderung des Hämoglobins (g/dl) von Baseline bis Woche 16 in APL2-302
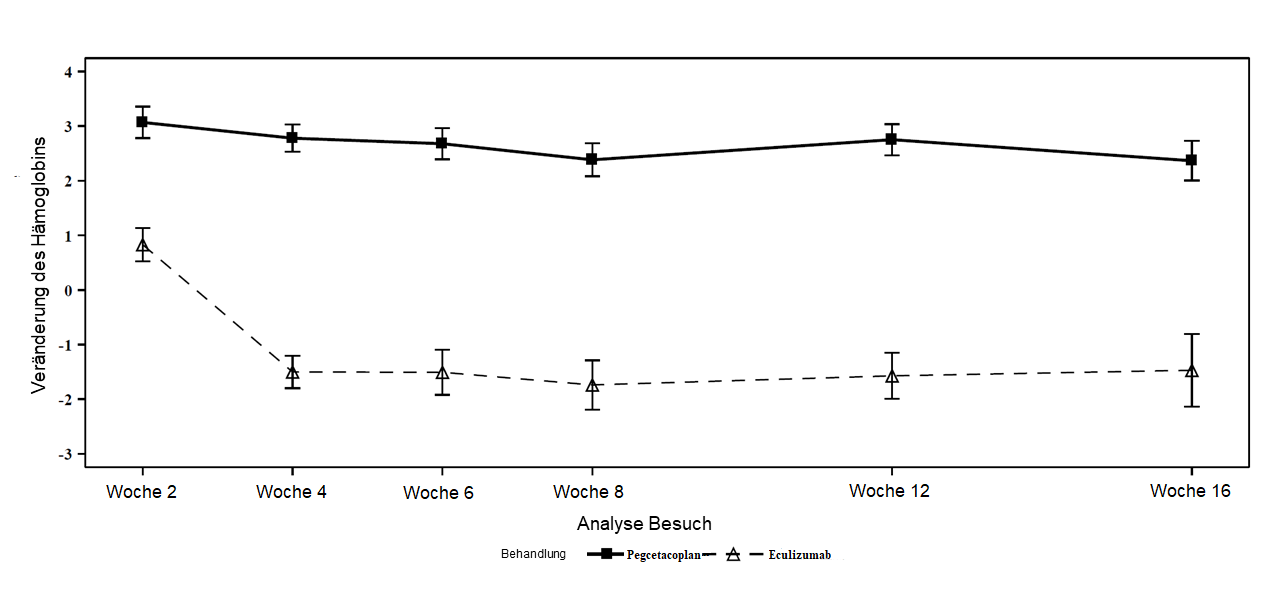
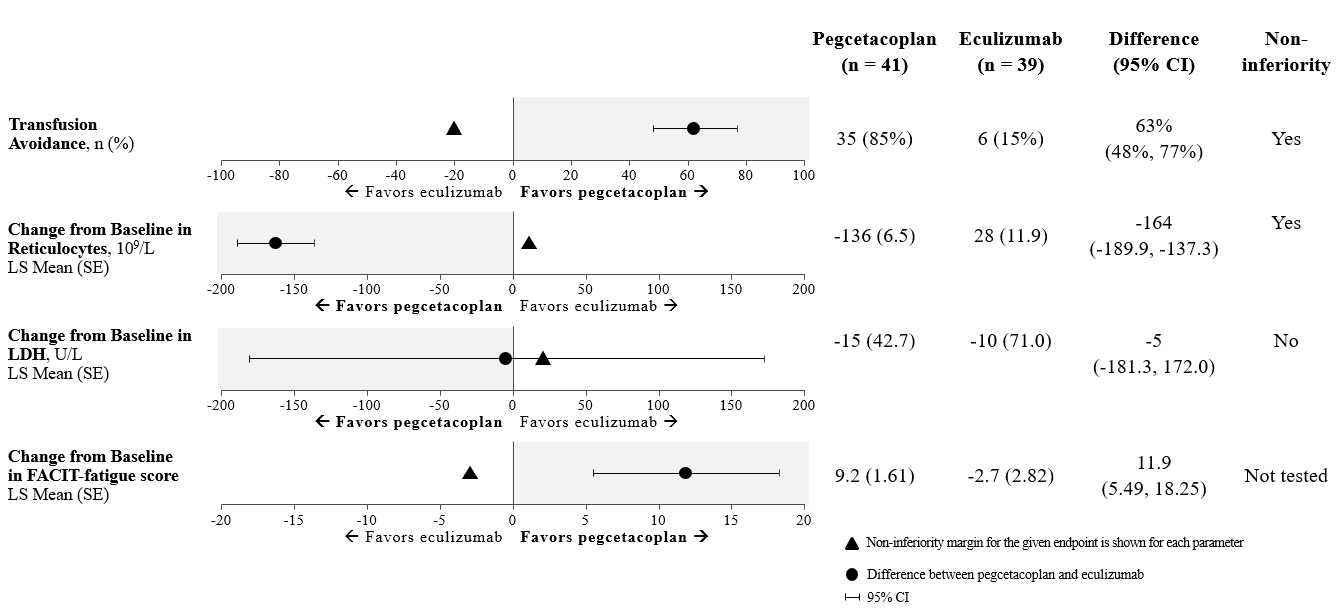
Die Nicht-Unterlegenheit (non-inferiority) für die wichtigen sekundären Endpunkte Transfusionsvermeidung und ARC im Vergleich zu den Ausgangswerten wurde nachgewiesen. Bei 85 % der Patienten in der mit Pegcetacoplan behandelten Gruppe konnten Transfusionen vermieden werden, gegenüber 15 % in der Eculizumab-Gruppe.
Die Nicht-Unterlegenheit wurde bei der Veränderung der LDH gegenüber dem Ausgangswert nicht nachgewiesen.
Aufgrund des hierarchischen Testablaufs wurde die Veränderung des FACIT-Fatigue-Scores gegenüber dem Ausgangswert nicht formal getestet.
Die korrigierten Mittelwerte, der Behandlungsunterschied, die Konfidenzintervalle und die statistischen Analysen, die für die wichtigsten sekundären Endpunkte durchgeführt wurden, sind in Abbildung 2 dargestellt.
Abbildung 2: Analyse wichtiger sekundärer Endpunkte in APL2-302
