CompositionPrincipes actifs
Epoetinum beta (érythropoïétine humaine recombinante) (rHEPO) produit par génie génétique dans des cellules CHO.
Excipients
Ureum, natrii chloridum, natrii dihydrogenphosphas dihydricus, dinatrii phosphas dodecahydricus, calcii chloridum dihydricum, polysorbatum 20 (produit à partir de maïs génétiquement modifié), glycinum, L-leucinum, L-isoleucinum, L-threoninum, acidum L-glutamicum, L-phenylalaninum, aqua ad iniectabile.
Recormon PS 2000/3000/4000/5000 0.48 mg de sodium par seringue préremplie.
Recormon PS 10 000 0.96 mg de sodium par seringue préremplie.
Recormon PS 30 000 1.00 mg de sodium par seringue préremplie.
Indications/Possibilités d’emploiAnémie chez des patients souffrant de néphropathie chronique
Traitement de l'anémie rénale symptomatique nécessitant une transfusion, dans l'insuffisance rénale chronique chez l'adulte, dialysé et non dialysé, et chez l'enfant de plus de 2 ans (en ce qui concerne les taux d'Hb limites, voir «Posologie/Mode d'emploi»).
Affections tumorales
Traitement de l'anémie symptomatique chez le patient adulte présentant une affection tumorale non myéloïde et traité par chimiothérapie (en ce qui concerne les taux d'Hb limites, voir «Posologie/Mode d'emploi»).
Prélèvement autologue différé (Programme de prélèvement autologue différé)
Augmentation du volume de sang autologue chez un patient participant à un programme de prélèvement autologue différé destiné à éviter les inconvénients de la transfusion homologue. L'utilisation dans cette indication doit être évaluée par rapport au risque accru d'événements thromboemboliques qui a été rapporté. Le traitement est indiqué chez les patients atteints d'une anémie modérée (Hb de 10 à 13 g/dl (de 6,21 à 8,07 mmol/l) sans carence en fer), si les méthodes d'épargne sanguine ne sont pas disponibles ou sont insuffisantes, soit lorsque l'importante intervention prévue nécessite un volume de sang important (au moins 4 unités de sang chez la femme ou au moins 5 unités chez l'homme), soit lorsque la période requise pour obtenir le volume de sang autologue nécessaire est trop courte.
Posologie/Mode d’emploiLe traitement par Recormon doit être instauré par des médecins possédant une expérience dans les indications susmentionnées.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Anémie chez des patients souffrant de néphropathie chronique
Les patients doivent faire l'objet d'une surveillance étroite afin de s'assurer que la plus faible dose efficace autorisée de Recormon soit utilisée pour contrôler convenablement les symptômes de l'anémie tout en maintenant un taux d'hémoglobine inférieur ou égal à 12 g/dl (7,5 mmol/l). La prudence est de mise lors des augmentations de la dose de Recormon chez les patients présentant une insuffisance rénale chronique. Chez les patients présentant une réponse faible du taux d'hémoglobine à Recormon, d'autres causes de cette faible réponse doivent être recherchées (voir «Mises en garde et précautions» et «Propriétés/Effets»). Les symptômes d'une anémie et ses manifestations secondaires peuvent varier selon l'âge, le sexe et le degré de sévérité général de la maladie; une évaluation médicale de l'évolution clinique et de l'état du patient respectif est donc nécessaire. Toutes les formulations de Recormon peuvent être administrées aussi bien par voie sous-cutanée que par voie intraveineuse pour augmenter le taux d'hémoglobine à une valeur maximale de 12 g/dl (7,5 mmol).
L'injection sous-cutanée permet d'atteindre l'objectif d'un taux maximal d'hémoglobine de 12 g/dl (7,5 mmol) au moyen d'une dose réduite d'environ 20-25% par rapport à l'injection intraveineuse.
En raison des variations intraindividuelles rencontrées chez les patients, on peut observer occasionnellement des taux d'hémoglobine supérieurs ou inférieurs aux concentrations souhaitées. De telles variations de l'hémoglobine peuvent être compensées par un ajustement de la dose, en tenant compte de la fourchette cible de l'hémoglobine de 10 g/dl (6,2 mmol/l) à 12 g/dl (7,5 mmol/l). Il faut éviter la persistance d'un taux d'hémoglobine supérieur à 12 g/dl (7,5 mmol/l); les directives relatives à l'ajustement adéquat de la dose en cas de survenue d'un taux d'hémoglobine supérieur à 12 g/dl (7,5 mmol/l) sont décrites ci-après.
Il faut éviter une augmentation de l'hémoglobine de plus de 2 g/dl (1,25 mmol/l) en l'espace de quatre semaines. Au cas où cela se produirait, il faut procéder comme prévu à un ajustement approprié de la dose. Si l'augmentation du taux d'hémoglobine est supérieure à 2 g/dl (1,25 mmol/l) en un mois ou si le taux d'hémoglobine augmente et approche les 12 g/dl (7,45 mmol/l), la dose doit être réduite d'environ 25%. Si le taux d'hémoglobine continue d'augmenter, le traitement doit être interrompu jusqu'à ce que le taux d'hémoglobine recommence à baisser, puis le traitement doit être repris à une dose réduite d'environ 25% par rapport à la dose précédemment administrée.
Pour stabiliser le taux d'hémoglobine cible, il faut s'assurer que l'on utilise la dose efficace la plus faible de Recormon qui permet d'obtenir le contrôle des symptômes de l'anémie.
En présence d'une hypertension ou d'affections cardiovasculaires, cérébrovasculaires ou vasculaires périphériques, l'augmentation hebdomadaire de l'hémoglobine et le taux cible d'hémoglobine devront être déterminés individuellement en tenant compte du tableau clinique.
Le traitement par toutes les formulations de Recormon comporte deux phases distinctes.
Phase de correction
Administration sous-cutanée
La dose initiale est de 3× 20 UI/kg de poids corporel par semaine. Si l'élévation de la valeur de l'hémoglobine est insuffisante (< 0,25 g/dl par semaine), on peut augmenter la dose toutes les 4 semaines de 3× 20 UI/kg/semaine. La posologie hebdomadaire peut aussi être fractionnée en doses quotidiennes.
