CompositionPrincipes actifs
Octreotidum* ut Octreotidi acetas.
Excipients
Copoly (DLlactidum-glycolidum), Mannitolum.
Solvant: Carboxymethylcellulosum natricum (correspond à 1.512 mg de sodium/ml); Mannitolum, Poloxamère 188; Aqua ad iniectabilia.
*DCI rec.
Indications/Possibilités d’emploiAcromégalie
Traitement des patients acromégaliques pour lesquels la chirurgie ou la radiothérapie sont inappropriées ou inefficaces, ou pendant la période de latence avant que la radiothérapie soit pleinement efficace.
Tumeurs du système endocrinien gastroentéropancréatique (GEP)
Traitement des patients présentant les symptômes de tumeurs endocrines gastroentéropancréatiques fonctionnelles:
·tumeurs carcinoïdes caractéristiques d'un syndrome carcinoïde,
·VIPomes,
·glucagonomes,
·gastrinomes/syndrome de Zollinger-Ellison,
·insulinomes pour contrôle préopératoire de l'hypoglycémie et traitement d'entretien,
·GRFomes.
Traitement des patients atteints de tumeurs neuroendocrines avancées bien différenciées (G1, G2) de l'intestin moyen (intestin grêle, cæcum ou appendice).
Posologie/Mode d’emploiSandostatine LAR ne doit être administré qu'en injection intramusculaire profonde dans le muscle fessier. En cas d'administrations répétées, l'injection doit être effectuée alternativement dans le muscle fessier droit et gauche (voir «Remarques concernant la manipulation - Instructions pour l'injection i.m. de Sandostatine LAR»).
Afin de garantir une posologie correcte, le kit d'injection Sandostatine LAR doit être réchauffé à température ambiante avant la reconstitution (voir «Remarques particulières», rubrique «Remarques concernant la manipulation»).
Acromégalie
Le traitement doit être initié avec une dose de 20 mg de Sandostatine LAR toutes les quatre semaines pendant 3 mois. La posologie sera éventuellement ajustée ultérieurement en fonction des taux sériques d'hormone de croissance (GH) et de somatomédine-C (IGF-1) ainsi que des symptômes cliniques conformément aux recommandations ci-dessous.
Les patients suivant un traitement à base d'octréotide en injection sous-cutanée peuvent débuter le traitement à base de Sandostatine LAR le lendemain de la dernière injection sous-cutanée de Sandostatine.
·Chez les patients dont les symptômes cliniques et les paramètres biochimiques (GH; IGF-1) ne peuvent pas être contrôlés pleinement (taux sériques de GH encore supérieurs à 2.5 µg/l) après 3 mois de traitement, la dose peut être augmentée à 30 mg toutes les quatre semaines.
·Chez les patients dont les concentrations de GH sont continuellement inférieures à 1 µg/l, dont les taux sériques d'IGF-1 se sont normalisés et chez lesquels la plupart des signes réversibles de l'acromégalie ont disparu après un traitement de 3 mois avec 20 mg, la dose peut être réduite à 10 mg de Sandostatine LAR. Dans le cas d'une dose aussi faible de Sandostatine LAR, il est toutefois recommandé de surveiller attentivement les taux sériques de GH et d'IGF-1 ainsi que les symptômes cliniques.
·Chez les patients qui sont toujours traités avec la même dose de Sandostatine LAR, un contrôle de la GH et de l'IGF-1 doit être effectué tous les 6 mois.
Tumeurs du système endocrinien gastroentéropancréatique
Tumeurs fonctionnelles du système neuroendocrinien gastroentéropancréatique ou de l'intestin moyen (carcinoïdes, VIPomes)
Le traitement doit être initié avec une dose de 20 mg de Sandostatine LAR toutes les quatre semaines. Chez les patients déjà traités par injection sous-cutanée d'octréotide, le dosage d'octréotide sous-cutané préalablement efficace doit être également poursuivi jusqu'à 2 semaines après la première injection de Sandostatine LAR.
·Chez les patients dont les symptômes et les marqueurs biologiques ont pu être contrôlés de manière satisfaisante après un traitement de 3 mois, la posologie peut être réduite à 10 mg de Sandostatine LAR toutes les 4 semaines.
·Chez les patients dont les symptômes n'ont pu être contrôlés que partiellement après un traitement de 3 mois, la posologie peut être augmentée à 30 mg de Sandostatine LAR toutes les quatre semaines.
Sous traitement par Sandostatine LAR, il se peut que les symptômes propres à des tumeurs du système gastroentéropancréatique puissent s'aggraver certains jours. Dans ce cas, il est recommandé d'administrer en complément de l'octréotide par voie sous-cutanée à la dose administrée avant le traitement par Sandostatine LAR. Ceci peut être nécessaire, surtout pendant les 2 premiers mois de traitement, jusqu'à ce que les taux thérapeutiques d'octréotide soient atteints.
Tumeur neuroendocrine de l'intestin moyen
La dose recommandée de Sandostatine LAR est de 30 mg toutes les 4 semaines. En cas d'absence de progression tumorale, le traitement par Sandostatine LAR doit être poursuivi afin de contrôler la tumeur.
Instructions posologiques particulières
Patients âgés
Une posologie spéciale chez les patients âgés au début d'un traitement par Sandostatine ou par Sandostatine LAR n'est pas nécessaire.
