CompositionPrincipe actif: antithrombine III, préparée à partir de plasma humain.
Excipients: glycine, chlorure de sodium, citrate de sodium.
Forme galénique et quantité de principe actif par unitéSubstance lyophilisée et solution à diluer pour injection/perfusion iv.
·Flacon avec substance lyophilisée (lyophilisat blanc): 500 UI ou 1000 UI d'antithrombine III
·Flacon avec solution à diluer: 10 ml ou 20 ml d'eau pour injection
Après la reconstitution avec 10/20 ml, Kybernin P contient 50 UI d'antithrombine III par ml.
L'activité (UI) est définie d'après le dosage chromogène de la pharmacopée européenne. L'activitié spécifique de Kybernin P est d'environ 5 UI/mg de protéine.
Excipient particulier dont l'efficacité est connue:
Un flacon de Kybernin P 500 contient jusqu'à 44,76 mg de sodium, ce qui correspond à 2% de la dose journalière maximale recommandée de sodium pour un adulte.
Un flacon de Kybernin P 1000 contient jusqu'à 89,52 mg de sodium, ce qui correspond à 4,5% de la dose journalière maximale recommandée de sodium pour un adulte.
Indications/Possibilités d’emploiTraitement de substitution chez les patients souffrant d'une déficience héréditaire en Antithrombine III:
·prophylaxie antithrombotique dans les situations où l'administration d'anticoagulants oraux ou d'héparine est contre-indiquée, particulièrement en période périopératoire et péripartale;
·traitement antithrombotique en cas de thromboses étendues, en association à l'héparine, ainsi qu'en cas de coagulopathie de consommation.
Posologie/Mode d’emploiLa solution reconstituée d'antithrombine III est injectée ou perfusée lentement.
En cas de déficience congénitale, la posologie doit être adaptée individuellement à chaque patient. C'est pourquoi, il convient de tenir compte des antécédents familiaux relatifs aux événements thromboemboliques, des facteurs de risque clinique réels et des examens biologiques.
Posologie usuelle
En cas de déficience héréditaire, la posologie et la durée du traitement de substitution dépendent du taux plasmatique de l'antithrombine, des symptômes d'une utilisation accrue, de la maladie sous-jacente et de la sévérité des symptômes cliniques. La dose et la fréquence de l'administration doivent toujours être adaptées individuellement, en fonction de l'efficacité clinique et des résultats de laboratoire.
Le nombre d'unités d'antithrombine administrées est exprimé en unités internationales (UI), conformément à la norme OMS actuelle applicable aux produits à base d'antithrombine. L'activité de l'antithrombine dans le plasma est exprimée soit en pourcentage (par rapport au plasma humain normal) soit en unités internationales (conformément à la norme internationale de l'antithrombine dans le plasma).
Une unité internationale (UI) de l'activité de l'antithrombine correspond à la teneur en antithrombine dans 1 ml de plasma normal humain. Le calcul de la posologie requise est basé sur le résultat empirique suivant lequel 1 UI d'antithrombine/kg de poids corporel augmente l'activité de l'antithrombine dans le plasma d'environ 1,5%.
La dose initiale est calculée selon la formule suivante:
Dose nécessaire = poids corporel [kg] × (100 – l'activité d'Antithrombine actuelle [%] × 2/3.
La valeur cible initiale de l'activité de l'antithrombine dépend de l'état clinique. Pour la substitution de l'antithrombine, la posologie doit être choisie de manière à atteindre et maintenir la concentration d'antithrombine souhaitée. Elle doit être surveillée en fonction des valeurs biologiques de l'activité de l'antithrombine. Ces déterminations de l'antithrombine doivent être faites au minimum deux fois par jour jusqu'à la stabilisation du patient, puis avec un suivi d'une fois par jour et en particulier, immédiatement avant la prochaine administration.
