CompositionPrincipes actifs
Ertapénème sous forme d'ertapénème sodique.
Excipients
Bicarbonate de sodium, hydroxyde de sodium pour l'ajustement du pH à 7,5.
Chaque flacon contient environ 137 mg de sodium.
Indications/Possibilités d’emploiInvanz est indiqué pour le traitement de patients atteints d'infections modérées à sévères ci-après, dues à des micro-organismes sensibles:
·infections intra-abdominales compliquées
·infections de la peau et des parties molles dans le pied diabétique après la démonstration de la sensibilité microbienne de l'agent pathogène (voir le tableau des agents pathogènes sensibles, ainsi que sous «Mises en garde et précautions»)
·lors de pneumonies communautaires
·lors d'infections compliquées des voies urinaires, y compris la pyélonéphrite, après la démonstration de la sensibilité microbienne de l'agent pathogène (voir le tableau des agents pathogènes sensibles)
·lors des infections gynécologiques suivantes: endométrite post-partum et avortement septique. Remarque: des chlamydies sont souvent impliquées dans les infections gynécologiques et en sont parfois la seule cause. Étant donné qu'Invanz n'est pas efficace contre les chlamydies, il convient de n'utiliser Invanz dans ce domaine d'indications qu'en association avec un médicament complémentaire efficace contre les chlamydies.
Il convient de tenir compte des recommandations officielles concernant l'utilisation appropriée des antibiotiques, en particulier des recommandations d'emploi destinées à empêcher l'augmentation des résistances aux antibiotiques.
Posologie/Mode d’emploiPosologie usuelle
Patients de 13 ans et plus âgés
La dose usuelle pour les patients de 13 ans et plus âgés est de 1 gramme (g) d'Invanz une fois par jour.
Enfants dès 3 mois jusqu'à 12 ans
La dose usuelle d'Invanz chez les patients âgés de 3 mois à 12 ans est de 15 mg/kg deux fois par jour (sans dépasser 500 mg deux fois par jour).
Invanz n'est pas recommandé chez les enfants de moins de 3 mois car il n'existe aucune donnée sur la sécurité et l'efficacité d'Invanz chez cette tranche d'âge.
Invanz est administré en perfusion par voie intraveineuse. Invanz devrait être perfusé en l'espace de 30 minutes.
Normalement, la durée du traitement par Invanz est de 3 à 14 jours mais elle peut varier selon le type et la sévérité de l'infection et des pathogènes en cause (voir «Indications/Possibilités d'emploi»). Après une amélioration satisfaisante de l'état clinique, il est possible de poursuivre le traitement par un antibiotique oral approprié s'il existe une indication clinique.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucune adaptation de la dose n'est recommandée chez les patients atteints d'une insuffisance hépatique (voir «Pharmacocinétique, Cinétique pour certains groupes de patients, Troubles de la fonction hépatique»).
Patients présentant des troubles de la fonction rénale
Adultes:
Invanz peut être utilisé pour le traitement des infections chez des patients adultes souffrant d'insuffisance rénale. Chez les patients dont la clairance de la créatinine est >30 ml/min/1,73 m2, aucune adaptation de la dose n'est nécessaire. Il convient que les patients adultes souffrant d'une insuffisance rénale avancée (clairance de la créatinine ≤30 ml/min/1,73 m2, y compris les patients sous hémodialyse) reçoivent une dose journalière de 0,5 g.
Si seul le taux sérique de créatinine est disponible, on peut utiliser la formule suivante (Cockcroft) pour déterminer la clairance de la créatinine. Il convient que le taux sérique de créatinine reflète la fonction rénale à l'état stationnaire.
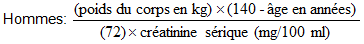
Femmes: (valeur pour les hommes) x (0,85)
Enfants et adolescents:
On ne dispose d'aucune donnée chez les enfants et les adolescents atteints d'insuffisance rénale.
Patients sous hémodialyse
Adultes:
Dans le cadre d'une étude clinique, près de 30 % de la dose ont été retrouvés dans le dialysat après l'administration par voie intraveineuse d'une dose unique de 1 g immédiatement avant une hémodialyse. Lorsque des patients hémodialysés adultes reçoivent la dose journalière recommandée de 500 mg d'Invanz en l'espace de 6 heures avant l'hémodialyse, une dose supplémentaire de 150 mg après l'hémodialyse est recommandée. Lorsque l'administration d'Invanz remonte à plus de 6 heures avant le début de l'hémodialyse, aucune dose supplémentaire n'est nécessaire. On ne dispose d'aucune donnée sur des patients soumis à une dialyse péritonéale ou à une hémofiltration.
Enfants et adolescents:
On ne dispose d'aucune donnée sur les enfants et les adolescents sous hémodialyse.
Patients âgés
A la dose recommandée, aucune adaptation de la dose n'est généralement nécessaire pour les patients âgés (≥65 ans). Toutefois, étant donné que l'ertapénème est principalement éliminé par voie rénale et que les patients âgés présentent souvent une diminution de la fonction rénale, il convient d'exclure la présence d'une insuffisance rénale avancée (voir «Posologie chez les patients présentant des troubles de la fonction rénale», voir également la dépendance en fonction de l'âge de la formule de Cockcroft).
Contre-indicationsL'ertapénème est contre-indiqué chez les patients présentant une hypersensibilité connue vis-à-vis de l'un des composants de ce médicament ou vis-à-vis d'autres médicaments de la même classe ainsi que chez les patients ayant déjà développé une réaction anaphylactique à d'autres antibiotiques bêta-lactames.
Mises en garde et précautionsDes réactions d'hypersensibilité (anaphylaxie) sévères et parfois fatales ont été rapportées chez des patients traités aux antibiotiques bêta-lactames. La survenue de ces réactions est plus probable chez des patients ayant des antécédents de multiples allergies. On dispose de rapports individuels chez des patients présentant une hypersensibilité à la pénicilline qui ont développé des réactions sévères d'hypersensibilité à un antibiotique bêta-lactame. Avant le début du traitement à l'ertapénème, il convient de rechercher avec beaucoup de soin des antécédents de réactions d'hypersensibilité aux pénicillines, aux céphalosporines, à d'autres antibiotiques bêta-lactames ainsi qu'à d'autres allergènes. L'apparition de réactions allergiques à l'ertapénème impose l'arrêt immédiat du traitement. Des réactions anaphylactiques sévères nécessitent l'instauration immédiate d'un traitement d'urgence.
Des réactions d'hypersensibilité peuvent également entraîner le syndrome de Kounis, une réaction allergique grave qui peut aboutir à un infarctus du myocarde. Les premiers symptômes de telles réactions peuvent inclure des douleurs thoraciques qui apparaissent en lien avec une réaction allergique aux antibiotiques bêta-lactamines.
