Propriétés/EffetsCode ATC
L04AJ01
Soliris est un anticorps humanisé monoclonal recombinant IgG2/4k qui se lie à la protéine C5 du complément et inhibe l’activation de la voie terminale du complément. L’anticorps Soliris contient des régions constantes humaines et des régions murines déterminant la complémentarité greffées sur les régions variables humaines des chaînes légères et lourdes. Soliris est composé de deux chaînes lourdes de 448 acides aminés et de deux chaînes légères de 214 acides aminés; son poids moléculaire est d’environ 148 kDa.
Soliris est produit dans un système d’expression de myélome murin (lignée cellulaire NS0) et purifié par chromatographie d’affinité et d’échanges d’ions. Le procédé de fabrication de la substance médicamenteuse comprend également des étapes spécifiques d’inactivation et de suppression virale.
Mécanisme d’action
L’eculizumab, principe actif de Soliris, est un inhibiteur de la voie terminale du complément qui se lie de manière spécifique à la protéine C5 du complément avec une affinité élevée, inhibant ainsi son clivage en C5a et C5b et empêchant la formation du complexe terminal du complément C5b-9. L’eculizumab préserve les protéines de la voie proximale du complément qui sont essentielles à l’opsonisation des microorganismes et à la clairance des complexes immuns.
Chez les patients atteints d’HPN, Soliris inhibe l’activation non contrôlée de la voie terminale du complément et l’hémolyse intravasculaire induite.
Chez la majorité des patients atteints d’HPN, des concentrations sériques d’eculizumab d’environ 35 µg/ml suffisent à une inhibition presque complète de l’hémolyse intravasculaire induite par la voie terminale du complément.
Dans l’HPN, l’administration chronique de Soliris a conduit à une réduction rapide et durable de l’activité hémolytique induite par le complément. Chez les patients atteints de SHU atypique, Soliris inhibe l’activation non contrôlée de la voie terminale du complément et la microangiopathie thrombotique induite.
Tous les patients atteints de SHU atypique traités par Soliris à la posologie recommandée ont montré une diminution rapide et durable de l’activité de la voie terminale du complément. Chez tous les patients atteints de SHU atypique, des concentrations sériques d’eculizumab d’environ 50 à 100 µg/ml suffisent à une inhibition quasi complète de l’activité de la voie terminale du complément.
Dans le SHU atypique, l’administration chronique de Soliris a conduit à une réduction rapide et durable de la microangiopathie thrombotique induite par le complément.
Chez les patients atteints de MAg réfractaire, l’activation non contrôlée de la voie terminale du complément provoque une lyse médiée par le complexe d’attaque membranaire (CAM) et une inflammation médiée par la protéine C5a au niveau de la jonction neuromusculaire, ce qui entraîne un défaut de transmission neuromusculaire. L’administration chronique de Soliris induit une inhibition immédiate, complète et maintenue de l’activité de la voie terminale du complément (concentrations sériques d’eculizumab ≥116 µg/ml).
Chez les patients atteints de NMOSD, l’activation non contrôlée de la voie terminale du complément provoquée par des auto-anticorps dirigés contre l’AQP4 provoque la formation du CAM et une inflammation médiée par la protéine C5a, qui entraîne une nécrose astrocytaire et une augmentation de la perméabilité de la barrière hémato-encéphalique ainsi que la mort des oligodendrocytes et des neurones environnants. L’utilisation à long terme de Soliris entraîne une inhibition immédiate, complète et durable de l’activité de la voie terminale du complément (concentrations sériques d’eculizumab ≥ 116 µg/ml).
Pharmacodynamique
Voir «Mécanisme d’action»
Efficacité clinique
Hémoglobinurie paroxystique nocturne
La sécurité et l’efficacité de Soliris chez les patients atteints d’HPN présentant une hémolyse ont été évaluées au cours d’une étude de 26 semaines en double aveugle, randomisée et contrôlée contre placebo (C04-001). Les patients atteints d’HPN ont été également traités par Soliris dans le cadre d’une étude de 52 semaines à un seul bras (C04-002) et dans une étude d’extension à long terme (E05-001). Les patients avaient reçu une vaccination méningococcique avant le début du traitement par Soliris. Dans toutes les études, la dose d’eculizumab était de 600 mg tous les 7 jours ± 2 jours pendant 4 semaines, suivie de 900 mg 7 jours 2 jours plus tard, puis 900 mg tous les 14 jours ± 2 jours pendant la durée de l’étude. Soliris a été administré par perfusion intraveineuse de 25 à 45 minutes (35 ± 10 minutes). Un registre observationnel non interventionnel des patients atteints d’HPN (M07-001) a également été mis en place pour caractériser l’évolution naturelle de l’HPN chez les patients non traités par Soliris et pour caractériser les résultats cliniques des patients traités par Soliris.
C04-001 (TRIUMPH), des patients atteints d’HPN ayant reçu au moins 4 transfusions au cours des 12 mois précédents, avec au moins 10 % de cellules HPN confirmé par une cytométrie en flux et dont la numération plaquettaire était d’au moins 100'000/microlitre, ont été randomisés soit dans le groupe Soliris (n=43) soit dans le groupe placebo (n=44). Avant la randomisation, tous les patients ont participé à une période d’observation initiale pour confirmer le besoin d’une transfusion de globules rouges et identifier la concentration d’hémoglobine (le point de référence) qui définirait les résultats de stabilisation de l’hémoglobine et de transfusion de chaque patient. Le point de référence de l’hémoglobine était inférieur ou égal à 9 g/dl chez les patients symptomatiques et inférieur ou égal à 7 g/dl chez les patients asymptomatiques. Les critères principaux d’efficacité étaient la stabilisation de l’hémoglobine (patients conservant une concentration en hémoglobine supérieure au point de référence de l’hémoglobine et n’ayant eu recours à aucune transfusion pendant les 26 semaines) et le recours à une transfusion sanguine. Les critères secondaires pertinents étaient la fatigue et la qualité de vie liée à l’état de santé. L’hémolyse était contrôlée principalement par la mesure des taux sériques de LDH et le pourcentage de globules rouges HPN était contrôlé par cytométrie en flux. Les patients recevant des anticoagulants et des corticostéroïdes systémiques à l’état basal ont poursuivi ces traitements. Les principales caractéristiques démographiques initiales étaient comparables dans les deux bras de traitement (voir tableau 2).
Dans l’étude non contrôlée C04-002 (SHEPHERD), des patients atteints d’HPN ayant reçu au moins une transfusion au cours des 24 mois précédents et dont la numération plaquettaire était d’au moins 30'000/microlitre ont reçu Soliris pendant 52 semaines. Les médicaments concomitants comportaient des agents antithrombotiques chez 63 % des patients et des corticostéroïdes systémiques chez 40 % des patients. Les paramètres initiaux sont présentés dans le tableau 2.
Tableau 2 : Données démographiques et paramètres des patients dans les études C04-001 et C04-002
|
|
C04-001
|
C04-002
| |
Paramètre
|
Placebo
N = 44
|
Soliris
N = 43
|
Soliris
N = 97
| |
Âge moyen (ET)
|
38,4 (13,4)
|
42,1 (15,5)
|
41,1 (14,4)
| |
Sexe - Féminin (%)
|
29 (65,9)
|
23 (53,5)
|
49 (50,5)
| |
Antécédent d’aplasie médullaire ou de SMD (%)
|
12 (27,3)
|
8 (18,7)
|
29 (29,9)
| |
Anticoagulants concomitants (%)
|
20 (45,5)
|
24 (55,8)
|
59 (61)
| |
Traitements concomitants par stéroïdes/immunosuppresseurs (%)
|
16 (36,4)
|
14 (32,6)
|
46 (47,4)
| |
Arrêt de traitement
|
10
|
2
|
1
| |
Concentrés érythrocytaires au cours des 12 mois précédents [médiane (1er trim., 3e trim.)]
|
17,0 (13,5/25,0)
|
18,0 (12,0/24,0)
|
8,0 (4,0/24,0)
| |
Taux moyen de Hb (g/dl) au point de référence (ET)
|
7,7 (0,75)
|
7,8 (0,79)
|
S/O
| |
Taux de LDH avant traitement (médiane, U/l)
|
2234,5
|
2032,0
|
2051,0
| |
Hémoglobine libre à l’état basal (médiane, mg/dl)
|
46,2
|
40,5
|
34,9
|
Dans l’étude TRIUMPH, les patients traités par Soliris ont présenté une réduction significative (p<0,001) de l’hémolyse, donnant lieu à des améliorations de l’anémie comme l’indiquent l’augmentation de la stabilisation de l’hémoglobine et la baisse du besoin en transfusions de globules rouges par rapport aux patients traités par placebo (voir tableau 3). Ces effets ont été observés chez des patients de chacune des trois strates de transfusions de concentrés érythrocytaires avant l’étude (4 à 14 unités ; 15 à 25 unités ; >25 unités). Après 3 semaines de traitement par Soliris, les patients ont signalé moins de fatigue et une amélioration de la qualité de vie liée à l’état de santé. En raison de la taille de l’échantillon et de la durée de l’étude, les effets de Soliris sur les événements thromboemboliques n’ont pas pu être déterminés. Dans l’étude SHEPHERD, 96 patients sur les 97 enrôlés ont terminé l’étude (un patient est décédé des suites d’un événement thromboembolique). La baisse de l’hémolyse intravasculaire, mesurée par les taux sériques de LDH, s’est maintenue pendant toute la durée du traitement et a entraîné une augmentation de l’évitement de transfusion, une diminution du besoin de transfusion de globules rouges et une baisse de la fatigue (voir tableau 3).