Lors de l'ajustement posologique, il convient aussi bien de tenir compte des résultats biologiques indiquant potentiellement une consommation accrue d'antithrombine ainsi que les paramètres cliniques. L'activité de l'antithrombine doit être d'au moins 80% de la norme pendant la durée du traitement, à moins que la situation clinique indique un niveau efficace différent.
La dose initiale habituelle en cas de déficience congénitale est de 30 à 50 UI/kg.
Par la suite, la posologie ainsi que la fréquence et la durée du traitement doivent être adaptées aux données biologiques et à la situation clinique.
Enfants et adolescents
40-60 UI d'antithrombine/kg de poids corporel/jour dépendant du statut de coagulation. Une plus haute dose peut s'avérer nécessaire dans des cas isolés et en fonction du tableau clinique. L'activité de l'antithrombine doit alors être souvent contrôlée et elle ne doit pas dépasser le taux de 120%.
Les expériences d'études cliniques montrent que l'utilisation de l'antithrombine n'est pas indiquée pour la traitement de l'IRDS (Infant Respiratory Distress Syndrome) chez les prématurés (voir «Mises en garde et précautions»).
Mode d'emploi
La solution sera injectée par voie i.v. ou lentement perfusée (max. 4 ml/min).
Voir indications pour la dissolution au chapitre «Remarques particulières» sous section «Remarques concernant la manipulation».
Contre-indicationsHypersensibilité à la substance active ou à l'un des excipients.
Mises en garde et précautionsLes expériences d'études cliniques et les évaluations systématiques de l'utilisation de l'antithrombine III pour le traitement des prématurés dans l'indication non homologuée de l'IRDS (Infant Respiratory Distress Syndrome) indiquent un risque accru d'hémorragies intracrâniennes et de mortalité, en l'absence d'un effet bénéfique démontré dans cette population de patients.
Le traitement des patients ayant une déficience héréditaire en antithrombine, ainsi que de leurs descendants, doit se faire individuellement, selon le besoin de chaque patient. Il doit être effectué par un spécialiste de la coagulation.
Le traitement de substitution est contrôlé par un test de fonction. Chez les patients ayant une consommation aiguë, la demi-vie de l'antithrombine peut être réduite à quelques heures. Chez ces patients, il devient alors nécessaire de déterminer plusieurs fois par jour l'activité de l'antithrombine. La détermination avec un substrat chromogène est la plus adéquate.
Des réactions d'hypersensibilité sont possibles en raison de la voie d'administration intraveineuse de la glycoprotéine. Il convient de suivre étroitement et soigneusement les patients pendant la perfusion afin de détecter d'éventuels symptômes. Il convient d'informer les patients des signes précoces de réactions d'hypersensibilité telles qu'éruption cutanée, urticaire généralisé, sentiment d'oppression thoracique, stridor, hypotension et anaphylaxie. En cas de survenue de ces syptômes après l'administration, les patients doivent immédiatement contacter leur médecin. Les directives médicales actuelles pour traiter le choc doivent être suivies.
Il est conseillé d'informer les patients atteints d'une déficience héréditaire en anthithrombine sur le caractère héréditaire de cette maladie ainsi que du risque de thrombose pendant la grossesse.
Sécurité virale
Lorsque des médicaments préparés à partir de sang ou de plasma humains sont utilisés, les maladies infectieuses causées par la transmission d'agents infectieux ne peuvent pas être totalement exclues. Ceci s'applique également aux agents pathogènes inconnus jusqu'à présent.
Les mesures habituelles de prévention du risque de transmission d'agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection clinique des donneurs, la recherche des marqueurs spécifiques d'infection (HbsAg et anticorps dirigés contre le VIH et le VHC) sur chaque don et sur les pools de plasma et l'inclusion dans le procédé de fabrication d'étapes efficaces et validées au moyen de virus enveloppés pour l'inactivation/l'élimination virale. Les mesures mises en place sont considérées comme efficaces vis-à-vis des virus enveloppés comme par exemple le virus de l'immunodéficience humaine (VIH), le virus de l'hépatite B (VHB) et le virus de l'hépatite C (VHC) ainsi que vis-à-vis des virus non enveloppés tels que le virus de l'hépatite (VHA) et le parvovirus B19.