Des réactions cutanées graves aux médicaments (SCAR) telles que le syndrome de Stevens-Johnson, la nécrolyse épidermique toxique, l'exanthème médicamenteux avec éosinophilie et symptômes systémiques (DRESS), l'érythème polymorphe et la pustulose exanthématique aiguë généralisée (AGEP) ont été signalées chez des patients traités par des antibiotiques bêta-lactames, y compris l'ertapénème (voir aussi «Effets indésirables»). Si de telles réactions se produisent, il faut cesser immédiatement Invanz et envisager une thérapie alternative.
Des rapports de cas dans la littérature ont montré que chez des patients traités à l'acide valproïque ou au divalproex sodique, une co-administration de carbapénèmes – dont l'ertapénème – entraîne une réduction de la concentration d'acide valproïque. Suite à cette interaction, les concentrations d'acide valproïque peuvent tomber sous la dose thérapeutique, avec un risque accru de convulsions inattendues. Une augmentation de la dose d'acide valproïque ou de divalproex sodique risque d'être insuffisante pour compenser efficacement cette interaction. D'une manière générale, l'administration d'ertapénème en association avec de l'acide valproïque ou avec du divalproex sodique n'est pas recommandée. On considérera l'utilisation d'autres antibiotiques au lieu des carbapénèmes pour le traitement des infections chez les patients dont les crises de convulsions sont traitées à l'acide valproïque ou au divalproex sodique. Si l'utilisation d'Invanz est nécessaire, on considérera l'ajout d'une thérapie anticonvulsivante supplémentaire (voir «Interactions»).
Comme pour les autres antibiotiques, l'utilisation à long terme d'ertapénème peut entraîner la prolifération excessive de germes résistants. L'état du patient doit absolument être contrôlé à plusieurs reprises. En cas de survenue d'une surinfection au cours du traitement, des mesures appropriées doivent être prises. L'influence de l'ertapénème sur la flore intestinale n'a pas fait l'objet d'études systématiques.
Une colite pseudomembraneuse a été rapportée avec presque tous les antibiotiques, y compris l'ertapénème; la sévérité peut varier d'une forme légère jusqu'à celle mettant en jeu de pronostic vital. C'est pourquoi ce diagnostic doit absolument être envisagé chez les patients présentant des diarrhées après l'administration d'antibiotiques. Des études laissent supposer qu'une toxine produite par Clostridium difficile est l'une des principales causes de colite associée aux antibiotiques.
Des crises convulsives ont été rapportées lors d'études cliniques chez des patients adultes traités par Invanz (1 g une fois par jour) pendant toute la durée de l'étude et une phase de suivi de 14 jours. Ces événements ont principalement touché des patients âgés, des patients atteints de maladies préexistantes du SNC (antécédents de lésions cérébrales ou crises convulsives) et/ou d'insuffisance rénale. Après la mise sur le marché, des observations similaires ainsi qu'une diminution du niveau de conscience associée à une perte de respiration spontanée et à la nécessité d'une respiration artificielle, et un cas isolé de coma ont été rapportés. Chez les patients présentant des effets indésirables significatifs affectant le SNC, il convient de demander une évaluation neurologique appropriée, d'assurer une prise en charge médicale et de réévaluer la posologie d'Invanz pour décider si celle-ci doit être réduite ou si Invanz doit être arrêté.
Invanz n'est pas recommandé chez les enfants de moins de 3 mois car il n'existe pas de données sur la sécurité et l'efficacité dans cette tranche d'âge.
L'efficacité de l'ertapénème dans le traitement d'infections du pied diabétique accompagnées d'ostéomyélite n'a pas été démontrée.
Ce médicament contient ca. 137 mg de sodium par flacon, ce qui équivaut à 6.85% de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte.
InteractionsLors de l'administration simultanée de probénécide, ce dernier inhibe l'élimination par voie rénale de l'ertapénème, car ces deux substances entrent en compétition pour le même mécanisme tubulaire actif de sécrétion. Cela entraîne de faibles élévations, toutefois statistiquement significatives, de l'exposition systémique (25%) et de la demi-vie d'élimination (19%). Aucune adaptation de la dose n'est cependant nécessaire lors de l'emploi simultané de probénécide. En raison de la faible influence sur la demivie, l'emploi simultané de probénécide pour prolonger la demi-vie de l'ertapénème n'est pas recommandé.
Des études in vitro laissent supposer que l'ertapénème n'inhibe pas le transport de la digoxine ou de la vinblastine par la glycoprotéine P et que l'ertapénème n'est pas un substrat pour le transport par la glycoprotéine P.
Des études in vitro avec des microsomes hépatiques humains indiquent que l'ertapénème n'est pas un inhibiteur des six principales isoformes du CYP P450 (CYP): 1A2, 2C9, 2C19, 2D6, 2E1 et 3A4. Des interactions avec des médicaments, dues à une inhibition par la glycoprotéine P ou à la clairance passant par le CYP sont peu probables (voir «Pharmacocinétique, Distribution, Métabolisme»).
A l'exception du probénécide, aucune substance n'a fait l'objet d'une étude clinique spécifique d'interaction.
Des rapports de cas dans la littérature ont montré que chez des patients traités à l'acide valproïque ou au divalproex sodique, une co-administration de carbapénèmes – dont l'ertapénème – entraîne une réduction de la concentration d'acide valproïque. Suite à cette interaction, les concentrations d'acide valproïque peuvent tomber sous la dose thérapeutique, avec un risque accru de convulsions inattendues. Bien que le mécanisme de cette interaction soit inconnu, les données obtenues dans des études in vitro et des essais sur l'animal suggèrent que les carbapénèmes peuvent inhiber l'hydrolyse transformant le dérivé glucuroconjugué de l'acide valproïque (VPA-g) en acide valproïque, et ainsi réduire la concentration sérique d'acide valproïque (voir «Mises en garde et précautions»).
Grossesse, allaitementGrossesse
Il n'existe pas d'étude contrôlée chez des femmes enceintes. Les études réalisées chez l'animal n'ont pas mis en évidence des effets directement ou indirectement nocifs sur le déroulement de la grossesse, le développement embryo-fœtal, le déroulement de l'accouchement ou le développement post-natal.
Il convient de n'utiliser l'ertapénème durant la grossesse que si son emploi est indispensable.
Allaitement
L'ertapénème passe dans le lait maternel. Il convient de décider, compte tenu de possibles effets indésirables chez l'enfant allaité et au vu de l'importance du médicament pour la mère, si celle-ci doit renoncer à allaiter durant le traitement ou si elle doit arrêter le médicament.
Effet sur l’aptitude à la conduite et l’utilisation de machinesDes étourdissements et une somnolence peuvent apparaître (voir «Effets indésirables»), et avoir un effet sur l'aptitude à la conduite ou l'utilisation de machines chez certains patients.
