CompositionPrincipes actifs
Prothrombinum multiplex humanum (Factor II coagulationis humanus, Factor VII coagulationis humanus, Factor IX coagulationis humanus, Factor X coagulationis humanus).
Excipients
Antithrombinum III, Heparinum natricum, Natrii citras, Natrii chloridum, Proteinum C humanum.
Teneur total en sodium: 68 mg/flacon.
Solvant: Aqua ad iniectabile.
Indications/Possibilités d’emploiProthromplex NF est indiqué pour le traitement et la prophylaxie des troubles de la coagulation provoqués par un déficit acquis ou congénital en facteurs II, VII, IX et X dépendants de la vitamine K:
·Hémorragies aiguës et prophylaxie peropératoire des saignements en cas de déficit acquis causé par:
·traitement anticoagulant oral;
·déficit en vitamine K (p.ex. malabsorption, alimentation parentérale prolongée);
·lésions hépatiques parenchymateuses (hépatite, cirrhose hépatique, lésion hépatique toxique sévère, etc.);
·coagulation intravasculaire disséminée (en cas de choc sévère, polytraumatisme, transfusions massives) après élimination de la coagulopathie intravasculaire disséminée sous-jacente.
·Hémorragies aiguës et prophylaxie peropératoire des saignements en cas de déficits congénitaux isolés ou associés en facteurs II, VII, IX et X. Étant donné l'effet thrombogène potentiel des préparations de complexe prothrombique, Prothromplex NF ne doit toutefois être utilisé que lorsque les facteurs isolés appropriés ne sont pas disponibles.
Posologie/Mode d’emploiL'utilisation de Prothromplex NF ne doit être effectuée que par un médecin spécialiste de la prise en charge des troubles de la coagulation.
La posologie et la durée du traitement de substitution dépendent de la gravité du trouble de la coagulation, de la localisation et de l'intensité du saignement ainsi que de l'état clinique du patient.
La dose et la fréquence d'administration doivent être calculées sur la base de chaque cas individuel. La dose nécessaire dépend de la demi-vie in vivo du facteur requis (voir «Pharmacocinétique») et du poids corporel du patient (voir «Pharmacocinétique»).
La posologie individuelle ne peut être déterminée que sur la base de dosages réguliers du taux plasmatique des facteurs de coagulation à corriger ou sur la base d'un test global évaluant le taux de complexe prothrombique (p.ex. temps de Quick/INR) et du suivi continu de l'état clinique du patient.
Déficit congénital en facteurs du complexe prothrombique
La dose calculée de traitement nécessaire repose sur la notion empirique qu'environ 1 UI de facteur IX par kg de poids corporel augmente l'activité plasmatique du facteur IX de 0,8% par rapport à l'activité normale, 1 UI de facteur VII par kg de poids corporel augmente l'activité plasmatique du facteur VII de 2% par rapport à l'activité normale, et 1 UI de facteurs II et X par kg de poids corporel augmente l'activité plasmatique des facteurs II et X de 1,5% par rapport à l'activité normale.
La dose requise est déterminée à l'aide de la formule suivante:
Dose initiale (p.ex. de facteur IX):
Nombre d'unités nécessaires = poids corporel (kg) × augmentation souhaitée du facteur IX (%) x 1,2.
La dose à administrer et la fréquence des injections doivent toujours être adaptées au cas particulier en fonction de l'efficacité clinique observée. Un déficit en facteur IX ne doit être traité par Prothromplex NF que lorsque l'on ne dispose pas de préparation spécifique de facteur IX.
Les patients nécessitant un traitement par un concentré de complexe prothrombique plasmatique humain pendant plus de 4 à 5 jours doivent être soigneusement surveillés afin de détecter tout signe de thrombose ou de coagulation intravasculaire disséminée (CIVD). Ces patients nécessitent un traitement spécial.
L'expérience dans le traitement de déficits congénitaux en facteurs II, VII et X est limitée. Etant donné la longue demi-vie du facteur II (40 – 60 heures) et du facteur X (30 – 60 heures), les patients présentant un déficit congénital en facteur II ou en facteur X nécessitent de plus faibles quantités de concentré de complexe prothrombique plasmatique humain.
