CompositionPrincipe actif
Rufinamide.
Excipients
Comprimés pelliculés:
Lactose monohydraté (100 mg: 20 mg, 200 mg: 40 mg, 400 mg: 80 mg), cellulose microcristalline, amidon de maïs, hypromellose, croscarmellose sodique (E466), stéarate de magnésium, silice colloïdale, laurilsulfate de sodium, macrogol 8000, dioxyde de titane (E171), talc, oxyde de fer rouge (E172). Teneur en sodium par comprimé: 100 mg: 0,49 mg, 200 mg: 0,98 mg, 400 mg: 1,96 mg.
Suspension buvable:
Solution à base de sorbitol cristallisant (sorbitol (E420) 175 mg/ml), propylène glycol (E1520) 25 mg/ml, cellulose microcristalline, carmellose sodique, acide citrique (E330), hydroxyéthylcellulose, sorbate de potassium (E202), diméticone, polysorbate 65, méthylcellulose, silicium dioxyde, stéarate de macrogol, acide benzoïque (E210) 0,005 mg/ml, acide sorbique (E200), acide sulfurique, poloxamère 188, arôme orange (contient de l'alcool benzylique), parahydroxybenzoate de méthyle (E218) 1,2 mg/ml, propyl-4-hydroxybenzoate (E216) 0,3 mg/ml, eau. Teneur en sodium par millilitre de suspension: 0,22 mg.
Indications/Possibilités d’emploiInovelon est indiqué comme traitement adjuvant dans des crises d'épilepsie associées au syndrome de Lennox-Gastaut chez les adultes et les enfants à partir d'un an.
Posologie/Mode d’emploiLe traitement par Inovelon doit être instauré par un médecin spécialisé en pédiatrie ou en neurologie et expérimenté dans la prise en charge de l'épilepsie.
Inovelon suspension orale et Inovelon comprimés pelliculés peuvent être échangés mutuellement à dose identique. Les patients doivent être surveillés pendant la phase de conversion.
Posologie usuelle
Utilisation chez les adultes, adolescents et enfants à partir d'1 an et pesant moins de 30 kg
- Patients ne recevant pas de valproate:
Le traitement doit être instauré à la posologie journalière de 10 mg/kg pour les comprimés pelliculés ou 0,25 ml/kg pour la suspension. La posologie est augmentée par paliers allant jusqu'à 10 mg/kg/jour pour les comprimés pelliculés ou 0,25 ml/kg/jour pour la suspension, tous les trois jours, jusqu'à une dose recommandée de 45 mg/kg/jour pour les comprimés pelliculés ou 1,125 ml/kg/jour pour la suspension. La dose maximale recommandée pour ces patients est de 1000 mg/jour pour les comprimés pelliculés ou 25 ml/jour pour la suspension.
- Patients recevant également du valproate:
Le valproate diminuant significativement la clairance du rufinamide, il est recommandé de diminuer la dose maximale d'Inovelon chez les patients recevant un traitement concomitant par le valproate. Le traitement doit être instauré à la posologie journalière de 10 mg/kg pour les comprimés pelliculés ou 0,25 ml/kg pour la suspension. La posologie est augmentée par paliers allant jusqu'à 10 mg/kg/jour pour les comprimés pelliculés ou 0,25 ml/kg/jour pour la suspension, tous les trois jours, jusqu'à une dose recommandée de 30 mg/kg/jour pour les comprimés pelliculés ou 0,75 ml/kg/jour pour la suspension. La dose maximale recommandée pour ces patients est de 600 mg/jour pour les comprimés pelliculés ou 15 ml/jour pour la suspension.
Utilisation chez les adultes et adolescents pesant au moins 30 kg
- Patients ne recevant pas de valproate:
Le traitement doit être instauré à la posologie journalière de 20 mg/kg pour les comprimés pelliculés ou 0,5 ml/kg pour la suspension. La posologie est augmentée par paliers allant jusqu'à 20 mg/kg/jour pour les comprimés pelliculés ou 0,5 ml/kg/jour pour la suspension, tous les deux jours, jusqu'à une dose recommandée de 45 mg/kg/jour pour les comprimés pelliculés ou 1,125 ml/kg/jour pour la suspension. La dose journalière ne devrait pas dépasser la dose maximale recommandée indiquée dans le tableau suivant pour la fourchette de poids.
|
Fourchette de poids
|
30,0–50,0 kg
|
50,1–70,0 kg
|
≥70,1 kg
| |
Dose maximale recommandée
|
1'800 mg/jour en comprimés pelliculés ou 45 ml/jour en suspension
|
2'400 mg/jour en comprimés pelliculés ou 60 ml/jour en suspension
|
3'200 mg/jour en comprimés pelliculés ou 80 ml/jour en suspension
|
- Patients recevant également du valproate:
Le traitement doit être instauré à la posologie journalière de 20 mg/kg pour les comprimés pelliculés ou 0,5 ml/kg pour la suspension. La posologie est augmentée par paliers allant jusqu'à 20 mg/kg/jour pour les comprimés pelliculés ou 0,5 ml/kg/jour pour la suspension, tous les deux jours, jusqu'à une dose recommandée de 30 mg/kg/jour pour les comprimés pelliculés ou 0,75 ml/kg/jour pour la suspension. La dose journalière ne devrait pas dépasser la dose maximale recommandée indiquée dans le tableau suivant pour la fourchette de poids.
|
Fourchette de poids
|
30,0–50,0 kg
|
50,1–70,0 kg
|
≥70,1 kg
| |
Dose maximale recommandée
|
1'600 mg/jour en comprimés pelliculés ou 40 ml/jour en suspension
|
2'000 mg/jour en comprimés pelliculés ou 50 ml/jour en suspension
|
2'400 mg/jour en comprimés pelliculés ou 60 ml/jour en suspension
|
Instructions posologiques particulières
Patients insuffisants hépatiques
Son emploi n'a pas été étudié chez les patients insuffisants hépatiques. Il est recommandé d'augmenter la dose avec prudence et précaution lors du traitement des patients présentant une insuffisance hépatique légère à modérée. L'utilisation chez les patients présentant une insuffisance hépatique grave n'est pas recommandée.