Die Ergebnisse waren in allen bestätigenden Analysen der primären und wichtigen sekundären Endpunkte konsistent, einschliesslich aller beobachteten Daten, die auch Daten nach der Transfusion enthielten.
Bei den mit Pegcetacoplan behandelten Patienten zeigten die primären und wichtigen sekundären Wirksamkeitsanalysen keine nennenswerten Unterschiede in Bezug auf Geschlecht, Rasse oder Alter.
Eine Hämoglobin-Normalisierung wurde in Woche 16 bei 34 % der Patienten in der Pegcetacoplan -Gruppe gegenüber 0 % in der Eculizumab-Gruppe erreicht. Eine Normalisierung des ARC wurde bei 78 % der Patienten in der mit Pegcetacoplan behandelten Gruppe gegenüber 3 % in der Eculizumab-Gruppe erreicht. Eine Normalisierung der LDH wurde bei 71 % der Patienten in der mit Pegcetacoplan behandelten Gruppe erreicht, gegenüber 15 % in der Eculizumab-Gruppe.
Insgesamt 77 Patienten nahmen an der 32-wöchigen OLP teil, während der alle Patienten Pegcetacoplan erhielten; die Gesamtexposition betrug bis zu 48 Wochen. Die Ergebnisse in Woche 48 stimmten im Allgemeinen mit denen in Woche 16 überein und belegen eine anhaltende Wirksamkeit.
