CompositionPrincipes actifs
Gadoteridolum
Excipients
Calteridolum calcicum, Trometamolum, Acidum hydrochloridum / Natrii hydroxidum (ad pH), Aqua ad iniectabile q.s. ad solutionem pro 1 ml.
Teneur en sodium: Max. 0.0575 mg/ml en tenant compte des ajouts variables d’hydroxyde de sodium.
Indications/Possibilités d’emploiEmployé en imagerie par résonance magnétique (IRM), ProHance permet le rehaussement du contraste du cerveau, du rachis et des tissus adjacents, avec une meilleure visualisation de lésions accompagnées d’anomalies vasculaires ou de lésions supposées être dues à une rupture de la barrière hématoencéphalique.
ProHance est aussi utilisée chez les adultes de 18 ans et plus lors de l’IRM corporelle, par exemple dans les maladies de la tête, du cou, du foie, du sein, du squelette, des muscles et des parties molles.
ProHance ne doit être utilisé que lorsque le diagnostic est nécessaire et que ce diagnostic ne peut pas être obtenu par imagerie par résonance magnétique (IRM) sans rehaussement de contraste.
Posologie/Mode d’emploiPosologie usuelle
La dose la plus faible permettant un rehaussement de contraste suffisant à des fins diagnostiques doit être utilisée. La dose doit être calculée en fonction de la masse corporelle du patient et ne doit pas dépasser la dose recommandée par kilogramme de masse corporelle, détaillée dans cette rubrique.
La dose recommandée chez les enfants et les adolescents (renforcement de contraste du cerveau, de la colonne et des tissus avoisinants), et chez les adultes est de 0,1 mmol/kg de la solution 0,5 M (0,2 mL/kg).
Dans quelques cas exceptionnels, comme la confirmation du caractère unique d'une métastase ou la détection de tumeurs leptoméningées, une deuxième injection de 0.2 mmol/kg peut être administrée.
Instructions posologiques particulières
Patients présentant des troubles de la fonction rénale
ProHance ne doit être administré aux patients présentant une insuffisance rénale sévère (clairance de la créatinine < 30 ml/min./1.73 m²) et aux patients durant la période pré-opératoire ou post-opératoire d’une transplantation hépatique qu’après une évaluation minutieuse du rapport bénéfice/risque et que si le diagnostic est nécessaire et ne peut être obtenu par IRM sans produit de contraste (voir «Mises en garde et précautions»). Si l’administration de ProHance est nécessaire, la dose ne doit pas dépasser 0.2 ml/kg (0.1 mmol/kg) de poids corporel et au cours d’un examen, il convient de ne pas administrer plus d’une dose. Etant donné qu’on ne dispose pas d’informations sur l’administration itérative, l’injection de ProHance ne doit pas être répétée à moins que l’intervalle entre les injections soit d’au moins de 7 jours.
Patients âgés
Aucune adaptation posologique n’est nécessaire. Malgré tout, utiliser avec prudence chez les patients à partir de 65 ans (voir «Mises en garde et précautions»).
Nouveau-nés jusqu'à l'âge de 4 semaines et nourrissons jusqu'à l'âge d'un an
En raison de l’immaturité de la fonction rénale chez le nouveau-né jusqu'à l'âge de 4 semaines et chez le nourrisson jusqu’à l’âge d’un an, ProHance ne doit être administré à ces patients qu’après un examen approfondi de la situation et à une dose n’excédant pas 0.2 ml/kg (0.1 mmol/kg) de poids corporel. Au cours d’un examen, ne pas administrer plus d’une dose. Etant donné qu’on ne dispose pas d’informations sur l’administration itérative, l’injection de ProHance ne doit pas être répétée à moins que l’intervalle entre les injections soit d’au moins de 7 jours.
Tout examen du corps entier est déconseillé chez l’enfant et l’adolescent de moins de 18 ans.
Mode d’administration
Le patient restera à jeun les deux heures précédant l’examen.
Pour assurer une injection complète du produit de contraste, rincer après la canule après l’injection avec 5 ml de solution physiologique. L’IRM à contraste rehaussé devrait être achevée dans l’heure suivant l’injection de ProHance.