Administration intraveineuse
La dose initiale est de 3× 40 UI/kg de poids corporel par semaine. On peut, au bout de 4 semaines, porter la dose à 80 UI/kg de poids corporel 3× par semaine, puis l'augmenter au besoin, tous les mois, de 20 UI supplémentaires par kg de poids corporel 3× par semaine.
Indépendamment du mode d'administration, la dose hebdomadaire maximale ne doit pas dépasser 720 UI/kg de poids corporel.
Phase d'entretien
Pour stabiliser le taux d'hémoglobine entre 10 et 12 g/dl, réduire la posologie tout d'abord à 50% de la dernière dose administrée en phase de correction, puis l'ajuster à 1 ou 2 semaines d'intervalle en fonction des besoins individuels du patient pour obtenir la dose d'entretien.
La dose d'entretien hebdomadaire peut être injectée par voie sous-cutanée en une seule fois ou répartie en trois à sept doses unitaires. Les patients dont l'état est stable lors d'une administration monohebdomadaire peuvent passer à un rythme d'administration d'une fois toutes les deux semaines. Dans ce cas, une augmentation de la dose peut s'avérer nécessaire. L'expérience enseigne que les patients dont la valeur de l'hémoglobine est initialement inférieure à 6 g/dl ont besoin de doses d'entretien plus importantes que ceux dont l'hémoglobine a une valeur plus élevée au départ. Ne pas dépasser un taux d'hémoglobine de 12 g/dl.
Utilisation chez les patients pédiatriques
Des essais cliniques chez des patients pédiatriques ont montré que les doses d'entretien sont en moyenne d'autant plus élevées que le sujet est plus jeune. On s'en tiendra toutefois au schéma posologique recommandé puisqu'on ne peut préjuger de la réponse individuelle.
Utilisation chez les patients âgés
Aucune étude spécifique n'a été menée. Les essais cliniques sur Recormon ont inclus une forte proportion de sujets âgés, mais rien ne suggérait qu'un ajustement posologique particulier soit nécessaire pour cette population de patients.
Patients présentant une insuffisance hépatique
Aucun essai clinique spécifique n'a été mené chez des sujets atteints d'insuffisance hépatique. Il n'y a pas d'instructions posologiques spéciales pour ces patients.
Durée du traitement
Le traitement par toutes les formulations de Recormon est conçu pour une longue durée. Il peut être interrompu à tout moment.
Affections tumorales
Recormon doit être administré par voie sous-cutanée aux patients anémiques (p.ex. taux d'hémoglobine ≤10 g/dl (6,2 mmol/l)). Les symptômes d'une anémie et ses manifestations secondaires peuvent varier selon l'âge, le sexe et le degré de sévérité général de la maladie; une évaluation médicale de l'évolution clinique et de l'état du patient respectif est donc nécessaire.
La dose hebdomadaire peut être injectée en une seule fois ou répartie en 3 à 7 doses unitaires.
La dose initiale recommandée est de 450 UI par kg de poids corporel et par semaine. On peut donner une dose fixe de 30 000 UI une fois par semaine, en se basant sur un poids moyen du patient de 67 kg, chez les patients ne présentant pas de grandes fluctuations de poids. En cas de réponse insuffisante imposant une augmentation de la dose, il faut cependant recourir à une posologie rapportée au poids.
En raison des variations intraindividuelles rencontrées chez les patients, on peut observer occasionnellement des taux d'hémoglobine supérieurs ou inférieurs aux concentrations souhaitées. De telles variations de l'hémoglobine peuvent être compensées par un ajustement de la dose. La fourchette cible de l'hémoglobine est comprise entre 10 g/dl (6,2 mmol/l) et 12 g/dl (7,5 mmol/l). Il faut éviter la persistance d'un taux d'hémoglobine supérieur à 12 g/dl (7,5 mmol/l).
Ajustements de la dose
Si au bout de 4 semaines de traitement, le taux d'hémoglobine a augmenté d'au moins 1 g/dl (0,62 mmol/l), il faut conserver la dose actuelle. La dose doit être doublée si le taux d'hémoglobine n'a pas augmenté d'au moins 1 g/dl (0,62 mmol/l) au bout de 4 semaines. Si la valeur de l'hémoglobine n'a pas augmenté d'au moins 1 g/dl (0,62 mmol/l) après huit semaines de traitement, il y a lieu de douter de l'efficacité ultérieure du traitement.
La dose maximale de 900 UI par kg de poids corporel et par semaine ne doit pas être dépassée.
Une fois l'objectif thérapeutique individuel atteint, il convient de réduire la dose de 25 à 50% afin de stabiliser le taux d'hémoglobine du patient. Une titration correspondante de la dose devra être envisagée. Si l'augmentation du taux d'hémoglobine est supérieure à 2 g/dl (1,3 mmol/l) en 4 semaines, la dose doit être réduite de 25 à 50%.
Si le taux d'hémoglobine dépasse 12 g/dl (7,5 mmol/l), il faut réduire la dose d'environ 25 à 50%. Il faut arrêter passagèrement le traitement par Recormon si le taux d'hémoglobine dépasse 13 g/dl (8,1 mmol/l). Une fois que le taux d'hémoglobine est à nouveau ≤12 g/dl (7,5 mmol/l) le traitement doit être repris à une dose réduite d'environ 25% par rapport à la dose précédemment administrée.
Au terme de la chimiothérapie, le traitement par Recormon doit être poursuivi pendant une période pouvant aller jusqu'à 3 semaines.
Pour stabiliser le taux d'hémoglobine cible, il faut s'assurer que l'on utilise la dose efficace la plus faible de Recormon qui permet d'obtenir le contrôle des symptômes de l'anémie.