Patients insuffisants rénaux
Une étude réalisée chez des patients présentant une insuffisance rénale sévère a indiqué qu'aucune adaptation posologique n'était nécessaire chez ces patients (voir «Pharmacocinétique»).
Patients hémodialysés: Une adaptation posologique n'est en général pas nécessaire chez les patients sous hémodialyse (voir «Pharmacocinétique»).
Sujets âgés
Il existe peu d'informations sur l'utilisation d'Inovelon chez les sujets âgés. Les paramètres pharmacocinétiques du rufinamide n'étant pas altérés chez les sujets âgés (voir «Pharmacocinétique»), il n'est pas nécessaire d'adapter la posologie chez les patients de plus de 65 ans.
Enfants et adolescents
L'analyse cinétique de population sur le rufinamide (voir «Pharmacocinétique») montre qu'aucune adaptation posologique en fonction de l'âge n'est nécessaire. La sécurité et l'efficacité du rufinamide chez les jeunes enfants âgés de moins d'un an n'ont pas encore été établies. Aucune donnée n'est disponible.
Effet des aliments
Inovelon doit être pris de préférence au cours des repas (voir «Pharmacocinétique»).
Schéma d'administration
Arrêt du traitement par Inovelon: L'arrêt du traitement doit être progressif. Dans les essais cliniques, le traitement par Inovelon était arrêté en réduisant la dose d'environ 25% tous les deux jours.
En cas d'oubli d'une ou de plusieurs doses, une évaluation clinique est nécessaire.
Mode d'administration
Inovelon est destiné à être pris par voie orale. Il est pris deux fois par jour, le matin et le soir, à intervalle de 12 heures, en deux doses égales. Un effet de l'alimentation ayant été observé, Inovelon doit être pris de préférence au cours des repas (voir «Pharmacocinétique»).
Comprimés:
Les comprimés pelliculés doivent être pris avec de l'eau. Le comprimé peut être divisé au niveau de la barre de sécabilité pour faciliter la prise ou pour prendre une dose fractionnée. Si le patient a des difficultés pour avaler, il est possible d'écraser les comprimés et de les administrer avec un demi-verre d'eau.
Suspension:
L'adaptateur à introduire dans le flacon (adaptateur sous pression PIBA) fourni dans l'emballage doit être inséré fermement dans le goulot du flacon avant l'utilisation et rester en place pendant toute la durée d'utilisation du flacon. Le capuchon s'insère aussi de manière adéquate une fois l'adaptateur en place. La seringue de dosage est introduite dans l'adaptateur et la dose prise une fois le flacon renversé. Le capuchon doit être revissé sur le flacon après chaque utilisation.
Les instructions d'utilisation de l'adaptateur et de la seringue sont présentées ci-dessous:
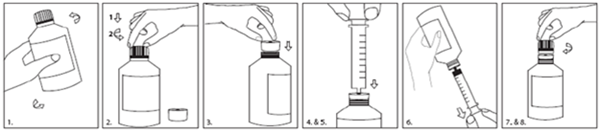
1.Agitez bien avant utilisation.
2.Appuyez (1) sur le bouchon en tournant (2) pour ouvrir le flacon.
3.Insérez l'adaptateur dans le goulot du flacon jusqu'à obtention d'un joint étanche.
4.Enfoncez complètement le piston de la seringue.
5.Insérez la seringue pour administration orale aussi loin que possible dans l'orifice de l'adaptateur.
6.Retournez l'ensemble et prélevez la quantité prescrite de suspension à partir du flacon.
7.Retournez le flacon et retirez la seringue pour administration orale.
8.Laissez l'adaptateur en place et refermez le flacon avec le bouchon.
9.Après administration de la dose, retirez le piston de la seringue et immergez-les dans de l'eau savonneuse chaude. Ensuite, placez le piston et le corps de la seringue dans de l'eau propre, rincez-les et laissez-les sécher à l'air libre. Ne les essuyez pas avec un linge.
La dose prescrite d'Inovelon sous forme de suspension buvable peut être administrée via une sonde d'alimentation entérale*. Suivre les instructions du fabricant de la sonde d'alimentation pour administrer le médicament. Pour garantir une posologie adéquate, après administration de la suspension buvable, la sonde d'alimentation entérale doit être rincée au moins une fois avec 1 ml d'eau. (*: sonde en chlorure de polyvinyle (PVC) de 40 cm de longueur au maximum et de CH 5 de diamètre.)
Contre-indicationsSyndrome du QT court familial (voir «Mises en garde et précautions»)
Hypersensibilité à la substance active, aux dérivés triazolés ou à l'un des excipients.