Chez les patients présentant une déficience congénitale qui reçoivent régulièrement un concentré d'antithrombine préparé à partir de plasma humain, il convient d'envisager une vaccination contre l'hépatite A et B. Il est fortement recommandé d'enregistrer le nom de la préparation et le numéro du lot lors de chaque administration de Kybernin P à un patient, pour établir un rapport entre le patient et le numéro du lot de la préparation.
Surveillance clinique et biologique en cas d'utilisation concomitante d'antithrombine et d'héparine
Pour ajuster la posologie de l'héparine et éviter une hypocoagulabilité excessive, des contrôles de l'étendue de l'anti-coagulation (TCA et le cas échéant activité anti-facteur xa) doivent être réalisés régulièrement, à intervalles rapprochés et en particulier dans les premières minutes et heures qui suivent le début de l'administration de l'antithrombine III.
Un traitement prolongé par l'héparine non fractionnée induisant un risque de diminution des taux d'antithrombine III, une mesure quotidienne de la concentration de l'antithrombine III devra être effectué dans ces circonstances et la dose individuelle adaptée si besoin.
InteractionsHéparine: le remplacement de l'antithrombine pendant l'administration d'héparine en doses thérapeutiques augmente le risque d'hémorragie. Par l'apport simultané d'héparine, l'effet inhibiteur de coagulation de l'antithrombine est considérablement augmenté. Cela a pour conséquence de diminuer fortement la demi-vie de l'antithrombine en raison de la potentialisation de l'activité de l'antithrombine. C'est pourquoi l'administration simultanée d'héparine avec de l'antithrombine aux patients présentant un risque accru d'hémorragie doit faire l'objet d'une surveillance clinique et biologique.
Grossesse/AllaitementLes données relatives à la sécurité des produits contenant de l'antithrombine pendant la grossesse sont limitées.
Aucune étude clinique n'a été menée à ce jour quant à la sécurité de Kybernin P pendant la grossesse et l'allaitement. Les examens effectués chez l'animal sont jusqu'ici peu significatifs et ne permettent pas de juger des effets sur l'aptitude de reproduction, sur le développement de l'embryon ou du fœtus, sur le cours de la grossesse ainsi que sur le développement pré- ou postnatal. Aucun effet négatif n'a été reporté jusqu'à présent lors de traitement pendant la grossesse ou l'allaitement. Kybernin P peut ainsi être administré aux femmes enceintes ou allaitant présentant une déficience en antithrombine en cas d'indication majeure uniquement en tenant compte du risque augmenté de thromboses.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune restriction sur l'aptitude à la conduite ou à l'utilisation de machines n'est connue à ce jour.
Effets indésirablesHypersensibilité ou réactions allergiques (y compris angioedème, brûlures et piqûres au point de perfusion, frissons, rougeur de la peau, urticaire généralisé, céphalées, éruption cutanée de type urticaire, hypotension, léthargie, nausées, agitation, tachycardie, sentiment d'oppression thoracique, fourmillements, vomissements, éternuements) sont rarement observés. Dans des cas isolés, ils peuvent conduire à une anaphylaxie sévère y compris un choc. Une fièvre est rarement observée.
Les effets indésirables suivants sont basés sur l'expérience d'administration après de la mise sur le marché. Sur base des résultats disponibles les fréquences ont été évaluées selon les critères suivants:
Très fréquent: ≥1/10
Fréquent: ≥1/100 à <1/10
Peu fréquent: ≥1/1000 à <1/100
Rare: ≥1/10'000 à <1/1000
Très rare: ≥1/10'000
|
Classe MedDra de systèmes d'organes
|
Effet indésirable
|
Fréquence
| |
Affections du système immunitaire
|
Hypersensibilité/réactions anaphylactiques (y compris anaphylaxie sévère et choc)
|
Rare
| |
Troubles généraux et anomalies au site d'administration
|
Fièvre
|
Rare
|
Information sur la sécurité virale: voir également le chapitre «Mises en garde et précautions».