La demi-vie du facteur VII est très courte (3–6 heures). Les patients présentant un déficit congénital en facteur VII peuvent donc nécessiter de plus grandes quantités de concentré de complexe prothrombique plasmatique humain. Ces patients doivent être soigneusement surveillés afin de détecter tout signe de thrombose ou de coagulopathie intravasculaire disséminée.
En cas d'intervention chirurgicale majeure, une surveillance attentive du traitement de substitution par des tests de la coagulation est essentielle.
L'efficacité et la sécurité de l'utilisation de Prothromplex NF dans la population pédiatrique n'ont pas été établies jusqu'à présent dans les essais cliniques de Takeda.
Déficit acquis en facteurs du complexe prothrombique
En cas de déficit acquis en facteurs du complexe prothrombique, p.ex. suite à un traitement par des antagonistes de la vitamine K, la préparation ne doit être utilisée que si une correction rapide est nécessaire (p.ex. en cas d'hémorragie sévère ou d'intervention). Dans les autres cas, l'administration de vitamine K et/ou de plasma ou une réduction de la dose d'antagoniste de la vitamine K peut être suffisante.
En cas d'hémorragie sévère et avant une intervention à haut risque de saignement, les patients doivent recevoir un concentré de complexe prothrombique humain afin d'obtenir un temps de prothrombine normal.
Instructions posologiques particulières
·Posologie en cas de déficit acquis en facteurs du complexe prothrombiqueLa formule suivante peut être utilisée pour le calcul de la dose:Dose (en UI) = augmentation souhaitée du temps de Quick (en %) × kg de poids corporel × 1,2.
·Traitement anticoagulant oral, déficit en vitamine K:Prophylaxie des saignements:En général, 1200-2400 UI* de Prothromplex NF suffisent pour prévenir les saignements (p.ex. après un surdosage d'anticoagulants oraux).Hémorragies aiguës:L'expérience montre que 2400 UI* de Prothromplex NF permettent de maîtriser les hémorragies. En cas d'hémorragie sévère, l'obtention de valeurs normales du temps de Quick doit être recherchée. Si nécessaire, des doses supplémentaires de 1200-2400 UI* de Prothromplex NF peuvent être administrées toutes les 6 à 12 heures.Interventions:Avant une intervention, l'obtention d'un temps de Quick d'au moins 50% (ou un thrombotest d'au moins 35%) doit être recherchée. En général, une dose de 2400 UI* de Prothromplex NF suffit en préopératoire. Après l'intervention, le temps de Quick doit être maintenu au-dessus de 35% pendant au moins 12 à 24 heures.
·Hémorragie par déficit en vitamine K chez le nourrisson:Le dosage doit être réalisé avec prudence étant donné le risque d'activation du système de la coagulation.Dans les hémorragies par déficit en vitamine K sévères chez le nourrisson (surtout celles du SNC), dans lesquelles on ne peut attendre que la vitamine K 1 agisse, il faut par principe s'efforcer de normaliser le temps de Quick. Dans la majorité des cas, 60 UI de Prothromplex NF/kg de poids corporel permettent de maîtriser l'hémorragie aiguë en attendant que la vitamine K 1 exerce avec certitude son action. Ce n'est que dans de rares cas qu'une nouvelle administration s'avère nécessaire au bout de 8 à 12 heures, il faut s'efforcer d'obtenir un temps de Quick d'au moins 50%.* Indication valable pour un patient adulte de poids normal.
·Affection hépatique sévère, lésion hépatique sévère:En cas d'hémorragie et avant une intervention chirurgicale, le temps de Quick doit être allongé à plus de 50%. Dans la plupart des cas, une dose unique de 25-30 UI/kg de poids corporel suffit pour contrôler les hémorragies aiguës et pour prévenir les hémorragies peropératoires. Si une dose unique ne suffit pas, il faut adapter la posologie individuelle en fonction des paramètres biologiques et du tableau clinique.
