CompositionPrincipes actifs
Paricalcitol.
Excipients
2 μg dans 1 ml de solution: éthanol 170 mg, propylène glycol 310 mg, eau pour préparation injectable.
5 μg dans 1 ml de solution: éthanol 170 mg, propylène glycol 310 mg, eau pour préparation injectable.
10 μg dans 2 ml de solution: éthanol 340 mg, propylène glycol 620 mg, eau pour préparation injectable.
Indications/Possibilités d’emploiTraitement de l'hyperparathyroïdie secondaire chez les patients atteints d'insuffisance rénale terminale et sous hémodialyse chronique.
Posologie/Mode d’emploiPosologie initiale
La dose initiale de Zemplar est calculée à partir du taux sérique de base de l'hormone parathyroïdienne (PTH) ou plus exactement de l'hormone parathyroïdienne intacte (PTHi) biologiquement active.
La dose initiale se calcule selon la formule suivante:
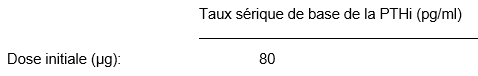
Les injections se font en mode intraveineux directe (en bolus) à un moment quelconque au cours de la dialyse, toutefois la fréquence des injections ne doit pas dépasser une injection tous les deux jours.
Ajustement de la posologie: si on n'observe pas de réaction satisfaisante au traitement avec le Zemplar, la dose peut être progressivement augmentée de 2 à 4 µg de paricalcitol à intervalles de deux à quatre semaines.
Pendant la phase d'adaptation posologique les taux sériques de calcium et de phosphore doivent être étroitement surveillés. Si l'on constate une augmentation du taux sérique de calcium au-delà de la valeur normale ou que le produit de la concentration sérique du calcium par celle du phosphore (Ca x P) est supérieur à 65 mg2/dl2, la dose doit immédiatement être réduite ou le traitement interrompu jusqu'à ce que ces paramètres se soient à nouveau normalisés. Le traitement avec le Zemplar pourra alors être repris à plus faible dose. Il est possible que le dosage puisse être réduit parce que le taux de PTH a diminué en réaction au traitement avec le Zemplar. L'augmentation progressive du dosage doit donc être adaptée au cas par cas.
Des doses supérieures à 0,24 µg/kg n'ont pas fait l'objet d'études cliniques suffisantes et ne doivent pas être administrées.
Le tableau 1 suivant comporte des explications sur la marche à suivre lors de l'ajustement de la posologie:
Tableau 1
|
Directives de posologie recommandées
| |
Concentration sérique de PTH
|
Dose de paricalcitol
| |
Pas de modification/augmentation
|
augmenter la dose
| |
Baisse de < 30%
|
augmenter la dose
| |
Baisse de > 30% à < 60%
|
maintenir la dose
| |
Baisse de > 60%
|
diminuer la dose
| |
Taux de 1,5 à 3 fois la limite supérieure de la norme
|
maintenir la dose
|
Insuffisance hépatique
La fraction de paricalcitol non liée est à peu près identique chez les patients atteints d'insuffisance hépatique faible (Child-Pugh A) et chez les sujets sains. Par conséquent une adaptation posologique pour ce groupe de patients n'est pas indiquée. On ne dispose pas de données pharmacocinétiques afférentes pour des patients atteints d'insuffisance hépatique grave. L'utilisation n'est pas recommandée.
Patients âgés (≥65 ans)
On ne dispose que d'expériences limitées de traitement de patients de ≥65 ans avec le paricalcitol dans des études de phase III. Au cours de ces études aucune différence générale n'a été constatée du point de vue de la sécurité et de l'efficacité du paricalcitol chez des patients plus jeunes et chez ceux de ≥65 ans.
Enfants
L'efficacité et la sécurité de paricalcitol chez l'enfant n'ont pas fait l'objet d'études. Les expériences concernant l'utilisation de paricalcitol en solution pour injection chez les patients de moins de 18 ans sont limitées.
Contre-indicationsLe Zemplar ne doit pas être administré aux patients atteints d'hypercalcémie ou présentant des signes d'intoxication à la vitamine D.
