Propriétés/EffetsCode ATC
R03DX05
Mécanisme d'action
L'omalizumab est un anticorps monoclonal humanisé qui se fixe de manière sélective aux immunoglobulines E (IgE) humaines. L'anticorps est une IgG1 kappa obtenue par la fusion d'une région d'origine humaine avec des régions de complémentarité se fixant aux IgE humaines et provenant d'un anticorps murin.
Pharmacodynamique
Patients souffrant d'asthme allergique
En se liant aux IgE libres, l'omalizumab inhibe la liaison des IgE au récepteur FcεRI de haute affinité (récepteur aux IgE de haute affinité). La quantité d'IgE libres pouvant déclencher la chaîne de réactions allergiques est réduite. Le traitement des sujets atopiques par l'omalizumab a entraîné la diminution du nombre de récepteurs FcεRI présents à la surface des basophiles. Par ailleurs, la libération d'histamine in vitro à partir de basophiles isolés chez des patients traités par Xolair a été réduite d'environ 90% après stimulation par un allergène par rapport aux valeurs pré-thérapeutiques.
Le traitement par Xolair entraîne une diminution du nombre d'éosinophiles dans le sang et les tissus ainsi qu'une diminution des médiateurs inflammatoires dont les interleukines (IL-4, IL-5 et IL-13) font également partie.
Dans les études cliniques, les taux sériques d'IgE libres ont subi une diminution dose-dépendante dans l'heure suivant la première dose et se sont maintenus entre les différentes doses.
Lors de l'utilisation des doses recommandées, la réduction moyenne des IgE libres a été supérieure à 96%. Les taux sériques d'IgE totales (c.-à-d. liées et non liées) ont augmenté après la première dose en raison de la formation de complexes omalizumab-IgE. Les complexes omalizumab-IgE ont un taux d'élimination plus lent que les IgE libres. 16 semaines après la première dose, les taux d'IgE totales ont été 5 fois plus élevés que ceux observés lors du traitement préalable, des tests standards ayant été utilisés pour leur mesure. Après l'interruption du traitement par Xolair, l'augmentation des IgE totales et la réduction des IgE libres ont été réversibles, sans qu'un effet rebond des taux d'IgE n'ait été observé après l'élimination de l'omalizumab. Les taux d'IgE totales existant avant le traitement n'ont pas été atteints dans l'année suivant l'interruption du traitement par Xolair.
Les effets de l'omalizumab sur les cellules B porteuses d'IgE et sur la régulation à long terme de la synthèse des IgE spécifiques de l'allergène ne sont pas clairement connus.
Patients atteints de polypes nasaux
Dans les études cliniques menées auprès de patients atteints de polypes nasaux, le traitement par Xolair a entraîné une réduction des taux sériques d'IgE libres et une augmentation des taux sériques d'IgE totales; ces effets sont similaires à ceux observés chez les patients souffrant d'asthme allergique.
Patients avec urticaire chronique spontanée (UCS)
Plusieurs théories sont proposées à propos de l'étiologie de l'UCS. L'une d'entre elles postule l'existence d'un mécanisme auto-immun. Des auto-anticorps anti IgE et contre le récepteur FcεRI de ceux-ci ont été isolés dans le sérum de certains patients atteints d'UCS. Ces auto-anticorps sont capables d'activer des granulocytes basophiles ou des mastocytes avec pour effet une libération d'histamine.
Une hypothèse relative au mécanisme d'action de l'omalizumab dans l'UCS avance une diminution des taux d'anticorps IgE libres dans le sang et par conséquent aussi dans la peau. Ceci induirait une régulation négative des récepteurs de surface des IgE avec diminution de la transmission des signaux descendants par la voie FcεRI et inhibition de l'activation cellulaire et de la réaction inflammatoire. Il en résulterait une diminution de la fréquence et de la sévérité de symptômes de l'UCS. Une autre hypothèse repose sur une diminution des taux d'IgE libre qui aurait pour conséquence une désensibilisation non spécifique rapide des mastocytes cutanés. La régulation négative de la voie FcεRI pourrait favoriser le maintien de cette réaction.
Dans les essais cliniques conduits auprès de patients atteints d'UCS, le traitement d'omalizumab a induit une diminution dose-dépendante des taux d'IgE libre et une augmentation des taux sériques d'IgE totale, comme c'était le cas chez les patients avec asthme allergique. La suppression maximale de l'IgE libre a été observée 3 jours après la dose sous-cutanée initiale. Après des doses multiples une fois toutes les 4 semaines, les taux sériques d'IgE libre mesurés avant l'administration sont restés stables de la semaine 12 à la semaine 24. Les taux sériques d'IgE totale ont augmenté après la dose initiale à la suite de la formation de complexes omalizumab-IgE, dont la vitesse d'élimination est inférieure à celle de l'IgE libre. Après des doses multiples de 75 mg à 300 mg toutes les 4 semaines, les taux d'IgE totale moyens mesurés dans le sérum avant la dose de la semaine 12 étaient deux à trois fois supérieurs à ceux mesurés avant le début du traitement et sont restés stables de la semaine 12 à la semaine 24. À la fin du traitement par Xolair, les taux d'IgE libre ont augmenté au cours d'une période de follow-up de 16 semaines sans traitement, tandis que les taux d'IgE totale ont diminué, tous deux en direction des valeurs observées avant le traitement.