·Posologie en cas de déficits congénitaux en facteurs II, VII, IX et/ou XLa formule suivante peut être utilisée pour le calcul de la dose:Pour le facteur IX (lorsqu'aucune préparation de facteur IX n'est disponible - voir «Indications»):Dose (en UI) = augmentation souhaitée du taux plasmatique (en %) × kg de poids corporel × 1,2.Pour les facteurs II et X: Dose (en UI) = augmentation souhaitée du taux plasmatique (en %) × kg de poids corporel × 0,6.Pour le facteur VII (lorsqu'aucune préparation de facteur VII n'est disponible - voir «Indications»):Dose (en UI) = augmentation souhaitée du taux plasmatique (en %) × kg de poids corporel × 0,5.
On trouvera dans le tableau suivant les valeurs indicatives du taux plasmatique requis de facteur IX dans diverses situations hémorragiques ainsi que la durée moyenne du traitement de substitution. Ces indications sont également valables pour le traitement de déficits congénitaux rares en facteurs II, VII ou X.
La fréquence des injections mentionnée ci-dessous a fait ses preuves dans le traitement du déficit en facteur IX et est également recommandée pour le traitement des déficits en facteurs II et X. Le facteur VII ayant une courte demi-vie de 6 heures au plus, il faut choisir soit des doses initiales plus élevées soit des intervalles de substitution plus rapprochés.
Une dose unique suffit généralement pour contrôler des hémorragies légères. Le cas échéant, une deuxième dose doit être administrée au bout de 24 heures.
En cas d'hémorragie sévère, le traitement de substitution doit être poursuivi jusqu'à la résorption du saignement tissulaire ou jusqu'à la cicatrisation complète. L'expérience montre qu'une dose équivalant aux deux tiers de la dose initiale, administrée toutes les 24 heures, suffit pour le traitement d'entretien.
La prophylaxie des saignements peropératoires exige l'administration de la dose préopératoire environ une heure avant le début de l'intervention. L'intervalle moyen entre les injections est habituellement de 12 heures, et 24 heures sont en général suffisantes par la suite en postopératoire.
|
Valeurs indicatives du taux plasmatique requis de facteur IX (FIX) et de la durée du traitement de substitution dans différentes situations hémorragiques. Pour le calcul de la dose, se référer à la formule citée plus haut.
| |
Situation hémorragique
|
Taux plasmatique initial requis de FIX*
|
Taux plasmatique requis de FIX pendant le traitement d'entretien*
|
Durée moyenne du traitement
| |
Hémorragies légères
|
20%
|
-
|
Dose unique normalement suffisante
| |
Hémorragies modérées
|
40%
|
20-40%
|
3-4 jours ou jusqu'à la cicatrisation complète
| |
Hémorragies menaçant le pronostic vital
ou interventions majeures
|
≥60%
|
1ère semaine postopératoire:
40-60%
2e semaine postopératoire: 20-40%
|
10-14 jours ou jusqu'à la cicatrisation complète
|
* en % de l'activité plasmatique normale du FIX
Mode d'emploi
Reconstituer le produit comme décrit au paragraphe «Instructions relatives à la reconstitution de la poudre pour solution injectable» (voir «Remarques concernant la manipulation»). Prothromplex NF doit être administré lentement par voie intraveineuse. Il est recommandé de ne pas administrer plus de 1 ml/min (25-30 UI/min).
Contre-indicationsHypersensibilité au principe actif ou à l'un des composants.
Coagulopathie intravasculaire disséminée (CIVD) et/ou hyperfibrinolyse. Allergie connue à l'héparine ou antécédents de thrombopénie induite par l'héparine.
Pour traiter des hémorragies menaçant le pronostic vital, Prothromplex NF ne doit être administré qu'après avoir stoppé ces processus par des moyens appropriés.
Mises en garde et précautionsL'avis d'un spécialiste de la prise en charge des troubles de la coagulation devrait être demandé (voir «Posologie»).
Les patients présentant un déficit congénital en facteurs II, VII, IX ou X ne doivent être traités par Prothromplex NF que lorsqu'aucun concentré de facteurs isolés n'est disponible.
Le traitement de substitution à base de concentrés de complexe prothrombique humain, y compris le traitement par Prothromplex NF, peut entraîner la formation d'anticorps circulants inhibant un ou plusieurs des facteurs du complexe prothrombique humain. La formation de ces inhibiteurs se traduit par une mauvaise réponse clinique.