SurdosageAucun symptôme dû à un surdosage n'a été observé jusqu'à présent.
Propriétés/EffetsCode ATC: B01AB02
Kybernin P est produit à partir de pools de plasma humain de donneurs sains. L'information sur le dépistage du plasma et des lots de plasma se trouve au chapitre «Mises en garde et précautions».
Le procédé de fabrication de Kybernin P comporte plusieurs étapes contribuant à éliminer ou inactiver les virus. Pour l'inactivation virale, un traitement de la préparation par la chaleur en solution aqueuse pendant 10 heures à 60 °C a été mis en place.
Mécanisme d'action:
L'antithrombine, une glycoprotéine composée de 432 acides aminés dont le poids moléculaire est de 58 kD, appartient à la famille des serpines (inhibiteur de la protéase sérine). C'est l'un des inhibiteurs physiologiques les plus importants de la coagulation sanguine. Les facteurs les plus fortement inhibés sont la thrombine et le facteur Xa, mais aussi les facteurs d'activation de contact, du système intrinsèque, et du complexe facteur VIIa/facteur tissulaire. L'activité de l'antithrombine est largement potentialisée par l'héparine et les effets anticoagulants de l'héparine dépendent de la présence de l'antithrombine.
L'antithrombine contient deux domaines fonctionnellement importants.
Le premier renferme le centre réactif qui constitue le site de clivage pour les protéinases telles que la thrombine, condition préalable pour la formation d'un complexe inhibiteur de protéinase stable. Le second est un domaine de liaison aux glycosaminoglycanes responsable de l'interaction avec l'héparine et les substances apparentées, laquelle accélère l'inhibition de la thrombine.
Les complexes antithrombine et facteurs de coagulation sont dégradés par le système réticulo-endothélial. L'activité normale de l'antithrombine est de 80 à 120% chez l'adulte. Chez les nouveau-nés, le taux d'activité est de 40 à 60%.
PharmacocinétiqueKybernin P est appliqué par voie intraveineuse et est immédiatement disponible à une concentration plasmatique correspondant à la posologie. Il se lie à la thrombine, au facteur Xa et à d'autres facteurs activés de la coagulation.
La demi-vie biologique de l'antithrombine est de 2,5 jours, mais peut être réduite à quelques heures lors d'une consommation aiguë. Chez ces patients, il est conseillé de mesurer l'activité de l'antithrombine plusieurs fois par jour. Dans ce but, la méthode d'analyse par un substrat chromogène est appropriée. La demivie est également diminuée lors d'un traitement simultané à l'héparine (voir chap. «Interactions»). Kybernin P a un comportement comparable à l'inhibiteur AT III endogène en ce qui concerne la distribution et le métabolisme dans l'organisme.
Le taux de récupération in vivo était en moyenne de 65% chez 5 volontaires sains (mesuré à tmax = 1.15 heure).
Données précliniquesL'antithrombine humaine est un constituant physiologique du corps humain.
Des études de toxicité de la dose unique ne sont pas significatives et ne permettent pas déterminer la dose toxique ou létale. Des études dans le modèle animal n'ont démontré aucune toxicité aiguë.
Des études de toxicité après l'apport de doses répétées dans le modèle animal ne sont pas réalisables parce que les animaux développent des anticorps contre les protéines hétérologues (humaines).
Jusqu'à présent, l'expérience clinique n'a rapporté aucun indice d'effets de toxicité embryo-fœtale, ni d'effets oncogéniques ou mutagènes.