Suspension buvable: La suspension orale contenant du sorbitol, elle n'est pas adaptée aux personnes présentant une intolérance au fructose (intolérance au fructose héréditaire). Elle ne doit pas non plus être utilisée chez les personnes présentant une hypersensibilité aux parabènes (Parahydroxybenzoate de méthyle et parahydroxybenzoate de propyle E216, E218) et au propylène glycol (E1520).
Mises en garde et précautionsStatus epilepticus
Lors des études de développement clinique, il a été observé des cas d'état de mal épileptique sous rufinamide, alors qu'aucun cas n'a été observé avec le placebo. Ces événements ont conduit à l'arrêt du rufinamide dans 20% des cas. Le rapport bénéfice/risque doit être réévalué si de nouveaux types de crises d'épilepsie apparaissent et (ou) si la fréquence de l'état de mal épileptique augmente par rapport à l'état initial du patient.
Raccourcissement de l'intervalle QT
Dans le cadre d'une étude approfondie portant sur l'espace QT, le rufinamide a entraîné un raccourcissement de l'espace QTc, proportionnel à sa concentration. Le syndrome du QT court familial est associé à un risque accru de mort cardiaque subite ou d'arythmies ventriculaires, en particulier une fibrillation ventriculaire. Il est supposé que de tels évènements surviennent principalement lorsque l'intervalle QT corrigé est inférieur à 300 msec. Les études électrocardiographiques formelles menées avec le rufinamide n'ont mis en évidence ni un raccourcissement de l'intervalle QT inférieur à 300 msec ni de signes indiquant une mort cardiaque subite ou des arythmies ventriculaires induites par la substance. On ne connait cependant pas à l'heure actuelle la pertinence clinique du raccourcissement de l'intervalle QT proportionnel à la concentration, observé avec le rufinamide. Le rufinamide ne doit donc pas être utilisé chez les patients présentant un syndrome du QT court familial. La prudence est également de mise si l'on envisage d'administrer le rufinamide en concomitance avec d'autres substances raccourcissant l'espace QT.
Sevrage du rufinamide
Les antiépileptiques, dont Inovelon, doivent être arrêtés progressivement afin de réduire le risque possible de crises d'épilepsie à l'arrêt. Dans les études cliniques, le traitement était arrêté en réduisant la dose d'environ 25% tous les deux jours. Les données sur l'arrêt des traitement antiépileptiques associés, une fois l'épilepsie contrôlée par l'adjonction d'Inovelon, sont insuffisantes.
Effets sur le système nerveux central
Le traitement par le rufinamide a été associé à des vertiges, une somnolence, une ataxie et des troubles de la marche, qui pourraient augmenter la survenue de chutes accidentelles dans cette population (voir «Effets indésirables»). Les patients et les aidants doivent exercer leur vigilance jusqu'à ce qu'ils soient familiarisés avec les effets possibles de ce médicament.
Réactions d'hypersensibilité
Des cas de syndrome grave d'hypersensibilité aux médicaments antiépileptiques incluant des syndromes DRESS (éruption cutanée avec éosinophilie et symptômes systémiques) et de Stevens-Johnson (SJS) sont survenus en association avec le traitement par rufinamide. Les signes et symptômes étaient variés; cependant les patients présentaient généralement, mais pas exclusivement, de la fièvre et une éruption cutanée associés à une atteinte d'autres organes/systèmes. Parmi les autres manifestations on peut citer une lymphadénopathie, des anomalies des épreuves fonctionnelles hépatiques et une hématurie. L'expression du trouble étant variable, d'autres signes et symptômes d'organes/systèmes non observés jusqu'ici peuvent apparaître. Ce syndrome s'est produit peu de temps après l'instauration du traitement par le rufinamide et dans la population pédiatrique. En cas de suspicion de cette réaction, il convient d'arrêter le rufinamide et d'instaurer un autre traitement. Tous les patients développant une éruption cutanée sous rufinamide doivent être étroitement surveillés.
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par Inovelon. Les médecins doivent tenter de s'assurer de l'utilisation d'un moyen de contraception approprié et doivent exercer leur jugement clinique pour évaluer si les contraceptifs oraux, ou les doses des composants du contraceptif oral, sont adaptés à l'état clinique particulier de la patiente (voir «Interactions» et «Grossesse/Allaitement»).
Idées suicidaires et comportement suicidaire
Des idées et un comportement suicidaires ont été rapportés chez des patients traités par des antiépileptiques dans plusieurs indications. Une méta-analyse d'études randomisées et contrôlées contre placebo de médicaments antiépileptiques a également montré une légère augmentation du risque d'idées et de comportements suicidaires. Le mécanisme de ce risque n'est pas connu et les données disponibles ne permettent pas d'exclure la possibilité d'un risque accru avec Inovelon.
Il convient donc de surveiller les patients afin de détecter les signes d'idées et de comportement suicidaires et d'envisager un traitement approprié. Les patients (et leurs aidants) doivent être informés qu'ils doivent consulter un médecin en cas d'apparition de signes d'idées ou de comportements suicidaires.
Ingrédients présentant un intérêt particulier
Comprimés pelliculés:
Les comprimés contiennent 20 mg (Inovelon 100 mg), 40 mg (Inovelon 200 mg), ou 80 mg (Inovelon 400 mg) de lactose. Les patients atteints d'une intolérance héréditaire rare au galactose, d'un déficit congénital en lactase ou d'un syndrome de malabsorption du glucose-galactose ne doivent pas prendre ce médicament. Les comprimés contiennent moins de 1 mmol de sodium (23 mg) par comprimé, c'est-à-dire qu'ils sont pratiquement «sans sodium».