Une hypersensibilité au principe actif ou à l'un des excipients mentionnés dans la composition constitue également une contre-indication.
Mises en garde et précautionsUn surdosage aigu de paricalcitol peut entraîner une hypercalcémie et nécessite une prise en charge d'urgence.
Lors du traitement avec le Zemplar le taux sérique de calcium, celui de phosphore ainsi que le produit Ca-P peuvent augmenter.
Pendant la première phase de traitement au Zemplar, les taux sériques de calcium et de phosphore doivent être étroitement surveillés, par exemple deux fois par semaine. Après une adaptation posologique réussie au paricalcitol les taux sériques de calcium et de phosphore doivent être déterminés au moins une fois par mois. Il est recommandé de contrôler le taux sérique ou le taux plasmatique de la PTHi tous les trois mois. Une détermination plus fréquente des paramètres de laboratoire, y compris de la PTHi, peut être nécessaire pendant la phase d'adaptation posologique.
Lors de l'apparition d'une hypercalcémie cliniquement significative, la dose doit être diminuée ou le traitement de paricalcitol doit être interrompu. Lors de l'administration prolongée de paricalcitol, une hypercalcémie, une augmentation du produit de la concentration sérique du calcium par celle du phosphore (Ca x P) ou une calcification métastatique risquent de se développer. Une hypercalcémie chronique peut entraîner un dépôt calcaire généralisé dans les vaisseaux et autres tissus mous. S'agissant des symptômes et des signes d'une intoxication à la vitamine D associée à une hypercalcémie, ainsi que les mesures à prendre en pareil cas, se reporter au paragraphe «Surdosage».
Si les taux de PTH sont réduits de manière excessive lors du traitement avec le paricalcitol, des lésions osseuses adynamiques peuvent apparaître.
La solution de Zemplar pour injection contient 20% (v/v) d'alcool, c'est-à-dire jusque'à 538 mg éthanol à la dose journalière maximale, équivalent à 13,4 ml de bière ou 5,6 ml de vin. Risque pour la santé des patients souffrants d'alcoolisme. À prendre en compte chez les femmes enceintes ou allaitant et chez les patients présentant un risque accru en raison d'une maladie hépatique ou d' épilepsie.
Ce médicament contient 309,9 mg de propylène glycol par 2 µg ou 5 µg de paricalcitol dans un volume de dosage de 1 ml ou 619,8 mg de propylène glycol par 10 µg de paricalcitol dans un volume de dosage de 2 ml.
On ne dispose pas d'expérience chez les patients atteints d'insuffisance hépatique modérée à grave (Child-Pugh B+C). Pour ces patients, la prudence est particulièrement de mise.
InteractionsLe paricalcitol ne devrait pas inhiber la clairance des substances métabolisées par le CYP1A2, le CYP2A6, le CYP2B6, le CYP2C8, le CYP2C9, le CYP2D6, le CYP2E1 ou le CYP3A ni augmenter la clairance des substances métabolisées par le CYP2B6, le CYP2C9 ou le CYP3A.
Aucune étude spécifique d'interaction n'a pas été menée avec le Zemplar solution injectable.
Une étude d'interaction portant sur des doses multiples de kétoconazole et de capsules de paricalcitol a montré que le kétoconazole double approximativement l'ASC0-∞ du paricalcitol (voir «Propriétés/Effets», «Pharmacocinétique»). Comme le paricalcitol est partiellement métabolisé par le CYP3A et que le kétoconazole est un inhibiteur fort du CYP3A, il convient d'être prudent lors de l'administration simultanée de paricalcitol et de kétoconazole ou d'autres inhibiteurs forts du CYP3A.
Des médicaments contenant du phosphate ou des préparations analogues de la vitamine D ne doivent pas être pris en même temps que le Zemplar, car il existe alors un risque accru d'hypercalcémie et d'une élévation du produit Ca x P.
La toxicité digitalique est renforcée par l'hypercalcémie de toute origine, de sorte que la prudence est de mise lorsque des préparations digitaliques sont prescrites en même temps que le Zemplar.
Le propylène glycol neutralise l'effet de l'héparine. Le Zemplar contient du propylène glycol en tant qu'adjuvant et ne doit pas être administré en même temps que l'héparine.