Enfants et adolescents
On ne dispose d'aucune expérience sur l'utilisation de Sandostatine LAR chez les patients de moins de 18 ans.
Patients présentant des troubles de la fonction rénale
Il n'est pas nécessaire d'ajuster la dose (voir «Pharmacocinétique»).
Patients présentant des troubles de la fonction hépatique
L'élimination de l'octréotide peut être réduite chez les patients souffrant de cirrhose hépatique. Une adaptation de la posologie de Sandostatine LAR chez les patients atteints de cirrhose hépatique n'est pas nécessaire en raison de la grande marge thérapeutique de l'octréotide.
Contre-indicationsHypersensibilité connue à l'octréotide ou à l'un des excipients.
Mises en garde et précautionsGénéralités
Étant donné que les tumeurs hypophysaires sécrétant de la GH sont parfois expansives et qu'elles peuvent de ce fait causer de graves complications (p.ex. rétrécissement du champ visuel), il est impératif de surveiller le patient de près. Dès les premiers signes d'expansion tumorale, il est recommandé d'envisager le recours à d'autres mesures.
Métabolisme du glucose
Chez les patients présentant un diabète sucré de type 1 insulinodépendant, il est possible que Sandostatine LAR puisse influencer la régulation du glucose et le besoin en insuline peut diminuer. Des cas d'hypoglycémie ont été rapportés.
Chez les patients non diabétiques et ceux présentant un diabète de type 2 avec des réserves d'insuline partiellement intactes, l'administration de Sandostatine par voie s.c. peut conduire à une augmentation postprandiale de la glycémie. C'est pourquoi il est recommandé de surveiller le taux de glucose et, le cas échéant, d'adapter la thérapie antidiabétique.
Patients avec insulinomes: l'octréotide inhibant la sécrétion de l'hormone de croissance et du glucagon plus fortement que celle de l'insuline et inhibant la sécrétion de l'insuline à court terme, l'intensité et la durée d'une hypoglycémie peuvent être renforcées chez ces patients. Ces patients doivent être suivis étroitement.
Affections des voies biliaires
Les analogues de la somatostatine inhibent la contractilité de la vésicule biliaire et réduisent la sécrétion biliaire, ce qui peut conduire à des anomalies de la vésicule biliaire, à la formation de boue biliaire (sludge) ou à la formation de calculs biliaires. L'incidence d'une lithiase biliaire sous traitement par Sandostatine par voie s.c. a été évaluée à 15 à 30%, contre une incidence de 5 à 20% dans la population générale. Les données à long terme pour Sandostatine LAR chez les patients souffrant d'acromégalie ou de tumeurs du système endocrinien gastroentéropancréatique indiquent que le traitement par Sandostatine LAR, comparativement à Sandostatine par voie s.c., n'entraîne pas plus fréquemment la formation de calculs biliaires. Lorsque des calculs biliaires sont présents, ils sont asymptomatiques la plupart du temps.
En outre, une dilatation des voies biliaires ainsi que des cas de cholécystite ou de cholangite (complication de la lithiase biliaire) ont été rapportés lors de l'utilisation de Sandostatine. C'est pourquoi il est recommandé de réaliser un examen de la vésicule biliaire par échographie avant le début du traitement par Sandostatine LAR puis tous les 6 à 12 mois pendant le traitement.
Pancréatite
Dans de très rares cas, une pancréatite aiguë a été rapportée. En général, celle-ci survient au cours des premières heures ou jours d'un traitement par Sandostatine et elle régresse à l'arrêt du médicament. En outre, une pancréatite «induite par la lithiase biliaire» a été rapportée chez des patients suivant un traitement à long terme par Sandostatine.
Effets indésirables cardiovasculaires
Une bradycardie est un effet indésirable fréquent du traitement par analogues de la somatostatine. Un ajustement de la dose de médicaments tels que les bêtabloquants, les antagonistes du calcium ou les médicaments destinés au contrôle du bilan hydro-électrolytique peut s'avérer nécessaire.
Des modifications de l'ECG telles qu'un allongement de QT, une déviation de l'axe, une repolarisation précoce, un faible voltage, une transition R/S, une propagation précoce de l'onde R et des modifications non spécifiques des ondes ST-T ont été observées. Le lien entre ces événements et l'octréotide n'a pas été clairement démontré, car un grand nombre de ces patients souffraient d'une cardiopathie sous-jacente.
Après la commercialisation, une hypersensibilité et des réactions allergiques ont été rapportées. Celles-ci étaient essentiellement accompagnées de réactions cutanées; la bouche et/ou les voies respiratoires étaient rarement affectées. Des cas isolés de choc anaphylactique ont été rapportés.
Autres précautions
Des taux sanguins réduits de vitamine B12 et des résultats anormaux au test de Schilling ont été observés chez quelques patients traités par l'octréotide. Il est recommandé, pendant le traitement par Sandostatine LAR, de surveiller le taux sanguin de vitamine B12 chez les patients présentant des antécédents de carence en vitamine B12.
La fonction thyroïdienne doit être surveillée chez les patients traités à long terme par l'octréotide.
L'octréotide peut modifier l'absorption des graisses alimentaires chez certains patients. On peut enregistrer en particulier une augmentation de l'excrétion de graisses dans les selles, mais rien n'indique à ce jour que le traitement par l'octréotide, même à long terme, entraîne une carence nutritionnelle due à une malabsorption.