Effets indésirablesPatients adultes
Dans l'ensemble, plus de 1900 patients ont été traités à l'ertapénème dans le cadre d'études cliniques; parmi ceux-ci, plus de 1850 ont reçu l'ertapénème à la dose de 1 g. Des effets indésirables (c'est-à-dire pour lesquels l'investigateur a estimé qu'ils sont possiblement, probablement ou certainement dus au médicament) sont apparus chez près de 20% des patients traités à l'ertapénème. Le traitement a été arrêté en raison de la survenue d'effets indésirables chez 1,3% des patients.
Pour les patients n'ayant reçu qu'Invanz, les effets indésirables les plus fréquemment rapportés au cours du traitement et pendant une phase de suivi de 14 jours suivant l'arrêt ont été: diarrhée (4,8%), complications au niveau du site de perfusion (4,5%) et nausées (2,8%). Les anomalies les plus fréquentes dans les analyses de laboratoire chez cette population ont été: taux accru d'ALAT (alanine aminotransférase) (4,6%), d'ASAT (aspartate aminotransférase) (4,6%) et de phosphatase alcaline (3,8%); augmentation du nombre de thrombocytes (3,0%). Dans des études comparatives avec l'association pipéracilline/tazobactam, une réduction du nombre de neutrophiles en-dessous de 1800/µl (ou à moins de 50% des valeurs initiales si celles-ci étaient inférieures à 1800/µl) a été constatée chez 3,4% des patients traités par ertapénème. Cette réduction était 2,48 fois plus fréquente que chez les patients sous pipéracilline/tazobactam (1,4%, résultat significatif).
En outre, les effets indésirables suivants ont été observés dans les études susmentionnées:
Très fréquents (≥1/10); fréquents (≥1/100 à <1/10); occasionnels (≥1/1000 à <1/100); rares (≥1/10 000 à <1/1000); très rares (<1/10 000).
Infections et infestations
Occasionnels: Candidiase, infection fongique, bactériurie, levures dans l'urine, résultat positif au test de détection de la toxine de Clostridium difficile.
Affections hématologiques et du système lymphatique
Fréquents: Augmentation du nombre de thrombocytes.
Occasionnels: Réduction du taux d'hémoglobine, de l'hématocrite, des nombres de leucocytes, de granulocytes neutrophiles polynucléaires et des thrombocytes; augmentation des nombres de leucocytes, de granulocytes éosinophiles et de granulocytes neutrophiles polynucléaires; prolongation du temps de thromboplastine partielle activée (TTPa) (et du temps de prothrombine chez l'enfant).
Rares: Neutropénie, thrombocytopénie, réduction du nombre de lymphocytes, augmentation des nombres de monocytes, de granulocytes neutrophiles immatures, de myélocytes, de métamyélocytes, de lymphocytes et de lymphocytes atypiques.
Affections du système immunitaire
Rares: Réactions allergiques.
Expérience après la mise sur le marché: Anaphylaxie, y compris réactions anaphylactoïdes (très rare).
Troubles du métabolisme et de la nutrition
Occasionnels: Elévation de la glycémie.
Rares: Hypoglycémie, réduction des taux sériques de bicarbonate et de potassium, augmentation des taux sériques de phosphore et de potassium.
Affections psychiatriques
Occasionnels: Confusion mentale.
Rares: Agitation, anxiété, dépression.
Expérience après la mise sur le marché: Hallucinations et altération de l'état mental (y compris agitation, agression, délire, désorientation).
Affections du système nerveux
Fréquents: Céphalées.
Occasionnels: Étourdissements, somnolence, insomnie, crises de convulsions.
Rares: Tremblement, syncope.
Expérience après la mise sur le marché: Diminution de l'état de conscience associée à une perte de respiration spontanée et à la nécessité d'une respiration artificielle, et un cas isolé de coma, dyskinésie, myoclonie, trouble de la marche, encéphalopathie (le rétablissement peut prendre plus de temps chez les patients atteints d'insuffisance rénale).
Affections oculaires
Rares: Anomalies sclérales.
Affections cardiaques
Rares: Arythmie, tachycardie.
Affections vasculaires
Fréquents: Phlébite/thrombophlébite.
Occasionnels: Hypotension.
Rares: Hypertension, hémorragies.
Affections respiratoires, thoraciques et médiastinales
Occasionnels: Dyspnée, gêne pharyngée.
Rares: Embarras de la respiration nasale, toux, épistaxis, pneumonie, râles, sibilances.
Affections gastro-intestinales
Fréquents: Diarrhée, nausées, vomissements.
Occasionnels: Douleurs abdominales, anorexie, dyspepsie, constipation, sécheresse buccale, régurgitations acides, candidiase buccale, entérocolite pseudomembraneuse.
Rares: Dysphagie, incontinence fécale.
Expérience après la mise sur le marché: Dents tachées.
Affections hépatobiliaires
Fréquents: Augmentation des taux de transaminases (ALAT, ASAT), augmentation du taux de phosphatase alcaline.
Occasionnels: Augmentation des taux sériques de bilirubine totale, de bilirubine directe et de bilirubine indirecte.
Rares: Altérations de la fonction hépatique, cholécystite, ictère, augmentation du taux sérique de LDH, augmentation du taux d'urobilinogène.
Affections de la peau et du tissu sous-cutané
Fréquents: Eruption, prurit.
Occasionnels: Érythème.
Rares: Dermatite, dermatomycose, desquamation, infection de plaie post-opératoire.
Expérience après la mise sur le marché: pustulose exanthématique aiguë généralisée (AGEP), réaction médicamenteuse avec éosinophilie et symptômes systémiques (syndrome DRESS), urticaire, vascularite d'hypersensibilité. D'autres réactions cutanées graves aux médicaments (SCAR) ont été signalées chez des patients traités par des antibiotiques bêta-lactames (voir aussi «Mises en garde et précautions»).
Affections musculosquelettiques et du tissu conjonctif
Rares: Crampes musculaires, douleur de l'épaule.
Expérience après la mise sur le marché: Faiblesse musculaire.
Affections du rein et des voies urinaires
Occasionnels: Augmentation du taux sérique de créatinine, augmentation du taux sérique d'urée, détection accrue de cellules épithéliales, d'érythrocytes et de leucocytes dans l'urine.
Rares: Infection des voies urinaires, insuffisance rénale, insuffisance rénale aiguë, taux réduit de créatinine sérique.
Affections des organes de reproduction et du sein
Occasionnels: Vaginite.
Rares: Avortement, saignement génital.
Troubles généraux et anomalies au site d'administration
Fréquents: Complications au niveau du site de perfusion.
Occasionnels: Extravasation, asthénie/fatigue, fièvre, œdème/gonflement, douleur thoracique, altération du goût.
Rares: Indurations au point de perfusion, malaise, péritonite pelvienne.
Enfants et adolescents
Au cours des études cliniques, un total de 384 patients pédiatriques ont été traités à l'ertapénème. Le profil de sécurité chez les patients pédiatriques est similaire à celui chez les adultes. Des événements indésirables (c'est-à-dire des événements considérés par les investigateurs comme étant possiblement, probablement ou certainement associés au médicament) ont été observés chez 20,8% des patients pédiatriques traités à l'ertapénème. Le traitement a été interrompue chez 0,5% des patients en raison d'événements indésirables.