Tableau 3 : Résultats d’efficacité dans les études C04-001 et C04-002
|
|
C04-001
|
C04-002*
| |
|
Placebo
N = 44
|
SOLIRIS
N = 43
|
Valeur p
|
SOLIRIS
N = 97
|
Valeur p
| |
Pourcentage de patients dont les taux d’hémoglobine étaient stabilisés à la fin de l’étude
|
0
|
49
|
< 0,001
|
S/O
| |
Concentrés érythrocytaires transfusés pendant le traitement (médiane)
|
10
|
0
|
< 0,001
|
0
|
< 0,001
| |
Absence de recours à la transfusion pendant le traitement (%)
|
0
|
51
|
< 0,001
|
51
|
< 0,001
| |
Taux de LDH à la fin de l’étude (médiane, U/l)
|
2167
|
239
|
< 0,001
|
269
|
< 0,001
| |
Aire sous la courbe du taux de LDH à la fin de l’étude (médiane, U/l x jour)
|
411822
|
58587
|
< 0,001
|
-632264
|
< 0,001
| |
Hémoglobine libre à la fin de l’étude (médiane, mg/dl)
|
62
|
5
|
< 0,001
|
5
|
< 0,001
| |
FACIT-Fatigue (taille de l’effet)
|
|
1,12
|
< 0,001
|
1,14
|
< 0,001
|
* Les résultats de l’étude C04-002 font référence à des comparaisons avant et après traitement.
195 patients traités par Soliris provenant des études C04-001, C04-002 et d’autres études initiales ont été enrôlés dans une étude d’extension à long terme (E05-001). Tous les patients ont conservé une baisse de l’hémolyse intravasculaire pendant toute la durée de l’exposition à Soliris comprise entre 10 et 54 mois. Le traitement par Soliris a entraîné une réduction du taux d’événements thromboemboliques par rapport à la même période de temps précédant le traitement. Ce résultat est toutefois à interpréter dans le cadre d’une étude clinique non contrôlée.
Les données du registre HPN (M07-001) ont été utilisées afin d’évaluer l’efficacité de Soliris chez les patients atteints d’HPN sans antécédents transfusionnels de culots globulaires. Ces patients manifestaient une forte activité de la maladie définie par une hémolyse élevée (LDH ≥1,5x la limite supérieure de la normale) et la présence d’un ou de plusieurs des symptôme(s) clinique(s) associé(s), à savoir : fatigue, hémoglobinurie, douleurs abdominales, essoufflement (dyspnée), anémie (hémoglobine <100 g/l), événement vasculaire majeur (incluant les thromboses), dysphagie ou dysfonction érectile.
Dans le registre HPN, une réduction de l’hémolyse et des symptômes associés a été observée chez les patients traités par Soliris. A 6 mois, les patients sans antécédent transfusionnel de culots globulaires et traités avec Soliris avaient des taux de LDH significativement (p<0,001) réduits par rapport à l’inclusion (taux de LDH médian de 305 UI/l; tableau 4). De plus, 74 % des patients sans antécédents de transfusion traités par Soliris ont présenté des améliorations cliniques significatives du score FACIT-Fatigue (soit une augmentation de 4 points ou plus) et 84 % ont présenté des améliorations cliniques significatives du score de fatigue EORTC (soit une diminution de 10 points ou plus).
Tableau 4 : Résultats d’efficacité (taux de LDH et score FACIT-Fatigue) chez les patients atteints d’HPN sans antécédents transfusionnels dans l’étude M07-001
|
|
M07-001
| |
Paramètre
|
Soliris
Aucune transfusion
| |
Taux de LDH à l’inclusion
(médiane, UI/l)
|
N=43
1447
| |
Taux de LDH à 6 mois
(médiane, UI/l)
|
N=36
305
| |
Score de FACIT-Fatigue à l’inclusion
(médiane)
|
N=25
32
| |
Score de FACIT-Fatigue à la dernière évaluation disponible (médiane)
|
N=31
44
|
Le score FACIT-Fatigue a été mesuré sur une échelle de 0 à 52, les valeurs supérieures indiquant moins de fatigue.
Syndrome hémolytique et urémique atypique
L’efficacité de Soliris dans le traitement du SHU atypique a été évaluée au cours de quatre études prospectives contrôlées portant sur 100 patients – trois études chez les patients adultes et adolescents (C08-002A/B, C08-003A/B, C10-004), une étude chez les patients pédiatriques (C10-003) –, et une étude rétrospective (C09-001r) portant sur 30 patients.
L’étude C08-002A/B, prospective contrôlée, en ouvert, a inclus des patients à un stade précoce du SHU atypique présentant des manifestations de microangiopathie thrombotique avec une numération plaquettaire ≤150 x 109/l malgré une plasmaphérèse / un échange plasmatique ou une transfusion de plasma frais congelé, et une augmentation des LDH et de la créatininémie au-dessus des limites supérieures de la normale. L’étude C08-003A/B, prospective, contrôlée, en ouvert a inclus des patients présentant un SHU atypique évoluant depuis plusieurs années sans manifestation clinique apparente de microangiopathie thrombotique et recevant de façon chronique une plasmaphérèse (PP) / un échange plasmatique (EP) ou une transfusion de plasma frais congelé (PFC) (≥1 fois toutes les 2 semaines et sans dépasser 3 fois par semaine, pendant au moins 8 semaines avant l’administration de la première dose). La durée de traitement par Soliris dans les 2 études prospectives était de 26 semaines; la majorité de ces patients a été incluse dans l’étude d’extension à long terme, en ouvert. Tous les patients inclus dans les deux études prospectives avaient un taux d’ADAMTS-13 supérieur à 5 %.
Les patients avaient été vaccinés contre les infections à méningocoque avant le traitement par Soliris ou avaient reçu une antibioprophylaxie appropriée jusqu’à 2 semaines après la vaccination. Dans toutes les études, la dose de Soliris chez l’adulte et l’adolescent atteints de SHU atypique a été de 900 mg tous les 7 jours ± 2 jours pendant 4 semaines, suivie de 1200 mg 7 jours ± ± 2 jours plus tard, puis 1200 mg tous les 14 jours ± 2 jours pour la durée de l’étude. Soliris a été administré en perfusion intraveineuse pendant 35 minutes. Le schéma posologique chez les patients pédiatriques, enfants et adolescents, de moins de 40 kg a été défini sur la base d’une modélisation pharmacocinétique qui a permis de déterminer les doses recommandées et le rythme d’administration en fonction du poids corporel (voir «Posologie / Mode d’emploi»).
Les critères d’évaluation principaux portaient sur l’évolution du nombre des plaquettes par rapport à l’inclusion dans l’étude C08-002A/B et l’absence de signe de microangiopathie thrombotique (MAT) dans l’étude C08-003A/B. Les critères d’évaluation supplémentaires portaient sur le nombre d’interventions relatives à la MAT, la normalisation hématologique, la réponse complète de la MAT, la diminution des LDH, la fonction rénale et la qualité de vie liée à l’état de santé. L’absence de signe de MAT a été définie par l’absence pendant au moins 12 semaines des critères suivants : diminution > 25 % des plaquettes par rapport à l’inclusion ; plasmaphérèse (PP) / échange plasmatique (EP) outransfusion de plasma frais congelé (PFC) ; nouvelle dialyse. Les interventions relatives à une MAT ont été définies par la nécessité d’une plasmaphérèse / d’un échange plasmatique ou d’une transfusion de plasma frais congelé et d’une nouvelle dialyse. La normalisation hématologique a été définie par la normalisation du nombre des plaquettes et des concentrations de LDH, maintenue sur au moins 2 mesures consécutives et pendant au moins 4 semaines. La réponse complète de la MAT a été définie par la normalisation hématologique et la réduction d’au moins 25 % du taux sérique de créatinine, maintenues sur au moins 2 mesures consécutives et pendant au moins 4 semaines.
Les caractéristiques à l’inclusion dans les deux études sont présentées dans le tableau 5.