Suspension:
Sorbitol: ce médicament contient 175 mg de sorbitol par ml, correspondant à 150 mg/kg/jour. Le sorbitol peut provoquer des troubles gastro-intestinaux et exercer un effet légèrement laxatif. Le sorbitol est une source de fructose. Les patients atteints d'intolérance héréditaire au fructose (IHF) ne doivent pas prendre ce médicament. Chez les bébés et les jeunes enfants (de moins de 2 ans), il est possible qu'une intolérance héréditaire au fructose (IHF) ne soit pas encore diagnostiquée. Avant d'utiliser ce médicament, il convient de recueillir une anamnèse détaillée auprès de chaque patient au regard des symptômes d'IHF. Propylène glycol: ce médicament contient 25 mg de propylène glycol par ml, correspondant à 21,4 mg/kg. L'utilisation concomitante avec un substrat de déshydrogénase – comme l'éthanol – peut être à l'origine d'effets indésirables graves chez les nouveau-nés. Sodium: la suspension contient moins de 1 mmol de sodium (23 mg) par ml, c'est-à-dire qu'elle est pratiquement «sans sodium». Acide benzoïque: ce médicament contient 0,005 mg d'acide benzoïque par ml, correspondant à 0,004 mg/kg. Alcool benzylique: ce médicament contient 0,0003 mg d'alcool benzylique par ml. L'alcool benzylique peut provoquer des réactions allergiques. Parabènes: la suspension contient du parahydroxybenzoate de méthyle et du propyl-4-hydroxybenzoate, qui peuvent provoquer des réactions allergiques, même retardées.
InteractionsEffets possibles d'autres médicaments sur Inovelon
Autres médicaments antiépileptiques
Aucune modification cliniquement pertinente des concentrations de rufinamide n'a été observée lors de l'administration concomitante de médicaments antiépileptiques connus pour être des inducteurs enzymatiques. Chez les patients traités par Inovelon, l'instauration du valproate peut entraîner une augmentation significative des concentrations plasmatiques de rufinamide (voir les recommandations à la rubrique «Posologie/Mode d'administration»).
L'introduction ou l'arrêt de ces médicaments ou l'adaptation de leur posologie pendant le traitement par Inovelon peut nécessiter une adaptation de la posologie d'Inovelon.
Aucune modification significative de la concentration de rufinamide n'est observée après l'administration concomitante de lamotrigine, de topiramate ou de benzodiazépines.
Effet possible d'Inovelon sur d'autres médicaments
Autres médicaments antiépileptiques
Les interactions pharmacocinétiques entre le rufinamide et les autres agents antiépileptiques ont été évaluées chez les patients épileptiques en utilisant une modélisation pharmacocinétique dans la population. Le rufinamide ne semble pas avoir d'effet clinique pertinent sur les concentrations à l'équilibre de la carbamazépine, de la lamotrigine, du phénobarbital, de la phénytoïne, du topiramate ou du valproate.
Contraceptifs oraux
La co-administration pendant 14 jours de 800 mg de rufinamide deux fois par jour et d'un contraceptif oral combiné comprenant 35 µg d'éthinylestradiol et 1 mg de noréthindrone a entraîné une diminution de l'ASC0-24 moyenne de 22% pour l'éthinylestradiol et de 14% pour la noréthindrone. Aucune étude n'a été menée avec les autres contraceptifs oraux et avec les implants contraceptifs. Il convient de conseiller aux femmes en âge de procréer et sous contraceptif hormonal d'utiliser un moyen de contraception supplémentaire sûr et efficace (voir «Mises en garde et précautions» et «Grossesse/Allaitement»).
Enzymes du cytochrome P450
Le rufinamide est métabolisé par hydrolyse; il n'est pas métabolisé à un taux appréciable par les enzymes du cytochrome P450. En outre, le rufinamide n'inhibe pas l'activité des enzymes du cytochrome P450 (voir «Pharmacocinétique»). Ainsi, il est improbable que des interactions cliniquement significatives dues à l'inhibition du système du cytochrome P450 par le rufinamide se produisent. Il a été montré que le rufinamide induisait l'enzyme CYP3A4 du cytochrome P450 et pouvait ainsi réduire les concentrations plasmatiques des médicaments métabolisés par cette enzyme. L'effet était léger à modéré. L'activité moyenne du CYP3A4, évaluée par la clairance du triazolam, a été augmentée de 55% après 11 jours de traitement par le rufinamide à raison de 400 mg deux fois par jour. L'exposition au triazolam a été réduite de 36%. Des doses plus fortes de rufinamide peuvent entraîner une induction plus marquée. Il n'est pas exclu que le rufinamide puisse également diminuer l'exposition à des médicaments métabolisés par d'autres enzymes ou transportés par des protéines de transport telles que la glycoprotéine P.
Il est recommandé de surveiller attentivement les patients traités par des médicaments métabolisés par le système enzymatique CYP3A, pendant deux semaines, au début ou après l'arrêt du traitement par Inovelon ou après une variation importante de la posologie. Il peut s'avérer nécessaire d'envisager une adaptation de la posologie du médicament administré de façon concomitante. Ces recommandations doivent également être prises en considération lorsque le rufinamide est utilisé en même temps que des médicaments à indice thérapeutique étroit comme la warfarine ou la digoxine.