Prélèvement autologue différé (Programme de prélèvement autologue différé)
La solution de Recormon est administrée par voie intraveineuse en deux minutes. Si un don de sang est possible (hématocrite >33 vol.-%), Recormon doit être administré après le prélèvement. On ne dépassera pas un hématocrite de 48 vol.-% pendant toute la durée du traitement.
La posologie individuelle doit être déterminée par le médecin traitant en fonction du volume de sang autologue à prélever et de la réserve érythrocytaire endogène du patient en question.
1.Le volume de sang autologue à prélever dépend des pertes sanguines prévisibles, de la mise en œuvre de méthodes d'épargne sanguine et de la condition physique du patient. Ce volume doit être suffisant pour éviter le recours à une transfusion homologue. Il correspond généralement au nombre de conserves de sang requises. Le volume de sang autologue nécessaire s'exprime en unités, chaque unité correspondant à 450 ml de sang total avec un hématocrite de 40%.
2.La capacité d'un patient à donner du sang est surtout fonction de son volume sanguin et de la valeur initiale de l'hématocrite. Ces deux variables déterminent l'importance de la réserve érythrocytaire endogène, calculée de la manière suivante:
Réserve érythrocytaire endogène (en ml) = volume sanguin (en ml) × (hématocrite-33): 100.
Calcul du volume sanguin
Chez l'homme
Volume sanguin (en ml) = 44 (ml/kg) × poids corporel (en kg) + 1600 (ml).
Chez la femme
Volume sanguin (en ml) = 41 (ml/kg) × poids corporel (en kg) + 1200 (ml) (pour un poids corporel >45 kg).
Les nomogrammes suivants permettent de décider si un traitement par une formulation de Recormon est nécessaire et, si oui, de déterminer la dose unitaire, sur la base du volume de sang autologue à prélever et de la réserve érythrocytaire endogène.
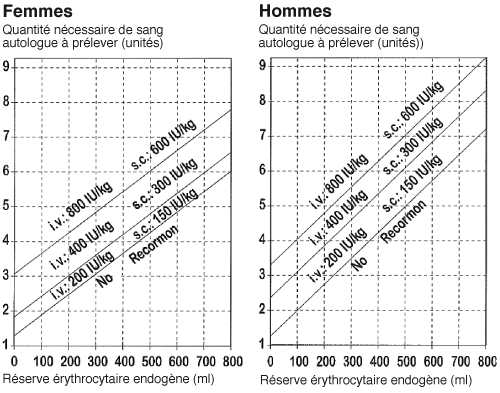
La dose unitaire ainsi déterminée est administrée en deux prises hebdomadaires pendant 4 semaines. On ne dépassera pas une dose maximale de 1600 UI/kg de poids corporel par semaine.
Instructions spéciales concernant l'administration
Des réactions anaphylactoïdes ayant été observées dans des cas isolés, la première administration ne doit avoir lieu que dans des conditions cliniques, en respectant un délai de surveillance minimale d'au moins 4 heures.
Anémie chez des patients souffrant de néphropathie chronique
Toutes les formulations de Recormon peuvent être administrées par voie sous-cutanée ou intraveineuse. Par voie intraveineuse, la solution doit être injectée en 2 min environ, aux hémodialysés p.ex. dans l'aiguille à fistule à la suite de la dialyse.
Chez le patient non hémodialysé souffrant d'une anémie d'origine rénale, préférer l'administration sous-cutanée pour éviter de trop solliciter les veines. L'injection sous-cutanée doit également se faire lentement, en veillant à ne pas dépasser si possible 1 ml par point d'injection pour réduire la douleur chez les patients qui y sont sensibles. Les volumes importants seront injectés en plusieurs points ou l'aiguille sera retirée sous la peau et l'injection poursuivie dans une autre direction.
Lorsque le patient s'administre lui-même le médicament par voie sous-cutanée, il convient de lui donner des instructions précises sur la technique d'auto-injection. Un contrôle médical régulier s'impose.
L'administration concomitante de fer implique, le cas échéant, une diminution de la dose de rhEPO afin d'éviter l'élévation trop rapide et trop importante du taux d'hémoglobine.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients conformément à la composition.
Recormon ne doit pas être utilisé en présence d'une hypertension artérielle non traitée ou difficilement contrôlable.
Recormon ne doit pas être utilisé pour l'obtention de sang autologue (programme de prélèvement autologue différé) chez les patients ayant présenté un infarctus du myocarde ou un accident vasculaire cérébral au cours du mois précédant le traitement, chez les patients avec angor instable ni chez les patients présentant un risque de thrombose veineuse profonde, par exemple ceux avec antécédent connu de thrombo-embolie veineuse.
En l'absence de données cliniques suffisantes, Recormon ne doit pas être administré aux femmes enceintes ou qui allaitent, ni aux enfants de moins de 2 ans.
Mises en garde et précautionsEn présence d'une anémie aiguë sévère nécessitant une correction immédiate, Recormon ne peut se substituer à une transfusion en urgence.
Recormon doit être utilisé avec prudence lors d'anémie réfractaire avec excès de blastes en transformation, d'affection épileptiforme, de thrombocytose, d'insuffisance hépatique chronique et de maladie vasculaire ischémique. Sont exclus du traitement les patients présentant des déficits en acide folique, en vitamine B12 et en fer, car ils réduisent l'efficacité de Recormon.
Patients présentant une insuffisance rénale chronique
La prudence est de mise lors des augmentations de la dose de Recormon chez les patients présentant une insuffisance rénale chronique, car des doses cumulées élevées d'époétine peuvent être associées à un risque accru de mortalité et à des événements cardiovasculaires et cérébrovasculaires sévères. Chez les patients présentant une faible réponse du taux d'hémoglobine aux époétines, d'autres causes de cette faible réponse doivent être recherchées (voir «Posologie/Mode d'emploi» et «Propriétés/Effets»).
Investigations
Pour assurer une érythropoïèse efficace, il faut, chez tous les patients, mesurer le taux de fer avant et pendant le traitement et, le cas échéant, procéder à une substitution martiale conformément aux directives thérapeutiques.