Tableau 5 : Données démographiques et caractéristiques des patients dans les études C08-002A/B et C08-003A/B
|
Paramètre
|
C08-002A/B
|
C08-003A/B
| |
Soliris
N = 17
|
Soliris
N = 20
| |
Délai entre le premier diagnostic et l’inclusion, médiane en mois (min, max)
|
10 (0,26/236)
|
48 (0,66/286)
| |
Délai entre la manifestation clinique actuelle de la MAT et l’inclusion, médiane en mois (min, max)
|
< 1 (< 1/4)
|
9 (1/45)
| |
Nombre de PP/EP ou transfusions de PFC pour les manifestations cliniques de la MAT en cours, médiane (min, max)
|
17 (2/37)
|
62 (20/230)
| |
Nombre de PP/EP ou transfusions de PFC dans les 7 jours avant la première dose d’eculizumab, médiane (min, max)
|
6 (0/7)
|
2 (1/3)
| |
Nombre de plaquettes à l’inclusion (x109/l), moyenne (DS)
|
109 (32)
|
228 (78)
| |
Taux de LDH à l’inclusion (UI/l),
moyenne (DS)
|
323 (138)
|
223 (70)
| |
Patient sans mutation identifiée,
n (%)
|
4 (24)
|
6 (30)
|
Les patients atteints de SHU atypique de l’étude C08-002A/B ont reçu Soliris pendant au moins 26 semaines. À l’issue de la période initiale de traitement de 26 semaines, la majorité des patients a continué à recevoir Soliris dans l’étude d’extension. La durée médiane de traitement par Soliris dans l’étude C08-002A/B était d’environ 100 semaines (entre 2 et 145 semaines).
Une réduction de l’activité de la voie terminale du complément et une augmentation du nombre des plaquettes par rapport à l’inclusion ont été observées après la mise sous Soliris. La réduction de l’activité de la voie terminale du complément a été observée chez tous les patients après la mise sous Soliris. Le tableau 6 présente les résultats d’efficacité de Soliris dans l’étude C08-002A/B. Tous les paramètres évaluant l’efficacité se sont améliorés ou maintenus pendant les 2 années de traitement. La réponse complète de la MAT a été maintenue chez tous les répondeurs. Chez les patients ayant poursuivi le traitement pendant plus de 26 semaines, 2 patients supplémentaires ont obtenu et maintenu une réponse complète de la MAT associée à la normalisation des LDH (1 patient) et à une diminution de la créatininémie (2 patients).
La fonction rénale, mesurée par le débit de filtration glomérulaire estimé (DFGe), a été améliorée et maintenue pendant le traitement par Soliris. Pour 4 des 5 patients qui nécessitaient une dialyse à l’entrée dans l’étude, il a été possible d’arrêter la dialyse pendant toute la durée du traitement par Soliris, et 1 patient a nécessité une nouvelle dialyse. Une amélioration de la qualité de vie liée à la santé a été notée chez les patients.
Dans l’étude C08-002A/B sur le SHU atypique, les patients avec ou sans mutation identifiée des gènes codant pour les protéines des facteurs de régulation du complément ont montré une réponse équivalente au traitement par Soliris.
Les patients atteints de SHU atypique de l’étude C08-003A/B ont reçu Soliris pendant au moins 26 semaines. À l’issue de la période initiale de traitement de 26 semaines, la majorité des patients a continué à recevoir Soliris dans l’étude d’extension. La durée médiane de traitement par Soliris dans l’étude C08-003A/B était d’environ 114 semaines (entre 26 et 129 semaines). Le tableau 6 présente les résultats d’efficacité de Soliris dans l’étude C08-003A/B.
Dans l’étude C08-003A/B sur le SHU atypique, les patients avec ou sans mutation identifiée des gènes codant pour les protéines des facteurs de régulation du complément ont montré une réponse équivalente au traitement par Soliris. Une réduction de l’activité de la voie terminale du complément a été observée chez tous les patients après la mise sous Soliris. Tous les paramètres évaluant l’efficacité se sont améliorés ou maintenus pendant les 2 années de traitement. La réponse complète de la MAT a été maintenue chez tous les répondeurs. Chez les patients ayant poursuivi le traitement pendant plus de 26 semaines, 6 patients supplémentaires ont obtenu et maintenu une réponse complète de la MAT associée à une diminution de la créatinine sérique. Aucun des patients n’a nécessité une nouvelle dialyse pendant le traitement par Soliris. La fonction rénale, mesurée par le DFGe médian, a été améliorée pendant le traitement par Soliris.
Tableau 6 : Résultats d’efficacité des études prospectives dans le SHU atypique C08-002A/B et C08-003A/B
|
|
C08-002A/B
N = 17
|
C08-003A/B
N = 20
| |
|
À 26 semaines
|
Après 2 ans1
|
À 26 semaines
|
Après 2 ans1
| |
Normalisation des plaquettes
Tous les patients, n (%) (IC à 95 %)
Patients avec un taux anormal à l’inclusion, n/n (%)
|
14 (82)
(57-96)
13/15 (87)
|
15 (88)
(64-99)
13/15 (87)
|
18 (90)
(68-99)
3/20 (15)
|
18 (90)
(68-99)
1/3 (33)
| |
Absence de signe de MAT,
n (%) (IC à 95 %)
|
15 (88)
(64-99)
|
15 (88)
(64-99)
|
16 (80)
(56-94)
|
19 (95)
(75-99)
| |
Nombre d’interventions relatives à la MAT, médiane par jour (min, max)
- avant traitement par eculizumab
- sous traitement par eculizumab
Valeur p
|
0,88
(0,04/1,59)
0 (0/0,31)
p<0,0001
|
0,88
(0,04/1,59)
0 (0/0,31)
p<0,0001
|
0,23
(0,05/1,09)
0
p<0,0001
|
0,23
(0,05/1,09)
0
p<0,0001
| |
Amélioration de l’IRC ≥1 stade,
n (%) (IC à 95 %)
|
10 (59)
(33-82)
|
12 (71)
(44-90)
|
7 (35)
(15-59)
|
12 (60)
(36-81)
| |
Modification du DFGe, ml/min/1,73m2 : médiane (limites)
|
20 (-1/-98)
|
28 (3/82)
|
5 (-1/20)
|
11(-42/30)
| |
Amélioration du DFGe ≥15 ml/min/1,73m2, n (%) (IC à 95 %)
|
8 (47)
(23-72)
|
10 (59)
(33-82)
|
1 (5)
(0-25)
|
8 (40)
(19-64)
| |
Modification de l’hémoglobine >20 g/l,
n (%) (IC à 95 %)
|
11 (65)
(38-86)2
|
13 (76)
(50-93)
|
9 (45)
(23-68)3
|
13 (65)
(41-85)
| |
Normalisation hématologique, n (%) (IC à 95 %)
|
13 (76)
(50-93)
|
15 (88)
(64-99)
|
18 (90)
(68-99)
|
18 (90)
(68-99)
| |
Réponse complète de la MAT,
n (%) (IC à 95 %)
|
11 (65)
(38-86)
|
13 (76)
(50-93)
|
5 (25)
(9-49)
|
11 (55)
(32-77)
|
1 À la date de point (20 avril 2012)
2 Étude C08-002 : 3 patients ont reçu des agents stimulants de l’érythropoïèse qui ont été arrêtés après l’initiation d’eculizumab.
3 Étude C08-003 : 8 patients ont reçu des agents stimulants de l’érythropoïèse qui ont été arrêtés chez 3 d’entre eux pendant le traitement par eculizumab.
L’étude C10-004 a inclus 41 patients qui présentaient des signes de microangiopathie thrombotique (MAT). Pour être inclus, les patients devaient avoir : un nombre de plaquettes au-dessous de la limite inférieure de la normale (LIN), des signes d’hémolyse comme une élévation du taux de LDH sérique, et une créatininémie au-dessus de la limite supérieure de la normale sans avoir recours à la dialyse chronique. L’âge médian des patients était de 35 ans (entre 18 et 80 ans). Tous les patients inclus dans l’étude C10-004 avaient un taux d’ADAMTS-13 au-dessus de 5 %. 51 % des patients avaient une mutation identifiée d’un facteur de régulation du complément ou des auto-anticorps. Au total, 35 patients ont reçu une plasmaphérèse / un échange plasmatique, une transfusion de plasma frais congelé, ou une administration de Soliris avant l’initiation de l’eculizumab. Le tableau 7 résume les caractéristiques cliniques et les caractéristiques liées à la maladie des patients à l’inclusion dans l’étude C10-004.
Tableau 7 : Caractéristiques des patients à l’inclusion dans l’étude clinique sur le SHU atypique C10-004
|
Paramètre
|
Étude SHU atypique C10-004
n = 41
| |
Délai entre le premier diagnostic de SHU atypique et la première dose administrée dans l’étude (mois), médiane (min, max)
|
0,79 (0,03 – 311)
| |
Délai entre la manifestation clinique actuelle de la MAT et la première dose administrée dans l’étude (mois), médiane (min, max)
|
0,52 (0,03 – 19)
| |
Nombre de plaquettes à l’inclusion (× 109/l), médiane (min, max)
|
125 (16 – 332)
| |
Taux de LDH à l’inclusion (U/l), médiane (min, max)
|
375 (131 – 3318)
| |
DFGe à l’inclusion (ml/min/1,73m2)
médiane (min, max)
|
10 (6; 53)
|
Les patients de l’étude C10-004 ont reçu Soliris pendant au minimum 26 semaines. Après la fin de la période initiale de traitement de 26 semaines, la plupart des patients ont choisi de poursuivre le traitement de façon chronique. À la date de point, la durée médiane de traitement par Soliris était approximativement de 50 semaines (entre 13 et 86 semaines).