L'administration simultanée de fortes doses de médicaments contenant du calcium ou de diurétiques thiazidiques avec le paricalcitol peut augmenter le risque d'hypercalcémie.
Les médicaments à base de magnésium (avant tout les antiacides) ne doivent pas être pris en même temps que des médicaments à base de vitamine D, car une hypermagnésémie peut survenir.
Les médicaments à base d'aluminium (avant tout antiacides et chélateurs de phosphate) ne devraient pas être pris constamment avec des médicaments contenant de la vitamine D, car il peut en résulter une hyperaluminémie sanguine et une toxicité osseuse liée à l'aluminium.
Grossesse, allaitementAucune donnée clinique n'est disponible sur l'utilisation pendant la grossesse.
Des études expérimentales chez l'animal indiqué une toxicité indirecte à des doses toxiques pour la grossesse, avec une influence sur la grossesse, le développement embryonnaire, le développement du fœtus et/ou le développement postnatal. Le risque potentiel pour l'homme n'est pas connu. Par conséquent, il n'est pas conseillé d'utiliser Zemplar au cours de la grossesse, sauf si cela est clairement nécessaire.
Comme aucune donnée n'est disponible concernant le passage éventuel du paricalcitol dans le lait maternel, l'allaitement doit être évité pendant le traitement par Zemplar.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude pertinente n'a été effectuée.
Effets indésirablesEffets indésirables des essais cliniques de phases II-IV contrôlés contre placebo et traitement actif.
Zemplar a été étudié chez 290 patients au cours d'essais cliniques de phases II-IV contrôlés contre placebo ou traitement actif.
L'effet indesirable le plus souvent observé en rapport avec Zemplar a été l'hypercalcémie (4,1% des patients). La survenue d'une hypercalcémie dépend de l'étendue de l'excès de suppression de la PTH qui peut être minimisé au moyen d'une titration adéquate de la posologie.
Les effets indésirables possiblement associés à l'administration de paricalcitol sont mentionnés au tableau 2 d'après leur fréquence.
Fréquences: très fréquents (≥10%), fréquents (>1%, ≤10%), occasionnels (>0,1%, ≤1%), rares (≥0,01%, <0,1%), très rares (<0,01%), inconnus (ne pouvant être évalués d'après les données disponibles).
Tableau 2: Effets indésirables observés au cours des études cliniques
|
Système organique
|
Fréquence
|
Effets indésirables
| |
Infections et infestations
|
occasionnels
|
Pneumonie, nasopharyngite, grippe, infection des voies respiratoires supérieures
| |
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes)
|
occasionnels
|
Cancer du sein
| |
Affections hématologiques et du système lymphatique
|
occasionnels
|
Anémie
| |
Affections endocriniennes
|
occasionnels
|
Hypoparathyroïdisme
| |
Troubles du metabolism et de la nutrition
|
fréquents
|
Hypercalcémie
| |
occasionnels
|
Hyperphosphatémie, hypocalcémie, perte d'appétit
| |
Affections psychiatriques
|
occasionnels
|
Délire, confusion, agitation, insomnie, nervosité, inquiétude
| |
Affections du système nerveux
|
fréquents
|
Céphalées, dysgueusie
| |
occasionnels
|
Vertige, hypoesthésie, myoclonies, paresthésies, syncope, événements cérébrovasculaires
| |
Affections oculaires
|
occasionnels
|
Conjonctivite
| |
Affections cardiaques
|
occasionnels
|
Fibrillation auriculaire, palpitations*, arrêt cardiaque
| |
Affections vasculaires
|
occasionnels
|
Hypotension artérielle, hypertension artérielle
| |
Affections respiratoires, thoraciques et médiastinales
|
occasionnels
|
Toux, dyspnée, orthopnée, œdème pulmonaire
| |
Affections gastro-intestinales*
|
fréquents
|
Saignements gastro-intestinaux*, diarrhée, constipation
| |
occasionnels
|
Ischémie intestinale, sécheresse buccale, troubles abdominaux, saignements rectaux, vomissements
| |
Affections de la peau et du tissu sous-cutané
|
occasionnels
|
Prurit, alopécie, exanthème prurigineux (rash), prurit, sensation de brûlure cutanée, exanthème vésicobulleux
| |
Affections musculosquelettiques et du tissu conjonctif
|
occasionnels
|
Arthralgie, myalgie, raideur des articulations, soubresauts musculaires
| |
Affections des organes de reproduction et du sein
|
occasionnels
|
Trouble de la fonction érectile, douleurs au sein
| |
Troubles généraux et anomalies au site d'administration
|
fréquents
|
Fièvre, frissons*, douleurs au site d'injection
| |
occasionnels
|
Modifications de la marche, œdème, asthénie, malaise, fatigue, péjoration de l'état général
| |
Investigations
|
occasionnels
|
SGOT (aspartate aminotransférase) élevée, valeurs anormales, perte de poids
|
* Palpitations, saignements gastro-intestinaux et frissons sont des effets indésirables (l'investigateur les évalue comme «non associés») observés à plus haute fréquence que sous placebo.
