CompositionPrincipes actifs
diboterminum alpha*.
* La dibotermine alpha (Protéine-2 ostéogénique humaine recombinante ; rhBMP-2) est une protéine humaine dérivée d’une lignée de cellules recombinantes d’ovaire de hamster chinois (CHO).
Excipients
Poudre : Glycinum, Natrii chloridum*, Saccharum, Polysorbatum 80, Acidum glutamicum,Natrii hydroxidum*, pro vitro.
Solvant: Aqua ad iniectabilia.
Matrice: Collagenum nativum (de boeuf).
*Après reconstitution, InductOs contient moins de 0,05 mg/ml de sodium.
Indications/Possibilités d’emploiInductOs est indiqué en tant qu’alternative à l’autogreffe osseuse pour l’arthrodèse lombaire intersomatique sur un niveau chez les adultes présentant une discopathie dégénérative et ayant suivi un traitement non chirurgical pour cette pathologie pendant au moins 6 mois.
InductOs est indiqué dans le traitement des fractures aiguës du tibia chez l’adulte, en tant que complément au traitement standard comprenant la réduction de la fracture ouverte et la fixation par enclouage centromédullaire sans alésage.
Posologie/Mode d’emploiInductOs doit être utilisé exclusivement par un chirurgien ayant une expérience de l’ostéosynthèse.
InductOs doit être préparé en suivant scrupuleusement les instructions pour la préparation (voir rubrique « Remarques particulières »). La dose appropriée dépend du volume de matrice imbibée nécessaire pour l’indication prévue.
Selon la configuration chirurgicale, si seulement une partie du produit est nécessaire, couper la matrice imbibée à la taille désirée et jeter la partie inutilisée.
Tableau posologique d’InductOs, boîte de 4 mg
|
Matrice InductOs imbibée
(boîte de 4 mg)
|
Dimensions de la matrice imbibée
|
Volume de la matrice imbibée
|
Concentration de la matrice imbibée
|
Dose de dibotermine alpha
| |
1 matrice
|
2,5 cm x 5 cm
|
1,3 cm3
|
1,5 mg/cm3
|
2 mg
| |
2 matrices
|
2 x (2,5 cm x 5 cm)
|
2,7 cm3
|
1,5 mg/cm3
|
4 mg
|
Tableau posologique d’InductOs, boîte de 12 mg
|
Portion de matrice InductOs imbibée
(boîte de 12 mg)
|
Dimensions de la matrice imbibée
|
Volume de la matrice imbibée
|
Concentration de la matrice imbibée
|
Dose de dibotermine alpha
| |
1/6 de la matrice
|
2,5 cm x 5 cm
|
1,3 cm3
|
1,5 mg/cm3
|
2 mg
| |
1/3 de la matrice
|
2,5 cm x 10 cm
|
2,7 cm3
|
1,5 mg/cm3
|
4 mg
| |
2/3 de la matrice
|
5 cm x 10 cm
|
5,3 cm3
|
1,5 mg/cm3
|
8 mg
| |
Matrice complète
|
7,5 cm x 10 cm
|
8 cm3
|
1,5 mg/cm3
|
12 mg
|
InductOs est préparé immédiatement avant l’usage.
Ce médicament est administré par implantation. InductOs ne doit pas être utilisé à une concentration supérieure à 1,5 mg/ml (voir «Surdosage»).
Les données concernant l’efficacité et la sécurité d’InductOs chez les personnes âgées (>65 ans) sont très limitées.
En l’absence d’un recul clinique suffisant, l’utilisation répétée d’InductOs et d’autres produits du même groupe de substances n’est pas recommandée.
La sécurité et l’efficacité d’InductOs n’ont pas été établies chez l’enfant.
Aucune étude n’a été effectuée chez des patients ayant une insuffisance hépatique, rénale ou cardiaque.
Le non-respect des instructions pour la préparation de la matrice pour implantation peut nuire à la tolérance et l’efficacité d’InductOs.
InductOs doit être manipulé avec des pinces. Au cours de la manipulation et de l’implantation, limiter la perte de liquide de la matrice. Ne pas presser.
Arthrodèse antérieure lombaire
InductOs ne doit pas être utilisé seul dans cette indication, mais doit être utilisé avec un ou plusieurs implants d’arthrodèse lombaire intersomatique approuvés (marqués CE). La compatibilité a été démontrée avec le titane, le polyétheréthercétone (PEEK) et les implants osseux
allogéniques.
Le volume nécessaire d’InductOs dépend de l’espace intervertébral et de la taille, de la forme et du volume interne du ou des implants d’arthrodèse lombaire intersomatique employés. Une attention toute particulière doit être apportée pour éviter un remplissage excessif de l’implant d’arthrodèse lombaire intersomatique et/ou de la partie antérieure de l’espace intervertébral (voir « Mises en garde et précautions »).
Typiquement, 4 mg (2,7 cm3 de matrice imbibée) d’InductOs sont utilisés dans l’espace intervertébral. La dose maximale est de 8 mg (5,3 cm3 de matrice imbibée) d’InductOs dans l’espace intervertébral. InductOs doit être introduit dans le ou les implant(s) d’arthrodèse lombaire intersomatique ou dans la partie antérieure de l’espace intervertébral.
Avant l’implantation
Boîte de 4 mg :
La matrice est prédécoupée en 2 pièces de 2,5 x 5 cm chacune.
Boîte de 12 mg :
La matrice se présente sous la forme d’une pièce de 7,5 x 10 cm. La matrice imbibée d’InductOs doit être coupée en 6 parties égales (environ 2,5 x 5 cm) pour faciliter le choix de la dose. Ces morceaux pourront ensuite être recoupés si nécessaire.