Le bénéfice thérapeutique d'une baisse du taux de GH («Growth hormone») et la normalisation de la concentration de l'IGF-1 («Insulin-like growth factor») peuvent éventuellement rétablir la fertilité chez les patientes atteintes d'acromégalie. En cas d'indication, il faut recommander aux patientes en âge de procréer d'utiliser des méthodes contraceptives appropriées pendant un traitement par l'octréotide (voir «Grossesse, Allaitement»).
Ce médicament contient moins de 1 mmol de sodium (23 mg) par volume de dosage, c'est-à-dire qu'il est pratiquement «sans sodium».
InteractionsInteractions pharmacocinétiques
L'octréotide réduit l'absorption intestinale de la ciclosporine et retarde celle de la cimétidine.
L'administration concomitante d'octréotide et de bromocriptine augmente la biodisponibilité de la bromocriptine.
Un nombre limité de données publiées indiquent que les analogues de la somatostatine pourraient réduire la clairance métabolique de substances métabolisées par l'enzyme du cytochrome P450. Ceci a été attribué à l'inhibition de l'hormone de croissance. Comme il n'est pas exclu que l'octréotide présente aussi cet effet, les autres médicaments métabolisés essentiellement par le CYP3A4 et présentant une marge thérapeutique étroite (p.ex. quinidine, terfénadine), doivent être administrés avec précaution.
Interactions pharmacodynamiques
Un ajustement de la dose de médicaments tels que les bêtabloquants, les antagonistes du calcium ou les médicaments destinés au contrôle du bilan hydro-électrolytique peut s'avérer nécessaire en cas d'administration simultanée avec Sandostatine LAR (voir «Mises en garde et précautions»).
Les ajustements des doses d'insuline et d'antidiabétiques peuvent s'avérer nécessaires en cas d'administration simultanée de Sandostatine LAR (voir «Mises en garde et précautions»).
Utilisation simultanée d'un agent radiothérapeutique couplé à un analogue de la somatostatine (PRRT, traitement par radionucléide des récepteurs peptidiques)
La somatostatine et ses analogues, comme l'octréotide, se lient de manière compétitive aux récepteurs de la somatostatine et peuvent influencer l'efficacité des agents radiothérapeutiques correspondants (comme p.ex. le 177Luoxodotréotide). L'administration de Sandostatine LAR comme analogue de la somatostatine à longue durée d'action doit donc être interrompue 4 à 6 semaines avant le début du traitement par un PRRT. Si nécessaire, les patients peuvent être traités jusqu'au plus tard 24 heures avant l'administration du PRRT avec des analogues de la somatostatine à courte durée d'action.
Dans les 4 à 24 heures après administration du PRRT, le traitement par Sandostatine LAR peut être poursuivi, mais il doit à nouveau être interrompu 4 à 6 semaines avant l'administration suivante de l'agent radiothérapeutique.
Grossesse, allaitementGrossesse
Mis à part un retard passager de croissance de la progéniture, les expérimentations animales avec l'octréotide n'ont révélé aucune incidence toxicologique de l'octréotide sur la reproduction (voir «Données précliniques»).
Il n'existe pas d'études adéquates et bien contrôlées chez la femme enceinte. Après la commercialisation, un nombre limité de patientes acromégaliques qui ont été enceintes lors d'un traitement par l'octréotide a été rapporté; toutefois, l'issue de la grossesse est inconnue dans la moitié de ces cas. La plupart des patientes ont reçu l'octréotide pendant le premier trimestre de la grossesse, à une dose comprise entre 100 et 300 µg/jour de Sandostatine par voie s.c. ou à une dose comprise entre 20 et 30 mg/mois de Sandostatine LAR. Dans environ deux tiers des cas où l'issue de la grossesse est connue, les femmes ont choisi de poursuivre le traitement par l'octréotide pendant leur grossesse. Dans la plupart des cas où l'issue est connue, les rapports font état de nouveau-nés sans particularité, mais aussi de quelques avortements spontanés pendant le premier trimestre. Aucune anomalie ou malformation congénitale n'a été observée.
Sandostatine LAR ne doit pas être utilisé chez la femme enceinte, sauf en cas de nécessité absolue.
Allaitement
On ignore si l'octréotide est excrété dans le lait maternel humain. Des études chez l'animal ont montré une excrétion de l'octréotide dans le lait maternel. Les patientes ne doivent pas allaiter pendant un traitement par Sandostatine LAR.
Fertilité
On ignore si l'octréotide a des incidences sur la fertilité humaine. L'octréotide en dose allant jusqu'à 1 mg/kg/jour n'a pas eu d'incidence sur la fertilité des rats mâles et femelles (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune expérience concernant l'influence de Sandostatine LAR sur l'aptitude à la conduite et à l'utilisation de machines n'est disponible.
Effets indésirablesLes effets indésirables les plus fréquemment rapportés dans les études cliniques après l'administration d'octréotide ont été: diarrhée, douleurs abdominales, nausées, distension, céphalées, lithiase biliaire, hyperglycémie et constipation.
Les autres effets indésirables fréquemment rapportés ont été: vertiges, douleur localisée, boue biliaire, trouble de la fonction thyroïdienne (p.ex. diminution de la TSH, diminution de la T4 totale et diminution de la T4 libre), défécations involontaires, tolérance au glucose réduite, vomissements, asthénie et hypoglycémie.