Effets secondaires supplémentaires provenant d'essais cliniques de phase IV, d'autres essais cliniques ou de la pharmacovigilance
Infections et infestations
Septicémie, infection vaginale.
Affections hématologiques et du système lymphatique
Lymphadénopathie.
Affections du système immunitaire
Hypersensibilité, angiœdème, œdème du larynx.
Affections endocriniennes
Hyperparathyréoïdisme.
Troubles du métabolisme et de la nutrition
Hyperkaliémie.
Affections du système nerveux
Seuil de stimulation plus élevé.
Affections oculaires
Glaucome, hyperémie dans l'œil.
Affections de l'oreille et du labyrinthe
Troubles de l'oreille.
Affections cardiaques
Arythmie.
Affections respiratoires, thoraciques et médiastinales
Sifflement (wheezing).
Affections gastro-intestinales
Dysphagie, gastrite, nausée.
Affections de la peau et du tissu sous-cutané
Hirsutisme, sudation nocturne, exanthème (rash), urticaire.
Troubles généraux et anomalies au site d'administration
Malaise thoracique, douleurs thoraciques, œdèmes, sensation modifiée, extravasation au site d'injection, œdème périphérique, douleurs, soif.
Investigations
Temps de saignement prolongé, arythmie cardiaque.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageJusqu'à présent aucun cas de surdosage accidentel aigu n'a été rapporté.
Un surdosage aigu de paricalcitol peut provoquer une hypercalcémie, une hypercalciurie, une hyperphosphatémie, ainsi qu'une suppression excessive de la parathormone (voir «Mises en garde et précautions»).
Les signes ou les symptômes d'une intoxication à la vitamine D associée à une hypercalcémie sont:
Symptômes précoces: faiblesse, céphalées, somnolence, nausées, vomissements, sécheresse buccale, constipation, douleur musculaires, douleurs osseuses, goût métallique.
Symptômes tardifs: anorexie, perte de poids, conjonctivite (calcifiante), pancréatite, photophobie, rhinorrhée, prurit, hyperthermie, perte de la libido, taux d'azote uréique sanguin élevé (BUN), hypercholestérolémie, augmentation de l'alanine aminotransférase sérique (ALAT) ainsi que de l'aspartate aminotransférase sérique (ASAT), calcification ectopique, hypertension, arythmie cardiale, somnolence, mort et, dans des cas rares, psychose.
Le traitement des patients avec une hypercalcémie cliniquement significative consiste en une réduction immédiate de la dose ou en une interruption du traitement avec le paricalcitol. Par ailleurs, un régime pauvre en calcium doit être prescrit et la prise éventuelle d'un supplément calcique arrêtée. Le patient doit être motivé et on doit le surveiller afin d'éviter un déséquilibre hydro-électrolytique. Particulièrement chez les patients sous digitaline, l'électrocardiogramme doit être observé de manière suivie, l'hémodialyse ou la dialyse péritonéale doit être remplacée au besoin par un dialysat appauvri en calcium. La concentration de paricalcitol dans l'organisme n'est toutefois pas réduite de manière significative par la dialyse.