Efficacité clinique
Asthme allergique
Adultes et adolescents (≥12 ans)
L'efficacité et la tolérance de Xolair ont été démontrées dans une étude pivot de 28 semaines, contrôlée contre placebo (étude 5) conduite chez 419 patients atteints d'asthme allergique sévère, âgés de 12 à 79 ans, ayant une réduction de la fonction pulmonaire (volume expiratoire maximal pendant la 1ère seconde: VEMS 40–80% des valeurs prédites) et dont les symptômes de l'asthme étaient mal contrôlés en dépit d'un traitement de > 1000 µg de dipropionate de béclométasone (ou équivalent) et de bêta2-agonistes à longue durée d'action. Les patients éligibles avaient présenté de multiples exacerbations de l'asthme ayant nécessité une corticothérapie systémique, avaient été hospitalisés ou s'étaient présentés dans un service d'urgences en raison d'une exacerbation sévère de l'asthme au cours de l'année précédente malgré un traitement continu par corticothérapie inhalée à fortes doses et de bêta2-agonistes à longue durée d'action. Xolair ou un placebo a été administré par voie sous-cutanée en addition à un traitement par > 1000 μg de dipropionate de béclométasone (ou équivalent) et de bêta2agonistes à longue durée d'action. Les patients ont de plus reçu des traitements de fond par corticoïde oral (22% des patients), théophylline (27%) et anti-leucotriènes (35%). Durant la phase de traitement, les thérapies additives contre l'asthme sont restées inchangées.
Le critère primaire représente le taux d'exacerbations de l'asthme ayant nécessité un traitement aigu par des corticostéroïdes systémiques. L'omalizumab a réduit le taux d'exacerbations de l'asthme de 19% (p = 0.153). Les autres évaluations ayant présenté une significativité statistique (p = 0.05) en faveur de Xolair, comprennent la réduction des exacerbations sévères (dans lesquelles la fonction pulmonaire du patient a été réduite à moins de 60% de la meilleure valeur personnelle et des corticostéroïdes systémiques ont été nécessaires), les consultations au service des urgences en relation avec l'asthme (y compris hospitalisation, consultation au service des urgences et consultations médicales non prévues) ainsi que l'amélioration de l'évaluation générale par le médecin de l'efficacité du traitement, de la qualité de vie en rapport avec l'asthme (AQL), des symptômes de l'asthme et de la fonction pulmonaire.
Dans une analyse de sous-groupe, la probabilité de tirer un bénéfice cliniquement significatif du traitement par Xolair a été plus élevée chez les patients avec des taux pré-thérapeutiques d'IgE totales ≥76 UI/ml. Chez ces patients de l'étude 1, Xolair a réduit de 40% (p = 0.002) la fréquence des exacerbations de l'asthme. Par ailleurs, les patients de la population avec des IgE totales ≥76 UI/ml de l'ensemble du programme de Xolair dans l'asthme sévère ont été plus nombreux à présenter des réponses cliniquement significatives (cf. tableau 6).
Quatre autres larges études secondaires contrôlées contre placebo d'une durée de 28 à 52 semaines conduites chez 1722 adultes et adolescents (études 3, 4, 5, 6) ont évalué l'efficacité et la tolérance de Xolair chez des patients atteints d'asthme persistant sévère. La plupart des patients étaient insuffisamment contrôlés mais ils recevaient un traitement antiasthmatique concomitant plus léger que les patients des études 1 ou 2. Les études 3–5 ont utilisé les exacerbations comme critère principal d'évaluation, tandis que l'étude 6 a principalement évalué l'épargne des corticoïdes inhalés.
L'étude 2 a évalué l'efficacité et la tolérance de l'omalizumab dans une population de 312 patients atteints d'asthme allergique sévère présentant des caractéristiques proches de celles de la population de l'étude 1. Le traitement par Xolair dans cette étude en ouvert a entraîné une réduction de 61% de la fréquence des exacerbations de l'asthme, réduction cliniquement significative par rapport au traitement antiasthmatique en cours administré seul.
Dans les études 3, 4 et 5, les patients traités par Xolair ont présenté une réduction de la fréquence des exacerbations de l'asthme respectivement de 37.5% (p = 0.027), 40.3% (p < 0.001) et 57.6% (p < 0.001) par rapport au placebo.
Dans l'étude 6, un nombre significativement supérieur de patients atteints d'asthme allergique sévère ont pu réduire leur dose de fluticasone à ≤500 μg/jour avec Xolair (60.3%) sans détérioration du contrôle de l'asthme, et ce par rapport aux patients du groupe placebo (45.8%, p < 0.05).
|
Tableau 6: résultats de l'étude
| |
|
Ensemble de la population de l'étude
| |
|
Xolair (N = 209)
|
Placebo (N = 210)
| |
Exacerbations de l'asthme
| |
Taux par période de 28 semaines
|
0.74
|
0.92
| |
% de réduction, valeur de p pour le rapport des taux
|
19.4%, p = 0.153
| |
Exacerbations sévères de l'asthme
| |
Taux par période de 28 semaines
|
0.24
|
0.48
| |
% de réduction, valeur de p pour le rapport des taux
|
50.1%, p = 0.002
| |
Visites d'urgence
| |
Taux par période de 28 semaines
|
0.24
|
0.43
| |
% de réduction, valeur de p pour le rapport des taux
|
43.9%, p = 0.038
| |
Évaluation globale du médecin
| |
% de répondeurs*
|
60.5%
|
42.8%
| |
Valeur de p**
|
< 0.001
| |
Amélioration à l'AQL***
| |
% de patients ≥0.5 d'amélioration
|
60.8%
|
47.8%
| |
Valeur de p
|
0.008
| |
* amélioration marquée ou contrôle complet
** valeur de p pour la distribution globale de l'évaluation
*** Asthma Quality of Life
|
Enfants âgés de 6 à < 12 ans
Les principales données concernant la sécurité d'emploi et l'efficacité de Xolair dans le groupe d'âge de 6 ans à < 12 ans sont issues d'une étude multicentrique randomisée, en double aveugle et contrôlée contre placebo (étude 7).