L'efficacité de Recormon peut être atténuée par une accumulation d'aluminium consécutive au traitement de l'insuffisance rénale chronique ou par d'autres causes d'anémie telles qu'une perte de sang occulte, une maladie du système hématopoïétique (thalassémie, myélodysplasie, par exemple) ou une ostéite fibro-kystique.
La kaliémie doit être régulièrement contrôlée pendant le traitement par Recormon. Une hyperkaliémie a été observée chez quelques patients insuffisants rénaux traités par Recormon, sans qu'un lien direct n'ait toutefois pu être établi. En cas d'hyperkaliémie ou d'augmentation de la kaliémie, on devra envisager l'arrêt du traitement par Recormon, jusqu'à la correction de l'hyperkaliémie.
Néphrosclérose
L'utilisation de Recormon chez un patient en prédialyse pour une néphrosclérose doit être décidée au cas par cas, une progression accélérée de l'insuffisance rénale ne pouvant être totalement exclue chez ces patients.
Absence de réponse
Une érythroblastopénie (PRCA = pure red cell aplasia) provoquée par des anticorps anti-érythropoïétine neutralisants a été rapportée en rapport avec un traitement par des ASE, y compris avec l'administration de Recormon. Une réaction croisée avec toutes les protéines érythropoïétiques a été retrouvée pour ces anticorps. Les patients chez qui l'on suspecte des anticorps anti-érythropoïétine neutralisants ou chez qui de tels anticorps ont été décelés, ne doivent pas être mis sous Recormon (voir «Effets indésirables»).
Tension artérielle
Chez les patients atteints d'une néphropathie chronique ou d'un cancer, une augmentation de la tension artérielle ou l'accentuation d'une hypertension déjà existante peut survenir, notamment en cas d'élévation rapide de l'hématocrite. Ces hypertensions relèvent d'un traitement médicamenteux.
Si le traitement médicamenteux ne permet pas de contrôler l'hypertension, une interruption passagère du traitement par Recormon est recommandée. Par ailleurs, un contrôle régulier de la tension artérielle est recommandé, surtout au début du traitement, ainsi qu'entre les dialyses. Dans des cas isolés, même des patients initialement normotendus ou hypotendus peuvent présenter une crise hypertensive avec des symptômes à type d'encéphalopathie nécessitant une prise en charge médicale immédiate en soins intensifs. La survenue brutale de céphalées lancinantes à type de migraine peut constituer un signal d'alarme à prendre très au sérieux (voir «Effets indésirables»).
SCAR
Des réactions graves d'hypersensibilité cutanée (SCAR; severe cutaneous adverse reactions), telles que le syndrome de Stevens-Johnson (SSJ) et la nécrolyse épidermique toxique (NET, syndrome de Lyell), qui peuvent mettre en jeu le pronostic vital ou avoir une issue fatale, ont été rapportées chez des patients traités par de l'époétine. Des cas particulièrement graves ont été observés lors de l'administration de formes retard.
Il convient d'attirer l'attention des patients sur les signes et symptômes au moment de la prescription et d'assurer une surveillance étroite quant à la survenue de réactions cutanées chez eux. Les signes typiques sont la fièvre, une éruption cutanée douloureuse qui s'étend, souvent précédée de fièvre et de symptômes grippaux, la formation de cloques et une inflammation des yeux et des muqueuses. Si des signes et symptômes évocateurs de telles réactions d'hypersensibilité apparaissent, arrêter immédiatement le traitement par Recormon.
Si un patient a présenté une réaction cutanée grave comme un SSJ ou une NET en raison de l'utilisation de Recormon, le traitement par Recormon ne doit en aucun cas être repris chez lui.
Thrombocytes
Chez les patients atteints d'une néphropathie chronique, il peut se produire une élévation modérée et dose-dépendante du nombre de plaquettes dans les limites de la normale, notamment en cas d'administration intraveineuse. Elle régresse au cours du traitement. Il est recommandé de contrôler régulièrement le nombre de plaquettes dans les 8 premières semaines du traitement. Le nombre de plaquettes et l'hématocrite/le taux d'hémoglobine doivent être systématiquement mesurés à intervalles réguliers, chez tous les patients.
Concentration d'hémoglobine d'entretien
Chez les patients atteints d'une néphropathie chronique, le taux d'hémoglobine d'entretien ne doit pas dépasser la limite supérieure du taux d'hémoglobine cible recommandé sous «Posologie/Mode d'emploi». Dans les études cliniques, un risque accru de décès et d'événements cardio-vasculaires sévères a été observé lors de l'utilisation d'agents stimulant l'érythropoïèse (ASE) destinés à obtenir un taux cible d'hémoglobine supérieur à 12 g/dl (7,5 mmol/l).
Les études cliniques contrôlées n'ont pas montré d'avantages significatifs lorsque le taux d'hémoglobine a été augmenté au-delà de ce qui est nécessaire pour maîtriser les symptômes de l'anémie et éviter une transfusion sanguine.
Patients cancéreux
Les ASE sont des facteurs de croissance qui stimulent principalement la production de globules rouges. Des récepteurs de l'érythropoïétine peuvent être produits à la surface de diverses cellules tumorales. On soupçonne que les ASE pourraient, comme tous les facteurs de croissance, stimuler la croissance de tumeurs. Aucune preuve d'une modification de la survie globale ou du risque de progression tumorale chez des patients atteints d'une anémie liée à un cancer n'a pu être fournie dans plusieurs études cliniques.
Les observations suivantes ont été faites dans des études cliniques contrôlées lors de l'utilisation de Recormon et d'autres ASE:
- Un raccourcissement du délai jusqu'à la progression tumorale chez les patients atteints de tumeurs avancées de la tête et du cou et traités par radiothérapie, lorsque le médicament a été administré jusqu'à atteindre un taux d'hémoglobine supérieur à 14 g/dl (8,7 mmol/l).
- Une survie globale plus courte et plus de décès imputables à la progression de la maladie au bout de 4 mois chez des patientes atteintes d'un cancer du sein métastatique et traitées par une chimiothérapie, lorsque le médicament a été administré jusqu'à atteindre un taux d'hémoglobine supérieur à 12-14 g/dl (7,5-8,7 mmol/l).