Affections gastro-intestinales et nutrition
Dans de rares cas, les effets secondaires gastro-intestinaux peuvent prendre l'apparence d'une occlusion intestinale aiguë, avec distension abdominale croissante, douleur épigastrique intense et défense abdominale douloureuse.
Réactions au site d'injection
Les réactions au site d'injection qui ont été rapportées chez des patients ayant reçu Sandostatine LAR sont des douleurs, des rougeurs, des hémorragies, des démangeaisons, des gonflements ou des durcissements. Dans la plupart des cas, ces évènements n'ont cependant pas nécessité d'intervention clinique.
Thrombopénie
Après la commercialisation, des cas de thrombopénie ont été rapportés, notamment durant le traitement intraveineux par Sandostatine chez des patients ayant une cirrhose hépatique et durant le traitement par Sandostatine LAR. La thrombopénie était réversible après l'arrêt du traitement.
Ci-dessous, les effets indésirables qui ont été observés dans les études cliniques réalisées avec l'octréotide ou pendant la surveillance du marché, sont énumérés par classes de systèmes d'organes MedDRA et par fréquence. Les fréquences sont régies par la convention suivante:
très fréquents (≥1/10); fréquents (≥1/100 à < 1/10); occasionnels (≥1/1000 à < 1/100); rares (≥1/10 000 à < 1/1000); très rares (< 1/10 000); fréquence inconnue (basés essentiellement sur les signalements spontanés de l'observation du marché, la fréquence exacte ne pouvant être déterminée).
Affections hématologiques et du système lymphatique
Fréquence inconnue: thrombopénie.
Affections du système immunitaire
Fréquence inconnue: réactions d'hypersensibilité, y compris les réactions anaphylactiques.
Affections endocriniennes
Fréquents: hypothyroïdie, trouble de la fonction thyroïdienne (p.ex. diminution de la TSH, diminution de la T4 totale et diminution de la T4 libre).
Troubles du métabolisme et de la nutrition
Très fréquents: hyperglycémie (10.8%).
Fréquents: hypoglycémie, tolérance au glucose réduite, anorexie.
Occasionnels: déshydratation.
Affections du système nerveux
Très fréquents: céphalées (12.4%).
Fréquents: vertiges.
Affections cardiaques
Fréquents: bradycardie.
Occasionnels: tachycardie.
Fréquence inconnue: arythmies.
Affections respiratoires, thoraciques et médiastinales
Fréquents: dyspnée.
Affections gastro-intestinales
Très fréquents: diarrhée (26.1%), douleurs abdominales (24.2%), nausées (14.3%), flatulences (14.2%), constipation (12.7%).
Fréquents: dyspepsie, vomissements, distension abdominale, stéatorrhée, coloration des selles.
Fréquence inconnue: pancréatite aiguë.
Affections hépatobiliaires
Très fréquents: lithiase biliaire (12.0%).
Fréquents: transaminases élevées, hyperbilirubinémie, cholécystite.
Fréquence inconnue: phosphatase alcaline sanguine augmentée, gamma-glutamyltransférase augmentée, ictère, cholestase, ictère cholestatique, hépatite cholestatique, hépatite aiguë sans cholestase.
Affections de la peau et du tissu sous-cutané
Fréquents: prurit, rash cutané, alopécie.
Fréquence inconnue: urticaire.
Troubles généraux et anomalies au site d'administration
Très fréquents: réactions localisées au site d'injection (10 à 30%, selon la dose et l'intervalle des injections, p.ex. douleurs, paresthésies, érythème).
Fréquents: asthénie.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSymptômes
Un nombre limité de cas de surdosages accidentels de Sandostatine LAR a été rapporté. Les doses allaient de 100 mg à 163 mg/mois de Sandostatine LAR. Le seul effet indésirable annoncé a été des bouffées de chaleur.
Des rapports font état de patients cancéreux ayant reçu des doses de Sandostatine LAR allant jusqu'à 60 mg par mois et jusqu'à 90 mg toutes les 2 semaines. Ces doses ont été généralement bien tolérées, les effets indésirables suivants ayant toutefois été rapportés: pollakiurie, fatigue, dépression, anxiété, difficultés de concentration.
Traitement
Le traitement du surdosage de Sandostatine LAR est symptomatique.
Propriétés/EffetsCode ATC
H01CB02
Mécanisme d'action
L'octréotide est un dérivé octapeptidique synthétique de la somatostatine naturelle; ses effets pharmacologiques sont similaires à ceux de l'hormone naturelle, mais sa durée d'action est considérablement plus longue. Elle inhibe l'hypersécrétion pathologique de l'hormone de croissance (GH) ainsi que des peptides et de la sérotonine produits au sein du système endocrinien gastroentéropancréatique (GEP).
Chez l'animal, l'octréotide inhibe la libération d'hormone de croissance, de glucagon et d'insuline de façon plus marquée que la somatostatine. Il présente de plus une plus grande sélectivité en ce qui concerne la suppression de l'hormone de croissance et du glucagon.
Il a été constaté chez le sujet sain que l'octréotide, comme la somatostatine, produisait les effets suivants:
·inhibition de la libération d'hormone de croissance (GH) stimulée par différents facteurs (arginine, effort ou hypoglycémie induite par l'insuline),
·inhibition de la libération postprandiale d'insuline, de glucagon, de gastrine et d'autres peptides du système gastroentéropancréatique (GEP), de même qu'inhibition de la sécrétion d'insuline et de glucagon stimulée par l'arginine,
·inhibition de la libération d'hormone thyréotrope (TSH [thyroid stimulating hormone]) induite par la protiréline (TRH [thyrotropin releasing hormone]).