Chez les patients pédiatriques traités uniquement par Invanz, les effets indésirables les plus fréquemment rapportés au cours du traitement et pendant une phase de suivi de 14 jours suivant l'arrêt du traitement ont été des douleurs au site de perfusion (6,1%) et des diarrhées (5,2%).
Pour cette population pédiatrique, les anomalies dans les analyses de laboratoire les plus souvent signalées ont été: augmentation des taux d'ALAT (2,9%) et d'ASAT (2,8%) et nombre réduit de neutrophiles (3,0%).
Chez les patients pédiatriques n'ayant reçu qu'Invanz, les effets indésirables observés au cours du traitement et pendant une phase de suivi de 14 jours suivant l'arrêt du traitement ont été:
Affections hématologiques et du système lymphatique
Fréquents: Réduction du nombre de neutrophiles.
Occasionnels: Augmentation du nombre de thrombocytes, prolongation du temps de thromboplastine partielle activée (TTPa) et du temps de prothrombine , réduction du taux d'hémoglobine.
Affections psychiatriques
Expérience après la mise sur le marché: Hallucinations et altération de l'état mental (y compris agression).
Affections du système nerveux
Occasionnels: Céphalées.
Affections vasculaires
Occasionnels: Bouffées congestives, hypertension, pétéchies.
Affections gastro-intestinales
Fréquents: Diarrhées.
Occasionnels: Coloration des selles, méléna.
Affections hépatobiliaires
Fréquents: Augmentation des taux d'ALAT et d'ASAT.
Affections de la peau et du tissu sous-cutané
Fréquents: Dermatite du siège.
Occasionnels: Érythème, éruption.
Troubles généraux et anomalies au site d'administration
Fréquents: Douleur au niveau du site de perfusion.
Occasionnels: Rougeur, sensation de chaleur, prurit, sensation de brûlure au site de perfusion.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucune donnée spécifique n'est disponible sur le traitement d'un surdosage par l'ertapénème. Le surdosage délibéré d'ertapénème est peu probable. L'administration intraveineuse d'ertapénème à une dose quotidienne de 3 g pendant 8 jours à des volontaires adultes n'a pas entraîné d'effet toxique significatif. Au cours des études cliniques chez les adultes, l'administration involontaire pouvant aller jusqu'à 3 g par jour n'a pas provoqué d'effet indésirable cliniquement significatif. Comme pour tous les autres bêta-lactames, il faut s'attendre, lors d'un surdosage d'ertapénème, à l'apparition de convulsions cérébrales. Lors d'études cliniques chez des enfants et des adolescents, une dose i.v. unique de 40 mg/kg jusqu'à 2 g au maximum n'a pas provoqué de toxicité.
Dans le cas d'un surdosage, l'ertapénème doit être arrêté, et des mesures de soutien générales doivent être prises, jusqu'à l'élimination rénale du médicament.
L'ertapénème peut être éliminé par hémodialyse; il n'existe cependant aucune donnée sur le recours à l'hémodialyse pour le traitement d'un surdosage.
Propriétés/EffetsCode ATC
J01DH03 (carbapénème, ertapénème)
Mécanisme d'action
Invanz (ertapénème destiné à l'injection) est un 1-β-méthylcarbapénème parentéral, stérile, synthétique, à longue durée d'action, dont la structure est apparentée à celle d'antibiotiques bêta-lactames tels que les pénicillines et les céphalosporines. L'ertapénème est efficace in vitro contre un large spectre de bactéries aérobies et anaérobies à Gram positif et à Gram négatif. L'effet bactéricide de l'ertapénème repose sur une inhibition de la synthèse de la paroi cellulaire après la fixation de l'ertapénème aux protéines de liaison des pénicillines (PBPs). Pour Escheria coli, l'affinité est élevée pour les PBPs 1a, 1b, 2, 3, 4 et 5 et de préférence pour les PBPs 2 et 3. L'ertapénème présente une stabilité élevée à l'hydrolyse par la plupart des classes de bêta-lactamases, notamment les pénicillinases et les céphalosporinases ainsi que les bêta-lactamases à spectre élargi (ESBL), à l'exception des métallo-bêta-lactamases.
Pharmacodynamique
Sensibilité microbiologique
Les concentrations limites CMI du Clinical and Laboratory Standards Institute (CLSI) sont les suivantes:
·Entérobactéries et staphylocoques: S ≤2 mg/l et R ≥8 mg/l
·S. pneumoniae: S ≤1 mg/l et R ≥4 mg/l
·Streptococcus spp. (bêta-hémolytique seulement): S ≤1 mg/l
·Haemophilus spp.: S ≤0,5 mg/l
·Anaérobies: S ≤4 mg/l et R ≥16 mg/l
(Remarque: Les concentrations critiques pour les staphylocoques et S. pneumoniae ne sont applicables qu'aux staphylocoques sensibles à la méthicilline et aux pneumocoques sensibles à la pénicilline respectivement.)
La prévalence de résistances peut varier en fonction de la région géographique et du temps pour certaines espèces. Il est donc souhaitable de disposer de données locales sur la résistance, surtout pour le traitement d'infections sévères. Le tableau ci-après ne peut donner qu'une indication de la probabilité si les micro-organismes sont sensibles ou non à l'ertapénème.
Spectre antibactérien
Micro-organismes contre lesquels Invanz est efficace in vitro:
Tableau 1:
|
Organisme
|
Ertapénème
| |
CMI90 µg/ml
| |
Organismes sensibles
| |
Aérobies Gram-positifs
| |
Staphylococcus aureus (MET S)*
|
0,25
| |
Staphylococcus, coagulase négative (MET S)
|
2
| |
Streptococcus agalactiae*
|
0,06
| |
Streptococcus pneumoniae (PEN S)*
|
0,03
| |
Streptococcus pyogenes*
|
0,016
| |
Viridans streptococci
|
2
| |
Aérobies Gram-négatifs
| |
Citrobacter freundii
|
0,25
| |
Enterobacter aerogenes
|
0,5
| |
Enterobacter cloacae
|
1
| |
Escherichia coli (ESBL +)
|
0, 25
| |
Escherichia coli* (ESBL -)
|
0,016
| |
Haemophilus influenzae*
|
0,06
| |
Haemophilus parainfluenzae
|
0,125
| |
Klebsiella oxytoca
|
0,03
| |
Klebsiella pneumoniae (ESBL +)
|
0,25
| |
Klebsiella pneumoniae* (ESBL -)
|
0,016
| |
Moraxella catarrhalis*
|
0,016
| |
Morganella morganii
|
0,06
| |
Proteus mirabilis*
|
0,03
| |
Proteus vulgaris
|
0,125
| |
Serratia marcescens
|
0,25
| |
Anaérobies
| |
Bacteroides fragilis*
|
2
| |
Clostridium spp. (sans C. difficile)*¹
|
2
| |
Eubacterium spp.*²
|
2
| |
Fusobacterium spp.³
|
2
| |
Peptostreptococcus spp.*4
|
0,5
| |
Porphyromonas asaccharolytica*
|
0,03
| |
Prevotella spp.*5
|
0,25
| |
Organismes résistants
| |
Aérobies Gram-positifs
| |
Corynebacterium jeikeium
|
>32
| |
Enterococcus faecalis
|
16
| |
Enterococcus faecium
|
64
| |
Staphylococcus aureus (MET R)
|
32
| |
Staphylococcus epidermidis (MET R)
|
32
| |
Staphylococcus, coagulase négative (MET R)
|
>16
| |
Aérobies Gram-négatifs
| |
Aeromonas hydrophila
|
16
| |
Acinetobacter spp.6
|
16
| |
Burkholderia cepacia
|
32
| |
Pseudomonas aeruginosa
|
>16
| |
Stenotrophomonas maltophilia
|
32
| |
Anaérobies
| |
Lactobacillus spp.7
|
32
| |
Autres:
| |
Chlamydia spp.