La calcémie sérique doit être observée très attentivement jusqu'à ce qu'une normocalcémie soit rétablie (se reporter au paragraphe «Mises en garde et précautions»).
Lorsque les taux sériques de calcium se sont normalisés, le paricalcitol peut être réinitialisé à plus faible dose. Si des taux de calcium devaient s'avérer durablement nettement plus élevés, il convient d'envisager des alternatives thérapeutiques.
Propriétés/EffetsCode ATC
H05BX02
Mécanisme d'action
Pharmacodynamique
Le paricalcitol est un analogue synthétique biologiquement actif de la vitamine D de calcitriol comprenant des modifications chimiques aux chaînes latérales (D2) et au cycle A (19 nor). Il a pu être démontré que la vitamine D et le paricalcitol diminuent les concentrations de la parathormone (PTH) dans l'organisme.
Efficacité clinique
Patients adultes
L'efficacité du Zemplar à baisser la concentration de la parathormone intacte et biologiquement active (taux de la PTHi) dans le plasma ou dans le sérum a été étudiée sur plus de 600 patients avec une insuffisance rénale terminale (IRT) sous hémodialyse dans des études de phase II et III. En outre, trois études de phase III multicentriques, pivotales, en double aveugle, contrôlées avec un placebo, d'une durée allant jusqu'à 12 semaines ont été menées avec une administration trois fois par semaine de paricalcitol avec une dose initiale de 0,04 µg/kg, qui a été augmentée jusqu'à une dose maximale de 0,24 µg/kg. Des baisses plus importantes statistiquement significatives de la parathormone active biologiquement intacte (PTHi) ont été constatées chez les patients traités avec le Zemplar (baisse moyenne totale de 379 pg/ml) par rapport aux patients traités avec le placebo (baisse moyenne totale de 70 pg/ml). Comparé au placebo, une baisse d'au moins 30% du taux sérique de base de la PTHi (88% contre 61%) a été atteinte pendant l'étude, chez un nombre significativement plus important de patients. Pendant et après la première apparition d'une baisse supérieure ou égale à 30% de la PTHi, entre le groupe placebo et le groupe verum, aucune différence significative concernant l'apparition d'hypercalcémie et/ou d'un produit accru de la concentration sérique du calcium par celle du phosphore (Ca x P > 70) n'a été observée.
Sur la base de deux études de phase III multicentriques pivotales, comparatives menées en double aveugle, le paricalcitol administré par voie intraveineuse a été comparé avec le calcitriol administré par voie intraveineuse à 460 patients sous hémodialyse atteints d'une insuffisance rénale terminale (IRT). Les deux préparations ont été administrées chacune deux à trois fois par semaine. Le Zemplar a été dosé avec une dose initiale de 0,04 µg/kg avec une augmentation de dose de 0,04 µg/kg toutes les 4 semaines (jusqu'à un maximum de 0,24 µg/kg). Le calcitriol a été administré avec une dose initiale de 0,01 µg/kg avec une augmentation de dose de 0,01 µg/kg (jusqu'à une dose maximale de 0,06 µg/kg). Les doses ont été augmentées jusqu'à ce qu'une diminution supérieure ou égale à 50% du taux sérique de base de la PTHi ait été atteinte, ou jusqu'à ce que la cinquième augmentation de dose ait été atteinte. La proportion de patients présentant une baisse au moins égale ou supérieure à 50% du taux sérique de base de la PTHi était sensiblement semblable pour les deux traitements. Toutefois, pour une proportion légèrement supérieure de patients traités avec le paricalcitol, on a observé une baisse durable des valeurs du taux sérique de base de la PTHi. Le Zemplar baisse plus rapidement le taux de la PTHi que le Calcitriol et ce, de manière statistiquement significative.