Une étude spécifique d'interaction chez des sujets sains n'a mis en évidence aucune influence du rufinamide à une dose de 400 mg deux fois par jour sur les paramètres pharmacocinétiques de l'olanzapine, un substrat du CYP1A2.
Il n'existe aucune donnée relative à l'interaction entre le rufinamide et l'alcool.
Grossesse, allaitementGrossesse
Risque lié à l'épilepsie et aux médicaments antiépileptiques en général:
Il a été montré que dans la descendance de femmes épileptiques, la prévalence de malformations est deux à trois fois plus élevée que le taux d'environ 3% dans la population générale. Dans la population traitée, on a observé une augmentation des malformations sous polythérapie. Toutefois, l'ampleur de la responsabilité du traitement et (ou) de la maladie n'a pas été élucidée.
De plus, un traitement antiépileptique efficace ne doit pas être interrompu de manière soudaine, car l'aggravation de la maladie est préjudiciable à la mère et au fœtus.
Risque lié au rufinamide:
Les études chez l'animal n'ont mis en évidence aucun effet tératogène, mais une fœtotoxicité à des doses induisant une toxicité maternelle (voir «Données précliniques»). Le risque potentiel en clinique n'est pas connu.
Il n'existe pas de données cliniques sur les femmes enceintes exposées au rufinamide.
En prenant en considération ces données, le rufinamide ne doit pas être administré pendant la grossesse ainsi que chez les femmes en âge de procréer n'utilisant pas de moyen de contraception à moins d'une nécessité absolue.
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par Inovelon. Les médecins doivent tenter de s'assurer de l'utilisation d'un moyen de contraception approprié et doivent exercer leur jugement clinique pour évaluer si les contraceptifs oraux, ou les doses des composants du contraceptif oral, sont adaptés à l'état clinique particulier de la patiente (voir «Interactions»).
Si les femmes traitées par le rufinamide planifient une grossesse, l'indication de ce produit doit être pesée minutieusement. Pendant la grossesse, un traitement antiépileptique efficace par le rufinamide ne doit pas être interrompu car l'aggravation de la maladie est préjudiciable à la mère et au fœtus.
Allaitement
Il n'existe pas de données sur l'excrétion du rufinamide dans le lait maternel. En raison de ses effets délétères possibles sur le nourrisson nourri au sein, il convient d'éviter l'allaitement pendant le traitement de la mère par le rufinamide.
Effet sur l’aptitude à la conduite et l’utilisation de machinesInovelon peut provoquer vertiges, somnolence et vision trouble. L'effet d'Inovelon sur l'aptitude à conduire des véhicules et à utiliser des machines peut être léger à important, selon la sensibilité individuelle. Il convient de conseiller aux patients de se montrer prudents au cours des activités nécessitant une grande vigilance, comme la conduite de véhicules ou l'utilisation de machines.
Inovelon suspension buvable peut en outre provoquer des symptômes semblables à ceux apparaissant après la consommation d'alcool. Il est recommandé aux patients de ne pas conduire de véhicules ou d'utiliser des machines s'ils ressentent une somnolence, des vertiges ou ont la vision trouble.
Effets indésirablesLe programme de développement clinique a inclus plus de 1900 patients avec différents types d'épilepsie, exposés au rufinamide. Maux de tête, vertiges, fatigue et somnolence ont été globalement les effets indésirables les plus fréquemment rapportés. Les effets indésirables les plus fréquemment observés à une incidence plus élevée que pour le placebo chez des patients atteints de syndrome de Lennox-Gastaut ont été la somnolence et les vomissements. L'intensité des effets indésirables était généralement légère à modérée. Le taux d'interruption pour effets indésirables chez les patients souffrant de syndrome de Lennox-Gastaut a été de 8,2% pour les patients recevant du rufinamide et de 0% pour les patients recevant un placebo. Les effets indésirables les plus fréquents entraînant un arrêt du traitement dans le groupe traité par le rufinamide ont été les éruptions cutanées et les vomissements.
Les effets indésirables rapportés avec une incidence supérieure à celle du placebo, au cours des études en double aveugle dans le syndrome de Lennox-Gastaut ou dans la population globale exposée au rufinamide sont énumérés dans le tableau ci-dessous par terme de préférence MedDRA, classe de système/organe et par fréquence.
La fréquence est indiquée comme suit: très fréquent (≥1/10), fréquent (≥1/100 et <1/10), peu fréquent (≥1/1'000 et <1/100), rares (≥1/10 000 à <1/1000), très rares (<1/10 000).
Infections et infestations
Fréquent: Pneumonie, grippe, rhinopharyngite, infection auriculaire, sinusite, rhinite.
Affections du système immunitaire
Rare: Hypersensibilité*.
Troubles du métabolisme et de la nutrition
Fréquent: Anorexie, troubles de l'alimentation, diminution de l'appétit.
Affections psychiatriques
Fréquent: anxiété, insomnie.
Affections du système nerveux
Très fréquent: Somnolence*, céphalées, vertiges*.
Fréquent: Etat de mal épileptique*, convulsions, troubles de la coordination*, nystagmus, hyperactivité psychomotrice, tremblement.
Affections oculaires
Fréquent: Diplopie, vision trouble.
Affections de l'oreille et du labyrinthe
Fréquent: Vertige.