- Un risque accru de décès lorsque le médicament a été administré jusqu'à atteindre un taux cible d'hémoglobine de 12 g/dl (7,5 mmol/l) chez des patients atteints d'affections malignes évolutives qui ne recevaient ni chimiothérapie ni radiothérapie. Les ASE ne sont pas indiqués pour le traitement de cette population de patients.
Le nombre de plaquettes et le taux d'hémoglobine doivent être contrôlés à intervalles réguliers chez les patients cancéreux.
Avant l'utilisation de préparations d'époétine chez les patientes et les patients atteints de tumeur et symptomatiques d'une anémie sous chimiothérapie myélosuppressive, il faut procéder avec la patiente/le patient à une évaluation soigneuse et individuelle du rapport risque/bénéfice en tenant particulièrement compte du pronostic. Les préparations d'époétine ne doivent pas être administrées dans le cadre d'un traitement curatif.
Héparine
Chez les patients atteints d'une néphropathie chronique, le taux élevé d'hématocrite nécessite fréquemment au cours du traitement par Recormon une augmentation de la dose d'héparine pendant l'hémodialyse. Si l'héparinisation n'est pas optimale, une obstruction du dialyseur peut se produire. Des thromboses du shunt peuvent survenir, surtout chez les patients ayant tendance à avoir une hypotension ou présentant des complications au niveau de leur fistule artério-veineuse (p.ex. sténoses, anévrysmes, etc.). Une révision précoce du shunt ainsi qu'une prophylaxie antithrombotique par l'acide acétylsalicylique sont recommandées chez ces patients.
L'expérience acquise dans le traitement de l'anémie par Recormon lors de myélome multiple, de lymphome non hodgkinien et de leucémie lymphoïde chronique est limitée aux patients adultes.
Prélèvement autologue différé
Chez les patients participant à un programme de prélèvement autologue différé, une augmentation du nombre de plaquettes, en général dans les limites de la normale, peut survenir. Il est donc recommandé de contrôler au moins une fois par semaine le nombre de plaquettes. Le traitement par Recormon doit être interrompu si l'élévation est supérieure à 150 x 109/l ou si le nombre de plaquettes est supérieur à la normale.
L'utilisation de Recormon chez un patient participant à un programme de prélèvement autologue différé doit respecter les directives officielles régissant le don de sang, notamment les suivantes:
·Les patients dont l'hématocrite est >33 vol.-% (hémoglobine >11 g/dl (6,83 mmol/l)) peuvent donner leur sang.
·Les patients dont le poids corporel est inférieur à 50 kg doivent faire l'objet d'une surveillance particulière.
·Le volume d'un don de sang individuel ne doit pas dépasser approximativement 12% du volume sanguin du patient.
Abus
L'usage abusif auquel pourraient recourir des sujets sains (à des fins de dopage notamment) peut entraîner une augmentation excessive de l'hématocrite, d'où un risque de complications cardiovasculaires menaçant le pronostic vital (risque de thrombose par polyglobulie avec concentration élevée d'hémoglobine).
Phénylalanine
Recormon contient de la phénylalanine comme excipient. Ceci doit être pris en considération chez les patients présentant des formes sévères de phénylcétonurie.
Sodium
Ce médicament contient moins de 1 mmol de sodium (23 mg) par seringue préremplie, c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsDonnées in vivo
L'expérimentation animale a montré que Recormon ne majore pas la toxicité médullaire exercée à l'égard des neutrophiles et des thrombocytes par des cytostatiques tels que l'étoposide, le cisplatine, le cyclophosphamide et le fluorouracil.
Autres interactions
L'action de Recormon peut être retardée ou atténuée par une intoxication à l'aluminium ou une infection.
La concentration d'hémoglobine augmentée par l'action de Recormon peut potentialiser l'effet des antiangineux.
Effet de Recormon sur d'autres médicaments
Les antihypertenseurs administrés simultanément peuvent avoir une efficacité réduite.
La poursuite concomitante d'un traitement par des anticonvulsivants peut aggraver la tendance aux convulsions.
Etant donné que la cyclosporine A se lie aux hématies, une interaction avec celle-ci reste possible. Si Recormon est administré en association avec la cyclosporine A, les taux sanguins de cette dernière doivent être surveillés. Le cas échéant, la dose de cyclosporine A sera ajustée en conséquence.
Effet d'autres médicaments sur Recormon
L'effet érythropoïétique de la rhEPO peut être potentialisé par l'administration concomitante de substances hématopoïétiquement actives destinées à pallier un état de carence, telles que le sulfate ferreux, la cyanocobalamine ou l'acide folique.
Grossesse, allaitementLes études de reproduction chez l'animal n'ont pas démontré de risque fœtal, mais on ne dispose d'aucune étude chez la femme enceinte.
En l'absence d'études chez la femme enceinte ou en période d'allaitement, Recormon est contre-indiqué pendant la grossesse et la période d'allaitement (voir «Contre-indications»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesRecormon n'a aucune influence ou une influence négligeable sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirablesLes effets indésirables ci-après ont été observés:
Toutes les indications
Des réactions graves d'hypersensibilité cutanée (SCAR), telles que le syndrome de Stevens-Johnson (SSJ), la nécrolyse épidermique toxique (NET, syndrome de Lyell), l'exfoliation cutanée et l'érythème polymorphe, ont été rapportées (voir «Mises en garde et précautions»). Leur fréquence n'est pas connue.
Anémie chez des patients souffrant de néphropathie chronique
L'effet indésirable le plus fréquent (21,9%) sous traitement par Recormon est une augmentation dose-dépendante de la pression artérielle ou l'aggravation d'une hypertension artérielle préexistante, notamment en cas d'augmentation rapide du taux d'hématocrite (voir «Mises en garde et précautions»). Dans des cas isolés, même des patients initialement normotendus ou hypotendus peuvent présenter une crise hypertensive avec symptômes à type d'encéphalopathie (céphalées, états confusionnels, troubles sensori-moteurs tels que troubles du langage et de la marche, etc. jusqu'à des crises tonico-cloniques généralisées) nécessitant une prise en charge en soins intensifs (voir «Mises en garde et précautions»).