Patients pédiatriques
La sécurité et l'efficacité du paricalcitol en injection ont été étudiées au cours d'une étude randomisée, contrôlée par placebo et en double aveugle pendant 12 semaines sur 29 patients pédiatriques de 5 à 19 ans atteints d'insuffisance rénale terminale sous hémodialyse. Presque tous les patients ont reçu une forme de vitamine D avant le début de l'étude. 76% des patients étaient masculin, 52% étaient caucasiens et 45% afro-américains. La dose initiale de paricalcitol était de 0,04 µg/kg, trois fois par semaine pour une concentration de PTHi <500 pg/ml, ou de 0,08 µg/kg trois fois par semaine pour une concentration de PTHi ≥500 pg/ml. La dose de paricalcitol a été adaptée par paliers de 0,04 µg/kg en fonction des concentrations sériques de PTHi, de calcium et de calcium x phosphate. Les doses ont été titrées comme suit: en cas de concentration de PTHi inférieure à 150 pg/ml ou plus de 60% inférieure à la valeur initiale, la dose a été diminuée ou le traitement a été temporairement arrêté. En cas de concentration de PTHi moins de 30% inférieure à la valeur initiale, de concentration sérique de calcium ≤11,0 mg/dl et de calcium x phosphate ≤75 mg²/dl², la dose a été augmentée. En cas de concentration de PTHi de 30% à 60% inférieure à la valeur initiale, de concentration sérique de calcium ≤11,0 mg/dl et de calcium x phosphate ≤75 mg²/dl², la dose a été inchangée. De plus, la dose a été réduite en cas de concentration de calcium x phosphate >75 mg²/dl². En cas de concentration sérique de calcium >11 mg/dl, l'administration a été interrompue jusqu'à ce que la valeur de calcium soit descendue à nouveau à un niveau ≤10,5 mg/dl. Ensuite, le traitement a été repris à la dose la plus faible suivante.
Les concentrations initiales moyennes de PTHi étaient de 841 pg/ml pour les 15 patients traités par le paricalcitol et de 740 pg/ml pour les 14 patients sous placebo. La dose moyenne administrée de paricalcitol était de 4,6 µg (fourchette: 0,8 µg–9,6 µg). 10 (67%) des 15 patients traités par le paricalcitol et 2 (14%) des 14 patients sous placebo ont terminé l'étude. 10 (71%) des patients sous placebo sont sortis de l'étude en raison d'une élévation excessive de la PTHi, définie comme deux concentrations successives de la PTHi supérieures à 700 pg/ml, et à cause de valeurs supérieures comparées aux valeurs initiales après quatre semaines de traitement. Dans l'analyse d'efficacité principale, 9 (60%) des 15 patients du groupe paricalcitol ont présenté deux diminutions successives de 30% de la PTHi contre 3 (21%) des 14 patients du groupe placebo (IC 95% pour la différence entre les groupes – 1%, 63%). Aucune hypercalcémie avec une concentration supérieure à 11,2 mg/dl n'a été constatée dans le groupe paricalcitol ni dans le groupe placebo.
PharmacocinétiqueLes propriétés pharmacocinétiques du paricalcitol ont été étudiées chez les patients atteints d'insuffisance rénale chronique nécessitant une dialyse.
Absorption
[Non pertinent]
Distribution
Le paricalcitol est administré par injection intraveineuse directe (en bolus).
Le paricalcitol présente une forte liaison, non saturable, aux protéines plasmatiques dans la plage de doses thérapeutiques (>99%). Le volume de distribution à l'état d'équilibre chez le sujet sain est d'environ 23,8 l après une dose de 0,24 µg/kg. Chez les patients atteints d'insuffisance rénale terminale exigeant une hémodialyse et une dialyse péritonéale, le volume de distribution moyen atteint 31 à 35 l après une dose de 0,24 µg/kg de paricalcitol administrée par voie intraveineuse
Métabolisme
Différents métabolites ont été détectés dans l'urine et les selles. Le paricalcitol n'a pas pu être détecté dans l'urine.
Les données in vitro montrent que le paricalcitol est métabolisé par différentes enzymes hépatiques et non hépatiques, y compris le CYP24 mitochondrial comme le CYP3A4 et UGT1A4. Les métabolites détectés incluent les produits de la 24-R-hydroxylation (concentrations plasmatiques faibles) ainsi que de la 24,28-dihydroxylation et de la glucuronidation directe. À des concentrations inférieures à 50 nM (21 ng/ml), le paricalcitol n'est pas un inhibiteur du CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 ou CYP3A. Aux concentrations de paricalcitol citées, on n'observe pas d'induction significative du CYP2B6, CYP2C9 et CYP3A4 (induction inférieure à un doublement).