Le volume d’InductOs correspondant au volume interne de l’implant d’arthrodèse lombaire doit être introduit (avec précaution et en évitant tout remplissage excessif) dans le creux de l’implant.
Veiller à ne pas comprimer le produit ou à ne pas remplir excessivement le volume prévu pour la formation du nouveau tissu osseux (voir « Mises en garde et précautions »).
Implantation
Conformément à la pratique standard, la substance discale et les parties cartilagineuses des plateaux vertébraux doivent être retirées tout en préservant la partie corticale, et l’hémostase doit être réalisée (voir « Interactions »).
Se référer à la notice du fabricant pour les instructions d’implantation de l’implant d’arthrodèse lombaire intersomatique.
InductOs ne doit pas être placé postérieurement à l’implant d’arthrodèse lombaire intersomatique lorsqu’un accès direct au canal rachidien et/ou à la (aux) racine(s) nerveuse(s) est possible. En cas de fuite possible dans le canal rachidien et la racine.
Après l’implantation
L’intérieur de l’espace intervertébral ne doit pas être irrigué après implantation d’InductOs et du ou des implants d’arthrodèse lombaire intersomatique. À l’extérieur de l’espace intervertébral, le champ chirurgical doit être irrigué si besoin, et tout liquide s’écoulant de la matrice imbibée doit être rincé.
Si un drain chirurgical est requis, celui-ci doit être placé loin du site d’implantation ou, de préférence, dans une zone superficielle par rapport au site d’implantation.
Traitement chirurgical des fractures aiguës du tibia
Le volume d’InductOs à implanter est déterminé par l'anatomie de la fracture et la possibilité de fermer la plaie sans trop tasser ou comprimer le produit. D'une façon générale, chaque site de fracture est traité avec le contenu d’une boîte. La dose maximale est de 24 mg (2 matrices entières de boîtes de 12 mg).
Avant l’implantation
La réduction définitive, la fixation et l’hémostase de la fracture doivent être réalisées avant d’implanter InductOs.
InductOs doit être plié ou coupé avant l’implantation.
Implantation
InductOs sera implanté une fois que les traitements standards de la fracture et de la plaie seront achevés (c’est-à-dire à la fermeture des tissus mous).
La surface accessible de la fracture (lignes de fracture et défauts osseux) doit être autant que possible recouverte d’InductOs. InductOs doit être placé de sorte qu’il recouvre la zone de la fracture et assure un bon contact avec les fragments proximaux et distaux principaux.
La matrice implantable peut être placée dans une cavité (sans la tasser), être pliée, roulée ou enveloppée, en fonction de la géométrie de la fracture. InductOs n’assure pas de stabilité mécanique et ne doit pas être utilisé pour remplir des espaces en présence de forces de compression.
Après l’implantation
Ne pas irriguer la plaie, une fois qu’InductOs est implanté.
Si un drain chirurgical est requis, celui-ci doit être placé loin du site d’implantation ou, de préférence, dans une zone superficielle par rapport au site d’implantation.
Pour assurer le maximum de l’efficacité potentielle, il est important de recouvrir complètement InductOs par les tissus mous après son implantation.
InductOs contient du polysorbate 80, connu pour augmenter le degré d’extraction de di-(2-ethylhexyl) phthalate (DEHP) à partir du polyvinylchloride (PVC). C’est pourquoi il est conseillé de ne pas laisser la préparation reconstituée dans l’emballage de PVC plus longtemps qu’indiqué dans la rubrique « Remarques concernant la manipulation ».
Contre-indicationsInductOs est contre-indiqué chez les patients présentant :
·une hypersensibilité connue à la dibotermine alpha, au collagène bovin de type I ou à n’importe quel autre excipient du médicament,
·une immaturité du squelette,
·toute malignité active ou tout patient subissant un traitement pour une tumeur maligne,
·une grossesse,
·une infection active au site de l’opération,
·un syndrome des loges persistant ou des séquelles neurovasculaires d’un syndrome des loges,
·des fractures pathologiques comme celles observées dans la maladie de Paget (mais non limitées à celle-ci) ou dans l’os métastatique.
Mises en garde et précautionsLe non-respect des instructions pour la préparation (rubrique « Remarques particulières - Remarques concernant la manipulation ») et du mode d’administration (rubrique « Posologie/Mode d’emploi ») peut nuire à la tolérance et à l’efficacité d’InductOs.
Chirurgie cervicale
La sécurité et l’efficacité d’InductOs lors d’une chirurgie cervicale n’ont pas été établies. InductOs ne doit pas être utilisé dans ce cas. Un oedème localisé associé à l’utilisation d’InductOs a été rapporté chez des patients ayant subi une chirurgie cervicale. L’apparition de l’oedème peut être retardée et, dans certains cas, il peut être suffisamment important pour entraîner une obstruction des voies aériennes et/ou une dysphagie.
Tumeurs malignes
InductOs ne doit pas être utilisé chez les patients ayant des antécédents ou une suspicion clinique de tumeur maligne au site d’application (voir « Contre-indications »).
Ossification ectopique
L'utilisation d’InductOs peut provoquer une ossification ectopique au niveau du site d’implantation et/ou dans les tissus environnants pouvant être responsable de complications. Une formation osseuse excessive sur le site d’implantation et une formation osseuse ectopique ont été observées. Il peut s’avérer nécessaire d’enlever cette formation osseuse par voie chirurgicale.
Augmentation de la résorption osseuse
InductOs peut causer une résorption initiale de l’os trabéculaire environnant mise en évidence par radiotransparence. Par conséquent, en l’absence de données cliniques, le produit ne doit pas être utilisé pour des applications directes sur l’os trabéculaire où une résorption passagère de l’os peut créer un risque de fragilité osseuse (voir « Effets indésirables »).