Affections respiratoires, thoraciques et médiastinales
Fréquent: Epistaxis.
Affections gastro-intestinales
Très fréquent: nausées, vomissements.
Fréquent: Douleurs abdominales hautes, constipation, dyspepsie, diarrhée.
Affections hépatobiliaires
Rare: Elévation des enzymes hépatiques.
Affections de la peau et du tissu sous-cutané
Fréquent: Eruption cutanée*, acné.
Affections musculosquelettiques et du tissu conjonctif
Fréquent: Mal de dos.
Affections des organes de reproduction et du sein
Fréquent: Oligoménorrhée.
Troubles généraux et anomalies au site d'administration
Très fréquent: Fatigue.
Fréquent: Trouble de la marche*.
Investigations
Fréquent: Perte de poids.
Lésions
Fréquent: Traumatisme crânien, contusion.
* Se référer à la rubrique «Mises en garde et précautions».
Effets indésirables après commercialisation
Population pédiatrique (1 à 4 ans): Dans une étude ouverte, multicentrique à groupes parallèles, des patients âgés d'un an à moins de 4 ans atteints d'un syndrome de Lennox-Gastaut insuffisamment contrôlé ont reçu du rufinamide à une posologie allant jusqu'à 45 mg/kg/jour (n=25), ou d'autres antiépileptiques choisis par le médecin traitant (n=11), comme traitement complémentaire. Le profil des effets indésirables du rufinamide dans cette population était semblable à celui observé dans les études menées chez les enfants à partir de 4 ans, adolescents et adultes. Dans le groupe des patients traités au rufinamide, l'incidence était supérieure à celle constatée dans le groupe recevant les traitements comparateurs en ce qui concerne les vomissements (24%), la somnolence (16%), la bronchite (12%), la constipation (12%), la baisse de l'appétit (12%), la toux (12%), l'éruption cutanée (12%), la perte de poids (8%), la pneumonie (8%), l'otite moyenne (8%), la gastroentérite (8%), la congestion nasale (8%) et la pneumopathie d'inhalation (8%) (voir aussi la rubrique «Propriétés/Effets»).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSignes et symptômes
L'administration répétée de 7'200 mg/jour n'a été associée à aucun signe ou symptôme majeur.
Traitement
Après un surdosage aigu, l'estomac peut être vidé par lavage gastrique ou vomissement provoqué. Il n'existe pas d'antidote spécifique pour le rufinamide. Un traitement symptomatique doit être instauré et peut comprendre une hémodialyse (voir «Pharmacocinétique»).
Propriétés/EffetsCode ATC
N03AF03
Mécanisme d'action
Le rufinamide module l'activité des canaux sodiques, prolongeant leur état inactif. Le rufinamide est actif chez divers modèles animaux d'épilepsie.
Efficacité clinique
Adultes, adolescents et enfants à partir de 4 ans
Inovelon comprimés pelliculés a été administré dans le cadre d'une étude en double aveugle, contrôlée contre placebo, à des doses pouvant atteindre 45 mg/kg/jour pendant 84 jours, à 139 patients dont les crises d'épilepsie associées au syndrome de Lennox-Gastaut (comprenant à la fois des absences atypiques et des chutes brutales sans perte de conscience) sont mal contrôlées. Des patients (de sexe masculin ou féminin, âgés de 4 à 30 ans) recevant 1 à 3 antiépileptiques concomitants à dose fixe ont été inclus. Chaque patient devait avoir présenté au moins 90 crises dans le mois ayant précédé l'entrée dans l'étude. On a observé une amélioration significative des trois critères principaux d'efficacité: la modification du pourcentage de la fréquence totale des crises d'épilepsie après 28 jours de la phase d'entretien, par rapport à sa valeur initiale (-35,8% sous Inovelon contre 1,6% sous placebo, p = 0,0006), le nombre de crises toniques et atoniques (-42,9% sous Inovelon contre 2,2% sous placebo, p = 0,0002) et le score d'intensité des crises résultant de l'évaluation globale effectuée par le parent/tuteur à la fin de la phase en double aveugle (forte ou très forte amélioration chez 32,2% des patients sous Inovelon contre 14,5% dans le bras placebo, p = 0,0041). Chez un nombre limité de patients, il a été appliqué des doses atteignant jusqu'à 4'000 mg/jour (pour un poids de 30-50 kg) ou 4'800 mg/jour (pour un poids supérieur à 50 kg).
La modélisation pharmacocinétique/pharmacodynamique de la population a démontré que la réduction de la fréquence des crises totales et des crises toniques et atoniques, l'amélioration de l'évaluation globale de l'intensité des crises et l'augmentation de la probabilité de réduction de la fréquence des crises étaient dépendantes des concentrations du rufinamide.
Enfants âgés d'un an à moins de 4 ans
Une étude ouverte, multicentrique, contrôlée par substance active a comparé le rufinamide comme traitement complémentaire à 1-3 autres antiépileptiques quelconques (Antiepileptic Drugs, AED), choisis par l'investigateur chez des patients pédiatriques âgés d'1 an à moins de 4 ans atteints du syndrome de Lennox-Gastaut mal maîtrisé. 25 patients ont reçu du rufinamide en traitement complémentaire pendant 106 semaines, à une posologie allant jusqu'à 45 mg/kg/jour en deux prises. 12 patients ont reçu un autre AED.