Studie an erwachsenen Patienten, die mit keinem Komplementinhibitor vorbehandelt wurden (APL2-308)
Bei der Studie APL2-308 handelte es sich um eine offene, randomisierte, kontrollierte Studie, an der Patienten mit PNH teilnahmen, die in den letzten 3 Monaten vor Studienbeginn nicht mit einem Komplementinhibitor behandelt worden waren und deren Hämoglobin-Wert unter der unteren Grenze des Normwerts (LLN) lag. Geeignete Patienten wurden im Verhältnis 2:1 randomisiert und erhielten entweder Pegcetacoplan oder eine supportive Behandlung (z. B. Transfusionen, Kortikosteroide, Supplementierung mit z. B. Eisen, Folsäure und Vitamin B12), die im Folgenden als Kontrollgruppe bezeichnet wird, während des gesamten 26wöchigen Behandlungszeitraums.
Die Randomisierung wurde anhand der Anzahl der Transfusionen von gepackten roten Blutkörperchen (PRBC) innerhalb der letzten 12 Monate vor Tag -28 (< 4; ≥ 4) stratifiziert. Zu jedem Zeitpunkt während der Studie konnte ein Patient, der der Kontrollgruppe zugewiesen wurde und dessen Hämoglobin-Wert ≥ 2 g/dl unter dem Ausgangswert lag oder der ein PNH-assoziiertes thromboembolisches Ereignis hatte, gemäß Prüfplan für den Rest der Studie auf Pegcetacoplan umgestellt werden.
Insgesamt wurden 53 Patienten randomisiert: 35 Patienten in die Pegcetacoplan-Gruppe und 18 Patienten in die Kontrollgruppe. Die demografischen Daten und die Krankheitsmerkmale beim Ausgangswertwaren im Allgemeinen zwischen den Behandlungsgruppen ausgewogen. Das Durchschnittsalter betrug 42,2 Jahre in der Pegcetacoplan-Gruppe und 49,1 Jahre in der Kontrollgruppe. Die durchschnittliche Anzahl von PRBC-Transfusionen in den 12 Monaten vor der Voruntersuchung betrug 3,9 in der Pegcetacoplan-Gruppe und 5,1 in der Kontrollgruppe. Fünf Patienten in jeder Gruppe (14,3 % in der Pegcetacoplan-Gruppe und 27,8 % in der Kontrollgruppe) wiesen eine aplastische Anämie in der Anamnese auf. Weitere Ausgangswerte waren: mittlerer Hämoglobin-Ausgangswert (Pegcetacoplan-Gruppe: 9,4 g/dl vs. Kontrollgruppe: 8,7 g/dl), ARC (Pegcetacoplan-Gruppe: 230,2 × 109/l vs. Kontrollgruppe: 180,3 × 109/l), LDH-Wert (Pegcetacoplan-Gruppe: 2 151,0 E/l vs. Kontrollgruppe: 1 945,9 E/l) und Thrombozytenzahl (Pegcetacoplan-Gruppe: 191,4 × 109/l vs. Kontrollgruppe: 125,5 × 109/l). 11 der 18 Patienten, die der Kontrollgruppe zugewiesen wurden, wechselten zu Pegcetacoplan, weil ihr Hämoglobin-Wert um ≥ 2 g/dl unter den Ausgangswert sank. Von den 53 randomisierten Patienten erhielten 52 (97,8 %) eine prophylaktische Antibiotikatherapie gemäß den örtlichen Verschreibungsleitlinien.
Der primäre und die sekundären Wirksamkeitsendpunkte wurden in Woche 26 bewertet. Die beiden ko-primären Wirksamkeitsendpunkte waren die Stabilisierung des Hämoglobin-Werts, definiert als Vermeidung eines Abfalls der Hämoglobin-Konzentration > 1 g/dl gegenüber dem Ausgangswert ohne Transfusion, und die Veränderung der LDH-Konzentration gegenüber dem Ausgangswert.
In der mit Pegcetacoplan behandelten Gruppe erreichten 30 von 35 Patienten (85,7 %) eine Stabilisierung des Hämoglobin-Werts gegenüber 0 Patienten in der Kontrollgruppe. Die bereinigte Differenz zwischen Pegcetacoplan und der Kontrollgruppe betrug 73,1 % (95%KI: 57,2 % bis 89,0 %; p < 0,0001).