Collection liquidienne
La formation d’une collection liquidienne (par exemple pseudokyste, oedème localisé, effusion au niveau du site de l’implant) parfois encapsulée, pouvant entraîner, dans certains cas, une compression nerveuse et une douleur, a été associée à l’utilisation d’InductOs. Une intervention médicale (aspiration et/ou enlèvement du matériel chirurgical) peut être nécessaire si les symptômes persistent (voir « Effets indésirables »).
Influence sur la grossesse et l’allaitement
Les femmes en âge de procréer doivent être informées que la formation d’anticorps antidibotermine alpha ou leur influence sur le développement du foetus n’ont pas encore été évaluées.
Au cours de l’essai clinique destiné à confirmer l’innocuité et l’efficacité d’InductOs, 2/277 (0,7%) patients traités au composant de transplantation osseuse InductOs et 1/127 (0,8%) patients traités par autogreffes osseuses ont développé des anticorps antidibotermine alpha. L’effet sur l’enfant à naître des anticorps maternels antidibotermine alpha, qui peuvent être présents pendant plusieurs mois après l’implantation du dispositif, n’est pas encore connu. De plus, on ignore si l’expression foetale de BMP-2 pourrait réexposer les mères qui étaient au préalable positives aux anticorps et provoquer ainsi une réponse immunitaire plus forte au BMP-2 avec des conséquences négatives pour le foetus.
L’innocuité et l’efficacité d’InductOs chez la mère allaitante n’ont pas été établies. On ignore si le BMP-2 est excrété dans le lait maternel.
Il est conseillé aux femmes en âge de procréer d’éviter une grossesse pendant l’année suivant le traitement par InductOs.
Populations particulières
Il n’existe aucune donnée relative à la sécurité d’emploi et à l’efficacité d’InductOs chez des patients porteurs d’une maladie auto-immune, notamment la polyarthrite rhumatoïde, le lupus érythémateux disséminé, la sclérodermie, le syndrome de Sjögren et la dermatomyosite/polymyosite.
L’efficacité et la sécurité d’InductOs n’ont pas été démontrées chez des patients présentant une ostéopathie métabolique.
Aucune étude n’a été effectuée chez des patients avec fonction hépatique, rénale ou cardiaque réduite.
Réponse immunitaire
De manière générale, la dibotermine alpha et le collagène bovin de type I peuvent provoquer des réactions immunitaires chez certains patients.
Anticorps antidibotermine alpha
Dans les études sur l’arthrodèse, 1,3% des patients ayant reçu InductOs ont développé des anticorps antidibotermine alpha, contre 0,8% des patients traités par autogreffe osseuse.
Dans les études sur les fractures d’os longs, 6,3% des patients ayant reçu de la dibotermine alpha avec une matrice à base de collagène bovin de Type I ont développé des anticorps antidibotermine alpha, contre 1,3% dans le groupe contrôle. Toutes les recherches d’anticorps neutralisant la protéine-2 ostéogénique ont été négatives.
Anticorps anti-collagène bovin de Type I
Dans les études sur l’arthrodèse, 13,5% des patients ayant reçu InductOs ont développé des anticorps anti-collagène bovin de Type I, contre 14,3% des patients traités par autogreffe osseuse.
Dans les études sur les fractures des os longs, 13,0% des patients ayant reçu de la dibotermine alpha avec une matrice à base de collagène bovin de Type I ont développé des anticorps anti-collagène bovin de Type I, contre 5,3% des patients du groupe contrôle. Aucun patient présentant un titre positif de collagène bovin de Type I n’a présenté d’anticorps anti-collagène humain de Type I à réaction croisée.
Bien qu’au cours des études cliniques aucune association n’ait été observée entre le résultat clinique et l’apparition d’effets indésirables, le développement potentiel d’anticorps neutralisants ou de réaction de type hypersensibilité ne peut être exclu. La possibilité d'une réaction immunitaire au produit devra être évaluée dans les cas où l’on suspecte un effet indésirable de type immunologique. Une attention particulière doit être apportée au rapport bénéfice/risque chez les patients ayant déjà reçu du collagène injectable (voir aussi « Contre-indications »). En l’absence d’expérience, l’utilisation répétée d’InductOs est déconseillée.
Mises en garde et précautions spéciales en relation avec l’arthrodèse lombaire intersomatique
La sécurité et l’efficacité d’InductOs n’ont pas été démontrées dans les conditions suivantes :
•utilisation avec des implants d’arthrodèse intersomatique fabriqués à partir de matériaux autres que le titane, le PEEK ou l’os
•implantation au niveau d’autres sites que la colonne vertébrale lombaire
•utilisation dans d’autres techniques chirurgicales que l’arthrodèse lombaire intersomatique.
Afin d’éviter tout effet pharmacologique excessif d’InductOs, une attention toute particulière doit être apportée pour éviter un remplissage excessif de l’implant d’arthrodèse lombaire intersomatique et/ou de la partie antérieure de l’espace intervertébral.
Ossification ectopique
Une formation osseuse à l’extérieur de l’espace intervertébral n’est pas souhaitable car elle pourrait avoir des conséquences délétères sur les structures neurovasculaires locales.
Lors des études cliniques menées sur l’arthrodèse lombaire intersomatique par voie postérieure avec de la dibotermine alpha dans le traitement de la discopathie dégénérative, une formation osseuse postérieure a été observée à la TDM. Celle-ci pourrait dans certains cas entraîner une compression nerveuse nécessitant une intervention chirurgicale (voir « Effets indésirables »). Par mesure de précaution, une barrière physique entre la matrice et le tissu neurologique doit être recréée (voir rubrique « Posologie/Mode d’emploi »).
Migration de l’implant
Une migration de l’implant susceptible de nécessiter une reprise chirurgicale est possible après l’utilisation d’InductOs dans le cadre d’une arthrodèse rachidienne (voir « Effets indésirables »).