L'objectif principal de l'étude était d'évaluer l'effet sur le développement cognitif et le comportement dans la population pédiatrique âgée d'1 an à moins de 4 ans avec syndrome de Lennox-Gastaut. Une caractérisation de la PK spécifique à l'âge a été effectuée en complément, et l'efficacité a été comparée. Le développement cognitif, le comportement et le développement du langage des participants traités par rufinamide étaient comparables à ceux des participants du groupes traités par les autres AED. Au fil du temps, aucune tendance homogène ne s'est dessinée dans aucun des deux groupes de traitement, en présence pourtant de petites tailles d'échantillon, d'un tableau clinique lourd au début de l'étude chez un grand nombre de participants, et d'un taux d'arrêt élevé dans les deux groupes de traitement, de sorte que les résultats n'ont pas pu être évalués de manière concluante.
La PK du rufinamide évaluée dans le cadre d'une modélisation de la population était indépendante de la dose, et était, compte tenu du poids corporel, non significativement dépendante de l'âge, ni comme covariable continue (1 à 35 ans), ni comme covariable catégorielle (catégories d'âge: 1 à moins de 4 ans, et 4 ans et plus). De même, l'exposition dans le groupe des 1 à moins de 2 ans était comparable à celle chez les participants de 2 à moins de 4 ans.
L'administration concomitante de valproate a diminué la clairance du rufinamide chez les participants âgés de 1 à moins de 4 ans, en fonction de la concentration, de même que chez les patients âgés de 4 ans et plus.
En raison des tailles limitées d'échantillons dans cette étude, les données n'étaient pas suffisantes pour apporter des indications sur les différences d'efficacité en matière de contrôle des crises entre les deux groupes de traitement.
PharmacocinétiqueAbsorption
Les concentrations plasmatiques maximales sont atteintes environ 6 heures après l'administration. Le pic de concentration (Cmax) et l'ASC plasmatique du rufinamide augmentent moins que proportionnellement avec les doses chez des sujets sains et chez les patients à jeun ou non, probablement en raison d'un comportement d'absorption limitée par la dose. Après des doses uniques, les aliments augmentent la biodisponibilité (ASC) du rufinamide d'environ 34% et le pic de concentration plasmatique de 56%.
La suspension orale et les comprimés pelliculés d'Inovelon sont bioéquivalents.
Distribution
Dans les études in vitro, seule une petite fraction de rufinamide (34%) était liée aux protéines sériques humaines, l'albumine représentant environ 80% de cette liaison. Ceci indique un risque minimum d'interactions médicamenteuses par déplacement à partir des sites de liaison lors de l'administration concomitante d'autres médicaments. Le rufinamide était distribué de façon homogène entre les érythrocytes et le plasma.
Métabolisme
Le rufinamide est presque exclusivement éliminé par métabolisme. La principale voie de métabolisme est l'hydrolyse du groupe carboxylamide en dérivé acide pharmacologiquement inactif, le CGP 47292. Le métabolisme via le cytochrome P450 est très minime. La formation de petites quantités de conjugués au glutathion ne peut être totalement exclue.
Pour les enzymes du P450 humain CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 ou CYP4A9/11-2, le rufinamide a démontré in vitro peu ou aucune capacité significative à agir comme inhibiteur par compétition ou de par son mécanisme d'action.
Elimination
La demi-vie d'élimination plasmatique est d'environ 6 à 10 heures chez les sujets sains et les patients épileptiques. Administré deux fois par jour à 12 heures d'intervalle, le rufinamide s'accumule dans la limite prédite par sa demi-vie terminale, indiquant que les paramètres pharmacocinétiques du rufinamide sont indépendants du temps (pas d'auto-induction du métabolisme).
Dans une étude de radiomarquage chez trois volontaires sains, la molécule mère (rufinamide) était le principal composant radioactif dans le plasma, représentant environ 80% de la radioactivité totale et le métabolite CGP 47292 ne constituant que 15% environ. L'excrétion rénale était la voie prédominante d'élimination des produits liés au médicament, représentant 84,7% de la dose.
Linéarité/non linéarité
La biodisponibilité du rufinamide est dépendante de la dose. Lorsque la dose augmente, la biodisponibilité diminue.
Cinétique pour certains groupes de patients
Sexe
La modélisation pharmacocinétique de la population a été utilisée pour évaluer l'influence du sexe sur les paramètres pharmacocinétiques du rufinamide. Ces évaluations indiquent que le sexe n'a pas d'effet sur la pharmacocinétique du rufinamide qui soit pertinent sur le plan clinique.
Insuffisance hépatique
Aucune étude n'a été réalisée chez les patients insuffisants hépatiques. Inovelon ne doit donc pas être administré chez les patients présentant une insuffisance hépatique grave.
Insuffisance rénale
La pharmacocinétique d'une dose unique de 400 mg de rufinamide n'a pas été modifiée chez les sujets présentant une insuffisance rénale chronique et sévère par rapport aux volontaires sains. Cependant, les concentrations plasmatiques étaient réduites d'environ 30% lorsqu'une hémodialyse était pratiquée après l'administration de rufinamide, ce qui laisse penser que cette technique peut se révéler utile en cas de surdosage (voir «Posologie/Mode d'emploi» et «Surdosage»).