Avant l’administration, les préparations parentérales doivent faire l’objet d’un contrôle visuel de la limpidité et de la coloration dans Ia mesure où la solution et le contenant le permettent. On détruira les restes de produit de contraste non utilisés.
Contre-indicationsSujets porteurs d’un stimulateur cardiaque ou d’un clip vasculaire.
Hypersensibilité au principe actif ou à l’un des excipients conformément à la composition.
Mises en garde et précautionsLes règles et principes généraux de sécurité usuels en IRM valent également pour l’utilisation du produit de contraste ProHance. Les méthodes diagnostiques mises en œuvre sous produit de contraste doivent l’être sous la direction d’un médecin ayant subi une formation spéciale et parfaitement au courant des procédures à suivre.
Les patients présentant une allergie connue aux médicaments ou d’autres réactions d’hypersensibilité doivent être observés étroitement pendant le traitement ainsi que l’administration du produit de contraste et même au-delà, si le médecin le juge nécessaire compte tenu du patient.
Comme avec les autres produits de contraste contenant du gadolinium, des réactions d’hypersensibilité anaphylactiques/anaphylactoïdes ont été également rapportées avec ProHance. Ces réactions présentaient des degrés de gravité distincts, y compris choc anaphylactique et décès. Elles concernaient un ou plusieurs systèmes ou organes, notamment le système respiratoire, cardiovasculaire et mucocutané. Symptômes fréquemment observés: sécheresse de la gorge, irritation de la gorge, dyspnée, sensation d’oppression thoracique, sensation de chaleur, dysphagie, brûlure, œdème pharyngé ou laryngé, hypotension.
Les cas de choc anaphylactique rapportés lors de l’usage de ProHance sont très rares. Les médicaments et appareils nécessaires doivent être disponibles pour le cas où des réactions graves se produiraient.
Comme avec les autres produits de contraste contenant du gadolinium, des mesures de précaution particulières doivent être prises chez les patients dont le seuil épileptogène est abaissé. Elles incluent une surveillance étroite de ces patients et la disponibilité et la proximité de tout matériel et médicament nécessaires pour un traitement immédiat des convulsions éventuelles.
Risques associés à l'utilisation intrathécale
Le gadotéridol ne doit pas être administré par voie intrathécale. Des réactions graves, pouvant engager le pronostic vital et ayant entraîné la mort dans certains cas, principalement des réactions neurologiques (p. ex., coma, encéphalopathie, convulsions), ont été signalées en cas d’administration intrathécale.
Patients présentant des troubles de la fonction rénale
Avant l’administration de ProHance, il est recommandé de vérifier la présence d’une insuffisance rénale chez tous les patients par des analyses de laboratoire.
Le gadotéridol étant éliminé par filtration glomérulaire, on usera de prudence en cas d’insuffisance rénale sévère (voir aussi «Pharmacocinétique» et «Instructions posologiques particulières»).
Des cas de fibrose néphrogénique systémique (FNS) ont été rapportés après utilisation de certains produits de contraste contenant du gadolinium chez des patients ayant une insuffisance rénale sévère aiguë ou chronique (clairance de la créatinine <30 ml/min./1,73 m²). Les patients devant bénéficier d’une transplantation hépatique sont particulièrement à risque, étant donné que l’incidence de l’insuffisance rénale aiguë est élevée dans ce groupe. Etant donné qu’il est possible que des cas de FNS surviennent avec ProHance, ce produit ne doit être administré aux patients présentant une insuffisance rénale sévère et aux patients durant la période pré-opératoire ou post-opératoire d’une transplantation hépatique qu’après une évaluation minutieuse du rapport bénéfice/risque et que si le diagnostic est nécessaire et ne peut être obtenu par IRM sans produit de contraste.
La réalisation d’une hémodialyse peu de temps après l’administration de ProHance pourrait faciliter l’élimination de ce produit de l’organisme. Il n’est pas établi que l’instauration d’une hémodialyse puisse prévenir ou traiter la FNS chez les patients qui ne sont pas déjà hémodialysés (cf. «Pharmacocinétique: Cinétique pour certains groupes de patients»).