Mises en garde et précautions spéciales en relation avec les fractures aiguës du tibia
InductOs est destiné à être utilisé chez des patients présentant les caractéristiques suivantes :
•une réduction et une stabilisation de fracture adéquates pour assurer la stabilité mécanique,
•un statut neurovasculaire adéquat (p.ex. absence de syndrome de loge, faible risque d’amputation),
•une hémostase adéquate (c.-à-d. fournissant un site d’implantation relativement sec),
•une absence de réparation défectueuse de large segment de l’os long, dans lequel la compression importante du tissu mou peut se produire.
L’implant ne peut être employé au site de fracture que dans de bonnes conditions de visibilité du site et avec très grand soin (voir aussi « Posologie/Mode d’emploi »).
Les données d’efficacité dans le cas d’une fracture du tibia n’ont été obtenues qu’à partir d’essais cliniques contrôlés dans lesquels les fractures ouvertes de tibia ont été stabilisées au moyen d’un enclouage centromédullaire (voir « Propriétés/Effets », « Pharmacodynamique »).
Dans une étude clinique où le canal médullaire a été alésé jusqu’au contact de l’os cortical, une augmentation du taux d’infection a été observée dans le groupe traité par InductOs comparativement au groupe témoin recevant le traitement standard (voir « Effets indésirables »). L’utilisation d’InductOs avec la technique d’enclouage avec alésage dans la réduction d’une fracture ouverte du tibia n’est pas recommandée.
InductOs n’assure pas la stabilité mécanique et ne doit pas être utilisé pour remplir un espace vide en présence de forces de compression. Les techniques de soins des fractures d’os long et des tissus mous doivent être basées sur des pratiques reconnues, y compris pour le contrôle de l’infection.
Excipients revêtant un intérêt particulier (tous les emballages)
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose maximale (2 boîtes de 12 mg), c.-à-d. qu’il est essentiellement « sans sodium ».
InteractionsAucune étude de métabolisme n’a été effectuée. La dibotermine alpha étant une protéine qui n’a pas été identifiée dans la circulation générale, il est peu probable qu’InductOs donne lieu à des interactions médicamenteuses de type pharmacocinétique.
Les données issues des études cliniques sur les fractures aiguës du tibia montrent que l’utilisation d’InductOs chez des patients recevant des glucocorticoïdes n’a été associée à aucun effet indésirable apparent.
Dans les études précliniques, l’administration concomitante de glucocorticoïdes a diminué la réparation osseuse (mesurée en pourcentage de changement par rapport au groupe contrôle), mais l’efficacité d’InductOs n’a pas été modifiée.
Une étude in vitro a démontré que la dibotermine alpha se lie aux agents hémostatiques et colles à base de fibrine. L’utilisation de ces produits à proximité d’InductOs n’est pas recommandée car elle pourrait entraîner une formation osseuse au niveau du site d’implantation de l’agent hémostatique ou de la colle à base de fibrine (voir « Posologie/Mode d’emploi »).
Dans les essais cliniques sur les fractures aiguës du tibia, des effets indésirables d’intensité modérée ou moyenne en rapport avec la cicatrisation de plaie (p.ex. drainage de plaie) ont été davantage observés chez les patients traités par InductOs et recevant de façon conjointe des AINS pendant 14 jours consécutifs, que chez les patients traités par InductOs n’ayant pas reçu d’AINS. Bien que le succès du traitement n’ait pas été compromis chez ces patients, une interaction entre les AINS et InductOs ne peut pas être exclue.
Grossesse, AllaitementGrossesse
Il n’existe aucune donnée relative à l’innocuité et à l’efficacité d’InductOs chez la femme enceinte.
Les études chez l’animal ont montré une toxicité pour la fonction reproductive (voir « Données précliniques »). Le risque potentiel chez l’homme n’est pas connu.
Comme les risques sur le foetus sont inconnus, dus au développement potentiel d’anticorps neutralisants antidibotermine alpha, InductOs est contre-indiqué en cas de grossesse (voir aussi « Contre-indications » et « Mises en garde et précautions »).
En outre, les femmes en âge de procréer doivent être informées que l’impact de la formation d’anticorps à la dibotermine alpha sur le développement du foetus n’a pas encore été évalué (voir « Mises en garde et précautions »).
Allaitement
Il n’est pas connu si la dibotermine alpha est excrétée dans le lait maternel et il n’existe aucune donnée relative à la sécurité et à l’efficacité d’InductOs chez les femmes qui allaitent. La prudence est de rigueur dans le cadre d’une prescription pendant l’allaitement (voir « Mises en garde et précautions »).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n’a été effectuée, mais puisque InductOs n’a pas d’effet systémique, il est peu probable qu’il occasionne une gêne au niveau de l’aptitude à la conduite et l’utilisation de machines.
Effets indésirablesPlus de 1700 patients ont été traités par InductOs lors des essais cliniques, parmi lesquels environ 500 patients avec des fractures ouvertes du tibia, environ 600 patients avec discopathie dégénérative de la colonne vertébrale et environ 600 patients dans des études cliniques portant sur des indications non approuvées en Suisse.
Les observations liées à l’utilisation d’InductOs après la mise sur le marché ont été inclus dans la liste des effets indésirables.
La fréquence des effets indésirables observés chez les patients exposés au traitement par InductOs est indiquée ci-dessous. Les fréquences sont indiquées comme très fréquents (≥ 1/10) ou fréquents (≥ 1/100, < 1/10). Les effets indésirables peu fréquent (≥ 1/1000, < 1/100), rare (≥ 1/10'000, < 1/1 000) ou très rare (< 1/10’000) ne sont pas connus.