Des thromboses du shunt peuvent survenir, surtout chez les patients ayant tendance à avoir une hypotension ou présentant des complications au niveau de leur fistule artério-veineuse (p.ex. sténoses, anévrysmes, etc.) (voir «Mises en garde et précautions»). Parallèlement à l'augmentation du taux d'hématocrite, il se produit dans la plupart des cas une diminution de la ferritinémie (voir «Mises en garde et précautions»). En outre, une augmentation transitoire de la kaliémie et de la phosphatémie a été observée dans des cas isolés (voir «Mises en garde et précautions»).
En particulier lors d'administration intraveineuse de Recormon, on peut observer une augmentation modeste, dose-dépendante, du nombre des plaquettes, qui reste dans les limites de la normale et qui régresse au fil du traitement. Il est très rare de voir s'installer une thrombocytose (voir «Mises en garde et précautions»).
Dans de très rares cas, des insuffisants rénaux chroniques traités par la rHEPO ont développé des anticorps anti-érythropoïétine neutralisants avec ou sans érythroblastopénie (PRCA). En présence d'un diagnostic de PRCA, le traitement par l'époétine doit être arrêté. En aucun cas, les patients ne doivent être mis sous une autre époétine (voir «Mises en garde et précautions»).
Les fréquences des effets indésirables rapportés au cours des études cliniques avec Recormon sont mentionnées ci-dessous. A l'intérieur de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Les effets indésirables sont rangés par classe de système d'organes de la classification MedDRA et par fréquence selon la convention suivante: «très fréquents» (≥1/10), «fréquents» (≥1/100 à <1/10), «occasionnels» (≥1/1000 à <1/100), «rares» (≥1/10'000 à <1/1000), «très rares» (<1/10'000), «fréquence inconnue» (ne peut être estimée sur la base des données disponibles).
Affections vasculaires
Fréquent: hypertension artérielle.
Occasionnel: crise hypertensive.
Affections du système nerveux
Fréquent: céphalées.
Affections hématologiques et du système lymphatique
Rare: thrombose du shunt.
Très rare: thrombocytose.
Affections tumorales
Des céphalées et une hypertension artérielle, causées par un traitement par l'époétine bêta et relevant d'un traitement médicamenteux, ont été observées dans 1 à 10% des cas (voir «Mises en garde et précautions»).
Chez quelques patients, une baisse des paramètres du fer sérique a été observée (voir «Mises en garde et précautions»).
Au cours d'études cliniques, une incidence légèrement accrue d'événements thrombo-emboliques a été observée chez des cancéreux traités par Recormon par rapport à des patients non traités ou recevant un placebo. De tels événements ont été observés chez 7% des patients traités par Recormon, contre 4% des patients du groupe témoin; cette différence par rapport au groupe témoin n'a toutefois été liée à aucune augmentation de la mortalité due à des événements thrombo-emboliques.
Les fréquences des effets indésirables rapportés au cours des études cliniques avec Recormon sont mentionnées ci-dessous. A l'intérieur de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Affections vasculaires
Fréquent: hypertension artérielle.
Affections hématologiques et du système lymphatique
Fréquent: événements thrombo-emboliques.
Affections du système nerveux
Fréquent: céphalées.
Prélèvement autologue différé (Programme de prélèvement autologue différé)
Chez les patients participant à un programme de prélèvement autologue différé, une légère augmentation des événements thromboemboliques a été rapportée, sans qu'un lien de causalité avec le traitement par Recormon n'ait toutefois pu être établi.
Dans les études contrôlées contre placebo, une carence martiale transitoire plus marquée a été observée chez les patients traités par Recormon que dans le groupe témoin (voir «Mises en garde et précautions»).
Les fréquences des effets indésirables rapportés au cours des études cliniques avec Recormon sont mentionnées ci-dessous. A l'intérieur de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Affections du système nerveux
Fréquent: céphalées.
Toutes les indications
Dans de rares cas, des réactions cutanées telles qu'éruptions, démangeaisons, urticaire ou réactions au point d'injection peuvent se produire. Dans de très rares cas (< 0,01%), des réactions anaphylactoïdes ont été rapportées en rapport avec un traitement par l'époétine bêta. Dans le cadre d'études cliniques contrôlées, aucune augmentation des réactions d'hypersensibilité n'a toutefois été mise en évidence.
Des symptômes pseudo-grippaux tels que fièvre, frissons, céphalées, courbatures, sensation de malaise et/ou douleurs osseuses ont été signalés dans de très rares cas, en particulier au début du traitement. Ces réactions étaient d'intensité légère à modérée et ont disparu en quelques heures ou en quelques jours.
Effets indésirables identifiés après la mise sur le marché
Les effets indésirables suivants ont été décrits après la commercialisation de Recormon:
Affections hématologiques et du système lymphatique
Fréquence inconnue: érythroblastopénie (PRCA = pure red cell aplasia).1,2
Affections de la peau et du tissu sous-cutané
Fréquence inconnue: syndrome de Stevens-Johnson (SSJ)/nécrolyse épidermique toxique (NET, syndrome de Lyell).1,2
1 Voir rubrique «Mises en garde et précautions».
2 Les données disponibles ne permettent pas d'évaluer le taux d'incidence ni la catégorie de fréquence.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLa réponse à la rhEPO est individuelle et dose-dépendante. Si une surdose unique ne risque pas de déclencher d'effets indésirables, il n'en est pas de même de surdoses répétées, qui peuvent entraîner hypertension et polyglobulie.
Propriétés/EffetsCode ATC
B03XA01
Mécanisme d'action
L'érythropoïétine humaine recombinante (rhEPO) est une glycoprotéine purifiée qui stimule l'érythropoïèse.