Nouveau-nés et nourrissons
En raison de l’immaturité de la fonction rénale des nouveau-nés jusquʼà lʼâge de 4 semaines et des nourrissons jusqu’à l’âge d’un an, ProHance ne doit être administré à ces patients qu’après un examen approfondi de la situation.
Des données pharmacocinétiques sur l’exposition au gadotéridol sont disponibles chez les enfants à partir de l’âge de 5 ans.
Patients âgés
Etant donné que la clairance rénale du gadotéridol peut être diminuée chez le sujet âgé, il est particulièrement important d’examiner les patients de plus de 65 ans concernant une éventuelle insuffisance rénale.
Lorsque ProHance n’est pas injecté au moyen d’une seringue à usage unique, on veillera très scrupuleusement à ne pas contaminer la seringue avec des résidus de détergent.
En cas d’examens séquentiels ou itératifs prévus suite à l’examen clinique du médecin, on ménagera des intervalles de temps suffisants entre les injections de manière à ce que la substance puisse être éliminée normalement.
L’utilisation de produits de contraste peut entraîner des réactions anaphylactoïdes menaçant en partie le pronostic vital, de type cardiovasculaire (choc) ou respiratoire (œdème laryngé, bronchospasme), ainsi que des symptômes abdominaux, de l’urticaire, un œdème de Quincke ou des complications neurologiques.
À chaque examen, il convient donc de disposer du personnel nécessaire pour une réanimation cardiopulmonaire d’urgence ainsi que des moyens correspondants (oxygène, adrénaline, matériel de perfusion, possibilités d’intubation et de ventilation, entre autres).
Il est indispensable d’être parfaitement au courant des mesures d’urgence.
Après administration du produit de contraste, le patient devra rester sous surveillance pendant au moins 30 à 60 minutes, puisque l’expérience montre que la plupart des accidents graves interviennent durant cette période.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c’est-à-dire qu’il est pratiquement «sans sodium».
InteractionsJusqu’à présent, aucune interaction médicamenteuse avec le gadotéridol n’a été signalée.
Ni les essais cliniques ni les examens biologiques n’ont fourni d’indications significatives quant à de possibles interactions.
Grossesse, AllaitementGrossesse
Les produits de contraste contenant du gadolinium peuvent traverser la barrière placentaire et conduire à une exposition fœtale. On ne dispose pas de données suffisantes chez l’être humain permettant une évaluation définitive du lien entre les produits de contraste contenant du gadolinium et les conséquences délétères pour le fœtus.
Il n’existe pas d’expériences sur l’administration de ProHance chez la femme enceinte.
Les études effectuées chez l’animal n’ont pas mis en évidence d’effets délétères directs ou indirects sur la reproduction (voir «Données précliniques»). Le gadotéridol ne doit pas être utilisé pendant la grossesse, à moins que la situation clinique de la patiente ne nécessite son administration.
Allaitement
Une très faible excrétion des produits de contraste contenant du gadolinium s’effectue dans le lait maternel (voir «Données précliniques»). À des doses cliniques, en raison de la faible quantité éliminée dans le lait et de la faible résorption dans le tractus intestinal, aucun effet n’est prévu chez le nourrisson. Le médecin et la mère allaitante doivent décider s’il faut poursuivre l’allaitement ou le suspendre pendant les 24 heures suivant l’administration de ProHance.
Effet sur l’aptitude à la conduite et l’utilisation de machinesCompte tenu du profil pharmacocinétique et pharmadynamique, aucun effet sur l’aptitude à la conduite et à l’utilisation de machines n’a été constaté du fait de l’emploi du gadotéridol, ou un effet négligeable.
Effets indésirablesLes effets indésirables suivants ont été rapportés suite à l’administration de ProHance. Les effets indésirables déclarés spontanément sont listés avec la fréquence «inconnu».
Les fréquences sont indiquées comme suit: «très fréquent» (≥1/10); «fréquent» (≥1/100, <1/10); «occasionnel» (≥1/1000, <1/100); «rare» (≥1/10 000, <1/1000); «très rare» (<1/10 000), «inconnu» (fréquence non estimable sur la base des données disponibles).