L'étude 7 était une étude contrôlée contre placebo comprenant un sous-groupe spécifique (N = 235) de patients dont le diagnostic correspondait à l'indication actuelle et qui étaient traités par des corticostéroïdes inhalés à forte dose (≥500 μg d'équivalent fluticasone/jour) et des bêtaagonistes de longue durée d'action.
Une exacerbation cliniquement significative était définie selon le jugement clinique de l'investigateur comme une aggravation des symptômes asthmatiques nécessitant le doublement de la dose initiale du corticostéroïde inhalé pendant au moins 3 jours et/ou un traitement d'urgence par des corticostéroïdes systémiques (oraux ou intraveineux) pendant au moins 3 jours.
Dans le sous-groupe spécifique de patients traités par des corticostéroïdes inhalés à forte dose, le groupe omalizumab a présenté une incidence des exacerbations de l'asthme statistiquement inférieure à celle dans le groupe placebo. À la semaine 24, le taux d'incidence chez les patients traités par l'omalizumab a été inférieur de 34% à celui sous placebo (rapport des taux 0.662; p = 0.047). Pendant la seconde période de traitement en double aveugle de 28 semaines, le taux d'incidence chez les patients traités par l'omalizumab a été inférieur de 63% à celui sous placebo (rapport des taux 0.37; p < 0.001).
Pendant la période de traitement en double aveugle de 52 semaines (comprenant la phase de corticothérapie à posologie fixe de 24 semaines et la phase d'adaptation de la posologie des corticoïdes de 28 semaines), les différences des taux d'incidence entre les groupes de traitement ont révélé une diminution des exacerbations de 50% chez les patients traités par l'omalizumab (rapport des taux 0.504; p < 0.001).
Par rapport au groupe placebo, le groupe omalizumab s'est caractérisé à la fin de la période de traitement de 52 semaines par une diminution plus importante de l'utilisation de bêtaagonistes en tant que traitement d'urgence, bien que la différence entre les groupes de traitement n'ait pas été statistiquement significative. En ce qui concerne l'évaluation globale de l'efficacité du traitement à la fin de la période de traitement en double aveugle de 52 semaines et en ce qui concerne le sous-groupe des patients sévères recevant des corticoïdes inhalés à forte dose et des bêta-agonistes à longue durée d'action, la proportion des patients chez lesquels l'efficacité du traitement a été jugée «excellente» a été plus élevée dans le groupe omalizumab que dans le groupe placebo. Les proportions des patients chez lesquels l'efficacité du traitement a été jugée «modérée» ou «faible» ont été plus faibles dans le groupe omalizumab que dans le groupe placebo. Les différences entre les groupes ont été statistiquement significatives (p < 0.001). Aucune différence n'a été constatée entre les groupes omalizumab et placebo en ce qui concerne les évaluations subjectives faites par les patients au sujet de leur qualité de vie.
Polypes nasaux
La sécurité et l'efficacité de Xolair ont été évaluées dans deux études cliniques randomisées, multicentriques, en double aveugle et contrôlées contre placebo (étude 1, N = 138; étude 2, N = 127) réalisée auprès de patients atteints de rhinosinusite chronique et de polypes nasaux. Les patients ont reçu Xolair ou un placebo par voie sous-cutanée toutes les 2 ou 4 semaines, la posologie et la fréquence d'utilisation correspondant aux données des tableaux 7 et 8 (cf. «Posologie/Mode d'emploi»). Par ailleurs, tous les patients ont reçu tout au long de l'étude un traitement de fond par de la mométasone administrée par voie intranasale. Une opération sino-nasale précédente ou un traitement systémique préalable par des corticostéroïdes n'étaient pas requis pour l'admission dans l'étude. Les participants à l'étude ont reçu Xolair ou un placebo pendant 24 semaines, et une phase de suivi sans traitement d'une durée de 4 semaines a ensuite eu lieu. Les données démographiques et les caractéristiques de référence incluant les comorbidités allergiques sont représentées dans le tableau 7.
|
Tableau 7: Données démographiques et caractéristiques de référence dans les études de polypes nasaux
| |
Paramètres
|
Étude 1 des polypes nasaux
N = 138
|
Étude 2 des polypes nasaux
N = 127
| |
Âge moyen en années (SD)
|
51.0 (13.2)
|
50.1 (11.9)
| |
% patients de sexe masculin
|
63.8
|
65.4
| |
Patients ayant utilisé des corticostéroïdes systémiques l'année précédente (%)
|
18.8
|
26.0
| |
Score NPS endoscopique bilatéral moyen (SD), intervalle: 0–8
|
6.2 (1.0)
|
6.3 (0.9)
| |
Score moyen de congestion nasale (NC)* (SD), intervalle: 0–3
|
2.4 (0.6)
|
2.3 (0.7)
| |
Score olfactif moyen* (SD), intervalle: 0–3
|
2.7 (0.7)
|
2.7 (0.7)
| |
Score total SNOT-22 * (SD), intervalle: 0–110
|
60.1 (17.7)
|
59.5 (19.3)
| |
Nombre moyen d'éosinophiles dans le sang (cellules/µl) (SD)
|
346.1 (284.1)
|
334.6 (187.6)
| |
IgE totales moyennes en UI/ml (SD)
|
160.9 (139.6)
|
190.2 (200.5)
| |
Asthme (%)
|
53.6
|
60.6
| |
Léger (%)
|
37.8
|
32.5
| |
Modéré (%)
|
58.1
|
58.4
| |
Sévère (%)
|
4.1
|
9.1
| |
Affection respiratoire exacerbée par l'aspirine (%)
|
19.6
|
35.4
| |
Rhinite allergique
|
43.5
|
42.5
| |
SD = écart-type; NPS = score de polypes nasaux (nasal polyps score);
SNOT 22 = questionnaire sur le test de résultats sino-nasaux comportant 22 questions;
IgE = immunoglobulines E; UI = unités internationales.
|
Dans le NPS, le NCS et les scores olfactifs, de syndrome de rhinorrhée postérieure et d'écoulement nasal ainsi que dans le score SNOT-22, un nombre de points plus élevé indique une gravité accrue de la maladie.