Élimination
Le paricalcitol est principalement éliminé par excrétion hépato-biliaire, 74% d'une dose radiomarquée sont excrétés dans les selles et 16% dans l'urine.
La plus grande part de la radioactivité détectée dans le plasma correspond à du paricalcitol radiomarqué inchangé. Deux métabolites non significatifs comparativement au paricalcitol ont été détectés dans le plasma humain. Un métabolite a été identifié comme étant du 24-R-9-hydroxyparicalcitol; ce métabolite a été moins actif que le paricalcitol dans un modèle de rat in vivo de suppression de la PTH.
En l'espace de deux heures après l'administration de doses de 0,04 à 0,24 µg/kg les concentrations de paricalcitol diminuent rapidement. Ensuite, les concentrations de paricalcitol diminuent de façon logarithmique-linéaire avec une demi-vie d'environ 15 heures chez les patients sous hémodialyse, et de 5 à 7 heures chez les patients sains. Aucune accumulation de la substance active n'a été observée.
Tableau 3
|
Paramètres pharmacocinétiques (moyenne ± écart type) du paricalcitol chez les patients atteints d'insuffisance rénale terminale et chez les sujets sains
(Dose de 0,24 µg/kg)
| |
Paramètres
|
Patients atteints d'insuffisance rénale terminale
(N=12)
|
Sujets sains
(N=24)
| |
Cmax (ng/ml)
|
1,680 ± 0,511
|
1,670 ± 0,529
| |
ASC0-∞ (ng x h/mL)
|
14,51 ± 4,12
|
7,77 ± 2,73
| |
T½ (h)
|
13,9 ± 7,3
|
6,7 ± 3,1
| |
CL (L/h)
|
1,49 ± 0,60
|
2,6 ± 1,1
| |
Vdβ (L)
|
30,8 ± 7,5
|
29,9 ± 17,9
|
Cinétique pour certains groupes de patients
Age, sexe, race
Concernant les paramètres pharmacocinétiques, aucune différence liée à l'âge, au sexe ou à la race n'a été constatée chez les patients adultes examinés.
Insuffisance hépatique
La fraction de paricalcitol non liée est sensiblement identique chez les patients avec une insuffisance hépatique faible et chez les sujets sains. Par conséquent, une adaptation posologique chez ce groupe de patients n'est pas indiquée. On ne dispose pas de données pharmacocinétiques afférentes pour des patients atteints d'insuffisance hépatique grave.
Insuffisance rénale
La pharmacocinétique du paricalcitol a été étudiée chez les patients atteints d'insuffisance rénale terminale ayant besoin d'une hémodialyse et d'une dialyse péritonéale. Comparativement aux sujets sains, les patients atteints d'insuffisance rénale terminale ont présenté une clairance réduite (51%) et un doublement approximatif de la demi-vie et de l'ASC (voir tableau 3).
Interactions
Une étude in vitro a montré que, à des concentrations inférieures à 50 nM (21 ng/ml, soit 20 fois plus environ que les concentrations atteintes avec la dose la plus élevée testée), le paricalcitol n'est pas un inhibiteur du CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2D6, CYP2E1 ni du CYP3A. Sur des hépatocytes frais directement cultivés, à des concentrations de paricalcitol atteignant au maximum 50 nM, une induction du CYP2B6, du CYP2C9 ou du CYP3A inférieure à un doublement a été observée, tandis qu'avec des contrôles actifs une induction 6 à 19 fois supérieure a été atteinte. Ces résultats indiquent que le paricalcitol ne devrait pas inhiber ni induire de manière cliniquement significative la clairance de substances métabolisées par ces enzymes.
Les interactions avec le paricalcitol solution par injection n'ont pas été étudiées in vivo.