Affections du système immunitaire
Occasionnel: réactions anaphylactiques/anaphylactoïdes.
Inconnu: angiooedema
Affections psychiatriques
Rare: anxiété.
Affections du système nerveux
Occasionnel: céphalées, paresthésie, vertiges, modifications du goût.
Rare: baisse des performances intellectuelles, troubles de la coordination, convulsions.
Inconnu: perte de connaissance, coma, réaction vasovagale.
Affections oculaires
Occasionnel: augmentation du flux lacrymal.
Affections de l’oreille et du labyrinthe
Rare: acouphènes.
Affections cardiaques
Rare: intervalle P-R prolongé, fréquence cardiaque élevée, troubles de la conduction AV, arrêt cardiaque, bradycardie
Inconnu: arrêt cardiaque.
Affections vasculaires
Occasionnel: rougeur cutanée, hypotension, hypertension.
Affections respiratoires, thoraciques et médiastinales
Rare: spasme du larynx, dyspnée, rhinite, toux, apnée, respiration sifflante.
Inconnu: arrêt respiratoire, œdème pulmonaire, syndrome de détresse respiratoire aiguë.
Affections gastro-intestinales
Fréquent: nausées.
Occasionnel: xérostomie, vomissements.
Rare: diarrhée, douleurs abdominales, œdème de la langue, prurit oral, gingivite, selles molles, dysphagie, augmentation de la salivation.
Affections de la peau et du tissu sous-cutané
Occasionnel: prurit, éruption cutanée, urticaire.
Rare: œdème facial.
Affections musculo-squelettiques et systémiques
Rare: rigidité.
Affections du rein et des voies urinaires
Inconnu: défaillance rénale aiguë.
Troubles généraux et anomalies au site d’administration
Occasionnel: douleurs au point d’injection, réaction au site de l’injection (notamment due à l’extravasation du produit de contraste) asthénie.
Rare: douleurs thoraciques, pyrexie.
Examens
Occasionnel: augmentation du rythme cardiaque.
Description de certains effets indésirables
Réactions vasovagales
Des réactions vasovagales entraînant, dans des cas rares, une syncope vasovagale, ont été rapportées pendant ou immédiatement après l’administration de gadotéridol. Cet état est souvent lié à des sentiments d’anxiété ou à des stimuli douloureux ou désagréables (p. ex. piqûre d’aiguille pour mise en place IV). D’autres symptômes fréquents sont les nausées, les vertiges et la diaphorèse.
Dans les cas graves, des syncopes peuvent se produire, les patients sont alors habituellement pâles et diaphorétiques avec conscience modifiée et présentent une bradycardie. De plus, les patients peuvent fréquemment être sujets à des pressentiments désagréables, à de l’agitation, à de la faiblesse et à un renforcement de la salivation. Une identification claire de cette réaction et un diagnostic différentiel par rapport aux réactions d’hypersensibilité ou anaphylactiques sont vitaux afin que les mesures de traitement adaptées puissent être appliquées afin de renverser le stimulus vagal.
Réactions anaphylactiques/anaphylactoïdes
Comme avec d’autres produits de contraste contenant du gadolinium , des réactions d’hypersensibilité anaphylactiques/anaphylactoïdes ont été rapportées avec le gadotéridol. Ces réactions présentaient des degrés de gravité distincts, y compris choc anaphylactique et décès. Elles concernent un ou plusieurs systèmes ou organes, dans la plupart des cas le système respiratoire, cardiovasculaire et/ou mucocutané. Les symptômes fréquemment rapportés incluent: sensation de resserrement dans la gorge, irritation de la gorge, dyspnée, troubles thoraciques, sensation de chaleur, dysphagie, brûlure, œdème pharyngé ou laryngé, hypotension.
Défaillance rénale aiguë
Des cas de défaillance rénale aiguë ont été rapportés chez des patients présentant une insuffisance rénale grave préexistante.
Fibrose néphrogénique systémique
Des cas isolés de fibrose néphrogénique systémique (FNS) ont été rapportés suite à l’utilisation du gadotéridol, la plupart chez des patients auxquels d’autres produits de contraste contenant du gadolinium étaient administrés simultanément (cf. «Mises en garde et précautions»).