Die kleinste quadratische (LS) mittlere (SE) Veränderung der LDH-Konzentration gegenüber dem Ausgangswert in Woche 26 betrug -1 870 E/l in der mit Pegcetacoplan behandelten Gruppe gegenüber -400 E/l in der Kontrollgruppe (p < 0,0001). Die Differenz zwischen Pegcetacoplan und der Kontrollgruppe betrug -1 470 (95%-KI: -2 113 bis -827). Die Behandlungsunterschiede zwischen der Pegcetacoplan-Gruppe und der Kontrollgruppe zeigten sich in Woche 2 und blieben bis Woche 26 bestehen (Abbildung 3). Die LDH-Konzentrationen in der Kontrollgruppe verblieben erhöht.
Abbildung 3: Mittlere (±SE) LDH-Konzentration (E/l) im Zeitverlauf nach Behandlungsgruppe in der Studie APL2-308
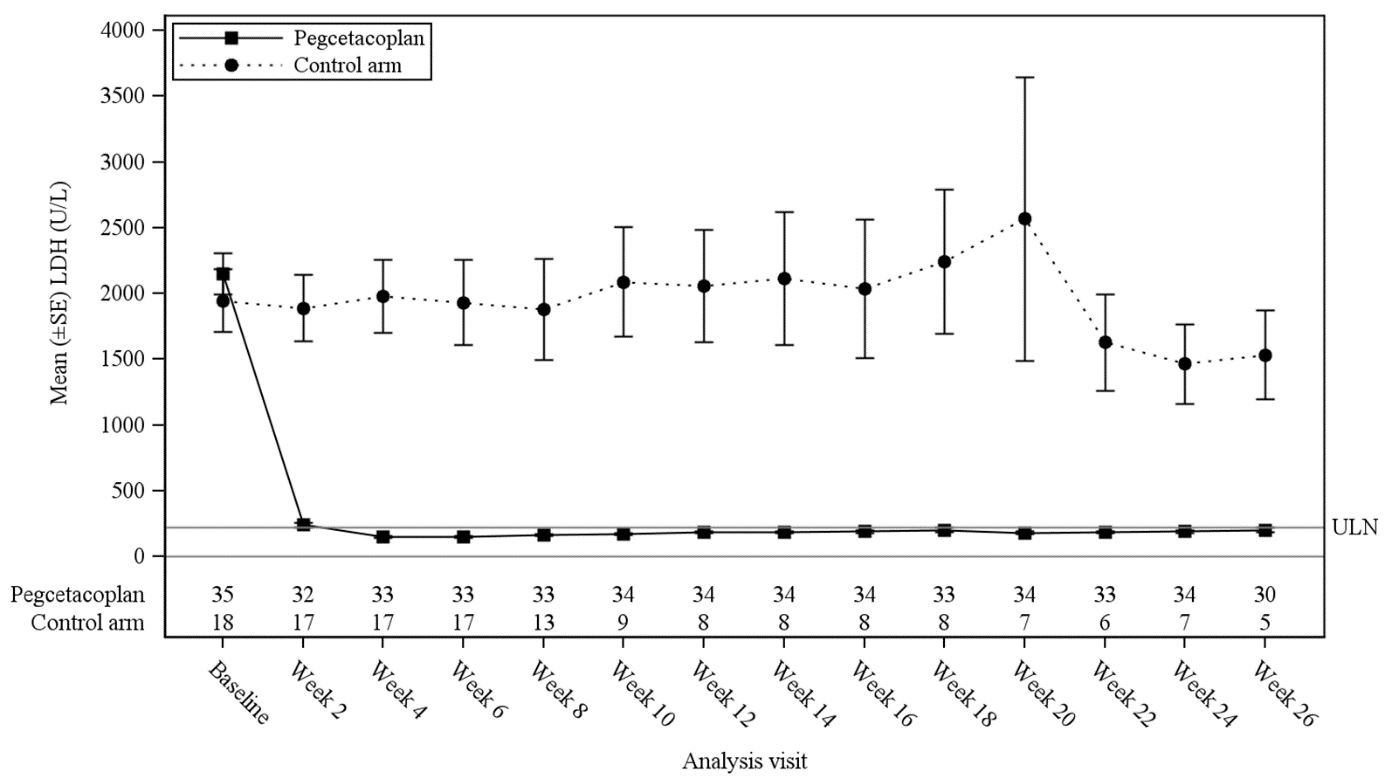
Bei den ausgewählten wichtigen sekundären Wirksamkeitsendpunkten Hämoglobin-Ansprechen ohne Transfusion, Veränderung des Hämoglobinspiegels und Veränderung der ARC zeigte sich in der mit Pegcetacoplan behandelten Gruppe ein signifikanter Behandlungsunterschied gegenüber der Kontrollgruppe (Tabelle 3).