Contrairement à la somatostatine, l'octréotide inhibe l'hormone de croissance de préférence par l'intermédiaire de l'insuline, et son administration n'entraîne pas de rebond d'hypersécrétion hormonale (p.ex. GH chez le patient acromégalique).
Pharmacodynamique
Efficacité clinique
Acromégalie
Sandostatine LAR fournit des concentrations sériques constantes et thérapeutiquement efficaces d'octréotide, ce qui entraîne une diminution des taux sériques de GH et une normalisation des taux sériques d'IGF-1 de manière constante. Chez la plupart des patients, Sandostatine LAR réduit considérablement les symptômes cliniques de la maladie tels que les céphalées, la transpiration, les paresthésies, la fatigue, les ostéoarthralgies et le syndrome du canal carpien.
Même si lors d'un traitement par des analogues de la somatostatine, il y a lieu de s'attendre à une diminution de la taille de la tumeur chez une partie des patients, tous les patients recevant ce traitement doivent être surveillés régulièrement (voir «Mises en garde et précautions»).
Chez des patients acromégaliques atteints d'un adénome hypophysaire sécrétant de la GH et non traités auparavant, un traitement par Sandostatine LAR a conduit dans la moitié des cas à une diminution du volume de la tumeur de > 20%.
L'efficacité et la sécurité de Sandostatine LAR pour le traitement de l'acromégalie après une dose unique de 10, 20 ou 30 mg a été examinée dans deux études randomisées, en double aveugle, incontrôlées menées sur 93 patients. Le critère d'efficacité primaire était le taux sérique moyen de STH après 12 heures. Les doses de 20 et 30 mg de Sandostatine LAR ont été en mesure de réduire le taux de STH à < 5 µg/l du jour 14 au jour 42.
Lors d'une prolongation ouverte consécutive, les patients ont pu recevoir jusqu'à 28 injections (à intervalles de 28 jours respectivement); les données de 87 patients sont disponibles pour cette période de traitement. Ce faisant, la dose a pu être ajustée entre 10 et 30 mg (dans des cas exceptionnels jusqu'à 40 mg) en fonction de la réponse individuelle. Sandostatine LAR a conduit sur tout l'intervalle de dosage à une suppression persistente du taux de STH. Ceci s'est également accompagné d'une baisse significative du taux d'IGF-I et d'une atténuation durable des symptômes cliniques de l'acromégalie.
Dans ces études, la tolérance de Sandostatine LAR était comparable à celle de Sandostatine administré par voie sous-cutanée.
Tumeurs fonctionnelles du système endocrinien gastropancréatique
Le traitement par Sandostatine LAR permet de contrôler de façon continue les symptômes engendrés par la maladie sous-jacente. Ce faisant, l'octréotide influence les différentes formes de tumeurs gastroentéropancréatiques de la façon suivante.
Carcinoïdes
L'administration d'octréotide peut entraîner une amélioration des symptômes, notamment du flush et de la diarrhée, accompagnée dans certains cas d'une baisse du taux plasmatique de sérotonine et d'une réduction de l'excrétion urinaire d'acide 5-hydroxy-indol-acétique.
L'efficacité et la sécurité de Sandostatine LAR à des doses de 10, 20 et 30 mg à 4 semaines d'intervalle pour le traitement d'un syndrome carcinoïde malin ont été examinées dans une étude randomisée en double aveugle en comparaison avec Sandostatine sous-cutané chez n = 93 patients. Un taux de réussite considérant l'intensité et la durée de la suppression des symptômes carcinoïdes a été défini comme critère d'efficacité. Le succès du traitement impliquait qu'au cours des 4 dernières semaines et sur un total maximum de 5 jours, un maximum de deux traitements d'urgence avec de l'octréotide administré par voie sous-cutanée soit requis dans les groupes Sandostatine LAR. Dans le groupe Sandostatine sous-cutané, le succès du traitement était avéré lorsque dans la même période, un maximum de deux augmentations de la dose sur un total de 5 jours maximum était nécessaire.
L'efficacité de Sandostatine LAR était comparable à celle de Sandostatine sous-cutané. Au terme de l'étude, le traitement avec Sandostatine sous-cutané s'est montré efficace chez. 58% des patients, avec Sandostatine LAR 10, 20 et 30 mg, chez 55%, 50% et 56% des patients.
VIPomes
Du point de vue biochimique, ces tumeurs se caractérisent par une surproduction de peptide intestinal vasoactif (VIP [vasoactive intestinal peptide]). Le traitement par l'octréotide permet, dans la plupart des cas, de diminuer les diarrhées sécrétoires graves qui en sont la manifestation typique, ce qui se traduit par une amélioration de la qualité de vie. Cet effet s'accompagne d'une diminution des troubles de l'équilibre électrolytiques liés à la diarrhée, p.ex. l'hypokaliémie, ce qui permet de supprimer l'apport hydro-électrolytique par voie entérale et parentérale. Dans certains cas, notamment dans le cas de métastases hépatiques, l'examen par scanner suggère que l'évolution tumorale a été ralentie ou stoppée, voire même que la masse tumorale a diminué. L'amélioration clinique s'accompagne généralement d'une réduction du taux plasmatique de VIP, qui peut se normaliser.