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucun cas de surdosage n’ayant été rapporté à ce jour, les symptômes correspondants n’ont pu être identifiés. Le gadotéridol peut être éliminé par hémodialyse. Il n’est toutefois pas établi qu’une hémodialyse puisse prévenir une fibrose néphrogénique systémique (FNS).
Propriétés/EffetsCode ATC
V08CA04
Mécanisme d’action
ProHance est une solution aqueuse stérile claire, incolore à légèrement jaunâtre, destinée aux injections. Dans les conditions d’emploi, ProHance est une solution hypertonique dont l’osmolarité est approximativement le double de celle du plasma. ProHance ne contient pas de conservateurs.
Le principe actif de ProHance, le gadotéridol, est une substance dotée de propriétés paramagnétiques qui développe un moment magnétique lorsqu’elle est placée dans un champ magnétique, développant un magnétisme local relativement élevé capable de prolonger le taux de relaxation des protons H avoisinants. En résonance magnétique nucléaire, la visualisation des tissus cérébraux sains et pathologiques dépend en partie de l’intensité différentielle des signaux RF (onde de radiofréquence) due 1) à la densité des protons, 2) au temps de relaxation spinréseau longitudinal T1 des protons, et 3) au temps de relaxation spin-spin transversal T2 des protons. Dès que le gadotéridol est placé dans un champ magnétique, il raccourcit les temps de relaxation T1 dans les tissus cibles. Aux doses recommandées, I’effet le plus sensible est obtenu lors de séquences en spin-écho pondérées T1.
Une barrière hémato-encéphalique lésée ou une anomalie vasculaire peuvent être source d’accumulation de gadotéridol dans des lésions du type néoplasie, abcès ou infarctus subaigu.
Pharmacodynamique
Voir « Mécanisme d’action ».
Efficacité clinique
Données manquantes.
PharmacocinétiqueAbsorption
Données manquantes.
Distribution
Après administration IV, la pharmacocinétique du gadotéridol chez le sujet sain évolue en fonction d’un modèle ouvert à deux compartiments. La demi-vie moyenne de distribution est d’environ 12 minutes. Le volume de distribution d’env. 129 ml/kg correspond à celui du liquide extracellulaire. Chez le rat, on n’a trouvé aucun indice de liaison aux protéines.
Métabolisme
Aucune preuve de biotransformation n’a pu être apportée; le gadotéridol n’est pas métabolisé.
Élimination
La demi-vie moyenne d’élimination du gadotéridol est d’environ 1 heure et 34 minutes. La substance est exclusivement éliminée par voie rénale (le taux d’excrétion atteignant en 24 h env. 95 % de la dose injectée).
La clairance rénale du gadotéridol correspond pour l’essentiel à la clairance plasmatique (env. 1.41 ml/min./kg contre 1.50 ml/min./kg). Cette quasi-identité confirme la voie d’excrétion majeure dans les urines. La clairance correspond à celle de substances éliminées par filtration glomérulaire.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
On ne dispose d’aucune donnée quant à la pharmacocinétique en cas d’insuffisance hépatique.
Troubles de la fonction rénale
La demi-vie de distribution n’est guère affectée par une insuffisance rénale modérée à sévère.
En cas d’insuffisance rénale modérée, la demi-vie d’élimination, qui est d’1 heure et 34 minutes chez le sujet sain, augmente à 6 heures et 57 minutes, en cas d’insuffisance rénale sévère à 9 heures et 32 minutes. La clairance plasmatique est de 37.2 ml/min. en cas d’insuffisance rénale modérée, de 16.0 ml/min. en cas d’insuffisance rénale sévère.
Chez l’hémodialysé, ProHance est effectivement hémodialysé. Le taux de clairance du gadotéridol atteint 97 % de la clairance de la créatinine et 71 % de la clairance de l’azote uréique du sang. Au terme de 3 séances de dialyse, on décèle encore dans le sang env. 2 % de la dose de gadotéridol injectée.
Données précliniquesMutagénicité
Les propriétés mutagènes du gadotéridol ont été examinées lors d’une série de tests in vivo. Aucun indice parlant pour un potentiel mutagène n’a été mis en évidence.