Une réduction de l’activité de la voie terminale du complément et une augmentation du nombre de plaquettes par rapport à l’inclusion ont été observées après la mise sous Soliris. Soliris a réduit les signes de MAT médiée par le complément comme le montre l’augmentation du nombre moyen de plaquettes entre l’inclusion et la 26e semaine. Dans l’étude, le nombre moyen de plaquettes a augmenté de 119±66 x109/l à l’inclusion à 200±84 x109/l à 1 semaine; cet effet a été maintenu sur 26 semaines (nombre moyen de plaquettes à la semaine 26 : 252±70 x109/l). La fonction rénale, évaluée par le DFGe médian, a été améliorée lors du traitement par Soliris. 20 des 24 patients qui avaient besoin d’un traitement par dialyse à l’entrée dans l’étude ont pu arrêter la dialyse pendant la durée du traitement par Soliris. Le tableau 8 résume les résultats d’efficacité de l’étude C10-004.
Tableau 8 : Résultats d’efficacité de l’étude prospective C10-004 dans le SHU atypique
|
Paramètre
|
Étude SHU atypique C10-004
(n = 41)
A 26 semaines
| |
Variation du nombre de plaquettes entre l’inclusion et la 26e semaine (109/l)
|
111 (-122; 362)
| |
Normalisation hématologique, n (%)
Durée de la normalisation hématologique, médiane en semaines (min, max)
|
36 (88)
46 (10; 74)
| |
Réponse complète de la MAT, n (%)
Durée de la réponse complète de la MAT, médiane en semaines (min, max)
|
23 (56)
42 (6; 74)
| |
Absence de signe de MAT, n (%)
IC à 95 %
|
37 (90)
77; 97
| |
Nombre d’interventions relatives à la MAT, médiane par jour (min, max) :
-Avant le traitement par eculizumab
-Pendant le traitement par eculizumab
|
0,63 (0; 1,38)
0 (0; 0,58)
|
1 À la date de point (4 septembre 2012), avec une période médiane de traitement par Soliris de 50 semaines (intervalle : de 13 à 86 semaines)
Un traitement à plus long terme avec Soliris (médiane de 52 semaines, intervalle de 15 à 126 semaines) a été associé à un taux plus important d’améliorations cliniques significatives chez les patients adultes atteints de SHU atypique. Lors de la poursuite du traitement par Soliris au-delà de 26 semaines, 3 patients supplémentaires (63 % des patients au total) ont obtenu une réponse complète de la MAT et 4 patients supplémentaires (98 % des patients au total) ont obtenu une normalisation hématologique. Lors de la dernière évaluation, 25 des 41 patients (61 %) avaient obtenu une amélioration du DFGe ≥15 ml/min/1,73 m2 par rapport à l’inclusion.
Myasthénie acquise généralisée réfractaire
L’efficacité de Soliris dans le traitement des patients présentant une MAg réfractaire a été évaluée à partir des données de 139 patients inclus dans deux études prospectives contrôlées (études C08-001 et ECU-MG-301) et dans une étude d’extension en ouvert (étude ECU-MG-302).
L’étude ECU-MG-301 (REGAIN) était une étude de phase III multicentrique randomisée en double aveugle, contrôlée contre placebo d’une durée de 26 semaines de Soliris chez des patients en échec des traitements antérieurs et qui restaient symptomatiques. Cent dix-huit (118) des 125 patients (94 %) ont terminé la période de traitement de 26 semaines et 117 patients (94 %) ont été inclus ensuite dans l’étude ECU-MG-302, une étude d’extension de l’efficacité et de la sécurité à long terme multicentrique en ouvert, au cours de laquelle tous les patients ont reçu le traitement par Soliris.
Dans l’étude ECU-MG-301, des patients atteints de MAg ayant une sérologie positive pour les anticorps anti-Rach, un grade II à IV de la classification clinique de la MGFA (Myasthenia Gravis Foundation of America) et un score MG-ADL total ≥ 6 ont été randomisés pour recevoir Soliris (n = 62) ou le placebo (n = 63). Tous les patients inclus dans l’étude présentaient une MAg réfractaire et répondaient aux critères prédéfinis suivants :
1) Patients en échec d’un traitement par 2 agents immunosuppresseurs ou plus (en association ou en monothérapie) pendant au moins un an, c’est-à-dire patients continuant à présenter une altération de la capacité à effectuer les activités quotidiennes malgré les traitements immunosuppresseurs.
OU
2) Patients en échec d’au moins un traitement immunosuppresseur et nécessitant des échanges plasmatiques ou des perfusions d’immunoglobulines intraveineuses (IgIV) réguliers pour contrôler les symptômes, c’est-à-dire patients nécessitant des EP ou des transfusions d’IgIV à intervalles réguliers pour la prise en charge de la faiblesse musculaire, au moins tous les 3 mois au cours des 12 mois précédents.
Les patients avaient reçu un vaccin anti-méningococcique avant le début du traitement par Soliris ou ont reçu une antibioprophylaxie appropriée pendant une durée allant jusqu’à 2 semaines après la vaccination. Dans les études ECU-MG-301 et ECU-MG-302, la dose de Soliris chez les patients adultes atteints de MAg réfractaire était de 900 mg tous les 7 ± 2 jours pendant 4 semaines, suivie de 1200 mg à la semaine 5 ± 2 jours, puis de 1200 mg tous les 14 ± 2 jours pendant la durée de l’étude. Soliris était administré en perfusion intraveineuse de 35 minutes. Le tableau 9 présente les paramètres initiaux des patients atteints de MAg réfractaire inclus dans l’étude ECU-MG-301.
Tableau 9 : Données démographiques et paramètres des patients inclus dans l’étude ECU-MG-301
|
|
Soliris (n=62)
|
Placebo (n=63)
| |
Âge lors du diagnostic de MA (ans),
moyenne (min, max)
|
38,0 (5,9; 70,8)
|
38,1 (7,7; 78,0)
| |
Sexe féminin, n (%)
|
41 (66,1)
|
41 (65,1)
| |
Ancienneté de la MA (années),
moyenne (min, max)
|
9,9 (1,3; 29,7)
|
9,2 (1,0; 33,8)
| |
Score MG-ADL à l’inclusion
|
|
| |
Moyenne (ET)
|
10,5 (3,06)
|
9,9 (2,58)
| |
Médiane
|
10,0
|
9,0
| |
Score QMG à l’inclusion
|
|
| |
Moyenne (ET)
|
17,3 (5,10)
|
16,9 (5,56)
| |
Médiane
|
17,0
|
16,0
| |
≥3 traitements immunosuppresseurs* antérieurs depuis le diagnostic, n (%)
|
31 (50,0)
|
34 (54,0)
| |
Nombre de patients ayant présenté des exacerbations depuis le diagnostic, n (%)
|
46 (74,2)
|
52 (82,5)
| |
Nombre de patients ayant présenté des poussées de la MA depuis le diagnostic, n (%)
|
13 (21,0)
|
10 (15,9)
| |
Antécédents d’assistance respiratoire de tout type depuis le diagnostic, n (%)
|
15 (24,2)
|
14 (22,2)
| |
Antécédents d’intubation depuis le diagnostic (classe V de la MGFA), n (%)
|
11 (17,7)
|
9 (14,3)
|
* Les traitements immunosuppresseurs incluaient, mais sans s’y limiter: corticoïdes, azathioprine, mycophénolate, méthotrexate, ciclosporine, tacrolimus ou cyclophosphamide.
Le critère d’évaluation principal de l’étude ECU-MG-301 était la variation du score total de l’échelle d’évaluation du retentissement des symptômes de la MA sur les activités quotidiennes (MG Activities of Daily Living Profile, MG-ADL – un instrument d’évaluation par le patient validé dans la MAg) à la semaine 26 par rapport au score initial. L’analyse principale du score MG-ADL était une analyse de covariance du rang le plus défavorable (Worst-Rank ANCOVA) avec un rang moyen de 56,6 pour Soliris et de 68,3 pour le placebo, sur la base des 125 patients de l’étude (p = 0,0698).
Le principal critère d’évaluation secondaire était la variation du score QMG (Quantitative MG Scoring System, un instrument d’évaluation par le médecin validé dans la MAg) total à la semaine 26 par rapport au score initial. L’analyse principale du score QMG était une analyse Worst-Rank ANCOVA avec un rang moyen de 54,7 pour Soliris et de 70,7 pour le placebo, sur la base des 125 patients de l’étude (p = 0,0129).
Le tableau 10 présente les résultats d’efficacité dans les analyses selon un modèle pour mesures répétées prédéfinies des critères d’évaluation principal et secondaires.