Glucagonomes
L'administration d'octréotide entraîne dans la plupart des cas une amélioration notable de l'érythème nécrolytique migrateur qui caractérise ces tumeurs. L'octréotide influence peu le léger état de diabète souvent observé chez les patients atteints de glucagonomes. En règle générale, les besoins en insuline ou en antidiabétiques oraux ne sont pas diminués. L'octréotide entraîne une amélioration des diarrhées éventuellement présentes et donc une augmentation pondérale. L'octréotide provoque souvent une baisse immédiate du taux plasmatique de glucagon. Cependant, cette baisse ne se maintient pas lors d'une administration prolongée bien que l'amélioration des symptômes persiste.
Gastrinomes/syndrome de Zollinger-Ellison
Le traitement par les inhibiteurs de la pompe à protons ou les inhibiteurs des récepteurs H2 ne permet pas toujours de prévenir les ulcérations gastriques récurrentes dues à l'hypersécrétion chronique d'acide gastrique stimulée par la gastrine; de plus, dans certains cas, il reste sans effet sur la diarrhée, qui est parfois très prononcée. Dans ces cas-là, l'octréotide peut, seul ou en association avec des inhibiteurs de la pompe à protons ou des inhibiteurs des récepteurs H2, réduire l'hypersécrétion d'acide gastrique et améliorer les symptômes, y compris la diarrhée, chez 50% des patients. Il peut aussi améliorer d'autres symptômes éventuellement causés par les peptides tumoraux, tel le flush. Chez certains patients, l'octréotide réduit le taux plasmatique de gastrine.
Insulinomes
L'octréotide entraîne une baisse de l'insuline immunoréactive circulante. Chez les patients porteurs de tumeurs opérables, l'octréotide peut contribuer à rétablir et à maintenir la normoglycémie avant l'intervention. Chez les patients porteurs de tumeurs bénignes ou malignes inopérables, le contrôle de la glycémie peut être amélioré dans certains cas sans que l'on observe simultanément une baisse durable des taux d'insuline circulante.
GRFomes
Ces tumeurs rares produisent le GRF (growth hormone releasing factor [facteur de libération de l'hormone de croissance]) seul ou associé à d'autres peptides actifs. L'octréotide a conduit, dans un des deux cas étudiés, à une amélioration des symptômes de l'acromégalie, affection qui en résulte. Cet effet est probablement basé sur une inhibition de la sécrétion de GRF et d'hormone de croissance, qui peut être associée à une régression de l'hypertrophie hypophysaire.
Tumeurs neuroendocrines avancées bien différenciées de l'intestin moyen
Des patients atteints de tumeurs métastatiques neuroendocrines bien différenciées fonctionnelles ou non fonctionnelles de l'intestin moyen ont été inclus dans une étude de phase III contrôlée contre placebo (PROMID).
85 patients ont été randomisés pour recevoir un traitement par Sandostatine LAR 30 mg toutes les quatre semaines (n = 42) ou par placebo (n = 43).
Les principaux critères d'inclusion étaient: non traité, histologiquement confirmé, inopérable localement ou avancée métastatique, bien différencié, tumeurs neuroendocrines fonctionnelles ou non fonctionnelles, carcinomes neuroendocrines fonctionnels ou non fonctionnels, tumeur primaire localisée dans l'intestin moyen ou d'origine inconnue (mais origine supposée dans l'intestin moyen après exclusion d'une tumeur primaire du pancréas, du thorax ou d'autres localisations).
L'intervalle de temps jusqu'à la progression tumorale (TTP [time to progression]) constitue le critère d'évaluation primaire, basé sur une évaluation centrale des résultats radiologiques réalisée conformément aux critères de l'OMS.
Le temps médian jusqu'à la progression tumorale dans le groupe avec Sandostatine LAR était de 14.3 mois et 5.9 mois pour le groupe avec placebo (HR = 0.36; IC à 95%, 0.21–0.61; p = 0.0001).
L'action du traitement chez les patients atteints de tumeurs fonctionnelles (HR = 0.41; IC à 95%, 0.18 à 0.92) était similaire à celle des patients atteints de tumeurs non fonctionnelles (HR = 0.32; IC à 95%, 0.15 à 0.66).
Dans la mesure où un avantage clinique significatif de Sandostatine LAR a été démontré dans l'analyse intermédiaire prévue au préalable au 18e mois, le recrutement de patients a été interrompu. Dans le bras avec Sandostatine LAR, le traitement a pu être poursuivi jusqu'à l'apparition d'une progression; dans le bras avec placebo, le traitement a pu être remplacé par un traitement actif.
La survie globale a été évaluée après un suivi supplémentaire de 4.5 ans. Il n'existe aucune différence entre les deux bras d'études.
PharmacocinétiqueAbsorption
Le profil pharmacocinétique de l'octréotide après l'injection de Sandostatine LAR reflète la libération de la matrice polymère et la dégradation biologique de la substance. Après la libération dans le circuit systémique, l'octréotide se répand selon ses propriétés pharmacocinétiques connues et décrites dans l'application sous-cutanée. Après une injection i.m. unique de Sandostatine LAR, le taux sérique d'octréotide atteint un pic initial temporaire dans l'heure qui suit l'administration, puis il chute progressivement à un taux d'octréotide non détectable en 24 h.