Mycoplasma spp.
Rickettsia spp.
Legionella spp.
|
|
CMI = concentration minimale inhibitrice
MET R = résistant à la méthicilline
MET S = sensible à la méthicilline
PEN S = sensible à la pénicilline
ESBL - = bêta-lactamases à large spectre - non productrices
ESBL + = bêta-lactamases à large spectre - productrices
* L'efficacité clinique a été démontrée pour des isolats sensibles dans les indications cliniques autorisées.
1 y compris Clostridium bifermentans, C. butyricum, C. cadaveris, C. clostridioforme, C. innocuum, C. perfringens, C. ramosum, C. symbiosum, C. tertium et Clostridium spp.
2 y compris Eubacterium lentum et Eubacterium spp.
3 y compris Fusobacterium gonidiaformans, F. mortiferum, F. naviforme, F. necrophorum, F. russii, F. varium, F. nucleatum, Fusobacterium spp.
4 y compris Peptostreptococcus anaerobius, P. asaccharolyticus, P. magnus, P. micros, P. prevotii, P. tetradius, Peptostreptococcus spp.
5 y compris Prevotella bivia, P. corporis, P. disiens, P. enoeca, P. heparinolytica, P. melaninogenica, P. oralis, P. oris, Prevotella spp.
6 y compris Acinetobacter anitratus, A. baumannii, A. lwoffii, et Acinetobacter spp.
7 des organismes Lactobacillus n'ont pas été spécifiés.
Pour les souches de bactéries considérées comme sensibles à l'ertapénème, les cas de résistance ont été rares dans les études de surveillance conduites en Europe. Pour les isolats résistants, une résistance à d'autres antibiotiques de la classe des carbapénèmes a été observée avec certaines souches, mais pas pour toutes. Les entérobactéries résistantes aux céphalosporines ou aux antibiotiques bêta-lactamases à large spectre parce qu'elles produisent des céphalosporinases et/ou des ESBL sont généralement sensibles à l'ertapénème.
Le mécanisme d'action de l'ertapénème diffère de celui des autres classes d'antibiotiques, comme les quinolones, les aminosides, les macrolides et les tétracyclines. Il n'existe pas de résistance croisée entre l'ertapénème et ces substances par une modification de la structure cible.
Les agents pathogènes peuvent présenter une résistance à plus d'une classe d'antibiotiques lorsque le mécanisme de résistance est basé sur ou inclut des barrières de perméabilité et/ou un mécanisme de pompe à efflux.
Comme lors de toute antibiothérapie, les données épidémiologiques concernant la résistance des agents pathogènes suspectés doivent être prises en compte lorsque l'antibiotique est utilisé de manière empirique.
Tests de sensibilité
Test de dilution
Des méthodes quantitatives sont utilisées pour déterminer les concentrations minimales inhibitrices (CMI) des antibiotiques. Ces CMI permettent d'évaluer la sensibilité des bactéries vis-à-vis de substances antimicrobiennes. Les CMI devraient être déterminées à l'aide d'une méthode standard. Les méthodes standard reposent sur un test de dilution (bouillon ou Agar), comme celui du Clinical and Laboratory Standards Institute (CLSI) ou sur des méthodes de test équivalentes utilisant des concentrations standard d'inoculum et d'ertapénème.
Test de diffusion
Les méthodes quantitatives basées sur la mesure du diamètre des zones de diffusion fournissent des données reproductibles sur la sensibilité des bactéries aux agents antibactériens. Une de ces méthodes normalisées du CLSI repose sur l'utilisation de concentrations standard d'inoculum. Cette méthode utilise des disques de papier imprégnés de 10 µg d'ertapénème.
Techniques en anaérobiose
La sensibilité des bactéries anaérobies à l'ertapénème peut être évaluée par des méthodes normalisées (CLSI).
Efficacité clinique
Adultes
L'ertapénème a été évalué chez l'adulte pour le traitement des infections intra-abdominales compliquées dans le cadre d'une étude clinique. Dans cette étude auprès de 665 patients, on a comparé l'ertapénème (1 g une fois par jour par voie i.v.) à l'association pipéracilline/tazobactam (3,375 g toutes les 6 heures par voie i.v.) pendant 5 à 14 jours. Une à deux semaines après la fin du traitement, les taux de succès cliniques et microbiologiques étaient de 89,6% (190/212) pour l'ertapénème et de 82,7% (162/196) pour l'association pipéracilline/tazobactam. Quatre à six semaines après la fin du traitement (test de confirmation de guérison), les taux de succès étaient de 86,7% (176/203) pour l'ertapénème et de 81,3% (157/193) pour l'association pipéracilline/tazobactam.
Afin de compléter ces résultats, une deuxième étude clinique multicentrique randomisée, réalisée en double aveugle, a été menée chez des patients atteints d'une infection intra-abdominale compliquée. Cette étude a également comparé l'ertapénème (1 g par voie i.v. une fois par jour) à l'association pipéracilline/tazobactam (3,375 g i.v. toutes les 6 heures) chez 500 patients. Pour la population évaluable du point de vue microbiologique, les taux de succès cliniques 2 semaines après la fin du traitement étaient de 82,0% (100/122) pour l'ertapénème et de 81,3% (87/107) pour l'association pipéracilline/tazobactam, alors que les taux de succès 4 et 6 semaines après la fin du traitement étaient de 78,6% (88/112) pour l'ertapénème et de 78,7% (74/94) pour l'association pipéracilline/tazobactam.