Comme pour tout traitement à base de dérivés de plasma par voie intraveineuse, des réactions d'hypersensibilité (comme la fièvre, urticaire, nausée, vomissements, détresse respiratoire, pression artérielle diminuée, choc) peuvent survenir.
L'administration doit être immédiatement arrêtée en cas de survenue de réactions d'hypersensibilité. De légères réactions peuvent être maîtrisées par des antihistaminiques. Le traitement de réactions hypotensives sévères doit obéir aux règles du traitement médical de référence du choc.
Un risque élevé de coagulation intravasculaire disséminée (CIVD), de complications thromboemboliques, d'infarctus du myocarde, d'accident vasculaire cérébral (ictus) et d'embolie pulmonaire a été rapporté avec des concentrés de complexe prothrombique plasmatique humain, particulièrement en cas d'administration répétée.
Les patients recevant des concentrés de complexe prothrombique plasmatique humain doivent faire l'objet d'une surveillance attentive afin de détecter les signes évocateurs d'une coagulation intravasculaire disséminée ou d'une thrombose.
En raison du risque de complications thromboemboliques, une surveillance particulièrement étroite doit être mise en place lors de l'administration de concentrés de complexe prothrombique plasmatique humain chez les patients atteints de maladie coronarienne ou d'infarctus du myocarde, chez les patients atteints d'affections hépatiques, chez les patients en période postopératoire, chez les nouveau-nés et chez les patients présentant un risque de thrombose ou de coagulation intravasculaire disséminée. Dans chacune de ces situations, le bénéfice potentiel du traitement par un concentré de complexe prothrombique plasmatique humain doit être mis en balance avec le risque de complications de ce type.
Les patients recevant un antivitamine K peuvent présenter un état d'hypercoagulabilité sous-jacente qui peut être exacerbé par l'administration de complexe prothrombique humain.
Le risque peut être particulièrement élevé lors du traitement du déficit en facteur VII isolé, car les autres facteurs de coagulation dépendant de la vitamine K peuvent, du fait de leur demi-vie plus longue, s'accumuler avec un taux plasmatique considérablement plus élevé qu'à la normale.
La quantité de sodium pouvant dépasser 200 mg lors de l'administration de la dose journalière maximale, les personnes suivant un régime pauvre en sel peuvent subir des effets négatifs.
Prothromplex NF est produit à partir de plasma humain. Les mesures habituelles de prévention de la transmission d'agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection des donneurs, l'analyse de chaque don et des pools de plasma à la recherche de marqueurs d'infections spécifiques et l'inclusion dans le procédé de fabrication d'étapes capables d'inactiver ou d'éliminer les virus. Néanmoins, en cas d'administration de médicaments préparés à partir de sang ou de plasma humain, le risque de transmission d'agents infectieux ne peut pas être totalement exclu. Ceci s'applique également aux virus inconnus ou émergents ou à d'autres agents pathogènes.
Les mesures de sécurité prises sont considérées comme efficaces pour lutter contre les virus enveloppés, tels que le VIH, VHB et VHC, ainsi que contre les virus non enveloppés, tels que le virus de l'hépatite A (VHA).
Les mesures prises peuvent avoir une efficacité limitée contre le parvovirus B19. L'infection par le parvovirus B19 peut avoir des conséquences graves chez la femme enceinte (infection fœtale) et chez les individus atteints d'immunodéficience ou présentant une érythropoïèse augmentée (p.ex. anémie hémolytique).
Pour les patients qui reçoivent régulièrement des préparations dérivées de sang ou de plasmas humains, une vaccination contre l'hépatite A et l'hépatite B est recommandée par principe.
Ce médicament contient 68 mg de sodium par flacon, ce qui équivaut à 3,4 % de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte.
Ce médicament contient de l'héparine. L'héparine peut provoquer des réactions allergiques et une baisse de la numération sanguine pouvant influer sur la coagulation. Les patients présentant des antécédents de réactions allergiques induites par l'héparine doivent éviter l'utilisation de médicaments contenant de l'héparine.