L'époétine bêta, obtenue par génie génétique, présente une composition en acides aminés et en glucides identique à celle de l'érythropoïétine native extraite de l'urine de patients anémiques.
L'érythropoïétine humaine recombinante (rhEPO) a un poids moléculaire de 32000 à 40000 daltons. La protéine, qui représente environ 58% de la molécule, comprend 165 acides aminés. Les 4 chaînes glucidiques sont reliées à la protéine par 3 liaisons N-glycosidiques et une liaison O-glycosidique.
Chez le sujet sain, l'érythropoïétine est produite par les reins et déversée dans le flux sanguin en fonction de la saturation des tissus en oxygène. Avec le sang, l'érythropoïétine parvient à son organe cible, qui est la moelle osseuse.
Pharmacodynamique
Lors de l'administration de rhEPO à des anémiques, le taux d'hémoglobine commence généralement à remonter 2 à 6 semaines après le début du traitement. On n'a pas encore pu démontrer chez l'enfant d'accélération concomitante d'une croissance retardée.
Efficacité clinique
Etudes chez les patients cancéreux
L'érythropoïétine est un facteur de croissance qui stimule principalement la production de globules rouges. Des récepteurs de l'érythropoïétine peuvent être produits à la surface de diverses cellules tumorales.
Le temps de survie et la progression tumorale ont été évalués dans cinq études contrôlées de grande envergure, menées chez au total 2833 patients. Quatre de ces études étaient des études en double aveugle, contrôlées contre placebo et la cinquième étude était une étude ouverte. Deux études ont recruté des patients recevant une chimiothérapie. Le taux cible d'hémoglobine était >13 g/dl dans deux études et de 12-14 g/dl dans les trois autres études. Dans l'étude ouverte, aucune différence quant à la survie globale n'a été constatée entre les patients traités par un ASE et les témoins. Dans les quatre études contrôlées contre placebo, le hazard ratio pour la survie globale a été de 1,25 à 2,47 en faveur des témoins. Ces études ont montré, par rapport aux groupes témoins, une augmentation continue inexpliquée et statistiquement significative, de la mortalité chez les patients atteints d'une anémie liée à différents cancers fréquents et recevant un ASE. Les différences d'incidence des thromboses et des complications reliées à celles-ci entre les patients ayant reçu un ASE et les patients des groupes témoins n'ont pas permis d'expliquer de manière satisfaisante le résultat de la survie globale dans ces études.
Une méta-analyse (reposant sur des valeurs individuelles de patients) regroupant les données des 12 études cliniques contrôlées, réalisées avec Recormon chez des patients cancéreux anémiques (n = 2301), a montré un hazard ratio estimé ponctuellement pour la survie globale de 1,13 en faveur des témoins (IC 95%: 0,87, 1,46). Chez les patients avec un taux initial d'hémoglobine ≤10 g/dl (n = 899), le hazard ratio estimé ponctuellement pour la survie a été de 0,98 (IC 95%: de 0,68 à 1,40).
Un risque relatif accru d'événements thromboemboliques a été observé dans la population globale (RR 1,62, IC 95%: 1,13; 2,31).
Une analyse systématique de plus de 9000 patients cancéreux ayant participé à 57 études cliniques a été effectuée. Une méta-analyse des données de survie globale a montré un hazard ratio estimé ponctuellement pour la survie globale de 1,08 en faveur des témoins (IC 95%: 0,99; 1,18; 42 études et 8167 patients). Un risque relatif accru d'événements thromboemboliques (RR 1,67, IC 95%: 1,35; 2,06, 35 études et 6769 patients) a été observé chez les patients ayant été traités par un ASE. Il existe donc une preuve cohérente permettant de conclure à l'absence d'avantages en matière de survie globale pour les patients cancéreux traités par un ASE. On ignore dans quelle mesure ces résultats pourraient s'appliquer aux patients cancéreux traités par une chimiothérapie et recevant un ASE avec pour objectif un taux d'hémoglobine inférieur à 13 g/dl, car l'ensemble des données étudiées ne contenait que peu de patients présentant ces caractéristiques.
L'efficacité de Recormon lors de myélome multiple, de lymphome non hodgkinien de faible malignité ou de leucémie lymphoïde chronique a été démontrée chez des patients présentant une carence relative en érythropoïétine, définie comme suit:
Taux d'érythropoïétine sérique ≤100 mU/ml pour une valeur de l'hémoglobine de > 9 à < 10 g/dl (> 5,58 à < 6,2 mmol/l).
Taux d'érythropoïétine sérique ≤180 mU/ml pour une valeur de l'hémoglobine de > 8 à < 9 g/dl (> 4,96 à < 5,58 mmol/l).
Taux d'érythropoïétine sérique ≤300 mU/ml pour une valeur de l'hémoglobine ≤8 g/dl (≤4,96 mmol/l).
Etudes chez des patients insuffisants rénaux
Dans une étude randomisée (CREATE) avec l'époétine bêta, menée chez 603 patients avec anémie rénale, une tendance à une augmentation des événements cardiovasculaires a été observée dans le groupe avec normalisation du taux d'hémoglobine à 13 et 15 g/dl, par comparaison avec le groupe présentant des taux cibles plus faibles, de 10,5-11,5 g/dl. Cette tendance n'a toutefois pas été statistiquement significative (58 cas contre 47, p = 0,20).
La fréquence des événements thromboemboliques a été de 11,3% chez les patients traités avec des taux cibles d'hémoglobine élevés (13-15 g/dl) versus 7,3% chez les patients avec un taux cible d'hémoglobine bas (10,5-12,5 g/dl) (p = 0,06). Le taux de thromboses de l'abord artérioveineux chez les patients ayant subi une dialyse a également été plus élevé avec les taux cibles d'hémoglobine élevés (4% versus 3%) (p = 0,42).
Le délai jusqu'à l'instauration d'une dialyse a été raccourci dans le groupe avec taux d'hémoglobine cibles plus élevés (p = 0,034), bien qu'aucune différence n'ait été constatée sur le plan de la clairance médiane de la créatinine. Dans l'ensemble, les résultats plaident néanmoins plutôt en faveur d'une correction partielle que d'une normalisation de routine du taux d'hémoglobine par Recormon.