Kétoconazole: L'effet de doses multiples de kétoconazole (200 mg deux fois par jour pendant 5 jours) sur la pharmacocinétique des gélules de paricalcitol administrées par voie orale a été étudié chez des sujets sains. La Cmax du paricalcitol n'a été que faiblement modifiée. L'ASC0-∞ a été environ doublée en présence de kétoconazole. La demi-vie moyenne du paricalcitol est de 17,0 heures en association avec le kétoconazole, contre 9,8 heures lorsque le paricalcitol est administré seul. L'interaction avec le paricalcitol administré par voie parentérale n'a pas été étudiée (voir «Mises en garde et précautions»).
Données précliniquesAucun effet du paricalcitol sur la fertilité des rats (mâles ou femelles) n'a été constaté lors de l'administration intraveineuse de doses pouvant aller jusqu'à 20 µg/kg (correspondant à 13 fois la dose maximum recommandée chez l'homme [0,24 µg/kg] en incluant une posologie ajustée en fonction de la surface corporelle en mg/m2).
Il a été montré que le paricalcitol administré de manière quotidienne à des lapins et des rats provoque un préjudice minimal (5%) sur le développement du fœtus. La dose quotidienne chez le lapin était de 0,5 fois la dose (0,24 µg/kg) chez l'homme (calculé par rapport à la surface corporelle en mg/m2). La dose quotidienne chez le rat était de deux fois celle utilisée chez l'homme (0,24 µg/kg) (sur la base du taux plasmatique d'exposition). Parmi les plus fortes doses de paricalcitol étudiées chez les rats (20 µg/kg trois fois par semaine, correspondant à 13 fois la dose maximale chez l'homme calculée en fonction de la dose par rapport à la surface corporelle en mg/m2) chez lesquels des symptômes de toxicité maternelle étaient déjà apparus sous forme d'hypercalcémie, une augmentation significative de la mortalité chez les rats nouveau-nés a été observée. Aucune autre répercussion sur le développement des descendants n'a été rapportée. Le paricalcitol n'a pas d'effet tératogène pour les posologies étudiées.
Dans des études de cancérogénicité chez les rats et les souris, le paricalcitol n'avait pas d'influence sur l'incidence des tumeurs à l'exception des tumeurs provoquées par les effets de la calcémie chronique. Le paricalcitol n'a pas fait preuve de génotoxicité in vitro avec ou sans activation métabolite dans le test d'Ames, dans un test de mutagénicité portant sur des cellules de lymphome de souris (L5178Y) ou dans un test d'aberrations chromosomiques sur lymphocytes humains. Il n'y a pas non plus d'indication de toxicité génétique dans un test micronucleus in vivo chez la souris.
Remarques particulièresIncompatibilités
Vu qu'aucune étude de tolérance n'a été menée, le Zemplar ne doit pas être mélangé avec d'autres médicaments.
Influence sur les méthodes de diagnostic
Aucune influence de Zemplar sur les méthodes de diagnostic n'est connue.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Ne pas conserver au dessus de 30 °C.
Remarques concernant la manipulation
La solution injectable Zemplar est destinée à l'utilisation exclusive par un médecin.
Le Zemplar, en tant que préparation destinée à une utilisation parentérale, doit être vérifié visuellement avant toute administration afin de détecter toute matière particulière ou coloration éventuelles.
Zemplar ne contient pas d'agents conservateurs. La stabilité en cours d'utilisation n'a pas été contrôlée. Pour ces raisons, la préparation doit être utilisée aussitôt après son ouverture.
Il est recommandé d'éliminer les restes de médicament non utilisés ou les déchets selon les directives locales en vigueur.
Numéro d’autorisation56312 (Swissmedic).
PrésentationLa solution injectable Zemplar 2 μg paricalcitol pour 1 ml de solution est disponible en conditionnement de:
5 flacons à 1 ml (B).
La solution injectable Zemplar 5 μg paricalcitol pour 1 ml de solution est disponible en conditionnement de:
5 flacons à 1 ml (B).
La solution injectable Zemplar 10 µg paricalcitol pour 2 ml (5 µg pour 1 ml) de solution es disponible en conditionnement de:
5 flacons à 2 ml (B).
Titulaire de l’autorisationAbbVie AG, 6330 Cham.
Mise à jour de l’informationDécembre 2020.
|