Au premier jour, moins de 0.5% de la quantité de substance totale libérée est mesuré. Le taux d'octréotide reste, chez la plupart des patients, au niveau des valeurs sous-thérapeutique pendant les 7 jours suivant l'injection de Sandostatine LAR.
Les taux d'octréotide atteignent un plateau au 14e jour env., puis restent relativement constants pendant les 3 à 4 semaines suivantes.
Après env. 42 jours, le taux d'octréotide commence à diminuer lentement du fait de la dégradation terminale de la matrice polymère.
Distribution
Le volume de distribution de l'octréotide s'élève à 0.27 l/kg, la clairance corporelle totale à 160 ml/min. La liaison aux protéines plasmatiques s'élève à 65%. L'octréotide ne se lie que très peu aux cellules sanguines.
Métabolisme
Pas de données.
Élimination
Après utilisation sous-cutanée, la demi-vie d'élimination s'élève à 100 minutes. La majeure partie des peptides est excrétée avec les fèces, environ 32% d'entre eux sont excrétés en l'état par l'urine.
Cinétique pour certains groupes de patients
Troubles de la fonction rénale
Un dysfonctionnement rénal n'a eu aucune influence sur l'exposition totale (AUC) à l'octréotide lorsqu'il est administré par voie sous-cutanée.
Troubles de la fonction hépatique
Une cirrhose – mais pas une stéatose hépatique – diminue l'élimination (30%) de l'octréotide.
Données précliniquesMutagénicité
L'octréotide et/ou ses métabolites administrés par voie sous-cutanée n'ont montré aucun potentiel mutagène lors d'essais in vitro sur des souches validées de bactéries ou de cellules de mammifères. Dans une étude, une augmentation d'aberrations chromosomiques a été observée dans les cellules V79 du hamster chinois, mais uniquement à des concentrations élevées et cytotoxiques. Sur des lymphocytes humains incubés en présence d'acétate d'octréotide, aucune augmentation d'aberrations chromosomiques n'a cependant été observée. Aucune activité clastogénique n'a été observée in vivo dans la moelle osseuse de souris traitées par l'octréotide (test du micronucleus); de même, aucun indice de génotoxicité n'a été observé dans les têtes de spermatozoïdes de souris mâles (test de réparation de l'ADN). Le contrôle des microsphères dans des essais standard pour la génotoxicité n'a montré aucun potentiel mutagène.
Carcinogénicité/toxicité chronique
Aucun potentiel de toxicité chronique pour l'utilisation humaine n'a été révélé lors des études à long terme chez le rat, la souris et le chien.
Une étude de carcinogénicité de 116 semaines a été conduite chez le rat avec de l'octréotide administré par voie s.c. Des adénocarcinomes de l'endomètre, dont l'incidence chez les animaux ayant reçu la dose la plus élevée de 1.25 mg/kg/j par voie s.c. était statistiquement significative, ont été observés. Ces observations étaient apparemment liées à un trouble de l'équilibre hormonal. Les données à disposition laissent supposer que les tumeurs survenant chez le rat par cette voie endocrine, sont spécifiques à l'espèce et ne sont donc pas déterminantes pour l'homme.
Toxicité sur la reproduction
Des études sur la toxicité sur la reproduction et le développement ont été menées sur des rats et des lapins auxquels des doses allant jusqu'à 1 mg/kg de poids corporel par jour ont été administrées. L'octréotide n'a montré aucune incidence sur la fertilité chez les rats mâles et femelles. Aucun signe d'effet tératogène, embryonnaire/fœtal ni autre incidence sur la reproduction dus à l'octréotide n'ont été relevés chez la progéniture des rats. Un certain retard passager de la croissance physiologique chez les jeunes très certainement dû à l'inhibition de l'hormone de croissance en raison de l'effet pharmacodynamique marqué a été constaté. Des études sur le développement pré- et postnatal ont révélé une descente tardive des testicules chez la progéniture mâle des mères traitées pendant la gestation et l'allaitement. La fertilité des jeunes animaux F1 concernés était toutefois normale. Nous supposons que ces observations sont à mettre sur le compte de l'inhibition de la croissance due à l'octréotide.
Les microsphères sont restées sans effet toxicologique sur la reproduction après examen dans des analyses standard de la toxicité sur la reproduction chez le rat et le lapin.
Remarques particulièresIncompatibilités
Les microsphères Sandostatine LAR pour injection doivent être utilisées exclusivement pour la préparation d'une dose unique et ne doivent pas être diluées avec ou mélangées à d'autres substances. C'est pourquoi aucune donnée sur la compatibilité avec d'autres solutions ou substances n'a été collectée.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Pour l'entreposage prolongé, les flacons de Sandostatine LAR doivent être conservés au réfrigérateur entre 2 et 8°C, à l'abri du gel et à l'abri de la lumière.
Sandostatine LAR peut être conservé à une température ne dépassant pas 25°C pendant environ 24 h avant l'injection. Toutefois, la suspension à injecter doit être préparée extemporanément.
Les médicaments doivent être tenus hors de portée des enfants.
Remarques concernant la manipulation
Instructions pour l'injection i.m. de Sandostatine LAR
Sandostatine LAR ne doit être administré que par du personnel médical formé.
Sandostatine LAR ne doit être administré que par injection intraglutéale profonde, JAMAIS par voie intraveineuse.
Suivre les instructions ci-après scrupuleusement afin de garantir un mouillage parfait de la poudre et sa suspension homogène avant l'injection i.m.