Tabelle 3: Analyse wichtiger sekundärer Endpunkte in Studie APL2-308
|
Parameter |
Pegcetacoplan |
Kontrollgruppe |
Differenz |
|
Hämoglobin-Ansprechen ohne Transfusiona |
25 (71 %) |
1 (6 %) |
54 % (34 %; 74 %) |
|
Veränderung des Hämoglobinspiegels (g/dl) vom Ausgangswert bis Woche 26 |
2,9 (0,38) |
0,3 (0,76) |
2,7 (1,0; 4,4) |
|
Veränderung der ARC (109/l) vom Ausgangswert bis Woche 26 |
-123 (9,2) |
-19 (25,2) |
-104 (-159; -49) |
a Das Hämoglobin-Ansprechen war definiert als Anstieg des Hämoglobins um ≥ 1 g/dl vom Ausgangswert bis Woche 26. ARC = Absolute Retikulozytenzahl, KI = Konfidenzintervall, LS = Kleinste Quadrate (least square), SE = Standardfehler (standard error)
Pharmakokinetik
Absorption
Pegectacoplan wird subkutan verabreicht und allmählich in den systemischen Kreislauf aufgenommen, wobei die mediane Tmax zwischen 108 und 144 Stunden (4,5 bis 6,0 Tage) liegt. Steady-State-Serumkonzentrationen wurden bei PNH-Patienten nach zweimal wöchentlicher Verabreichung von 1080 mg etwa 4 bis 6 Wochen nach der ersten Dosis erreicht. Bei Patienten, die mit einem Komplementinhibitor vorbehandelt waren (Studie APL2-302), lag das geometrische Mittel(%CV) der Steady-State-Serumkonzentrationen bei Patienten, die 16 Wochen lang behandelt wurden, zwischen 655 µg/ml (18,6 %) und 706 µg/ml (15,1 %). Bei Patienten, die zuvor keinen Komplementinhibitor erhielten (APL2-308), betrug in Woche 26 das geometrische Mittel (%VK) der Steady-State-Serumkonzentration 744 µg/ml (25,5 %) bei zweimal wöchentlicher Gabe. Es wurde keine formelle Studie zur absoluten Bioverfügbarkeit durchgeführt; in einer Querschnittsstudie zum Vergleich der Exposition nach Verabreichung von SC- und IV-Formulierungen an gesunde Freiwillige wurde die Bioverfügbarkeit auf 87 % geschätzt.
Distribution
Der Mittelwert (%CV) des zentralen Verteilungsvolumens von Pegcetacoplan beträgt bei Patienten mit PNH etwa 3,98 l (32 %).
Metabolismus
Aufgrund seiner PEGylierten Peptidstruktur ist der Abbau von Pegcetacoplan über katabolische Wege in kleine Peptide, Aminosäuren und PEG zu erwarten.
Elimination
Nach mehrfacher subkutaner Verabreichung von Pegcetacoplan beträgt der geschätzte Mittelwert (CV%) der Clearance (CL) 0,015 l/Stunde (30 %) und die mediane effektive Eliminationshalbwertszeit (t1/2) 8,6 Tage bei Patienten mit PNH.
Die Ergebnisse einer radiomarkierten Studie an Cynomolgus-Affen deuten darauf hin, dass der primäre Ausscheidungsweg des markierten Peptidanteils über die Urinausscheidung erfolgt.