L'ertapénème a été examiné au cours d'une étude clinique contrôlée, randomisée, effectuée auprès de 586 patients adultes, sur le traitement des infections du pied diabétique. Cette étude a comparé l'ertapénème (1 g par voie i.v. une fois par jour) à l'association pipéracilline/tazobactam (3,375 g par voie i.v. toutes les 6 heures). Les deux régimes thérapeutiques offraient la possibilité d'un passage à un traitement oral par amoxicilline/acide clavulanique après au moins 5 jours du traitement intraveineux. La durée totale de traitement (voie parentérale et orale) était de 5 à 28 jours. Dix jours après la fin du traitement, le taux de succès clinique était de 87,4% (180/206) pour l'ertapénème et de 82,7% (162/196) pour l'association pipéracilline/tazobactam.
L'ertapénème a été évalué chez 866 patients adultes au total pour le traitement de la pneumonie communautaire dans le cadre de deux études cliniques. Dans les deux études, l'ertapénème (1 g une fois par jour par voie parentérale) a été comparé à la ceftriaxone (1 g une fois par jour par voie parentérale). Les deux régimes thérapeutiques ont permis l'option de passer à l'amoxicilline/acide clavulanique par voie orale avec une durée totale de traitement de 10 à 14 jours (parentéral et oral). Les taux de succès cliniques (données regroupées des deux études) 7 à 14 jours après la fin du traitement (test de confirmation de guérison) étaient de 92,0% (335/364) pour l'ertapénème et de 91,8% (270/294) pour la ceftriaxone.
L'ertapénème a été évalué chez un nombre total de 850 patients adultes souffrant d'infections urinaires compliquées, y compris la pyélonéphrite, dans le cadre de deux études cliniques. Dans les deux études, l'ertapénème (1 g une fois par jour par voie parentérale) a été comparé à la ceftriaxone (1 g une fois par jour par voie parentérale). Les deux régimes thérapeutiques ont permis l'option de passer à la ciprofloxacine (500 mg deux fois par jour) par voie orale avec une durée totale de traitement de 10 à 14 jours (parentéral et oral). Les taux de succès microbiologiques (données regroupées des deux études) 5 à 9 jours après la fin du traitement (test de confirmation de guérison) étaient de 89,5% (229/259) pour l'ertapénème et de 91,1% (204/224) pour la ceftriaxone.
L'ertapénème a été évalué dans le cadre d'une étude clinique chez 412 patientes adultes souffrant d'infections pelviennes aiguës, parmi lesquelles 350 patientes présentaient une infection obstétricale/du post-partum et 45 un avortement septique. Dans cette étude, on a comparé l'ertapénème (1 g une fois par jour par voie i.v.) à l'association pipéracilline/tazobactam (3,375 g toutes les 6 heures par voie i.v.), pendant 3 à 10 jours. Les taux de succès cliniques 2 à 4 semaines après la fin du traitement (test de confirmation de guérison) étaient de 93,9% (153/163) pour l'ertapénème et de 91,5% (140/153) pour l'association pipéracilline/tazobactam.
Sécurité et efficacité en pédiatrie
L'ertapénème a été évalué également chez des enfants et adolescents âgés de 3 mois à 17 ans, dans le cadre de deux études cliniques multicentriques randomisées.
La première étude a inclus 404 patients et a comparé l'ertapénème (15 mg/kg par voie i.v. toutes les 12 heures chez les patients âgés de 3 mois à 12 ans, ou 1 g par voie i.v. chez les patients âgés de 13 ans à 17 ans) avec la ceftriaxone (50 mg/kg par voie i.v. en deux doses par jour chez les patients âgés de 3 mois à 12 ans et 50 mg/kg par voie i.v. sous forme de dose unique journalière chez les patients âgés de 13 à 17 ans) pour le traitement d'infections compliquées des voies urinaires (UTI), des infections cutanées et du tissu conjonctif (SSTI) ou des pneumonies communautaires (CAP). Les deux options thérapeutiques ont donné la possibilité de passer à l'amoxicilline/clavulanate (parentérale et orale) pendant un maximum de 14 jours de traitement. Les taux de succès microbiologiques dans la population évaluable selon le protocole (EPP) étaient de 87,0% (40/46) pour l'ertapénème et de 90,0% (18/20) pour la ceftriaxone chez les patients atteints d'UTI. Les taux de succès cliniques de l'analyse EPP chez les patients atteints de SSTI étaient de 95,5% (64/67) pour l'ertapénème et de 100% (26/26) pour la ceftriaxone, et chez les patients atteints de CAP, ces taux étaient de 96,1% (74/77) pour l'ertapénème et de 96,4% (27/28) pour la ceftriaxone.
La deuxième étude avait inclus 112 patients et a comparé l'ertapénème (15 mg/kg par voie i.v. toutes les 12 heures chez les patients âgés de 3 mois à 12 ans, ou une fois par jour 1 g par voie i.v. chez les patients âgés de 13 ans à 17 ans) avec l'association ticarcilline/clavulanate (50 mg/kg pour un poids corporel inférieur à 60 kg ou 3,0 g chez les patients de plus de 60 kg, 4 ou 6 fois par jour) durant une période de 14 jours au maximum pour le traitement d'infections intra-abdominales compliquées (IAI) et d'infections pelviennes aiguës (API). Chez les patients présentant une IAI (principalement des patients souffrant d'une appendicite perforée ou compliquée), les taux de succès cliniques étaient de 83,7% (36/43) pour l'ertapénème et de 63,6% (7/11) pour l'association ticarcilline/clavulanate dans l'analyse EPP. Chez les patients traités en raison d'une API (endomyométrite postopératoire ou spontanée lors de l'accouchement ou avortement septique), les taux de succès cliniques étaient de 100% (23/23) pour l'ertapénème et de 100% (4/4) pour l'association ticarcilline/clavulanate d'après l'analyse EPP.
PharmacocinétiqueDistribution
Chez l'être humain, l'ertapénème présente une forte liaison aux protéines plasmatiques. On a observé chez de jeunes adultes en bonne santé que la liaison de l'ertapénème aux protéines diminue à mesure que la concentration plasmatique de l'ertapénème augmente, passant de 95% environ pour une concentration plasmatique de <100 microgrammes/ml à 85% environ pour une concentration plasmatique approximative de 300 microgrammes/ml.
Les concentrations plasmatiques moyennes de l'ertapénème après une perfusion de 30 minutes d'une dose unique de 1 g ou de 2 g chez de jeunes adultes sains sont présentées dans le Tableau 2.
Tableau 2:
Concentration plasmatique de l'ertapénème après l'administration d'une dose unique
|
Dose
|
Concentration plasmatique moyenne (microgrammes/ml)
| |
|
0,5 h
|
1 h
|
2 h
|
4 h
|
6 h
|
8 h
|
12 h
|
18 h
|
24 h
| |
1 g i.v.*
|
155
|
115
|
83
|
48
|
31
|
20
|
9
|
3
|
1
| |
2 g i.v.*
|
283
|
202
|
145
|
86
|
58
|
36
|
16
|
5
|
2
|
* Les doses intraveineuses ont été perfusées à une vitesse constante durant 30 minutes.