Effets indésirables spécifiques à l’utilisation pour l’arthrodèse lombaire antérieure
Affections du système nerveux
Fréquent : Evénements radiculopathiques (incluant la radiculite, la radiculopathie lombaire, la douleur radiculaire, la radiculite lombosacrée, la radiculopathie et la sciatique)
Affections musculo-squelettiques et du tissu conjonctif
Fréquent : L’ossification ectopique* (incluant l’exostose, l’ossification extrasquelettique, la calcification ectopique postopératoire, l’augmentation de la formation osseuse et la calcification au niveau du site de l’implant)
Troubles généraux et anomalies au site d’administration
Fréquent : migration de l‘implant*, collection liquidienne* (incluant l’oedème localisé, le pseudokyste et l’effusion au niveau du site de l’implant)
Effets indésirables identifiés après commercialisation (fréquence inconnue)
Affections musculo-squelettiques et du tissue conjonctif
Ostéolyse*, augmentation de la résorption osseuse*
Effets indésirables spécifiques à l’utilisation dans les fractures aiguës du tibia
Infections et infestations
Très fréquent : infection localisée*
Troubles généraux et anomalies au site d’administration
Fréquent : Migration de l’implant*, collection liquidienne* (incluant l’oedème localisé, le pseudokyste et l’effusion au niveau du site de l’implant)
Effets indésirables identifiés après commercialisation (fréquence inconnue)
Affections musculo-squelettiques et du tissue conjonctif
Ostéolyse, augmentation de la résorption osseuse
* voir ci-dessous pour les informations complémentaires
Formation de nouveaux tissus osseux et remodelage osseux
Le remodelage osseux fait partie du mécanisme d’action pharmacologique de la dibotermine alpha (voir rubrique « Propriétés/Effets – Mécanisme d’action/pharmacodynamique »). Ce processus fait intervenir à la fois une résorption et une formation osseuses. Dans certains cas, une exagération de ces phénomènes peut entraîner des complications telles qu’une compression nerveuse, due à une ossification ectopique, ou à une migration de l’implant, associé à la résorption osseuse ou à l’ostéolyse.
Pendant la période de suivi de deux ans des essais cliniques menés sur l’arthrodèse lombaire intersomatique utilisant une approche postérieure, une ossification ectopique observée par radiographie a touché davantage de patients traités avec InductOs que de patients ayant reçus une greffe autologue (voir « Mises en garde et précautions »). Cette observation radiologique pouvait être symptomatique ou non.
Collection liquidienne
En raison de l’action angiogénique d’InductOs, une collection liquidienne (pseudokyste, oedème localisé, effusion au niveau du site de l’implant), parfois encapsulée, peut survenir, entraînant dans certains cas une compression nerveuse et une douleur.
Les oedèmes localisés ont été fréquents lors de l’utilisation pour l’arthrodèse cervicale. Il a été observé un délai d’apparition de l’oedème et, dans certains cas, il a été suffisamment important pour entraîner une obstruction des voies aériennes. InductOs n’est pas approuvé pour une utilisation dans l’arthrodèse cervicale (voir « Mises en garde et précautions »).
Infection localisée
Dans une étude où le canal médullaire a été alésé jusqu’au contact de l’os cortical, une infection localisée spécifique au membre fracturé est survenue chez plus d’1 patient sur 10. Une augmentation du taux d’infection a été observée dans le groupe traité par InductOs par rapport au groupe témoin recevant seulement le traitement standard (respectivement 19% versus 9 %; voir « Mises en garde et précautions »). Lors de l’utilisation de la technique d’enclouage sans alésage, les taux d’infection étaient similaires entre le groupe traité et le groupe témoin (21 % dans le groupe InductOs, versus 23 % dans le groupe témoin).
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageL’utilisation d’InductOs à des quantités inférieures ou comparables à celles recommandées pour l’arthrodèse lombaire a été associée à des cas d’oedèmes localisés suffisamment graves pour entraîner une obstruction des voies respiratoires chez les patients ayant subi une chirurgie cervicale (voir « Mises en garde et précautions »).
Propriétés/EffetsCode ATC
M05BC01
Mécanisme d’action
La dibotermine alpha est une protéine ostéoinductrice qui induit la formation de nouveau tissu osseux au site d’implantation. La dibotermine alpha se lie à des récepteurs de surface des cellules mésenchymateuses et provoque la différenciation de ces cellules en cellules formant du cartilage et des os. Les cellules différenciées forment de l’os trabéculaire tandis que la matrice est dégradée et qu’une invasion vasculaire se produit en même temps. Le processus de formation osseuse se développe de l’extérieur de l’implant vers le centre, jusqu’à ce que tout l’implant d’InductOs soit remplacé par de l’os trabéculaire.
Le remodelage de l’os trabéculaire environnant se produit en accord avec les forces biomécaniques qui s’exercent sur lui. L’implantation d’InductOs dans de l’os trabéculaire a eu comme conséquence la résorption transitoire de l’os autour de l’implant, suivie de son remplacement par un nouvel os, plus dense. Le remodelage osseux dû à InductOs peut être responsable de l’intégration biologique et biomécanique du nouvel os induit par InductOs avec l’os environnant. L’évaluation radiographique, biomécanique et histologique de l’os induit indique qu’il fonctionne biologiquement et biomécaniquement comme l’os natif.
Par ailleurs, des études précliniques ont indiqué qu’en cas de fracture, l’os induit par InductOs peut se réparer de lui-même d’une façon équivalente à celle de l’os natif.
Des études précliniques ont suggéré que la formation d’os induite par InductOs est un processus auto-limité, formant un volume d’os bien défini. Cette auto-limitation est probablement due à la dégradation de la dibotermine alpha au site d’implantation, ainsi qu’à la présence d’inhibiteurs de protéines ostéogéniques (BMP) dans les tissus environnants. De plus, plusieurs études précliniques indiquent qu’un mécanisme de rétrocontrôle négatif au niveau moléculaire limite l’induction osseuse par les protéines ostéogéniques.