Lors de chaque administration de Prothromplex NF à un patient, il est recommandé de documenter le nom et le numéro de lot de la préparation afin de maintenir un lien entre le patient et le lot du produit.
InteractionsL'efficacité des antagonistes de la vitamines K est réduite.
Interactions avec d'autres médicaments:
Aucune étude d'interaction n'a été réalisée.
Grossesse, allaitementIl n'existe pas de données cliniques concernant l'utilisation chez la femme enceinte.
Le médicament ne doit être utilisé pendant la grossesse et l'allaitement qu'en cas de nécessité absolue et si le bénéfice potentiel justifie le risque encouru.
Voir «Mises en garde et précautions» pour des informations concernant des infections par le parvovirus B19.
Les effets de Prothromplex NF sur la fertilité n'ont pas été établis dans les essais cliniques contrôlés.
Effet sur l’aptitude à la conduite et l’utilisation de machinesRien n'indique que Prothromplex NF pourrait avoir une influence sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirablesLes effets indésirables listés ci-dessous ont été obtenus à partir de l'expérience après la mise sur le marché ou dans une étude clinique avec Prothromplex NF pour l'inversion de l'anticoagulation orale chez les patients présentant un déficit acquis en facteurs de coagulation du complexe prothrombique (II, VII, IX, X).
La fréquence a été évaluée selon la convention suivante: «très fréquent (≥1/10); «fréquent» (≥1/100 à <1/10), «occasionnel» (≥1/1000 à <1/100), «rare» (≥1/10'000 à <1/1000), «très rare» (<1/10'000), «fréquence inconnue» (ne peut être estimée sur la base des données disponibles).
Affections hématologiques et du système lymphatique
Fréquent: coagulation intravasculaire disséminée, anticorps inhibant un ou plusieurs facteurs du complexe prothrombique (facteur II, VII, IX, X) *
* Développement chez les patients présentant un déficit congénital en facteurs
Affections du système immunitaire
Fréquent: choc anaphylactique, réaction anaphylactique, réactions d'hypersensibilité
Fréquence inconnue: angioœdème
Affection du système nerveux
Fréquent: apoplexie, céphalée
Fréquence inconnue: léthargie, paresthésie, impatiences
Affections cardiaques
Fréquent: infarctus du myocarde, tachycardie, défaillance cardiaque
Affections vasculaires
Fréquent: thrombose veineuse, évènements thromboemboliques artériels, hypotension, bouffée congestive, choc, thrombophlébite
Affections respiratoires, thoraciques et médiastinales
Fréquent: embolie pulmonaire, dyspnée, bronchospasme
Affections gastro-intestinales
Fréquent: nausées, vomissements
Affections de la peau et du tissu sous-cutané
Fréquent: urticaire, éruption érythémateuse, prurit
Affections du rein et des voies urinaires
Fréquent: syndromes néphrotiques
Troubles généraux et anomalies au site d'administration
Fréquent: fièvre, réaction au site de perfusion
Des réactions d'hypersensibilité peuvent survenir (voir «Mises en garde et précautions»).
Le traitement de substitution par Prothromplex NF peut entraîner la formation d'anticorps circulants (voir «Mises en garde et précautions»).
Lors du traitement à base de concentrés de complexe prothrombique, des événements thromboemboliques peuvent survenir, en particulier après l'administration de fortes doses et/ou chez les patients présentant un risque de thrombose.
Pour de plus amples informations concernant la sécurité virale, voir «Mises en garde et précautions».
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageL'administration de doses élevées de concentré de complexe prothrombique plasmatique humain a été associée à des cas d'infarctus du myocarde, de coagulation intravasculaire disséminée, de thrombose veineuse et d'embolie pulmonaire. Par conséquent, le risque de survenue de complications thromboemboliques ou de coagulation intravasculaire disséminée augmente en cas de surdosage chez les patients présentant des facteurs de risque de ces complications.
Propriétés/EffetsCode ATC
B02BD01
Mécanisme d'action/Pharmacodynamique
Les facteurs II, IX et X sont des composants du système intrinsèque de la coagulation et le facteur VII est un composant du système extrinsèque. Ils sont synthétisés dans le foie par une voie vitamine K-dépendante. Ensemble, ils forment le complexe prothrombique. En cas de déficit en un ou plusieurs de ces facteurs, la coagulation sanguine est perturbée et un traitement de substitution approprié est nécessaire.