Les co-critères d'évaluation principaux étaient le score bilatéral de polypes nasaux (NPS) et le score quotidien moyen de congestion nasale (NCS), tous deux déterminés à la semaine 24. Le NPS a été déterminé par endoscopie au début de l'étude et à des moments déterminés au préalable (intervalle: 0–4 par narine), et le NPS total a été calculé à partir de ces valeurs (intervalle: 0 = meilleure valeur à 8 = valeur la plus mauvaise). La congestion nasale a été évaluée tous les jours à l'aide de l'échelle NCS (intervalle: 0 = meilleure valeur à 3 = valeur la plus mauvaise). Avant d'être randomisés, les patients devaient présenter un NPS ≥5 et un NCS hebdomadaire moyen > 1 malgré l'utilisation de mométasone par voie intranasale. Dans les deux études, les NPS moyens des deux groupes de traitement au début de l'étude étaient équilibrés.
Dans l'étude 1 comme dans l'étude 2 des polypes nasaux, les patients ayant reçu Xolair ont présenté à la semaine 24 une amélioration significativement plus importante d'un point de vue clinique du NPS comme de la moyenne du NCS sur la semaine par rapport à la valeur initiale que les patients ayant reçu le placebo (cf. tableau 8).
Les améliorations les plus importantes du NPS et du NCS dans le groupe traité par Xolair par rapport au groupe placebo ont déjà été observées lors de la première évaluation à la semaine 4, comme le montre la figure 8. À la semaine 4, la différence des moyennes des moindres carrés (LS) pour la modification du NPS par rapport à la valeur initiale dans le groupe traité par Xolair en comparaison avec le groupe placebo était de -0.92 (IC à 95%: -1.37, -0.48) dans l'étude 1 et de -0.52 (IC à 95%: -0.94, -0.11) dans l'étude 2. Pour le NCS, la différence des moyennes des LS pour la modification par rapport à la valeur initiale à la semaine 4 dans le groupe traité par Xolair en comparaison avec le groupe placebo était de -0.25 (IC à 95%: -0.46, -0.04) dans l'étude 1 et de -0.26 (IC à 95%: -0.45, -0.07) dans l'étude 2. Toutefois, les tests statistiques n'étaient pas pré-spécifiés à ce moment-là.
|
Tableau 8: Modification du score de polypes nasaux et du score moyen de congestion nasale sur 7 jours à la semaine 24 par rapport à la valeur initiale dans les études 1 et 2 des polypes nasaux
| |
|
Étude 1 des polypes nasaux
|
Étude 2 des polypes nasaux
| |
|
Placebo
|
Xolair
|
Placebo
|
Xolair
| |
N
|
66
|
72
|
65
|
62
| |
Score de polypes nasaux
| |
Moyenne de la référence
|
6.32
|
6.19
|
6.09
|
6.44
| |
Moyenne des LS pour la modification jusqu'à la semaine 24 par rapport à la valeur initiale
|
0.06
|
-1.08
|
-0.31
|
-0.90
| |
Différence des moyennes des LS par rapport au placebo
|
-1.14
|
-0.59
| |
IC à 95% de la différence
|
-1.59, -0.69
|
-1.05, -0.12
| |
Valeur de p
|
< 0.0001
|
0.0140
| |
Valeur moyenne sur 7 jours du score quotidien de congestion nasale
| |
Moyenne de la référence
|
2.46
|
2.40
|
2.29
|
2.26
| |
Moyenne des LS pour la modification jusqu'à la semaine 24 par rapport à la valeur initiale
|
-0.35
|
-0.89
|
-0.20
|
-0.70
| |
Différence des moyennes des LS par rapport au placebo
|
-0.55
|
-0.50
| |
IC à 95% de la différence
|
-0.84, -0.25
|
-0.80, -0.19
| |
Valeur de p
|
0.0004
|
0.0017
|
LS = moindres carrés (détermination de la moyenne selon la méthode des Least Squares = moindres carrés)
Figure 1: Modification moyenne par rapport à la valeur initiale des scores de congestion nasale et de polypes nasaux en fonction du groupe de traitement dans les études 1 et 2 des polypes nasaux

Un critère d'évaluation principal secondaire a été l'évaluation de la modification du score total des symptômes nasaux (total nasal symptom score, TNSS) à la semaine 24 par rapport à la valeur initiale. Le TNSS rapporté par les patients était un score qui correspondait à la somme de 4 scores de symptômes quotidiens équipondérés. Ceux-ci étaient: le NCS, le score olfactif, le score de rhinorrhée postérieure et le score de rhinorrhée antérieure. L'intervalle du TNSS était de 0 = meilleure valeur à 12 = valeur la plus mauvaise. Dans le cadre du traitement par Xolair, il y a eu une amélioration significative du TNSS quotidien moyen par rapport au placebo. La différence des moyennes des LS pour la modification par rapport à la valeur initiale jusqu'à la semaine 24 était de -1.91 points (IC à 95%: -2.85, -0.96; p = 0.0001) dans l'étude 1 et de -2.09 points (IC à 95%: -3.00, -1.18; p < 0.0001) dans l'étude 2.