Des données histologiques issues des études animales menées sur l’arthrodèse lombaire intersomatique utilisant une approche chirurgicale antérieure ou postérieure ont démontré que la dibotermine alpha était biocompatible avec les implants intersomatiques en titane, en PEEK ou allogéniques et qu’elle était associée à des taux élevés et constants de fusion indépendant de l’approche chirurgicale ou du matériau de l’implant, et à une formation de tissu fibreux moins importante que l’autogreffe.
Des études cliniques de pharmacologie démontrent que la matrice seule n’est pas ostéoinductrice et n’est plus présente dans des biopsies pratiquées aussi précocement que 16 semaines après l’implantation.
Pharmacodynamique
Se référer à « Mécanisme d’action ».
Efficacité clinique
Données pharmacodynamiques spécifiques aux études sur l’arthrodèse lombaire antérieure
L’efficacité et la sécurité d’InductOs ont été mises en évidence dans une étude randomisée, contrôlée, multi-centrique, de non-infériorité sur 279 patients âgés de 19 à 78 ans subissant une arthrodèse lombaire par voie antérieure ouverte. Les patients ont suivi un traitement non chirurgical pendant au moins six mois avant traitement pour l’arthrodèse lombaire antérieure. Les patients ont été randomisés pour recevoir un implant de fusion intersomatique en titane en association soit avec InductOs, soit avec une autogreffe d’os prélevé dans la crête iliaque.
24 mois après l’opération, il a été démontré qu’InductOs était statistiquement non-inférieur à l’autogreffe osseuse, avec un taux de réussite de fusion déterminé par radiographie de 94,4% pour InductOs contre 88,9% pour l’autogreffe osseuse (IC bilatéral à 95% pour la différence : -1,53, 12,46). Concernant la douleur et le handicap (score d’Oswestry), le taux de réussite était de 72,9% dans le groupe traité par InductOs contre 72,5% dans le groupe traité par autogreffe osseuse (IC bilatéral à 95% pour la différence : -11,2, 12,0).
Une méta-analyse post-hoc de 6 études cliniques contrôlées regroupant les données de patients traités par InductOs ou par autogreffe osseuse avec des implants de fusion intersomatique marqués CE ou des implants osseux allogéniques selon diverses approches chirurgicales ont montré que, 24 mois après l’intervention, InductOs était associé à un meilleur taux de réussite de la fusion (95 %, 241 patients sur 255) que l’autogreffe osseuse (85 %, 177 patients sur 209), avec un rapport des cotes de 3,26 (IC à 95% : 1,172 ; 9,075 ; p = 0,024). La différence absolue estimée en terme de taux de réussite de la fusion entre InductOs et l’autogreffe osseuse était de 11,7 % (IC à 95 % : 0,8 % ; 22,5 %, p = 0,035).
Une analyse regroupant les données de sécurité de 8 études cliniques 24 mois après l’intervention a montré que la fréquence des pseudarthroses était approximativement deux fois plus faible après un traitement par InductOs (4,8 %, 22 des 456 patients) en comparaison avec l’autogreffe osseuse (12,7 %, 31 des 244 patients).
Données pharmacodynamiques spécifiques aux études sur les fractures aiguës du tibia
L’efficacité d’InductOs a été démontrée dans un essai multinational, randomisé, contrôlé, en simple-aveugle de 450 patients (âgés de 18 à 87 ans ; 81% de sexe masculin) avec des fractures ouvertes du corps du tibia, nécessitant un traitement chirurgical. Les patients ont reçu (selon une distribution de type 1:1:1) soit les soins standard (groupe contrôle) comportant une fixation par enclouage centromédullaire et les soins de routine des tissus mous, soit les soins standard plus InductOs 0,75 mg/ml, soit les soins standard plus InductOs 1,5 mg/ml. Les patients ont été suivis pendant 12 mois après la fermeture des tissus mous.
Dans l’essai pivot sur la fracture aiguë du tibia, InductOs a augmenté la probabilité de guérison fracturaire. Les patients traités par InductOs 1,5 mg/ml ont eu un risque d’échec de traitement (intervention secondaire pour aider à la guérison de la fracture) réduit de 44% par rapport aux patients du groupe de soins standard (RR = 0,56; CI 95% = 0,40 à 0,78). Ces résultats ont été corroborés de façon indépendante et en insu par un ensemble de radiologues. Le nombre d’interventions secondaires et ultérieures a été réduit de façon significative chez les patients traités par InductOs, en particulier en ce qui concerne des interventions plus invasives telles que la greffe osseuse et le changement d’enclouage (p = 0,0326).
La proportion de patients guéris après traitement par InductOs 1,5 mg/ml était significativement plus élevée pour toutes les consultations post-opératoires à partir de 10 semaines jusqu’à 12 mois, ce qui suggère une guérison accélérée des fractures.
InductOs 1,5 mg/ml était significativement plus efficace (en comparaison avec le groupe contrôle) chez les patients avec ou sans antécédents de tabagisme.
Gravité des fractures : Le traitement par InductOs 1,5 mg/ml était significativement efficace pour toutes les classes de fracture, y compris les fractures sévères Gustilo IIIB (risque d’intervention secondaire réduit de 52% par rapport aux patients ayant eu des soins standard).
A la consultation de 6 semaines après le traitement, la proportion de patients présentant une cicatrisation des lésions des tissus mous était significativement plus élevée dans le groupe InductOs 1,5 mg/ml, que dans le groupe soins standard (83% contre 65%; p = 0,0010).
La proportion de patients avec un échec lié au matériel (vis de blocage pliées ou cassées) était significativement plus basse dans le groupe InductOs 1,5 mg/ml que dans le groupe soins standard (11% contre 22%; p = 0,0174).