Efficacité clinique
Aucune donnée.
PharmacocinétiqueLa demi-vie plasmatique du facteur II est de 40 – 60 heures, celle du facteur VII de 6 heures au plus, celle du facteur IX de 16 – 30 heures et celle du facteur X de 30 – 60 heures.
Données précliniquesLes facteurs du complexe prothrombique plasmatique humain (contenus dans le concentré) sont des composants normaux du plasma humain et se comportent comme des facteurs endogènes. On ne peut pas réaliser d'études probantes chez l'animal sur la toxicité aiguë et chronique. Les études de toxicité après une administration unique montrent seulement les effets d'un surdosage auxquels il y a lieu de s'attendre lors de l'utilisation chez l'homme et ont donc un intérêt limité.
Les études de toxicité après une administration répétée chez l'animal sont irréalisables en raison du développement d'anticorps aux protéines hétérologues, à l'origine d'interférences. Les expériences cliniques ne fournissant aucun indice d'effets cancérogènes et mutagènes des facteurs du complexe prothrombique plasmatique humain, la réalisation d'études expérimentales, chez des espèces hétérologues en particulier, n'a pas été jugée nécessaire.
Remarques particulièresIncompatibilités
Pour ne pas compromettre l'efficacité et la tolérance du produit, Prothromplex NF, comme tous les autres concentrés de facteurs, ne doit pas être mélangé avec d'autres médicaments avant l'administration et ne doit pas être dilué dans des solutions pour perfusion. Le cas échéant, il est recommandé de rincer la voie veineuse commune avec une solution saline isotonique avant et après l'utilisation de Prothromplex NF.
Influence sur les méthodes de diagnostic
En cas d'administration de fortes doses de Prothromplex NF, il y a lieu de tenir compte de l'héparine entrant dans la constitution du produit administré lors de la réalisation de tests de la coagulation sensibles à l'héparine. Le sulfate de protamine ou d'autres réactifs disponibles permettent, le cas échéant, de neutraliser in vitro l'effet de l'héparine sur les analyses de la coagulation.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2 – 8 °C). Ne pas congeler.
Conserver dans l'emballage d'origine pour le protéger de la lumière.
Tenir hors de portée des enfants.
Pendant la durée de conservation (2 – 8 °C) le produit peut être conservé à température ambiante (15–25 °C) une fois pendant 6 mois au maximum. Le cas échéant, la date de fin de cette période de 6 mois doit être indiquée comme nouvelle date de péremption sur l'emballage. Si le produit est conservé à 15 – 25 °C, il ne doit plus être placé au réfrigérateur à 2 – 8 °C mais doit être utilisé ou jeté.
Remarques concernant la manipulation
Après reconstitution, la solution prête à l'emploi ne doit pas être placée au réfrigérateur.
La stabilité chimique et physique de la solution reconstituée a été démontrée pendant 3 heures à 20-25 °C. Pour des raisons microbiologiques, Prothromplex NF doit être utilisé immédiatement après la reconstitution, sauf si la reconstitution se déroule dans des conditions aseptiques contrôlées et validées, car la préparation ne contient aucun conservateur.
Si cela n'est pas possible, le délai d'utilisation et les conditions de stockage relèvent de la responsabilité de l'utilisateur. Le produit reconstitué doit être inspecté visuellement à la présence de particules et à la turbidité avant l'administration.
Ne pas utiliser de solutions troubles ou présentant un dépôt. Eliminer dans les règles de l'art les solutions non utilisées ainsi que les déchets.
Instructions générales
·Reconstituer le médicament en utilisant exclusivement le kit de reconstitution fourni.
·Vérifier la date de péremption et s'assurer que la poudre Prothromplex NF et l'eau pour préparations injectables (solvant) sont à température ambiante avant la préparation. Ne pas utiliser après la date de péremption indiquée sur les étiquettes et l'emballage secondaire.
·Utiliser une technique aseptique (conditions de propreté et de présence microbienne réduite) et une surface de travail plane pendant la procédure de reconstitution. Se laver les mains et mettre des gants d'examen propres (le port de gants est facultatif).