Dans le cadre du traitement par Xolair, il y a eu une amélioration significative dans le SNOT-22 (Sino-Nasal OutcomeTest) qui combine des questions du domaine des symptômes sino-nasaux, de la psychologie et de la qualité du sommeil. Le SNOT-22 était compris entre 0 et 110 (0 = meilleure valeur, 110 = valeur la plus mauvaise). La différence des moyennes des LS pour la modification du SNOT-22 par rapport à la valeur initiale jusqu'à la semaine 24 dans le cadre du traitement par Xolair, en comparaison avec le placebo, était de -16.12 (IC à 95%: -21.86, -10.38; p < 0.0001) dans l'étude 1 et de -15.04 (IC à 95%: -21.26, -8.82; p < 0.0001) dans l'étude 2.
Dans le cadre du traitement par Xolair, il y a également eu une amélioration significative du score UPSIT (University of Pennsylvania Smell Identification Test) quotidien moyen par rapport au placebo. Le score UPSIT était compris entre 0 et 40 (0 = valeur la plus mauvaise, 40 = meilleure valeur). La différence des moyennes des LS pour la modification par rapport à la valeur initiale jusqu'à la semaine 24 dans le cadre du traitement par Xolair, en comparaison avec le placebo, était de 3.81 points (IC à 95%: 1.38, 6.24; p = 0.0024) dans l'étude 1 et de 3.86 points (IC à 95%: 1.57, 6.15; p = 0.0011) dans l'étude 2.
Dans les deux études, l'effet sur le TNSS et le SNOT-22 a déjà été observé dès la première évaluation à la semaine 4. En outre, l'effet sur le score UPSIT a été observé dans les deux études lors de la première évaluation à la semaine 8.
Des analyses supplémentaires des critères d'évaluation secondaires ont inclus les évaluations du NPS et du NCS à la semaine 16. Dans le cadre du traitement par Xolair, il y a eu une amélioration significative du NPS à la semaine 16 (0 = valeur la plus mauvaise, 8 = meilleure valeur) par rapport au placebo. La différence des moyennes des LS pour la modification par rapport à la valeur initiale jusqu'à la semaine 16 dans le cadre du traitement par Xolair, en comparaison avec le placebo, était de -1.01 (IC à 95%: -1.43, -0.60; p < 0.0001) dans l'étude 1 et de -0.91 (IC à 95%: -1.39, -0.44; p = 0.0002) dans l'étude 2 [XX]. Dans le cadre du traitement par Xolair, il y a eu une amélioration significative du NCS à la semaine 16 (0 = meilleure valeur, 3 = valeur la plus mauvaise) par rapport au placebo. La différence des moyennes des LS pour la modification du NCS quotidien moyen par rapport à la valeur initiale jusqu'à la semaine 16 dans le cadre du traitement par Xolair, en comparaison avec le placebo, était de -0.57 (IC à 95%: -0.83, -0.31; p < 0.0001) dans l'étude 1 et de -0.59 (IC à 95%: -0.87, -0.30; p < 0.0001) dans l'étude 2.
Urticaire chronique spontanée (UCS)
Le programme de développement clinique de phase III dans l'UCS comprenait trois études multicentriques randomisées, en double aveugle, contrôlées contre placebo, en groupes parallèles: Q4881g, Q4882g et Q4883g.
Les observations ont porté sur des adultes et des adolescents (âgés de 12 ans et plus) avec UCS depuis ≥6 mois (de 6 mois à 66 ans, 6 ans en moyenne) et souffrant de poussées récurrentes malgré les antihistaminiques aux doses maximales admises (UAS7 ≥16/42 durant ≥8 jours consécutifs).
Les études Q4881g et Q4882g avaient pour but de tester l'efficacité et la sécurité d'un traitement de 75 mg, 150 mg ou 300 mg de Xolair toutes les 4 semaines durant 24 resp. 12 semaines avec une période de followup sans traitement de 16 semaines chez des patients (12–75 ans) atteints d'une UCS réfractaire malgré l'administration d'antihistaminiques H1.
L'étude Q4883g avait pour objectif de tester l'efficacité et la sécurité de 300 mg de Xolair, administrés toutes les 4 semaines durant 24 semaines avec une période de followup sans traitement de 16 semaines chez des patients (12–75 ans) atteints d'une UCS réfractaire malgré l'administration d'antihistaminiques H1 et/ou H2 et/ou des antagonistes du récepteur des leucotriènes (LTRA).
|
Tableau 9
|
Critères d'évaluation de l'efficacité
| |
Modification de l'Itch Severity Score (ISS, échelle 0–21) hebdomadaire à la semaine 12 versus valeur initiale
|
Principal critère d'évaluation des études Q4881g et Q4882g
Critère d'évaluation secondaire de l'étude de sécurité Q4883g
| |
Délai jusqu'à une réponse MID a (diminution de ≥5 points versus valeurs initiales) du score ISS hebdomadaire jusqu'à la semaine 12
|
Critères d'évaluation secondaires des trois essais Q4881g, Q4882g et Q4883g
| |
Modification du score d'activité de l'urticaire (UAS7 b, échelle 0–42) mesuré sur une période de 7 jours à la semaine 12 versus valeur initiale
| |
Proportion des patients avec un score d'activité de l'urticaire ≤6 (UAS7b ≤6) mesuré sur 7 jours à la semaine 12
| |
Proportion des patients avec un score d'activité de l'urticaire = 0 (UAS7b = 0) mesuré sur 7 jours à la semaine 12 c
| |
Modification du score hebdomadaire des papules à la semaine 12 versus valeur initiale
| |
Modification du score global de l'indice de qualité de vie dermatologique (DLQI) à la semaine 12 versus valeur initiale
| |
Proportion des patients avec journées sans angio-œdème entre la semaine 4 et la semaine 12 d
| |
a
MID: différence minimale significative (Minimally Important Difference)
b UAS7: combinaison de l'intensité du prurit et du nombre de papules; somme des scores mesurés lors de 7 jours consécutifs
c Analyse post-hoc de l'étude Q4882g
d La proportion moyenne des jours sans angio-œdème entre la semaine 4 et la semaine 12 a été calculée pour l'ensemble de la population de l'étude, y compris les patients sans symptômes d'angio-œdème.