La suspension doit être préparée uniquement extemporanément.
En cas d'injections répétées, alterner entre le muscle fessier droit et gauche.
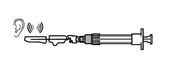
Contenu de l'emballage:
|
|

| |
a
|
Flacon de substance sèche Sandostatine LAR
| |
b
|
Seringue avec solvant
| |
c
|
Adaptateur pour flacon
| |
d
|
Aiguille de sécurité
|
La reconstitution doit être réalisée dans des conditions aseptiques.
|
Étape 1
| |
|

|
Sortir le kit d'injection Sandostatine LAR du réfrigérateur et le laisser atteindre la température ambiante. Pour ce faire, laisser le kit pendant au moins 30 à 60 min à température ambiante, mais pas plus de 24 h.
| |
Étape 2
| |
|

|
Enlever l'obturateur du bouchon du flacon de Sandostatine LAR.
Désinfecter le bouchon en caoutchouc du flacon avec un tampon d'alcool.
Retirer ensuite le film de protection de la plaquette thermoformée qui contient l'adaptateur pour le flacon. NE PAS retirer l'adaptateur de la plaquette thermoformée. Positionner l'adaptateur avec la plaquette thermoformée sur le flacon. Presser l'adaptateur sur le flacon jusqu'à ce qu'il s'enclenche de manière audible.
Une fois l'adaptateur enclenché, retirer la plaquette thermoformée verticalement vers le haut et l'éliminer.
| |
Étape 3
| |
|

|

|
Ôter le capuchon de protection de la seringue préremplie contenant le solvant et visser la seringue sur l'adaptateur du flacon.
Pousser lentement le piston vers le bas et injecter tout le solvant dans le flacon sans remuer la substance sèche de Sandostatine LAR.
| |
Étape 4
| |
|

|
Ne pas remuer le flacon pendant 5 min jusqu'à ce que le solvant ait mouillé entièrement la substance sèche de Sandostatine LAR. Préparer le patient pendant ce temps.
Sans retourner le flacon, contrôler la poudre au fond et sur les parois du flacon. S'il reste des endroits où la poudre n'est pas mouillée, laisser encore reposer le flacon afin d'obtenir un mouillage complet de la poudre.
Remarque: il est normal que le piston remonte légèrement car le flacon est soumis à une légère surpression.
| |
Étape 5
| |
|

|
Lorsque la substance sèche est complètement imbibée, enfoncer entièrement le piston dans la seringue préremplie.
Maintenir le piston enfoncé et remuer modérément le flacon par des mouvements circulaires horizontaux pendant au moins 30 s. Vérifier que la poudre au fond et sur les parois du flacon est complètement dissoute (une suspension homogène d'apparence laiteuse doit être obtenue). Si ce n'est pas le cas, remuer à nouveau modérément le flacon par des mouvements circulaires horizontaux pendant env. 30 s.
ATTENTION: ne pas agiter le flacon violemment, cela pourrait provoquer une floculation ce qui rendrait la suspension inutilisable.
| |
Étape 6
| |
|
Désinfecter le point d'injection avec un tampon d'alcool.
| |
|

|
Retourner la seringue préremplie avec le flacon à la verticale et tirer lentement le piston afin d'aspirer le contenu du flacon dans la seringue préremplie.
| |
|

|
Ensuite, dévisser immédiatement la seringue préremplie de l'adaptateur.
| |
Étape 7
| |
|

|
Visser l'aiguille de sécurité sur la seringue.
Déplacer doucement la seringue préremplie afin de maintenir une suspension laiteuse homogène.
| |
|

|
Retirer le capuchon de protection de l'aiguille.
Tapoter légèrement la seringue préremplie pour éliminer toutes les bulles d'air visibles. Vérifier que le site d'injection ne soit pas contaminé.
Poursuivre immédiatement avec l'étape 8.
| |
Étape 8
| |
|

|

|
Piquer l'aiguille à un angle de 90° par rapport à la peau dans le muscle fessier droit ou gauche et aspirer pour s'assurer que l'aiguille n'a pas pénétré dans un vaisseau sanguin. Sinon, la position de l'aiguille doit être modifiée.
Injecter lentement et profondément dans le muscle fessier avec une pression régulière jusqu'à ce que la seringue soit vide.
Une fois l'injection terminée, activer le dispositif de protection pour aiguilles, comme indiqué à l'étape 9.
| |
Étape 9
| |
|

|
Le dispositif de protection pour aiguilles est activé d'une seule main:
A) soit en pressant l'encoche du dispositif de protection sur une surface dure (figure A),
B) soit en poussant l'encoche vers l'avant avec l'index (figure B).
| |
|
|
Un «clic» audible indique que le dispositif de protection a été correctement activé.
Éliminer immédiatement l'adaptateur et la seringue préremplie dans un récipient de sécurité ou dans une poubelle stable.
|
Numéro d’autorisation53161 (Swissmedic).
PrésentationSandostatine LAR 10 mg en flacon: 1 [A]
Sandostatine LAR 20 mg en flacon: 1 [A]
Sandostatine LAR 30 mg en flacon: 1 [A]
Titulaire de l’autorisationNovartis Pharma Schweiz AG, Risch; Domicile: 6343 Rotkreuz.
Mise à jour de l’informationJanvier 2021.
|