·Réchauffer le flacon non ouvert contenant le solvant (eau pour préparations injectables) à température ambiante ou corporelle (maximum 37 °C).
·Prothromplex NF doit être administré immédiatement après sa reconstitution. La solution est limpide ou légèrement opalescente. La solution doit être jetée si elle est trouble ou contient un dépôt.
Instructions relatives à la reconstitution de la poudre pour solution injectable:
|
|
Étapes
|
Image
| |
1
|
·Retirer les capuchons de protection des flacons.
|
|

| |
2
|
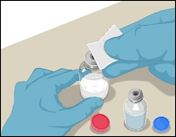
·Désinfecter chaque bouchon en le frottant avec un tampon alcoolisé stérile distinct (ou toute autre solution stérile adaptée conseillée) pendant plusieurs secondes.
·Laisser sécher le bouchon en caoutchouc. Poser les flacons sur une surface plane.
|
|
| |
3
|
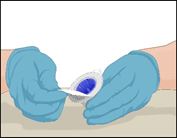
·Ouvrir l'emballage du dispositif Mix2Vial en retirant complètement l'opercule sans toucher l'intérieur de l'emballage.
·Ne pas retirer le dispositif Mix2Vial de l'emballage.
|
|
| |
4
|
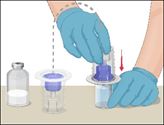
·Retourner l'emballage contenant le dispositif Mix2Vial et le placer par-dessus le flacon de solvant.
·Enfoncer fermement et bien droit le perforateur en plastique bleu du dispositif au centre du bouchon du flacon de solvant. Retirer l'emballage du dispositif Mix2Vial en le tenant par les rebords.
·Veiller à ne pas toucher le perforateur en plastique transparent.
·Le flacon de solvant est maintenant relié au dispositif Mix2Vial et il est prêt à être relié au flacon de Prothromplex NF.
|
|
| |
5
|
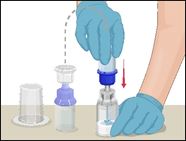
·Pour relier les deux flacons, retourner le flacon de solvant et le placer par-dessus le flacon contenant la poudre Prothromplex NF.
·Enfoncer fermement et bien droit le perforateur en plastique transparent dans le bouchon du flacon de Prothromplex NF. Cette opération doit être effectuée immédiatement, afin d'éviter toute contamination microbienne du liquide.
·Le solvant s'écoule dans le flacon de Prothromplex sous l'effet du vide. Vérifier que la totalité du solvant a été transférée.
·Ne pas utiliser si le flacon n'est plus sous vide et si le solvant ne s'écoule pas dans le flacon de Prothromplex NF.
|
|
| |
6
|
·Faire tourner délicatement les flacons reliés en un mouvement régulier ou laisser reposer le médicament reconstitué pendant 5 minutes puis le faire tourner délicatement pour dissoudre complètement la poudre.
·Ne pas agiter. Cela altèrerait le médicament. Ne pas le mettre au réfrigérateur après reconstitution.
|
|

| |
7
|
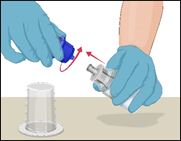
·Désolidariser les deux parties du dispositif Mix2Vial en tenant la partie en plastique transparent du dispositif Mix2Vial reliée au flacon de Prothromplex NF d'une main et la partie en plastique bleu du dispositif Mix2Vial reliée au flacon de solvant, désormais vide, de l'autre main.
·Séparer les deux flacons en dévissant la partie en plastique bleu dans le sens inverse des aiguilles d'une montre.
·Ne pas toucher l'extrémité du raccord en plastique relié au flacon contenant le Prothromplex NF dissous.
·Poser le flacon de Prothromplex NF sur une surface de travail plane. Jeter le flacon de solvant vide.
|
|
| |
8
|
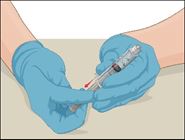
·Aspirer de l'air dans une seringue en plastique stérile à usage unique vide en tirant sur le piston.