PharmacocinétiqueAbsorption
InductOs développe son activité au site d’implantation.
Distribution
Lors de deux études préliminaires, des échantillons de sérum ont été collectés avant et après l’intervention chirurgicale chez quelques patients avec des fractures d’os longs. La dibotermine alpha n’était pas détectable dans le sérum.
Métabolisme
Lors d’études sur des animaux (rats) avec de l’InductOs contenant de la dibotermine alpha marquée de façon radioactive, le temps moyen de persistance au site d’implantation était de 4 – 8 jours. Les taux circulants maximaux de dibotermine alpha (0,1% de la dose implantée) ont été observés dans les 6 heures suivant l’implantation. Lors d’une injection intraveineuse, la demi-vie terminale de dibotermine alpha était de 16 minutes chez le rat et de 6,7 minutes chez le singe cynomolgus.
Élimination
On peut en conclure que la dibotermine alpha est libérée lentement de la matrice sur le site d’implantation, et est rapidement éliminée lorsqu’elle arrive dans la circulation systémique.
Données précliniquesLes données précliniques n’indiquent aucun risque spécial pour l’homme sur la base des études conventionnelles de pharmacologie de sécurité, de toxicité aiguë, de toxicité à dose répétée et de genotoxicité.
Des études sur des souris génétiquement modifiées ont indiqué que le BMP-2 est critique pour le développement foetal et que le manque d’activité BMP-2 peut causer une mort néonatale ou des malformations congénitales.
La dibotermine alpha a montré des effets variables sur les lignées cellulaires de tumeur humaine in vitro. Les données in vivo disponibles sur les lignées cellulaires de tumeur humaine ne suggèrent pas de potentiel pour la promotion de croissance tumorale ou des métastases. Le potentiel cancérogène d’InductOs n’a pas été testé in vivo, celui-ci étant un produit à usage unique (voir aussi « Contre-indications »).
Dans les études de toxicité de la reproduction chez le rat, où la dibotermine alpha a été administrée par voie intraveineuse pour maximiser l’exposition générale, une augmentation du poids foetal et de l’ossification foetale a été observée et un effet relatif au traitement ne peut être exclu.
Des anticorps antidibotermine ont été recherchés chez des lapins femelles gestantes après une immunisation importante avec la dibotermine alpha dans le but d’induire expérimentalement la production d’anticorps antidibotermine alpha. Quelques foetus de lapin ayant un poids diminué ont présenté une diminution de l’ossification des os frontaux et pariétaux (4 cas sur 151 foetus de lapin), généralement considérée comme réversible, et pour laquelle un lien avec la présence d’anticorps ne peut être exclu. Aucune autre altération de la morphologie foetale externe, viscérale ou squelettique n’a été observée.
Une étude sur l’implantation vertébrale d’InductOs a été réalisée sur un modèle canin. InductOs a été implanté directement sur la dure-mère exposée après laminectomie. Bien qu’un rétrécissement et une sténose du foramen vertébral aient été observés, aucune minéralisation de la dure-mère, sténose de la colonne vertébrale ou déficit neurologique n’ont été observés après application d’InductOs.
Remarques particulièresIncompatibilités
La matrice reconstituée ne doit pas être mélangée à d’autres produits pharmaceutiques.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention « EXP » sur l’emballage.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 30°C.
Ne pas congeler.
Conserver dans l’emballage d’origine à l’abri de la lumière.
Tenir hors de la portée des enfants.
Remarques concernant la manipulation
La dibotermine alpha doit être préparée uniquement après reconstitution avec le solvant et la matrice de collagène inclus dans la boîte d’InductOs.
Préparation d’InductOs, matrice implantable
Pour éviter de surcharger la matrice, il est important de reconstituer la dibotermine alpha et d’imbiber la matrice entière comme il est décrit ci-dessous.
Boîte de 4 mg :
Dans un champ non stérile
1. En utilisant une technique aseptique, placer une seringue, une aiguille et l’emballage interne des matrices sur le champ stérile.
2. Désinfecter les bouchons des flacons de poudre et de solvant.
3. En utilisant la seringue restante et l‘aiguille restante de la boîte, reconstituer la poudre dans son flacon avec 3,2 ml de solvant. Pour ce faire, injecter doucement le solvant dans le flacon contenant la poudre. Renverser doucement le flacon afin d’aider à la dissolution. Ne pas agiter. Jeter la seringue et l'aiguille après usage.
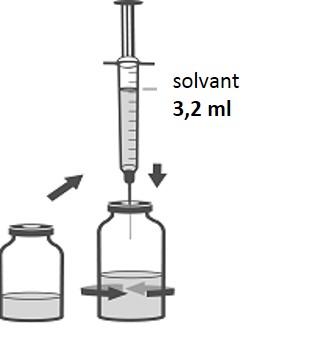
4. Désinfecter le bouchon du flacon reconstitué de dibotermine alpha.
Dans un champ stérile
5. Ouvrir l’emballage interne des matrices et laisser les matrices dans leur support.
6. À l’aide de la seringue et de l'aiguille mises en champ stérile à l'étape 1, prélever, de façon aseptique, 2,8 ml de solution reconstituée de dibotermine alpha du flacon placé dans le champ non stérile. Tenir le flacon à l’envers pour faciliter le prélèvement.
7. En laissant les matrices dans leur support, répartir UNIFORMÉMENT 1,4 ml de solution de dibotermine alpha sur chacune des deux matrices de 2,5 x 5 cm, selon la figure ci-dessous.