Remarques particulièresIncompatibilités
L'utilisation d'une solution d'hydroxyéthylamidon (HEA) comme diluant (pour perfusion) n'est pas à recommander, car on a pu observer une perte de l'activité de l'antithrombine.
Kybernin P ne doit pas être mélangé à d'autres médicaments dans la seringue ou dans le matériel de perfusion. La dopamine, la dobutamine et le furosémide ne doivent pas être administrés simultanément dans la même veine. Pour des indications sur la dissolution, voir sous «Remarques concernant la manipulation».
Stabilité
Kybernin P ne peut être utilisé au-delà de la date de péremption indiquée sur l'emballage et le récipient.
Remarques concernant le stockage
Kybernin P ne doit pas être conservé au-delà de +25 °C. Tenir à l'abri de la lumière. Ne pas surgeler! Tenir les médicaments hors de portée des enfants.
Une stabilité physico-chimique de 8 heures est justifiée après reconstitution à température ambiante (au maximum +25 °C). Du point de vue microbiologique et parce que Kybernin P ne contient aucun agent conservateur, il est impératif d'utiliser immédiatement le produit reconstitué. Ne pas le conserver au-delà de 8 heures à une température ambiante de +25 °C s'il n'est pas utilisé immédiatement.
Utiliser le contenu immédiatement après ouverture du récipient.
Remarques pour la manipulation
La substance lyophilisée doit être complètement dissoute, dans des conditions aseptiques, dans 10 ou 20 ml d'eau pour injection. On obtiendra une solution limpide à légèrement opalescente. Dans le cas de perfusion, une solution à 5% d'albumine humaine convient également comme solution de dilution du lyophilisat. Pour des dilutions jusqu'à 1:5, les solutions suivantes de perfusion peuvent être employées: une solution de lactate de Ringer, une solution physiologique de NaCl, une solution de glucose à 5% ou une solution de polygéline.
Le produit reconstitué doit être inspecté visuellement avant l'administration afin de détecter toute particule ou décoloration. La solution doit être transparente ou légèrement opalescente. Des solutions troubles ou des solutions contenant un sédiment ou des particules ne doivent pas être utilisées.
Préparation
Pour garantir la manipulation correcte du kit de transfert pour liquides stériles Transofix®, veuillez procéder comme suit:
|
|
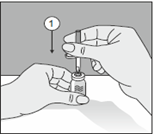
|
1. Après avoir retiré l'un des deux capuchons de protection, planter le perforateur mis à nu à la verticale à travers le bouchon en caoutchouc du flacon.
| |
|
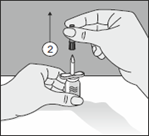
|
2. Retirer le capuchon de protection du second perforateur.
| |
|
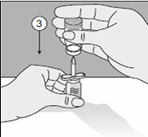
|
3. Enfoncer tête en bas le flacon contenant le produit sur ce même perforateur.
| |
|

|
4. Renverser le flacon à 180°.
| |
|
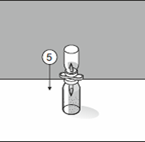
|
5. Placer le flacon contenant le produit sur une surface plane. La solution passe à présent dans le flacon qui contient le produit.
| |
|

|
6. Retirer du flacon contenant le produit le dispositif de transfert pour liquides stériles Transofix® avec le flacon de solution. Kybernin en poudre se dissout. Kybernin reconstitué peut maintenant être prélevé dans une seringue et administré.
|
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Numéro d’autorisation46928 (Swissmedic).
PrésentationKybernin P 500 [B]
1 flacon de substance lyophilisée à 500 UI,
1 flacon de 10 ml d'aqua ad iniect.,
1 set de transfusion. B
Kybernin P 1000 [B]
1 flacon de substance lyophilisée à 1000 UI,
1 flacon de 20 ml d'aqua ad iniect.,
1 set de transfusion. B
Titulaire de l’autorisationCSL Behring AG, Bern.
Mise à jour de l’informationJuillet 2017.
|