Dans une étude randomisée avec l'époétine alpha chez des patients en prédialyse (CHOIR), dans laquelle 1432 patients ont été répartis dans un bras avec un taux d'hémoglobine cible élevé (13,5 g/dl) ou dans un bras avec un taux d'hémoglobine cible bas (11,5 g/dl), le nombre d'événements cardio-vasculaires a été significativement plus élevé dans le groupe avec les taux cibles élevés que dans celui avec les taux cibles bas (17% vs 14%; 125 vs 97 cas; p = 0,03). La fréquence des événements thromboemboliques a été de 18% dans le bras de l'étude avec un taux d'hémoglobine élevé et de 17% dans celui avec un taux d'hémoglobine bas (p = 0,65).
Des analyses post-hoc poolées d'études cliniques avec des agents stimulant l'érythropoïèse (ASE) ont été réalisées chez les patients atteints d'insuffisance rénale chronique (patients dialysés, diabétiques et non diabétiques). Une tendance à l'augmentation du risque de mortalité globale et des événements cardiovasculaires et cérébrovasculaires a été observée à des doses cumulées d'ASE plus élevées, indépendamment du statut de dialysé ou de diabétique des patients (voir «Posologie/Mode d'emploi» et «Mises en garde et précautions»).
PharmacocinétiqueAbsorption
Après administration sous-cutanée d'époétine bêta à des patients avec insuffisance rénale, l'absorption retardée a mené à une concentration sérique en plateau. Les concentrations maximales ont été atteintes après 12 à 28 h en moyenne. Il n'y a pas de rapport direct entre la concentration sérique de la rhEPO et son action thérapeutique.
La biodisponibilité de l'époétine bêta en administration sous-cutanée atteint 23 à 42% par rapport à la voie intraveineuse.
Distribution
Le volume de distribution correspond à peu près à 1-2 fois le volume plasmatique.
On ignore si la rhEPO franchit la barrière placentaire et si elle passe dans le lait maternel; elle ne traverse toutefois pas la barrière hémato-encéphalique.
Métabolisme
Non applicable.
Élimination
Les études pharmacocinétiques réalisées sur des sujets sains et sur des patients avec insuffisance rénale montrent que la demi-vie de l'époétine bêta en injection intraveineuse est de 4 à 12 h.
La demi-vie terminale après administration sous-cutanée est plus longue qu'après injection intraveineuse, soit 13 à 28 h en moyenne.
Données précliniquesCarcinogénicité
L'époétine bêta n'influe ni sur la prolifération in vitro de lignées cellulaires non hématologiques saines ou malignes ni sur celle, in vivo, de «modèles de tumeurs» transplantables. Les résultats d'une étude de cancérogénicité chez la souris traitée par érythropoïétine homologue n'ont mis en évidence aucun potentiel prolifératif ou cancérogène.
Génotoxicité
L'époétine bêta s'est avérée non génotoxique dans le test de mutagénicité d'Ames, dans le test du micronoyau, dans le test de mutation ponctuelle HGPRT in vitro et dans le test d'aberration chromosomique sur cellules lymphocytaires humaines mises en culture.
Toxicité sur la reproduction
Des études chez le rat et le lapin n'ont mis en évidence aucun élément donnant à penser que l'époétine bêta pourrait avoir des propriétés embryotoxiques, fœtotoxiques ou tératogènes. Aucun trouble de la fertilité n'a été observé. Une étude sur la toxicité périnatale et postnatale n'a montré aucun effet nocif sur la grossesse et la lactation des femelles ni sur le développement prénatal et postnatal de leur descendance.
Remarques particulièresIncompatibilités
Se référer aux instructions suivantes pour éviter une perte d'efficacité du produit:
Ne pas mélanger la solution reconstituée avec d'autres médicaments ou à une solution pour perfusion.
Pour l'injection, n'utiliser que le matériel synthétique livré avec le médicament!
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Conserver le récipient dans son carton pour le protéger de la lumière.
La chaîne du froid ne doit pas être interrompue. Chez le distributeur, le grossiste et le pharmacien, Recormon PS doit être constamment conservé au réfrigérateur (2-8 °C). Le patient doit lui aussi conserver constamment le produit au réfrigérateur (2-8 °C). Dans le cadre d'un traitement ambulatoire, le produit peut être stocké une seule fois hors du réfrigérateur, à température ambiante (pas au-dessus de 25 °C), pour une durée ne dépassant pas 3 jours.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Instructions spéciales concernant l'administration
Les seringues de Recormon PS sont préremplies. Ne doivent être injectées que les solutions limpides ou légèrement opalescentes, incolores et exemptes de particules visibles. Les seringues préremplies de Recormon PS sont des produits stériles, mais sans agents conservateurs. N'administrer en aucun cas plus d'une dose par seringue préremplie.
Instructions concernant la préparation et l'injection des seringues préremplies de Recormon
Pour l'emploi des seringues préremplies de Recormon PS, voir le mode d'emploi à la fin de l'information destinée aux patients.
Numéro d’autorisation54766 (Swissmedic).
PrésentationRecormon PS 2000
Seringues préremplies avec solution injectable et 6 canules 27 G½: 6 [A]
Recormon PS 3000
Seringues préremplies avec solution injectable et 6 canules 27 G½: 6 [A]
Recormon PS 4000
Seringues préremplies avec solution injectable et 6 canules 27 G½: 6 [A]
Recormon PS 5000
Seringues préremplies avec solution injectable et 6 canules 27 G½: 6 [A]
Recormon PS 10 000
Seringues préremplies avec solution injectable et 6 canules 27 G½: 6 [A]
Recormon PS 30 000
Seringues préremplies avec solution injectable et 4 canules 27 G½: 4 [A]
Titulaire de l’autorisationRoche Pharma (Suisse) SA, Bâle.
Mise à jour de l’informationAvril 2022.
|