|
Dans les essais Q4881g et Q4882g, la dose de 75 mg n'a atteint de manière consistante ni le principal critère d'efficacité (modification de l'Itch Severity Score (ISS) hebdomadaire à la semaine 12 versus valeur initiale) ni plusieurs des critères secondaires. Cette dose a par conséquent été considérée comme non efficace et ces résultats ne sont pas présentés ci-dessous.
Le principal critère d'efficacité, la modification de l'Itch Severity Score hebdomadaire de la semaine 12 versus valeur initiale, a été atteint aussi bien avec la dose de 150 mg qu'avec la dose de 300 mg dans les études Q4881g et Q4882g et il a été atteint avec la dose de 300 mg dans l'étude Q4883g (critère d'évaluation secondaire; cf. Tableau 10).
|
Tableau 10: Modification de l'Itch Severity Score hebdomadaire à la semaine 12 versus valeur initiale, études Q4881g, Q4882g et Q4883g (population mITT *)
| |
|
Placebo
|
Omalizumab
150 mg
|
Omalizumab
300 mg
| |
Étude Q4881g
| |
N
|
80
|
80
|
81
| |
Moyenne (SD)
|
-3.63 (5.22)
|
-6.66 (6.28)
|
-9.40 (5.73)
| |
Différence des moyennes des LS vs placebo1
|
-
|
-2.95
|
-5.80
| |
IC à 95% pour la différence
|
-
|
-4.72,-1.18
|
-7.49,-4.10
| |
Valeur de p vs placebo2
|
-
|
0.0012
|
< 0.0001
| |
Étude Q4882g
| |
N
|
79
|
82
|
79
| |
Moyenne (SD)
|
-5.14 (5.58)
|
-8.14 (6.44)
|
-9.77 (5.95)
| |
Différence des moyennes des LS vs placebo1
|
-
|
-3.04
|
-4.81
| |
IC à 95% pour la différence
|
-
|
-4.85,-1.24
|
-6.49,-3.13
| |
Valeur de p vs placebo2
|
-
|
0.0011
|
< 0.0001
| |
Étude Q4883g
| |
N
|
83
|
-
|
252
| |
Moyenne (SD)
|
-4.01 (5.87)
|
-
|
-8.55 (6.01)
| |
Différence des moyennes des LS vs placebo1
|
-
|
-
|
-4.52
| |
IC 95% pour la différence
|
-
|
-
|
-5.97, -3.08
| |
Valeur de p vs placebo2
|
-
|
-
|
< 0.0001
| |
* Population Intent-to-Treat modifiée (mITT): englobe tous les patients randomisés ayant reçu au moins une dose du médicament à l'étude. La méthode BOCF (Baseline Observation Carried Forward) a été appliquée pour le calcul des valeurs manquantes.
1 La moyenne des LS a été calculée à l'aide d'un modèle d'ANCOVA. Les critères de stratification étaient la valeur initiale de l'Itch Severity Score hebdomadaire (< 13 vs ≥13) et le poids initial (< 80 kg vs ≥80 kg).
2 Les valeurs de p proviennent du t-test ANCOVA.
|
La figure 2 illustre l'évolution temporelle de l'ISS hebdomadaire moyen au cours de l'étude Q4881g. L'Itch Severity Score hebdomadaire moyen a significativement diminué dans les deux groupes de traitement. L'effet maximal a été atteint environ à la semaine 12, puis est resté constant durant la phase de traitement de 24 semaines. Dans les études Q4883g (300 mg durant une période de traitement de 24 semaines) et Q4882g (150 mg ou 300 mg durant une période de traitement de 12 semaines), les résultats étaient semblables à ceux de l'étude Q4881g.
Dans les trois essais (cf. figure 2 pour l'étude Q4881g), l'Itch Severity Score hebdomadaire moyen a augmenté dans les deux groupes de dosage de manière progressive et parallèlement aux symptômes au cours de la phase de follow-up sans traitement de 16 semaines. À l'issue de la phase de follow-up, les valeurs moyennes étaient comparables à celles du groupe placebo, mais inférieures aux valeurs moyennes initiales correspondantes.
Figure 2: Évolution temporelle de l'Itch Severity Score hebdomadaire moyen dans l'étude Q4881g (BOCF, population mITT)

BOCF = Baseline Observation Carried Forward; mITT = population Intent-to-Treat-modifiée
Délai jusqu'à l'apparition d'une réponse MID de l'ISS hebdomadaire jusqu'à la semaine 12
Dans les études Q4881g et Q4882g, le délai médian jusqu'à l'obtention d'une MID de 5 points de l'ISS hebdomadaire était de 2 semaines chez les patients du groupe recevant 150 mg (p = 0.0301 dans l'étude Q4881g; p = 0.0101 dans l'étude Q4882g) et de 1 semaine chez les patients sous 300 mg (p < 0.0001) versus 4 semaines chez les patients des groupes placebo. Des résultats comparables ont été rapportés dans l'étude Q4883g avec un délai médian de 2 semaines jusqu'à l'obtention d'une MID dans le groupe sous 300 mg (p < 0.0001) contre 5 semaines dans le groupe placebo.