Carcinogénicité
Aucune étude sur la cancérogénicité n’a été effectuée.
Toxicité sur la reproduction
Des études sur la fertilité, la toxicité embryonnaire et la phase périnatale et postnatale ont été menées chez le rat et le lapin avec le gadotéridol appliqué par voie IV à des doses situées entre 6 et 10 mmol/kg (60 à 100 fois supérieures à la dose humaine) et ont mis en évidence des effets de toxicité paternelle, maternelle et embryonnaire.
Remarques particulièresIncompatibilités
ProHance ne doit pas être mélangé avec d’autres médicaments.
Influence sur les méthodes de diagnostic
Aucune donnée disponible jusqu’à présent.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Conserver à température ambiante (15-25°C).
Conserver à l’abri de la lumière.
Tenir hors de la portée des enfants.
Remarques concernant la manipulation
Réservé à un usage unique. Toute quantité résiduelle et d’autres matériaux doivent être éliminés.
ProHance ne doit pas être congelé. Les ampoules congelées sont à jeter.
Etiquette détachable pour le dossier du patient
L’étiquette détachable de traçabilité doit être collée dans le dossier médical du patient afin d’assurer une documentation exacte du produit de contraste contenant du gadolinium utilisé. La dose utilisée doit également être indiquée.
ProHance seringue préremplie
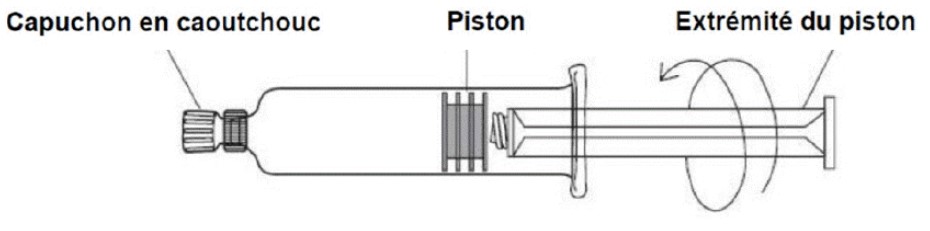
Suivez attentivement les indications ci-dessous :
1. Tournez la tige du piston dans le sens des aiguilles d’une montre pour visser son extrémité filetée dans le piston et poussez le piston de quelques millimètres vers l’avant afin d’éliminer toute résistance éventuelle entre le piston et le cylindre en verre de la seringue.
2. En tenant la seringue préremplie en position verticale, enlevez le capuchon en caoutchouc de la pointe de la seringue de façon aseptique et fixez une aiguille ou un cathéter stérile à usage unique doté d’un raccord compatible en exerçant une pression tout en tournant.
3. Tout en maintenant la seringue en position verticale, éliminez l’air en poussant le piston jusqu’à ce que le liquide apparaisse à l’extrémité de l’aiguille ou remplisse entièrement le cathéter. Réalisez l’injection après un test d’aspiration habituel. Pour vous assurer que toute la dose de produit de contraste a été injectée, l’injection doit être suivie d’un rinçage avec une solution saline physiologique.
Numéro d’autorisation52599, 52273 (Swissmedic)
PrésentationSeringues préremplies de 10 ml à 2.793 g (5.0 mmol): 1 et 5 [B].
Seringues préremplies de 15 ml à 4.1895 g (7.5 mmol): 1 et 5 [B].
Seringues préremplies de 17 ml à 4.7481 g (8.5 mmol): 1 et 5 [B].
Flacons de 5 ml à 1.3965 g (2.5 mmol): 1, 5, 10, 20 [B].
Flacons de 10 ml à 2.793 g (5.0 mmol): 1, 5, 10, 20 [B].
Flacons de 15 ml à 4.1895 g (7.5 mmol): 1, 5, 10, 20 [B].
Flacons de 20 ml à 5.586 g (10 mmol): 1, 5, 10, 20 [B].
Flacons de 50 ml à 13.965 g (25 mmol): 1, 10 [B].
Titulaire de l’autorisationBRACCO SUISSE SA, Cadempino
Mise à jour de l’informationJuin 2025
|