Tableau 10 : Étude ECU-MG-301 – Variation des paramètres d’efficacité entre l’inclusion et la semaine 26
|
Critères d’efficacité : variation du score total à la semaine 26 par rapport au score initial
|
Soliris
(n=62)
(ETM)
|
Placebo
(n=63)
(ETM)
|
Variation dans le groupe Soliris par rapport au groupe placebo – différence de la moyenne des MC (IC à 95 %)
|
Valeur p (selon des analyses sur mesures répétées)
| |
MG-ADL
|
-4,2 (0,49)
|
-2,3 (0,48)
|
-1,9
(-3,3; -0,6)
|
0,0058
| |
QMG
|
-4,6 (0,60)
|
-1,6 (0,59)
|
-3,0
(-4,6; -1,3)
|
0,0006
| |
MGC
|
-8,1 (0,96)
|
-4,8 (0,94)
|
-3,4
(-6,0; -0,7)
|
0,0134
| |
MG-QoL-15
|
-12,6 (1,52)
|
-5,4 (1,49)
|
-7,2
(-11,5; -3,0)
|
0,0010
|
ETM = Erreur type de la moyenne; IC = intervalle de confiance; MGC = Myasthenia Gravis Composite Score; MG-QoL15 = MG Quality of Life 15
Dans l’étude ECU-MG-301, un répondeur clinique pour le score MG-ADL total était défini comme un patient présentant une amélioration d’au moins 3 points du score. À la semaine 26, le pourcentage de répondeurs cliniques ne recevant pas de traitement de secours était de 59,7 % dans le groupe Soliris et de 39,7 % dans le groupe placebo (p = 0,0229). Dans l’étude ECU-MG-301, un répondeur clinique pour le score QMG total était défini comme un patient présentant une amélioration d’au moins 5 points du score. À la semaine 26, le pourcentage de répondeurs cliniques ne recevant pas de traitement de secours était de 45,2 % dans le groupe Soliris et de 19 % dans le groupe placebo (p = 0,0018).
Le tableau 11 présente une vue d’ensemble des patients mentionnant une détérioration clinique et des patients ayant besoin d’un traitement de secours pendant les 26 semaines.
Tableau 11 : Détérioration clinique et traitement de secours dans l’étude ECU-MG-301
|
Paramètre
|
Statistique
|
Placebo
(N=63)
|
Soliris
(N=62)
| |
Nombre total de patients mentionnant une détérioration clinique
|
n (%)
|
15 (23,8)
|
6 (9,7)
| |
Nombre total de patients ayant besoin d’un traitement de secours
|
n (%)
|
12 (19,0)
|
6 (9,7)
|
Sur les 125 patients inclus dans l’étude ECU-MG-301, 117 patients sont entrés ensuite dans une étude d’extension à long terme (étude ECU-MG-302), au cours de laquelle ils ont tous reçu Soliris. Les patients traités précédemment par Soliris dans l’étude ECU-MG-301 ont continué à présenter un maintien de l’effet de Soliris sur tous les paramètres (scores MG-ADL, QMG, MGC et MG-QoL15) pendant 130 semaines supplémentaires de traitement par l’eculizumab. Chez les patients qui avaient reçu le placebo dans l’étude ECU-MG-301 (bras placebo/eculizumab de l’étude ECU-MG-302), il a été observé une amélioration après l’instauration du traitement par l’eculizumab, qui s’est maintenue pendant plus de 130 semaines dans l’étude ECU-MG-302. La figure 1 présente la variation des scores MG-ADL (A) et QMG (B) par rapport aux scores initiaux après 26 semaines de traitement dans l’étude ECU-MG-301 et après 130 semaines de traitement (n = 80 patients) dans l’étude ECU-MG-302.
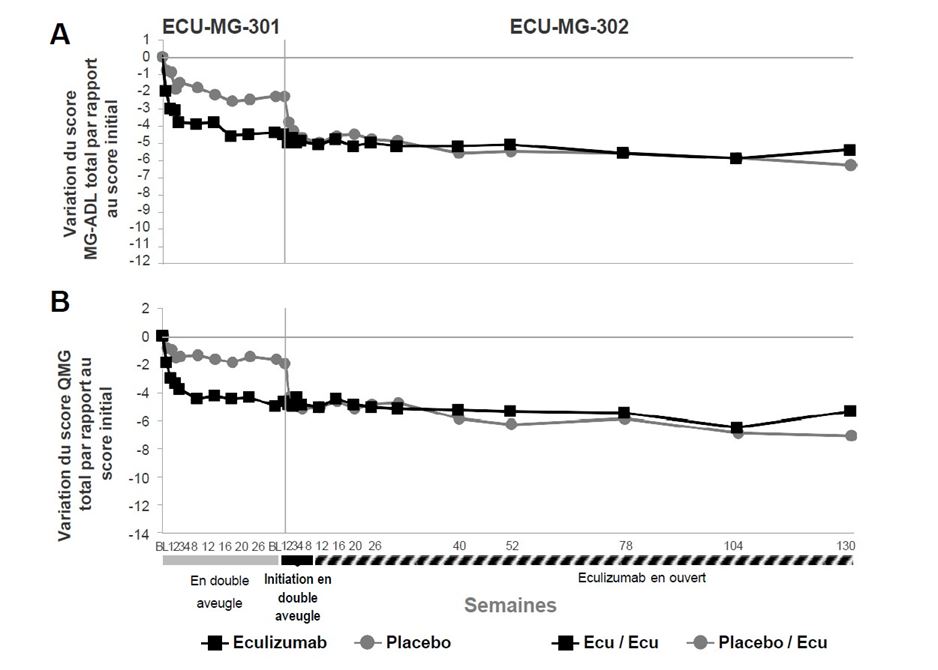
Figure 1 : Variations moyennes des scores MG-ADL (1A) et QMG (1B) par rapport aux scores initiaux dans les études ECU-MG-301 et ECU-MG-302
Dans l’étude ECU-MG-302, les médecins avaient la possibilité d’ajuster les traitements immunosuppresseurs de fond. Dans ce contexte, il a été observé une diminution de la dose quotidienne d’au moins un agent immunosuppresseur chez 65,0 % des patients ; le traitement immunosuppresseur en cours a été arrêté chez 43,6 % des patients. La raison la plus fréquente de modification du traitement immunosuppresseur était l’amélioration des symptômes de la myasthénie acquise généralisée.
Maladie du spectre de la neuromyélite optique
Les données de 143 patients dans une étude contrôlée (ECU-NMO-301) et de 119 patients qui ont poursuivi le traitement dans une étude d’extension en ouvert (ECU-NMO-302) ont été utilisées pour évaluer l’efficacité et la sécurité de Soliris dans le traitement des patients atteints de NMOSD.
L’étude ECU-NMO-301 était une étude de phase III multicentrique, en double aveugle, randomisée et contrôlée par placebo, portant sur des patients atteints de NMOSD traités par Soliris.
Dans l’étude ECU-NMO-301, les patients atteints de NMOSD ayant un test sérique positif pour les anticorps anti-AQP4, un historique d’au moins 2 poussées dans les 12 derniers mois ou 3 poussées dans les 24 derniers mois et au moins 1 poussée dans les 12 mois précédant la sélection, et un score «Expanded Disability Status Scale» (EDSS) ≤ 7 ont été randomisés pour recevoir Soliris (n = 96) ou le placebo (n = 47) selon un rapport 2:1.
Les patients ont été autorisés à recevoir un traitement de fond avec des médicaments immunosuppresseurs à dose stable pendant l’étude, à l’exception du rituximab et de la mitoxantrone.
Les patients ont reçu soit une vaccination contre le méningocoque au moins 2 semaines avant le début du traitement par Soliris, soit un traitement prophylactique avec des antibiotiques appropriés jusqu’à 2 semaines après la vaccination. Dans le programme de développement clinique de l’eculizumab dans la NMOSD, la dose de Soliris chez les patients adultes atteints de NMOSD était de 900 mg tous les 7 ± 2 jours pendant 4 semaines, puis de 1200 mg pendant la semaine 5 ± 2 jours et enfin de 1200 mg tous les 14 ± 2 jours pendant toute la durée de l’étude. Soliris a été administré en perfusion intraveineuse de 35 minutes.
La majorité (90,9 %) des patients étaient des femmes. Environ la moitié des patients (49,0 %) étaient caucasiens. L’âge moyen lors de la première administration du médicament à l’étude était de 45 ans.