·La quantité d'air doit être égale à la quantité de Prothromplex NF reconstitué qui sera prélevée dans le flacon.
|
|
| |
9
|
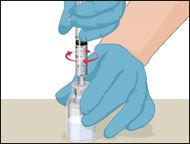
·Placer la seringue sur le raccord en plastique transparent du flacon contenant le Prothromplex NF reconstitué, resté en position verticale, et la visser dans le sens des aiguilles d'une montre.
|
|
| |
10
|
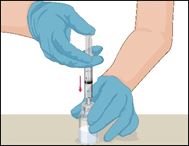
·Tenir le flacon d'une main et, de l'autre main, injecter la quantité total d'air de la seringue dans le flacon.
|
|
| |
11
|
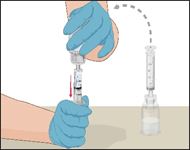
·Retourner la seringue raccordée au flacon de Prothromplex NF afin que le flacon soit désormais au-dessus. Veiller à ce que le piston de la seringue reste enfoncé. Prélever la solution Prothromplex NF dans la seringue en tirant lentement sur le piston.
·Ne pas prélever la solution dans la seringue puis la réinjecter dans le flacon car cela pourrait altérer au médicament.
|
|
| |
12
|
·Une fois prête pour la perfusion, détacher la seringue en la dévissant dans le sens inverse des aiguilles d'une montre. Inspecter visuellement la seringue pour s'assurer de l'absence de particules; la solution doit être limpide et légèrement opalescente.
·Ne pas utiliser la solution si elle est trouble ou contient un dépôt. Dans ce cas, il convient de contacter le médecin.
|
|

|
Instructions relatives à l'administration
Inspecter la solution préparée dans la seringue pour vérifier l'absence de particules et de décoloration avant l'administration (la solution doit être limpide, incolore et exempte de particules). Il n'est pas rare qu'il reste quelques flocons dans le flacon contenant le produit après la reconstitution. Le filtre inclus dans le dispositif Mix2Vial élimine totalement ces particules. La filtration n'influence pas les calculs de la dose à administrer. La solution présente dans la seringue ne doit pas être utilisée si elle est trouble ou contient des flocons ou des particules après la filtration.
1.Fixer une aiguille sur une seringue contenant la solution de Prothromplex NF. Pour plus de confort, il est recommandé d'utiliser un dispositif de perfusion à ailettes (papillon). Diriger l'aiguille vers le haut et chasser les bulles d'air en tapotant doucement sur la seringue avec les doigts et en faisant lentement et délicatement sortir l'air de la seringue et de l'aiguille.
2.Poser un garrot sur le bras et préparer le site de perfusion en frottant bien la peau avec un tampon alcoolisé stérile (ou toute autre solution stérile adaptée).
3.Insérer l'aiguille dans la veine et retirer le garrot. Perfuser lentement Prothromplex NF, sans dépasser un débit de 1 ml par minute. Détacher la seringue vide.Remarque: Ne pas retirer l'aiguille à ailettes tant que toutes les injections n'ont pas été réalisées et ne pas toucher le raccord entre l'aiguille et la seringue.
4.Retirer l'aiguille de la veine et utiliser un tampon stérile pour exercer une pression sur le site de perfusion pendant plusieurs minutes.
Ne pas replacer le capuchon sur l'aiguille. Placer l'aiguille, la seringue et tous les flacons vides dans un conteneur pour objets tranchants pour une élimination appropriée. Ne pas les jeter dans les ordures ménagères ordinaires.
Numéro d’autorisation41330 (Swissmedic)
PrésentationChaque emballage de Prothromplex NF 500 contient: 1 flacon de poudre pour solution injectable (pour administration intraveineuse), 1 flacon de solvant (eau pour préparations injectables Ph. Eur.) pour dissolution et 1 dispositif Mix2Vial.
Prothromplex NF 500:
Poudre contenant 375 – 708 UI de facteur II, 417 UI de facteur VII, 500 UI de facteur IX et 500 UI de facteur X et 17 ml d'eau pour préparations injectables
Catégorie de remise: B
Titulaire de l’autorisationTakeda Pharma AG, 8152 Opfikon
Mise à jour de l’informationSeptembre 2023
|