Les simulations effectuées à partir de données pharmacocinétiques de patients hémodialysés présentant une insuffisance rénale chronique et sévère indiquent qu'une hémodialyse (6 heures par jour lors de 3 jours par semaine) ne provoque pas de diminution du taux de rufinamide qui soit pertinente sur le plan clinique (diminution moyenne d'environ 12%). Si après l'instauration du traitement par hémodialyse chez un patient qui est, du point de vue clinique, stabilisé sous rufinamide, on observe une modification des crises épileptiques, par exemple, une augmentation de la fréquence des crises (voir «Posologie/mode d'emploi», une adaptation posologique doit être envisagée.
Sujets âgés
Une étude pharmacocinétique chez des volontaires sains âgés n'a pas mis en évidence de différence significative des paramètres pharmacocinétiques par rapport aux adultes plus jeunes.
Enfants et adolescents
La clairance du rufinamide est généralement plus faible chez les enfants que chez les adultes; cette différence est liée à la taille corporelle. L'analyse cinétique de population menée sur 115 patients, comprenant 85 enfants (24 entre 1 et 3 ans, 40 entre 4 et 11 ans et 21 entre 12 et 17 ans) a montré que la pharmacocinétique, après prise en compte du poids corporel, n'est pas significativement influencée par l'âge. Aucune étude avec des jeunes enfants de moins d'un an n'a été effectuée.
Données précliniquesLes études conventionnelles de toxicologie n'ont mis en évidence aucun risque particulier aux doses cliniques pertinentes. Les toxicités observées chez les chiens à des doses comparables à l'exposition humaine à la dose maximale recommandée ont été des anomalies hépatiques comprenant des thrombus biliaires, une cholestase et une élévation des enzymes hépatiques que l'on pense être dus à une sécrétion biliaire accrue dans cette espèce. Aucun signe de risque associé n'a été identifié dans les études de toxicité à doses répétées chez le rat et le singe.
Toxicité de reproduction
Dans les études de toxicité sur la reproduction et le développement, on a observé une diminution de la croissance fœtale et de la survie et certains cas de mort-nés secondaires à la toxicité maternelle. Cependant, aucun effet n'a été observé chez la descendance pour ce qui concerne la morphologie ou des fonctions telles que l'apprentissage ou la mémorisation. Inovelon ne s'est pas avéré tératogène pour les souris, les rats et les lapins.
Le profil de toxicité du rufinamide chez les jeunes animaux est comparable à celui des animaux adultes. De jeunes rats et chiens ont présenté une prise de poids réduite. Chez les jeunes rats on a observé, aux doses de 150 mg/kg, une hypertrophie hépatocellulaire centro-bulbaire adaptative, et aux doses de 50 et 150 mg/kg, une vacuolisation cytoplasmique hypophysaire. Chez les jeunes chiens, on a constaté des concentrations sériques plus élevées d'alanine-aminotransférase et des dépôts de pigments marron dans le foie. Les chiens ayant reçu 200 mg/kg dès l'âge de six semaines (correspondant à une exposition plasmatique (AUC) dans la fourchette de l'exposition thérapeutique), avaient des foies plus lourds et une croissance et prise de nourriture légèrement réduites, par comparaison au groupe témoin. Tous ces résultats étaient réversibles en l'espace de 4 à 10 semaines.
Le rufinamide ne s'est pas avéré génotoxique et n'a pas eu d'effet carcinogène. Dans l'étude de carcinogénèse chez la souris, on a observé une fibrose de la moelle osseuse à des niveaux d'exposition comparables à ceux de l'exposition clinique. Cet effet indésirable n'a pas été décrit lors des études cliniques mais pourrait s'avérer pertinent pour l'homme. Les tumeurs osseuses bénignes (ostéomes) et l'hyperostose observées chez la souris ont été considérées comme le résultat de l'activation d'un virus spécifique à la souris par les ions fluorure libérés au cours du métabolisme oxydatif du rufinamide. Concernant le pouvoir immunotoxique, on a observé un petit thymus et une involution thymique chez les chiens lors d'une étude sur 13 semaines, avec une réponse significative à la dose la plus élevée chez les mâles. Dans l'étude sur 13 semaines, des modifications affectant la moelle osseuse et des organes lymphoïdes ont été décrites chez les femelles avec une incidence faible. Chez les rats, on a observé un appauvrissement de la moelle osseuse et une atrophie du thymus uniquement dans l'étude du pouvoir carcinogène.
Remarques particulièresStabilité
Ne pas utiliser au-delà de la date de péremption figurant après la mention «EXP» sur l'emballage.
Remarques concernant le stockage
Comprimés pelliculés: à conserver à une température ne dépassant pas 30 °C.
Suspension buvable: à conserver à une température ne dépassant pas 30 °C. La suspension doit être utilisée dans les 90 jours après l'ouverture du flacon.
Numéro d’autorisation58097, 62066 (Swissmedic).
PrésentationInovelon 100 mg (sécable) (présentation actuellement non commercialisée) [B]
Inovelon 200 mg (sécable): 60 comprimés pelliculés (présentation à 50 comprimés pelliculés actuellement non commercialisée) [B]
Inovelon 400 mg (sécable): 100 comprimés pelliculés (présentations à 50, 60 et 200 comprimés pelliculés actuellement non commercialisées) [B]
Inovelon 40 mg/ml: 1 flacon avec fermeture sécurisée pour les enfants contenant 460 ml de suspension buvable, 2 seringues de dosage calibrées à 20 ml et un adaptateur à insérer dans le flacon. [B]
Titulaire de l’autorisationEisai Pharma SA, Zurich.
Mise à jour de l’informationSeptembre 2022.
|