Tableau 12 : Histoire de la maladie et paramètres initiaux des patients dans l’étude ECU-NMO-301
|
Variable
|
Statistiques
|
Placebo
(N = 47)
|
Eculizumab
(N = 96)
|
Total
(N = 143)
| |
Histoire de la NMOSD
| |
Âge à la première manifestation clinique de la MNSOD (années)
|
Moyenne (EC)
|
38,5 (14,98)
|
35,8 (14,03)
|
36,6 (14,35)
| |
Médiane
|
38,0
|
35,5
|
36,0
| |
Min, max
|
12 ; 73
|
5 ; 66
|
5 ; 73
| |
Délai entre la première manifestation clinique de la NMOSD et l’administration de la première dose du médicament étudié (années)
|
Moyenne (EC)
|
6,601 (6,5863)
|
8,156 (8,5792)
|
7,645 (7,9894)
| |
Médiane
|
3,760
|
5,030
|
4,800
| |
Min, max
|
0,51 ; 29,10
|
0,41; 44,85
|
0,41 ; 44,85
| |
Taux annuel de poussée «historique» dans les 24 mois précédant le dépistage
|
Moyenne (EC)
|
2,07 (1,037)
|
1,94 (0,896)
|
1,99 (0,943)
| |
Médiane
|
1,92
|
1,85
|
1,92
| |
Min, max
|
1,0 ; 6,4
|
1,0; 5,7
|
1,0 ; 6,4
| |
Paramètres initiaux
| |
Score EDSS initial
|
Moyenne (EC)
|
4,26 (1,510)
|
4,15 (1,646)
|
4,18 (1,598)
| |
Médiane
|
4,00
|
4,00
|
4,00
| |
Min, max
|
1,0 ; 6,5
|
1,0 ; 7,0
|
1,0 ; 7,0
| |
Pas d’utilisation de TSI au début de l’étude
|
n (%)
|
13 (27,7)
|
21 (21,9)
|
34 (23,8)
|
Abréviations : EDSS = Expanded Disability Status Scale ; TIS = traitement immunosuppresseur ; max = maximum ; min = minimum ; MNSOD = maladie du spectre de la neuromyélite optique ; ET = écart type
Le critère d’évaluation principal de l’étude ECU-NMO-301 était le délai d’apparition de la première poussée au cours de l’étude, confirmée par un comité indépendant en aveugle par rapport au traitement. Au cours de l’étude, un effet significatif sur le délai de première poussée confirmée a été observé avec l’eculizumab par rapport au placebo (réduction du risque relatif de 94 % ; rapport de risque de 0,058 ; p<0,0001) (figure 2). Les patients traités par Soliris ont montré une amélioration similaire du délai d’apparition de la première poussée confirmée au cours de l’étude avec ou sans TIS concomitant.
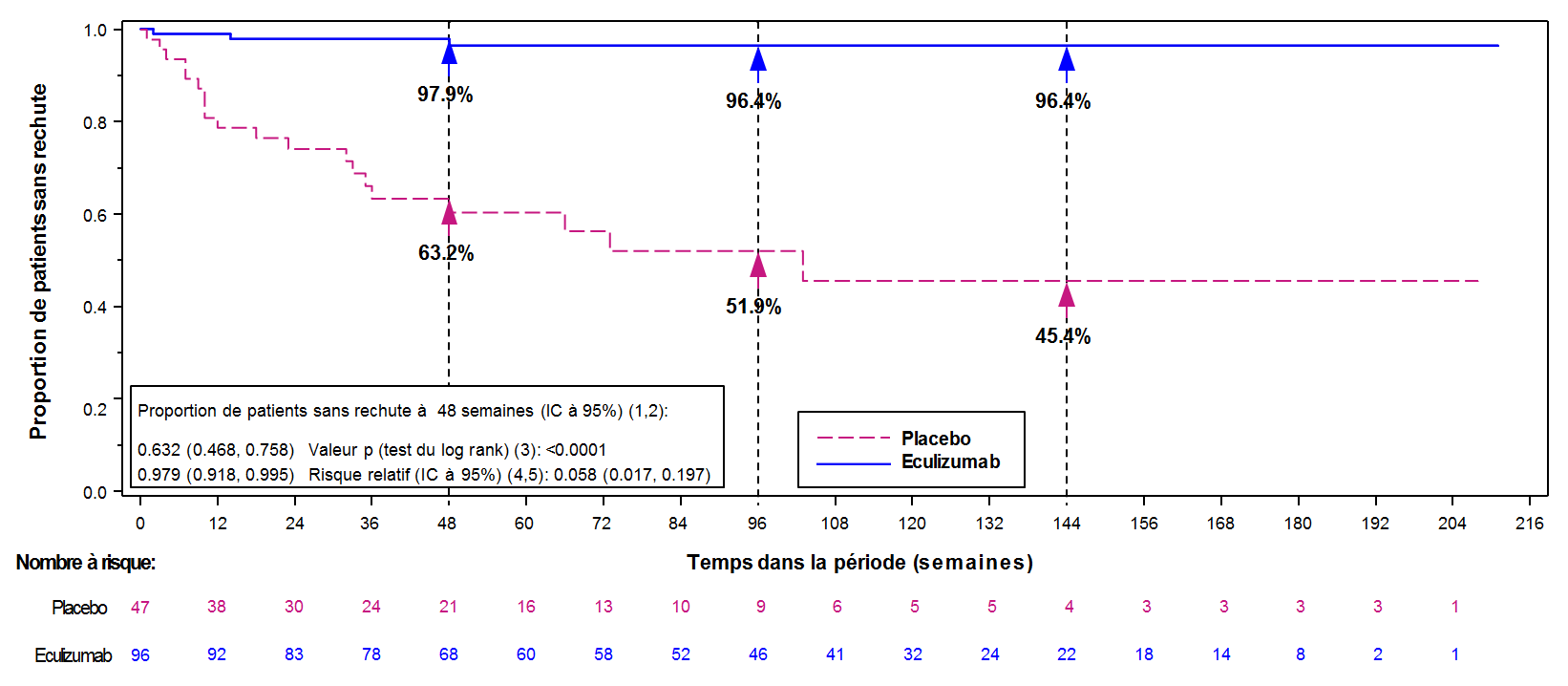
Figure 2 : Courbe de Kaplan-Meier du délai jusqu’à la première poussée confirmée au cours de l’étude ECU-NMO-301 – Population globale d’analyse des données
Remarque : les patients qui n’avaient pas présenté de poussée confirmée pendant l’étude ont été censurés à la fin de la période d’étude.
Les analyses stratifiées sont basées sur quatre strates de randomisation :
(i) faible score EDSS lors de la randomisation (<= 2,0), (ii) score EDSS élevé (> = 2,5 à <= 7) et pas de prétraitement lors de la randomisation, (iii) score EDSS élevé (> = 2,5 à <= 7) et maintien du ou des mêmes TIS depuis la dernière poussée lors de la randomisation, (iv) score EDSS élevé (> = 2,5 à <= 7) et changements du ou des TIS depuis la dernière poussée lors de la randomisation.
1 Basé sur la méthode de Kaplan-Meier.
2 Basé sur la transformation log-log complémentaire.
3 Basé sur un test du log-rank stratifié.
4 Basé sur un modèle modèle à risques proportionnels de Cox stratifié.
5 Basé sur un intervalle de confiance de Wald.
Abréviations : IC = Intervalle de confiance ; EDSS = Expanded Disability Status Scale ; TIS = traitement immunosuppresseur
Le rapport des taux annualisés de poussées (TAP) confirmé au cours de l’étude (IC à 95 %) entre le groupe traité par l’eculizumab et le groupe traité par placebo était de 0,045 (0,013 ; 0,151). Cela représente une réduction relative confirmée du TAP de 95,5 % au cours de l’étude chez les patients traités par l’eculizumab par rapport au groupe traité par placebo (p < 0,0001) (Tableau 13).
Tableau 13 : Taux annualisé de poussées confirmé au cours de l’étude – Population globale d’analyse des données
|
Variable
|
Statistique
|
Placebo
(n = 47)
|
Eculizumab
(n = 96)
| |
Nombre total de poussées
|
Somme
|
21
|
3
| |
Nombre total de patients-années pendant la période d’étude
|
n
|
52,41
|
171,32
| |
TAR ajusté confirméa
|
Taux
|
0,350
|
0,016
| |
IC à 95 %
|
0,199; 0,616
|
0,005; 0,050
| |
Effet du traitementa
|
Rapport des taux (eculizumab/placebo)
|
…
|
0,045
| |
IC à 95 %
|
…
|
0,013; 0,151
| |
Valeur p
|
…
|
<0,0001
| |
a
Basé sur une régression de Poisson ajustée pour les strates de randomisation et le TAP historique dans les 24 mois précédant la sélection.
Abréviations: TAP = taux annualisé de poussées; IC = intervalle de confiance.
|
Par rapport aux patients recevant un placebo, les patients traités par Soliris présentaient des taux annualisés d’hospitalisation inférieurs (0,04 pour Soliris contre 0,31 pour le placebo), une utilisation moindre de corticostéroïdes intraveineux pour les poussées aiguës (0,07 pour Soliris contre 0,42 pour le placebo) et moins de traitements par échange plasmatique (0,02 pour Soliris contre 0,19 pour le placebo).
L’évaluation des modifications, du début jusqu’à la fin de l’étude, concernant les critères d’évaluation secondaires que sont le handicap neurologique (score EDSS [valeur p = 0,0597] et mRS [valeur p nominale = 0,0154]), la déficience fonctionnelle (score HAI [valeur p nominale = 0,0002]), la qualité de vie (EVA EQ-5D [valeur p nominale = 0,0309] et l’index EQ-5D [valeur p nominale = 0,0077]) est favorable à l’eculizumab par rapport au placebo.
L’analyse finale de l’étude ECU-NMO-302 (durée médiane du traitement 20,00 semaines [0,1 ; 198,4 semaines]) a montré une diminution statistiquement et cliniquement significative du TAP au cours de l’étude (déterminée par le médecin traitant) sous traitement par eculizumab, sur la base de la modification médiane (min, max) (-1,825 [-6,38 ; 1,02], p <0,0001) par rapport au TAP historique (24 mois avant la sélection pour l’étude ECU-NMO-301).