Linearität/Nicht Linearität
Die Exposition von Pegcetacoplan steigt dosisproportional von 45 bis 1440 mg.
Kinetik spezieller Patientengruppen
Basierend auf den Ergebnissen der Populations-PK-Analyse wurde kein Einfluss des Alters, der ethnischen Zugehörigkeit oder des Geschlechts auf die Pharmakokinetik von Pegcetacoplan festgestellt.
Im Vergleich zu einem 70 kg schweren Referenzpatienten wird die mittlere Steady-State-Konzentration bei Patienten mit einem Körpergewicht von 50 kg um etwa 20 % höher vorhergesagt. Bei Patienten mit einem Körpergewicht von 40 kg wird von einer um 45 % höheren mittleren Konzentration ausgegangen. Es liegen nur wenige Daten zum Sicherheitsprofil von Pegcetacoplan bei Patienten mit einem Körpergewicht unter 50 kg vor.
Nierenfunktionsstörungen
In einer Studie mit 8 Patienten mit schwerer Nierenfunktionsstörung, definiert als Kreatinin-Clearance (CrCl) unter 30 ml/min nach der Cockcroft-Gault-Formel (mit 4 Patienten mit Werten unter 20 ml/min), hatte die Nierenfunktionsstörung keinen Einfluss auf die Pharmakokinetik von 270 mg Pegcetacoplan in Form einer Einzeldosis (siehe Dosierung/Anwendung). Es liegen nur wenige Daten zu Patienten mit PNH und Nierenfunktionsstörung vor, die zweimal wöchentlich die klinische Dosis von 1080 mg erhielten. Es liegen keine klinischen Daten über die Anwendung von Pegcetacoplan bei Patienten mit dialysepflichtiger terminaler Niereninsuffizienz vor.
Leberfunktionsstörungen
Es wurden keine spezifischen Studien durchgeführt, um die Auswirkungen von Leberfunktionsstörungen auf die Pharmakokinetik von Pegcetacoplan zu bestimmen. Da die Biotransformation hauptsächlich über den Katabolismus erfolgt, ist nicht zu erwarten, dass Leberfunktionsstörungen die Clearance von Pegcetacoplan beeinflussen (siehe Patienten mit Leberfunktionsstörungen).
Ältere Patienten
Auf der Grundlage einer populationspharmakokinetischen Analyse war die scheinbare Clearance (CL/F) bei älteren Patienten und Patienten unter 65 Jahren ähnlich und es wurden keine offensichtlichen altersbedingten Unterschiede beobachtet (siehe Ältere Patienten). Die Zahl der älteren Patienten war jedoch begrenzt.
Präklinische Daten
In vitro und in vivo Toxikologiedaten lassen keine besonders besorgniserregende Toxizität für den Menschen erkennen. Die bei Tieren beobachteten Wirkungen bei Expositionsniveaus, die den klinischen Expositionsniveaus ähnlich sind, werden im Folgenden beschrieben.
Toxizität bei wiederholter Gabe
Studien zur wiederholten Verabreichung von Pegcetacoplan an Kaninchen und Cynomolgus-Affen mit täglichen, subkutanen Dosen bis zum Siebenfachen der menschlichen Dosis (1080 mg zweimal wöchentlich) wurden durchgeführt. Zu den histologischen Befunden bei beiden Spezies gehörten dosisabhängige, epitheliale Vakuolisierung und Infiltrate von vakuolisierten Makrophagen in verschiedenen Geweben. Diese Ergebnisse wurden mit hohen kumulativen Dosen von langkettigem PEG in anderen zugelassenen PEG-haltigen Arzneimitteln in Verbindung gebracht, hatten keine klinischen Konsequenzen und wurden nicht als nachteilig angesehen.
Bei Expositionen (Cmax und AUC), die geringer oder vergleichbar mit denen der menschlichen Dosis waren, wurde mikroskopisch eine Degeneration der Nierentubuli bei beiden Spezies beobachtet nach 4 Wochen und 9 Monaten täglicher Pegcetacoplan-Gabe, welche minimal und nicht fortschreitend waren.