Chez les adultes, la surface sous la courbe de la concentration plasmatique (ASC) de l'ertapénème augmente presque proportionnellement à la dose sur l'intervalle des doses allant de 0,5 à 2 g.
Il n'y a pas d'accumulation d'ertapénème après l'administration de doses intraveineuses multiples allant de 0,5 à 2 g par jour chez les adultes.
Les concentrations plasmatiques moyennes (µg/ml) de l'ertapénème chez des enfants et des adolescents sont présentées dans le Tableau 3.
Tableau 3:
Concentration plasmatique de l'ertapénème chez des patients pédiatriques après une dose unique i.v.*
|
Âge (Dose)
|
Concentration plasmatique moyenne (μg/ml)
| |
0,5 h
|
1 h
|
2 h
|
4 h
|
6 h
|
8 h
|
12 h
|
24 h
| |
3 à 23 mois
| |
(15 mg/kg)†
|
103,8
|
57,3
|
43,6
|
23,7
|
13,5
|
8,2
|
2,5
|
-
| |
(20 mg/kg†
|
126,8
|
87,6
|
58,7
|
28,4
|
-
|
12,0
|
3,4
|
0,4
| |
(40 mg/kg)‡
|
199,1
|
144,1
|
95,7
|
58,0
|
-
|
20,2
|
7,7
|
0,6
| |
2 à 12 ans
| |
(15 mg/kg)†
|
113,2
|
63,9
|
42,1
|
21,9
|
12,8
|
7,6
|
3,0
|
-
| |
(20 mg/kg)†
|
147,6
|
97,6
|
63,2
|
34,5
|
-
|
12,3
|
4,9
|
0,5
| |
(40 mg/kg)‡
|
241,7
|
152,7
|
96,3
|
55,6
|
-
|
18,8
|
7,2
|
0,6
| |
13 à 17 ans
| |
(20 mg/kg)†
|
170,4
|
98,3
|
67,8
|
40,4
|
-
|
16,0
|
7,0
|
1,1
| |
(1 g)§
|
155,9
|
110,9
|
74,8
|
-
|
24,0
|
-
|
6,2
|
-
| |
(40 mg/kg)‡
|
255,0
|
188,7
|
127,9
|
76,2
|
-
|
31,0
|
15,3
|
2,1
|
* Les doses i.v. ont été perfusées à une vitesse constante durant 30 minutes.
† Jusqu'à la dose maximale de 1 g/jour
‡ Jusqu'à la dose maximale de 2 g/jour
§ Sur la base de trois patients examinés lors de deux études sur la sécurité et l'efficacité, qui se sont mis à disposition pour une évaluation pharmacocinétique et ont reçu 1 g d'ertapénème.
Le volume de distribution (Vdss) de l'ertapénème se situe entre 8,6 et 8,9 litres chez les hommes et entre 7,0 et 7,5 litres chez les femmes; il se monte à environ 0,2 litre/kg chez des enfants âgés de 3 mois à 12 ans et à environ 0,16 litre/kg chez les enfants et les adolescents âgés de 13 ans à 17 ans.
L'ertapénème pénètre les vésicules cutanées induites par succion. Les concentrations d'ertapénème atteintes dans le liquide vésiculaire au troisième jour après une dose de 1 g administrée par voie intraveineuse une fois par jour à tous les moments de prélèvement des échantillons sont présentées dans le Tableau 4. Le rapport entre l'ASC dans le liquide vésiculaire et l'ASC plasmatique se monte à 0,61.
Tableau 4:
Concentration d'ertapénème (microgrammes/ml) chez des adultes dans le liquide vésiculaire de tous les échantillons pris au troisième jour, après administration de doses de 1 g par jour par voie intraveineuse.
|
0,5 h
|
1 h
|
2 h
|
4 h
|
8 h
|
12 h
|
24 h
| |
7
|
12
|
17
|
24
|
24
|
21
|
8
|
Métabolisme
Après une perfusion i.v. de 1 g d'ertapénème radiomarqué chez de jeunes adultes en bonne santé, la plus grande partie (94%) de la radioactivité décelée dans le plasma correspondait à l'ertapénème inchangé. Le principal métabolite de l'ertapénème est formé dans les reins par hydrolyse du cycle bêta-lactame.
Élimination
L'ertapénème est éliminé principalement par les reins. La demi-vie plasmatique chez les jeunes adultes en bonne santé et chez les patients âgés de 13 à 17 ans est d'environ 3,5 heures pour les femmes et d'environ 4,2 heures pour les hommes, et elle se monte à environ 2,5 heures chez les enfants âgés de trois mois à 12 ans.
A la suite de l'administration par voie i.v. d'une dose de 1 g d'ertapénème radiomarqué à de jeunes adultes en bonne santé, environ 80% de la radioactivité de la dose ont été retrouvés dans l'urine et 10% dans les fèces; quant aux 80% d'ertapénème radiomarqué récupérés dans l'urine, environ 38% ont été excrétés sous forme inchangée, et environ 37% sous la forme du principal métabolite.
Chez de jeunes adultes en bonne santé ayant reçu une dose de 1 g par voie i.v., les concentrations moyennes d'ertapénème retrouvées dans l'urine dépassaient 985 microgrammes/ml 0 à 2 heures après la perfusion et 52 microgrammes/ml 12 à 24 heures après la perfusion.
Cinétique pour certains groupes de patients
Sexe
Les concentrations plasmatiques de l'ertapénème présentent de légères différences entre les hommes et les femmes mais ces différences ne sont pas cliniquement significatives (Cmax plus élevée et t1/2 plus courte chez les femmes, avec une clairance rénale plus élevée chez les femmes).
Troubles de la fonction hépatique
La pharmacocinétique de l'ertapénème n'a pas été établie chez les patients atteints d'insuffisance hépatique. Étant donné que le métabolisme de l'ertapénème par le foie est très faible, on ne s'attend pas à une altération significative de la pharmacocinétique de l'ertapénème chez les patients présentant une insuffisance hépatique (voir «Posologie/Mode d'emploi»).
Troubles de la fonction rénale
Après l'administration d'une dose unique
Après l'administration d'une dose unique de 1 g d'ertapénème par voie intraveineuse à des adultes atteints d'insuffisance rénale légère (clairance de la créatinine 60-90 ml/min/1,73 m2), les ASC de la concentration totale d'ertapénème (lié ou non) et d'ertapénème non lié étaient semblables à celles des adultes en bonne santé (âgés de 25 à 82 ans). Lors d'insuffisance rénale modérée (clairance de la créatinine 31-59 ml/min/1,73 m2), les ASC de la concentration totale et de l'ertapénème non lié sont environ 1,5 fois à 1,8 fois supérieures à celles des adultes en bonne santé. Lors d'insuffisance rénale avancée (clairance de la créatinine 5-30 ml/min/1,73 m2), les ASC de la concentration totale et de l'ertapénème non lié sont environ 2,6 fois à 3,4 fois plus élevées que celles des adultes en bonne santé. Chez les patients sous hémodialyse, les ASC de la concentration totale et de l'ertapénème non lié sont 2,9 à 6 fois plus élevées entre les dialyses que celles des adultes en bonne santé (voir «Posologie/Mode d'emploi»).