Globalement, seules des données limitées d’efficacité et de sécurité sont disponibles pour le traitement par Soliris sur 20 semaines.
Dans l’étude ECU-NMO-302, les médecins ont eu la possibilité d’adapter les traitements immunosuppresseurs de fond. Dans ce cadre, le changement le plus fréquent du traitement immunosuppresseur a été une réduction de la dose, qui a eu lieu chez 21,0 % des patients. En outre, 15,1 % des patients ont interrompu le TIS en cours.
Soliris (eculizumab) n’a pas été étudié pour le traitement des poussées aiguës chez les patients atteints de NMOSD.
Sécurité et efficacité chez les patients âgés.
MAg réfractaire
Vingt-deux (22) (17,6 %) patients âgés (>65 ans) présentant une MAg réfractaire ont été traités par Soliris dans les études cliniques. Il n’a pas été observé de différences importantes liées à l’âge en termes de sécurité et d’efficacité.
Maladie du spectre de la neuromyélite optique
Soliris n’a été étudié que sur un total de 9 patients âgés atteints de NMOSD (3 sous placebo, 6 sous eculizumab), de sorte qu’aucune déclaration fiable ne peut être faite sur la sécurité et l’efficacité au sein de cette population.
Sécurité et efficacité en pédiatrie
Hémoglobinurie paroxystique nocturne
Dans l’étude M07-005, un total de 7 patients pédiatriques atteints d’HPN, avec un poids médian de 57,2 kg (entre 48,6 et 69,8 kg) et âgés de 11 à 17 ans (âge médian : 15,6 ans), ont été traités par Soliris.
Le traitement par eculizumab à la posologie recommandée pour les enfants et adolescents a été associé à une réduction de l’hémolyse intravasculaire mesurée par les taux sériques de LDH. Une diminution significative ou une élimination du besoin en transfusion sanguine ont aussi été constatées, ainsi qu’une tendance à une amélioration globale de l’état général. L’efficacité du traitement par eculizumab chez les patients pédiatriques atteints d’HPN apparaît cohérente avec celle observée chez les patients adultes atteints d’HPN inclus dans les études pivots (C04-001 et C04-002) (tableaux 3 et 14).
Tableau 14 : Résultats d’efficacité dans la population pédiatrique HPN de l’étude M07-005
|
|
|
Valeur de p
| |
|
moyenne (DS)
|
Test des rangs signés de Wilcoxon
| |
Variation entre la valeur de base et la valeur à 12 semaines des LDH (UI/l)
|
-771 (914)
|
0,0156
| |
ASC LDH
(U/l x jour)
|
-60 634 (72 916)
|
0,0156
|
Syndrome hémolytique et urémique atypique
Dans l’étude C09-001r, un total de 15 patients pédiatriques (âgés de 2 mois à moins de 12 ans) ont été traités par Soliris. 50 % des patients avaient une mutation identifiée d’un facteur de régulation du complément ou des auto-anticorps. La durée médiane entre le diagnostic de SHU atypique et la première dose de Soliris était de 14 mois (de <1 à 110 mois). La durée médiane entre la manifestation clinique actuelle de la microangiopathie thrombotique (MAT) et la première dose de Soliris était d’un mois (de <1 à 16 mois). La durée médiane de traitement par Soliris était de 16 semaines (de 4 à 70 semaines) pour les enfants de moins de 2 ans (n=5) et de 31 semaines (de 19 à 63 semaines) pour les enfants de 2 ans à moins de 12 ans (n=10).
Dans l’ensemble, les résultats d’efficacité pour ces patients pédiatriques étaient en accord avec ceux observés chez les patients inclus dans les études pivots sur le SHU atypique C08-002 et C08-003 (tableau 6). Aucun des patients pédiatriques n’a nécessité une nouvelle dialyse.
Tableau 15 : Résultats d’efficacité dans la population pédiatrique de l’étude C09-001r
|
Paramètre
|
<2 ans
(n = 5)
|
2 à <12 ans
(n = 10)
|
<12 ans
(n = 15)
| |
Patients ayant une normalisation des plaquettes, n (%)
|
4 (80)
|
10 (100)
|
14 (93)
| |
Réponse complète de la MAT, n (%)
|
2 (40)
|
5 (50)
|
7 (50)
| |
Nombre d’interventions relatives à la MAT, médiane par jour (intervalle)
-Avant le traitement par eculizumab
-Pendant le traitement par eculizumab
|
1 (0/2)
<1 (0/<1)
|
< 1 (0,07/1,46)
0 (0/<1)
|
<1 (0/2)
0 (0/<1)
| |
Patients avec une amélioration du DFGe ≥15 ml/min/1,73 m², n (%)
|
2 (40)
|
6 (60)
|
8 (53)
|
Chez les patients pédiatriques présentant des manifestations récentes et sévères de la microangiopathie thrombotique (MAT) avant mise sous traitement, l’eculizumab a permis le contrôle de la MAT et une amélioration de la fonction rénale (tableau 15).
Chez les patients pédiatriques présentant des manifestations cliniques prolongées et sévères de la microangiopathie thrombotique (MAT) avant mise sous traitement, l’eculizumab a permis le contrôle de la MAT. Toutefois, la fonction rénale est restée inchangée en raison d’atteintes rénales irréversibles préalables (tableau 16).
Tableau 16 : Résultats d’efficacité dans la population pédiatrique de l’étude C09-001r en fonction de la durée des manifestations actuelles sévères de microangiopathie thrombotique (MAT)
|
|
Durée des manifestations actuelles sévères de la MAT
| |
< 2 mois
(N = 10) %
|
> 2 mois
(N = 5) %
| |
Patients ayant une normalisation des plaquettes, n (%)
|
9 (90)
|
5 (100)
| |
Absence de signe de MAT, n (%)
|
8 (80)
|
3 (60)
| |
Réponse complète de la MAT, n (%)
|
7 (70)
|
0
| |
Amélioration du DFGe
≥15 ml/min/1,73 m2, n (%)
|
7 (70)
|
0*
|
*Un patient a montré une amélioration du DFGe après transplantation rénale.
Au total, 22 enfants et adolescents (âgés de 5 mois à 17 ans) ont reçu un traitement par Soliris dans l’étude C10-003.
Dans l’étude, les patients inclus devaient avoir un nombre de plaquettes au-dessous de la limite inférieure de la normale (LIN), des signes d’hémolyse comme une élévation du taux de LDH sérique au-dessus de la limite supérieure de la normale (LSN) et un niveau de créatininémie ≥97e percentile par rapport à l’âge sans avoir recours à la dialyse chronique. L’âge médian des patients était de 6,5 ans (entre 5 mois et 17 ans). Les patients inclus dans l’étude SHU atypique C10-003 avaient un taux d’ADAMTS-13 au-dessus de 5 %. Cinquante pour cent des patients avaient une mutation identifiée de facteur de régulation du complément ou des auto-anticorps. Un total de 10 patients a reçu une plasmaphérèse/un échange plasmatique, une transfusion de plasma frais congelé ou une dose de Soliris avant la mise sous Soliris. Le tableau 17 résume les caractéristiques cliniques et les caractéristiques liées à la maladie chez les patients à l’inclusion dans l’étude C10-003.
Tableau 17 : Caractéristiques des patients pédiatriques à l’inclusion dans l’étude SHU atypique C10-003
|
Paramètre
|
1 mois à <12 ans
(n=18)
|
Tous les patients
(n=22)
| |
Délai entre le premier diagnostic de SHU atypique et la première dose administrée dans l’étude (mois),
|
0,51 (0,03 – 58)
|
0,56 (0,03-191)
| |
Délai entre la manifestation clinique actuelle de la MAT et la première dose administrée dans l’étude (mois),
|
0,23 (0,03 – 4)
|
0,2 (0,03-4)
| |
Nombre de plaquettes à l’inclusion (× 109/l), médiane (min, max)
|
110 (19-146)
|
91 (19-146)
| |
Taux de LDH à l’inclusion (U/l), médiane (min, max)
|
1510 (282-7164)
|
1244 (282-7164)
| |
DFGe à l’inclusion (ml/min/1,73 m2)
médiane (min, max)
|
22 (10, 105)
|
22 (10, 105)
|
Les patients de l’étude C10-003 ont reçu Soliris pendant au minimum 26 semaines. Après la fin de la période initiale de traitement de 26 semaines, la plupart des patients ont choisi de poursuivre le traitement de façon chronique.
Une réduction de l’activité de la voie terminale du complément a été observée chez tous les patients après la mise sous Soliris. Soliris a réduit les signes de MAT médiée par le complément comme le montre l’augmentation du nombre moyen de plaquettes entre l’inclusion et la 26e semaine. Dans l’étude, le nombre moyen de plaquettes a augmenté de 88±42 x109/l à l’inclusion à 281±123 x109/l à 1 semaine; cet effet a été maintenu sur les 26 semaines (taux moyen de plaquettes à la semaine 26 : 293±106 x109/l).