Modification de l'UAS7 à la semaine 12 versus valeur initiale
Dans les essais de phase III, les groupes traités par 150 mg et 300 mg d'omalizumab différaient de manière statistiquement significative du groupe placebo en termes de modification moyenne de l'UAS7 à la semaine 12 versus valeur initiale (figure 2 pour l'étude Q4881g). Le seuil de signification statistique (p < 0.0001) a été atteint dans les trois études pour le groupe recevant 300 mg et dans les études Q4881g (p = 0.0008) et Q4882g (p = 0.0001) dans le groupe recevant 150 mg.
La figure 3 représente l'évolution temporelle moyenne de l'UAS7 dans l'étude Q4881g. Celle-ci était marquée par une diminution significative par rapport à la valeur initiale dans les deux groupes de traitement avec un effet maximal aux alentours de la semaine 12. Cet effet est resté constant durant toute la période de traitement de 24 semaines. Dans les études Q4882g (150 mg et 300 mg au cours d'une période de traitement de 12 semaines) et Q4883g (300 mg au cours d'une phase de traitement de 24 semaines), les résultats étaient comparables à ceux observés dans l'étude Q4881g.
Dans les trois études (cf. figure 3 pour l'étude Q4881g), l'UAS7 a progressivement augmenté au cours des 16 semaines de follow-up sans traitement dans les deux groupes sous omalizumab avec une réapparition parallèle des symptômes. À la fin de la phase de followup, les valeurs moyennes étaient comparables à celles sous placebo, mais inférieures aux valeurs initiales correspondantes.
Figure 3: Évolution temporelle moyenne de l'UAS7, étude Q4881g (BOCF, population mITT)

BOCF = Baseline Observation Carried Forward;
mITT = population Intent-to-Treat modifiée;
UAS7 = score d'activité de l'urticaire sur une période de 7 jours
Proportion de patients avec un UAS7 ≤6 à la semaine 12
La proportion des patients ayant un UAS7 ≤6 à la semaine 12 est représentée dans la figure 4. Les taux de réponse étaient tous statistiquement significatifs, se situant entre 52% et 66% (dose de 300 mg; p < 0.0001), resp. entre 40% et 43% (dose de 150 mg; p < 0.001) contre 11–19% dans le groupe placebo.
Figure 4: Proportion des patients avec UAS7 ≤6 à la semaine 12, études Q4881g, Q4882g et Q4883g

Proportion des patients avec un UAS7 = 0 à la semaine 12
La proportion des patients ayant présenté une réponse complète, autrement dit un UAS7 = 0 à la semaine 12 était de 34–44% (dose de 300 mg, statistiquement significatif, tous p < 0.0001), resp. 15–22% (dose de 150 mg) versus 5–9% dans le groupe placebo (figure 5).
Figure 5: Proportion des patients avec un UAS7 = 0 à la semaine 12, études Q4881g, Q4882g et Q4883g
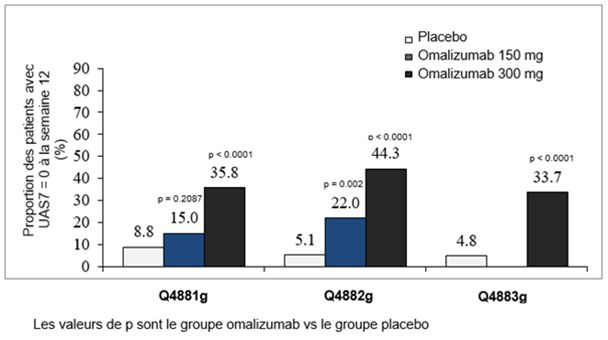
Analyse prospective dans les études Q4881g et Q4883g et analyse post-hoc dans l'étude Q4882g
Modification du score hebdomadaire relatif au nombre de papules à la semaine 12 versus valeur initiale
Dans les trois essais de phase III, le changement moyen du score hebdomadaire relatif au nombre de papules à la semaine 12 versus valeur initiale était statistiquement significatif dans le groupe traité par 300 mg (p < 0.001) avec une diminution du nombre de papules par rapport au placebo(-11.35 dans Q4881g, -11.97 dans Q4882g et -10.46 dans Q4883g versus -4.37, -5.22 resp. -4.49 dans les groupes placebo correspondants). Dans le groupe traité par 150 mg, le changement moyen était de -7.78 (p = 0.0017) dans Q4881g et de -9.75 (p < 0.0001) dans Q4882g.
Proportion des jours sans angio-œdème de la semaine 4 à la semaine 12
Les groupes traités par 300 mg ont bénéficié dans les trois essais de phase III de la plus grande proportion moyenne de jours sans angio-œdème entre les semaines 4 et 12 (91–96%). L'augmentation de la proportion de jours sans angio-œdème était statistiquement significative par rapport au placebo (p < 0.001) (fig. 6). Dans le groupe traité par 150 mg, la proportion moyenne de jours sans angio-œdème pendant le même laps de temps était de 89.6% dans l'étude Q4881g et de 91.6% dans l'étude Q4882g. Les valeurs correspondantes sous placebo étaient de 88.2% et 89.2% dans les deux essais concernés. Les différences entre les groupes sous 150 mg et sous placebo n'ont atteint le seuil de signification statistique dans aucune de ces deux études.