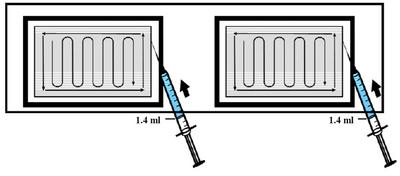
8. Attendre AU MOINS 15 minutes avant d'utiliser les matrices implantables reconstituées. Les matrices implantables doivent être utilisées dans un délai de 2 heures après préparation.
9. Suivre les instructions correspondant à l’opération prévue – arthrodèse antérieure lombaire ou réparation d’une fracture aiguë du tibia.
Boîte de 12 mg :
Dans un champ non stérile
1.En utilisant une technique aseptique, placer une seringue, une aiguille et l’emballage interne de la matrice sur le champ stérile.
2.Désinfecter les bouchons des flacons de dibotermine alpha et de solvant.
3.En utilisant la seringue restante et l‘aiguille restante de la boîte, reconstituer la poudre dans son flacon avec 8,4 ml de solvant. Pour ce faire, injecter doucement le solvant dans le flacon contenant la poudre. Renverser doucement le flacon afin d’aider à la dissolution. Ne pas agiter. Jeter la seringue et l'aiguille après usage.
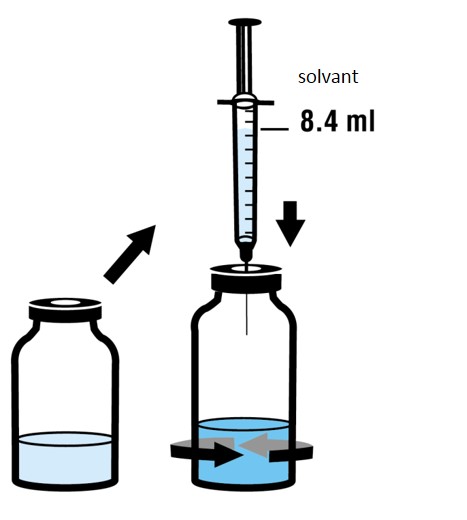
4.Désinfecter le bouchon du flacon reconstitué de dibotermine alpha.
Dans un champ stérile
5.Ouvrir l’emballage interne de la matrice et laisser la matrice dans son support.
6.A l’aide de la seringue et de l’aiguille mises en champ stérile à l’étape 1, prélever, de façon aseptique, 8.0 ml de la solution reconstituée de dibotermine alpha du flacon situé dans le champ non stérile. Tenir le flacon à l’envers pour faciliter le prélèvement.
7.En laissant la matrice sur son support, répartir UNIFORMÉMENT la solution de dibotermine alpha sur la matrice selon la figure ci-dessous.
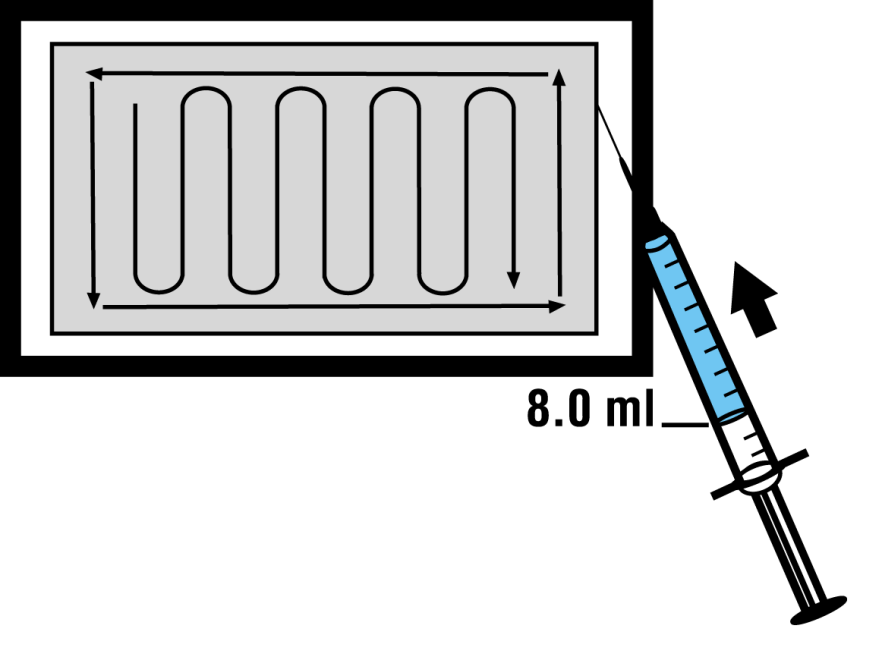
8.Attendre AU MOINS 15 minutes avant d’utiliser la matrice implantable reconstituée. La matrice implantable doit être utilisée dans un délai de 2 heures après préparation.
9.Suivre les instructions correspondant à l’opération prévue – arthrodèse antérieure lombaire ou réparation d’une fracture aiguë du tibia.
Numéro d’autorisation56828 (Swissmedic).
PrésentationChaque boîte de 4 mg d’InductOs 1.5 mg/ml poudre, solvant et matrice pour matrice pour implantation contient :
•1 flacon de poudre (Diboterminum alfa 4 mg)
•1 flacon de solvant (eau pour préparations injectables 10 ml)
•2 matrices stériles (2,5 cm x 5 cm)
•2 seringues stériles de 5 ml
•2 aiguilles stériles (20 gauge).
Chaque boîte de 12 mg d’InductOs 1.5 mg/ml poudre, solvant et matrice pour matrice pour implantation contient
•1 flacon de poudre (Diboterminum alfa 12 mg)
•1 flacon de solvant (eau pour préparations injectables 10 ml)
•1 matrice stérile (7,5 x 10 cm)
•2 seringues stériles de 10 ml
•2 aiguilles stériles (20 gauge)
[B]
Titulaire de l’autorisationMedtronic BioPharma Sàrl, Tolochenaz.
Mise à jour de l’informationFévrier 2025
|