Après l'administration de multiples doses
La pharmacocinétique à l'état stationnaire après administration de multiples doses par voie intraveineuse n'a pas été étudiée chez les insuffisants rénaux. Une accumulation d'ertapénème ne peut être exclue chez ces patients.
On ne dispose d'aucune donnée chez les enfants et les adolescents atteints d'insuffisance rénale.
Patients âgés
Après l'administration d'une dose de 1 g ou de 2 g d'ertapénème par voie i.v., les concentrations plasmatiques étaient légèrement plus élevées (d'environ 39% et 22%) chez les personnes âgées (≥65 ans) que chez les jeunes adultes (<65 ans) (voir «Posologie/Mode d'emploi, Patients âgés»).
Enfants et adolescents
Les concentrations plasmatiques mesurées chez les enfants et les adolescents âgés de 13 à 17 ans sont comparables à celles déterminées chez les adultes après une dose journalière unique i.v. de 1 g.
Après une dose de 20 mg/kg (jusqu'à une dose maximale de 1 g), les paramètres pharmacocinétiques mesurés chez les patients âgés de 13 à 17 ans étaient généralement comparables à ceux mesurés chez les jeunes adultes en bonne santé. Parmi les six patients âgés de 13 à 17 ans, trois ont reçu moins qu'une dose de 1 g. Afin d'estimer les données pharmacocinétiques dans le cas où tous les patients de ce groupe d'âge recevraient une dose de 1 g, les données pharmacocinétiques ont été adaptées mathématiquement à une dose de 1 g, en supposant une linéarité. Une comparaison des résultats montre qu'une dose journalière de 1 g d'ertapénème chez des patients âgés de 13 à 17 ans donne un profil pharmacocinétique comparable à celui observé chez les adultes. Les rapports (13 à 17 ans par rapport aux adultes) pour l'ASC, la concentration immédiatement après la fin de la perfusion et la concentration au milieu de l'intervalle des doses étaient de 0,99, 1,20 et 0,84.
Les concentrations plasmatiques au milieu de l'intervalle des doses après une dose i.v. unique de 15 mg/kg d'ertapénème chez des patients âgés de 3 mois à 12 ans sont comparables aux concentrations plasmatiques au milieu de l'intervalle des doses après une dose journalière i.v. de 1 g chez des adultes (voir «Distribution»). La clairance plasmatique (ml/min/kg) de l'ertapénème chez les patients âgés de 3 mois à 12 ans est à peu près égale au double de celle mesurée chez les adultes. Pour la dose de 15 mg/kg, l'ASC (doublée afin de modéliser une administration biquotidienne, soit l'effet de 30 mg/kg/jour) chez les patients âgés de 3 mois à 12 ans était comparable à l'ASC chez de jeunes adultes en bonne santé ayant reçu une dose de 1 g par voie intraveineuse.
Données précliniquesLes données précliniques issues des études conventionnelles de sécurité, de pharmacologie, de toxicité chronique, de génotoxicité, de mutagenèse et de toxicité sur la reproduction n'ont pas révélé de risque particulier pour l'homme.
Aucune étude à long terme n'a été réalisée chez l'animal afin d'évaluer le potentiel cancérogène de l'ertapénème.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité avec Invanz, ce médicament ne doit en aucun cas être mélangé à d'autres médicaments ou d'autres solutions de perfusion. Ne jamais utiliser de solvant ou de solution de perfusion contenant de l'alpha-D-glucose (dextrose) pour la reconstitution ou l'administration de l'ertapénème.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Stabilité après ouverture
Solution reconstituée (solution initiale) et solution pour perfusion: une fois reconstituée, la solution reconstituée (solution initiale) doit être diluée immédiatement dans une solution à 0,9% (isotonique) de chlorure de sodium (voir «Remarques concernant la manipulation») et peut alors être conservée à température ambiante (25°C) pour une utilisation dans les 6 heures qui suivent, ou au réfrigérateur (2-8°C) pendant 24 heures pour une utilisation dans les 4 heures qui suivent son retrait du réfrigérateur.
Les solutions d'Invanz ne doivent pas être congelées. Jeter les solutions congelées.
Remarques particulières concernant le stockage
Avant dissolution: Conserver à température ambiante (15 à 25°C).
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Invanz est administré par perfusion intraveineuse. Il doit être perfusé sur une période de 30 minutes.
Patients âgés de 13 ans et plus:
Préparation pour l'administration intraveineuse:
Invanz doit être dissous et dilué immédiatement avant son administration.
1.Dissoudre le contenu d'un flacon de 1 g d'Invanz avec 10 ml d'une des solutions suivantes: eau pour injection, chlorure de sodium à 0,9% (isotonique) pour injection ou eau pour injection contenant un agent conservateur (0,1 g/ml d'ertapénème).
2.Bien agiter pour dissoudre et transférer immédiatement le contenu du flacon dans 50 ml d'une solution isotonique de chlorure de sodium à 0,9%.
3.La perfusion doit être effectuée dans les 6 heures qui suivent la dissolution.
Enfants âgés de 3 mois à 12 ans:
Préparation pour l'administration intraveineuse:
Invanz doit être dissous et dilué immédiatement avant son administration.
1.Dissoudre le contenu d'un flacon de 1 g d'Invanz avec 10 ml d'une des solutions suivantes: eau pour injection, chlorure de sodium à 0,9% (isotonique) pour injection ou eau pour injection contenant un agent conservateur (0,1 g/ml d'ertapénème).
2.Bien agiter pour dissoudre et prélever immédiatement un volume correspondant à 15 mg/kg de poids corporel (ne pas dépasser 500 mg deux fois par jour) et diluer dans une solution isotonique à 0,9% de chlorure de sodium, jusqu'à une concentration finale de 20 mg/ml ou moins.
3.La perfusion doit être effectuée dans les 6 heures qui suivent la dissolution.
Les médicaments destinés à l'administration par voie parentérale doivent être contrôlés quant à la présence de particules visibles ou d'une coloration. Les solutions d'Invanz sont incolores à jaune pâle. Toute variation au sein de cette gamme de couleurs n'affecte pas l'efficacité de ce médicament.
Numéro d’autorisation55902 (Swissmedic).
PrésentationInvanz 1 g: flacon en verre de type I de 15 ml avec un bouchon gris en butyle et une capsule blanche en plastique sur une bande dorée scellée en aluminium, destiné à un usage unique.
Emballages à 1 flacon. (A)
Titulaire de l’autorisationMSD MERCK SHARP & DOHME AG, Lucerne.
Mise à jour de l’informationJuillet 2025
Implementation HMV4/RCN000024665-CH
|