Figure 6: Proportion des jours sans angio-œdème de la semaine 4 à la semaine 12, études Q4881g, Q4882g et Q4883g

Modification du score global de l'indice de la qualité de vie dermatologique (DLQI) à la semaine 12 versus valeur initiale
Dans les trois essais de phase III, le changement moyen du score global DLQI entre la semaine 12 et la valeur initiale était statistiquement significativement supérieur dans le groupe traité par 300 mg (p < 0.001) par rapport à celui du groupe placebo. Dans l'étude Q4882g, le groupe traité par 150 mg d'omalizumab présentait une différence statistiquement significative (p = 0.022) versus placebo (figure 7).
Figure 7: Modification du score global de l'indice de la qualité de vie dermatologique à la semaine 12 versus valeur initiale dans les études Q4881g, Q4882g et Q4883g

Efficacité à l'issue de 24 semaines de traitement
Le tableau 11 présente les résultats après 24 semaines de traitement. Les ordres de grandeur de la réponse sont comparables à ceux observés après 12 semaines de traitement.
|
Tableau 11: Efficacité après 24 semaines de traitement dans les études Q4881g et Q4883g (population mITT*)
| |
Paramètre de l'étude
|
Semaine
|
Placebo
|
Omalizumab
150 mg
|
Omalizumab
300 mg
| |
Variation vs ligne de base du Itch Severity Score (BOCF) hebdomadaire, valeur moyenne
| |
Étude Q4881g
|
Semaine 24
|
-5.41
|
-6.47
|
-9.84**
| |
Étude Q4883g
|
Semaine 24
|
-4.03
|
NA
|
-8.60**
| |
Variation vs ligne de base de l'UAS7 (BOCF), valeur moyenne
| |
Étude Q4881g
|
Semaine 24
|
-11.73
|
-14.21
|
-22.11**
| |
Étude Q4883g
|
Semaine 24
|
-8.85
|
NA
|
-19.15**
| |
Proportion de patients avec UAS7 ≤6, % de patients
| |
Étude Q4881g
|
Semaine 24
|
25.0
|
36.3
|
61.7**
| |
Étude Q4883g
|
Semaine 24
|
16.9
|
NA
|
55.6**
| |
Proportion de patients avec UAS7 = 0, % de patients
| |
Étude Q4881g
|
Semaine 24
|
12.5
|
20.0
|
48.1**
| |
Étude Q4883g
|
Semaine 24
|
3.6
|
NA
|
42.5**
| |
* Population Intent-to-Treat modifiée (mITT): englobe tous les patients randomisés ayant reçu au moins une dose du médicament à l'étude
** valeur de p ≤0.0001 dans le test statistique correspondant entre la substance active et le placebo
NA: ne s'applique pas (Not Applicable).
BOCF: Baseline Observation Carried Forward
|
Efficacité après 48 semaines de traitement
Dans une étude d'une durée de 48 semaines, 206 patients atteints d'UCS non contrôlée par un traitement par antihistaminique H1, âgés de 12 à 75 ans, ont participé à une phase de traitement en ouvert par l'omalizumab 300 mg toutes les 4 semaines, d'une durée de 24 semaines, en tant que traitement d'appoint. Les patients qui ont répondu au traitement pendant cette phase en ouvert ont ensuite reçu à l'aveugle de manière aléatoire, l'omalizumab 300 mg (81 patients) ou un placebo (53 patients), toutes les 4 semaines pendant 24 semaines supplémentaires.
Parmi les patients ayant été traités par l'omalizumab pendant un total de 48 semaines, 21% ont présenté une dégradation clinique (valeur UAS7 de 12 ou plus pendant au moins 2 semaines consécutives après la randomisation entre les semaines 24 et 48), par comparaison à 60.4% des patients traités par le placebo à la semaine 48 (La différence est de -39.4%, p <0.0001, IC à 95%: -54.5%, -22.5%).
L'étude prospective portant sur un registre de grossesse (EXPECT)
Une étude prospective portant sur un registre de grossesse menée de 2006 à 2018 aux États-Unis (EXPECT) a porté sur 250 femmes enceintes asthmatiques ayant été traitées par Xolair. 246 d'entre elles avaient été traitées par Xolair au cours du premier trimestre de grossesse et 78.4% (196/250) des femmes avaient été traitées au moins une fois par Xolair au cours de chacun des 3 trimestres de grossesse, la durée totale d'exposition étant de 8.7 mois (médiane). Les résultats de l'étude EXPECT dans les sous-groupes concernés de mères et de nourrissons ont été comparés aux fréquences ajustées en fonction de l'âge dans une cohorte externe de 1153 femmes enceintes asthmatiques (sans traitement par Xolair) ajustée en fonction de la maladie, la cohorte ayant été déterminée à partir de banques de données du domaine de la santé d'habitants de la province canadienne de Québec et désignée comme Quebec External Comparator Cohort (QECC).
Chez les nourrissons de l'étude EXPECT ayant été comparés à des nourrissons de la QECC (n = 223), la prévalence d'anomalies congénitales majeures était de 8.1% et était ainsi comparable à celle des nourrissons de la QECC (8.9%). Dans le cas des grossesses de l'étude EXPECT utilisées pour la comparaison avec la QECC (n = 230), 99.1% ont donné lieu à des naissances vivantes; il en a été de même pour 99.3% des grossesses de la QECC.
Dans une sous-étude d'EXPECT, les taux de plaquettes ont été examinés chez 51 nourrissons nés de mères exposées à Xolair; tous ces taux étaient dans les limites normales.
|