Obwohl bei den Tieren keine offensichtlichen Anzeichen einer Nierenfunktionsstörung beobachtet wurden, sind die klinische Bedeutung und die funktionellen Folgen dieser Resultate unbekannt.
Genotoxizität
Pegcetacoplan war nicht mutagen, wenn es in In-vitro-Assays mit bakterieller Rückmutation (Ames) getestet wurde, und es war nicht genotoxisch in einem in vitro Assay an menschlichen TK6-Zellen oder in einem in vivo Mikronukleustest an Mäusen.
Kanzerogenität
Es wurden keine Langzeitstudien zur Kanzerogenität von Pegcetacoplan an Tieren durchgeführt.
Reproduktionstoxizität
Tierexperimentelle Reproduktionsstudien mit Pegcetacoplan wurden an Cynomolgus-Affen durchgeführt. Die Behandlung trächtiger Cynomolgus-Affen mit Pegcetacoplan in einer subkutanen Dosis von 28 mg/kg/Tag (das 2,9-fache der menschlichen Steady-State-Cmax) von der Trächtigkeit bis zur Geburt führte zu einem statistisch signifikanten Anstieg der Aborte (31,6 %) oder Totgeburten (21,1 %) im Vergleich zu den Kontrollen (5,0 % bzw. 0 %).
Die Erhöhungen wurden als Pegcetacoplan-bedingt und nachteilig betrachtet. Aufgrund der erhöhten Inzidenz von Aborten und Totgeburten bei 28 mg/kg/Tag wurde der NOAEL in dieser Studie auf 7 mg/kg/Tag festgelegt.
Bei Nachkommen, die am berechneten Geburtstermin zur Welt kamen, wurden keine maternalen Toxizitäten oder teratogenen Wirkungen beobachtet. Ausserdem wurden bei Säuglingen bis zu 6 Monaten nach der Geburt keine Auswirkungen auf die Entwicklung beobachtet. Eine systemische Exposition gegenüber Pegcetacoplan wurde bei Föten von Affen, die mit 28 mg/kg/Tag behandelt wurden, während der Organogenese bis zum zweiten Trimenon festgestellt, aber die Exposition war minimal (weniger als 1 %, pharmakologisch nicht signifikant).
Fertilität
Spezifische Studien an Nagetieren zur Fruchtbarkeit und frühen Embryonalentwicklung mit Pegcetacoplan wurden nicht durchgeführt, da Pegcetacoplan nur bei Menschen und nichtmenschlichen Primaten pharmakologisch wirksam ist. Mikroskopische Untersuchungen der männlichen und weiblichen Geschlechtsorgane in den Studien zur Toxizität nach wiederholter Verabreichung an Affen zeigten keine schädlichen Wirkungen von Pegcetacoplan bei männlichen oder weiblichen Tieren.
Laktation
Bei Affen wurde weniger als 1 % Ausscheidung von Pegcetacoplan in der Milch nachgewiesen; daher wird die Wahrscheinlichkeit einer klinisch relevanten Exposition des gestillten Säuglings über die Muttermilch als minimal angesehen.
Sonstige Hinweise
Inkompatibilitäten
Nicht zutreffend.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit „EXP“ bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8 °C) lagern.
Den Behälter in der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Detaillierte Anweisungen zur Vorbereitung und Verabreichung von Aspaveli finden Sie in der Gebrauchsanweisung in der Packungsbeilage.
Nicht verwenden, wenn die Flüssigkeit trüb aussieht, Partikel enthält oder dunkelgelb ist.
Entsorgen Sie teilweise benutzte Durchstechflaschen und Einwegartikel entsprechend den örtlichen Vorschriften.
Zulassungsnummer
68674
Packungen
Aspaveli gibt es als gebrauchsfertige Lösung in Einwegfläschchen.
1 Durchstechflasche [A]
8 Durchstechflaschen [A]
Zulassungsinhaberin
Swedish Orphan Biovitrum AG, Basel
Stand der Information
März 2025