La fonction rénale, évaluée par le DFGe, a été améliorée lors du traitement par Soliris. 9 des 11 patients qui avaient recours à la dialyse à l’inclusion n’en avaient plus besoin au 15e jour du traitement par Soliris. Les réponses étaient similaires quel que soit l’âge des patients allant de 5 mois à 17 ans. Dans l’étude C10-003, les réponses au traitement par Soliris étaient similaires chez les patients avec ou sans mutation identifiée des gènes codant pour les protéines régulatrices du complément ou des auto-anticorps dirigés contre le facteur H.
Le tableau 18 résume les résultats d’efficacité de l’étude SHU atypique C10-003.
Tableau 18 : Résultats d’efficacité de l’étude prospective C10-003 dans le SHU atypique
|
Paramètre
|
1 mois à <12 ans
(n = 18)
A 26 semaines
|
Tous les patients
(n = 22)
A 26 semaines
| |
Normalisation hématologique complète, n (%)
Durée de la normalisation hématologique complète, médiane en semaines (min, max)
|
14 (78)
35 (13; 78)
|
18 (82)
35 (13; 78)
| |
Réponse complète de la MAT, n (%)
Durée de la réponse complète de la MAT, médiane en semaines (min, max)1
|
11 (61)
40 (13; 78)
|
14 (64)
37 (13; 78)
| |
Absence de signe de MAT, n (%)
IC à 95 %
|
17 (94)
S/O
|
21 (96)
77 ; 99
| |
Nombre d’interventions relatives à la MAT, médiane par jour (min, max) :
avant traitement par eculizumab,
-Pendant le traitement par eculizumab
|
S/O
S/O
|
0,4 (0; 1,7)
0 (0; 1,01)
| |
Amélioration du DFGe ≥15 ml/min/1,73•m2, n (%)
|
16 (89)
|
19 (86)
| |
Modification du DFGe (≥15 ml/min/1,73 m2) à la semaine 26, médiane (min, max)
|
64 (0; 146)
|
58 (0; 146)
| |
Amélioration de l’IRC ≥1 stade, n (%)
|
14/16 (88)
|
17/20 (85)
| |
Absence de PP/EP ou de prise de Soliris, en (%)
Absence de nouvelle dialyse, n (%)
IC à 95 %
|
16 (89)
18 (100)
S/O
|
20 (91)
22 (100)
85; 100
|
1À la date de point (12 octobre 2012) avec une durée médiane de traitement par Soliris de 44 semaines (intervalle de 1 dose à 88 semaines).
Un traitement à plus long terme avec Soliris (médiane de 55 semaines, intervalle de 1 jour à 107 semaines) a été associé à un taux plus important d’améliorations cliniques significatives chez les patients pédiatriques, enfants et adolescents, atteints de SHU atypique. Lors de la poursuite du traitement par Soliris au-delà de 26 semaines, 1 patient supplémentaire (68 % des patients au total) a obtenu une réponse complète de la MAT et 2 patients supplémentaires (91 % des patients au total) ont obtenu une normalisation hématologique. Lors de la dernière évaluation, 19 des 22 patients (86 %) avaient obtenu une amélioration du DFGe ≥15 ml/min/1,73 m2 par rapport à l’inclusion. Aucun patient n’a eu besoin d’une nouvelle dialyse sous Soliris.
Myasthénie acquise généralisée réfractaire
Au total, 11 patients pédiatriques atteints de MAg réfractaire dans l’étude ECU-MG-303 ont reçu Soliris. Le poids corporel moyen (fourchette) des patients traités au début de l’étude était de 59,7 kg (37,2 à 91,2 kg) et l’âge moyen (fourchette) à l‘inclusion était de 15 ans (12 à 17 ans). Tous les patients inclus dans l’étude étaient des patients présentant une ou plusieurs des caractéristiques suivantes :
1. Traitement sans résultat pendant ≥ 1 an avec au moins 1 traitement immunosuppresseur, défini comme : (i) faiblesse persistante avec limitation des activités de la vie quotidienne ou (ii) aggravation et/ou crise myasthénique pendant le traitement ou (iii) intolérance au TIS due à des effets secondaires ou à des maladies concomitantes.
2. Nécessité d‘un traitement d’entretien avec EP ou IVIg pour contrôler les symptômes (c’est-à-dire les patients ayant régulièrement besoin de PE ou d’IVIg pour traiter la faiblesse musculaire, au moins tous les 3 mois au cours des 12 derniers mois précédant la sélection).
Les paramètres initiaux des patients pédiatriques atteints de MAg réfractaire inclus dans l’étude ECU MG 303 sont présentés dans le tableau 19.
|
Tableau 19 : Données démographiques et paramètres des patients dans l’étude ECU-MG-303
| |
|
Eculizumab (n = 11)
| |
Sexe féminin
|
n (%)
|
9 (81,8 %)
| |
Durée de la MG (délai entre le diagnostic de la MG et le premier médicament à l’étude [années])
|
Moyenne (ET)
Médiane (min, max)
|
3,99 (2,909)
2,90 (0,1; 8,8)
| |
Valeur initiale score total MG
|
Moyenne (ET)
Médiane (min, max)
|
5,0 (5,25)
4,0 (0; 19)
| |
Valeur initiale score total QMG
|
Moyenne (ET)
Médiane (min, max)
|
16,7 (5,64)
15,0 (10; 28)
| |
Classification MGFA à l‘inclusion
IIa
IIb
IIIa
IIIb
IVa
IVb
|
n (%)
|
2 (18,2)
3 (27,3)
3 (27,3)
0
3 (27,3)
0
| |
Nombre de patients ayant eu des exacerbations de la MG, y compris crise de MG, depuis le diagnostic
Non
Oui
Exacerbation
Crise de MG
|
n (%)
|
4 (36,4)
7 (63,6)
6 (54,5)
3 (27,3)
| |
Traitement IVIg au long cours au début de l‘étude
Oui
Non
|
n (%)
|
6 (54,5)
5 (45,5)
| |
Nombre de traitements immunosuppresseurs au début de l’étude
0
1
2
|
n (%)
|
2 (18,2)
4 (36,4)
5 (45,5)
| |
Patients avec traitements immunosuppresseursa au début de l’étude n (%)
Corticoïdes
Azathioprine
Mycophénolate-Mofétil
Tacrolimus
|
n (%)
|
8 (72,7)
1 (9,1)
2 (18,2)
3 (27,3)
|
aLes traitements immunosuppresseurs comprenaient les corticostéroïdes, l’azathioprine, le cyclophosphamide, la ciclosporine, le méthotrexate, le mycophénolatemofétil ou le tacrolimus. Au début de l’étude, aucun des patients n’a reçu de cyclosporine, de cyclophosphamide ou de méthotrexate.
Abréviations : IVIg = immunglobuline intraveineuse ; max = maximum ; MG = myasthénie grave ; MG ADL = Myasthenia Gravis Activities of Daily Living (profil de la myathénie grave pour les activités de la vie quotidienne) ; MGFA = Myasthenia Gravis Foundation of America ; min = minimum ; QMG = Quantitative Myasthenia Gravis score for disease severity (score quantitatif de la myasthénie grave pour la sévérité de la maladie) ; ET = écart-type
Le critère d’évaluation principal de l’étude ECU-MG-303 était le changement du score total QMG par rapport à la valeur initiale au cours du temps, indépendamment du traitement d‘urgence. Les patients pédiatriques traités par Soliris ont présenté une amélioration statistiquement significative du score global QMG sur l’ensemble de la période de traitement primaire de 26 semaines. Les résultats pour les critères principal et secondaires de l’étude ECU-MG-303 sont présentés dans le tableau 20.
L’efficacité du traitement par Soliris chez les patients pédiatriques atteints de MAg réfractaire était identique à celle observée chez les patients adultes atteints de MAg réfractaire dans le cadre de l’étude d’autorisation ECU MG 301 (tableau 10).
Tableau 20 : Résultats d’efficacité de l‘étude ECU-MG-303
|
Critères d’efficacité : variation du pourcentage total à la semaine 26 par rapport à la valeur initiale
|
Différence des moyennes des moindres carrés (ETM)
IC à 95 %
| |
QMG
|
-5,8 (1,2)
(-8,40; -3,13)
na = 10
| |
Score total MG-ADL
|
-2,3 (0,6)
(-3,63; -1,03)
na = 10
| |
Score total MGC
|
-8,8 (1,9)
(-12,93; -4,69)
na = 9
|
na est le nombre de patients à la semaine 26
Abréviations : IC = intervalle de confiance LS = moindres carrés ; MG-ADL = Myasthenia Gravis Activities of Daily Living (profil de la myathénie grave pour les activités de la vie quotidienne) ; MGC = composite myasthénie grave ; QMG = Quantitative Myasthenia Gravis score for disease severity (score quantitatif de la myasthénie grave pour la sévérité de la maladie) ; ETM = écart-type de la moyenne;
Dans l’étude ECU-MG-303, une réponse clinique aux scores totaux QMG et MG-ADL était définie comme une amélioration d’au moins 5 et. 3 points par rapport à la valeur initiale. La proportion de répondants cliniques pour le score total QMG et MG-ADL à la semaine 26 était de 70 % et 50 % respectivement, indépendamment du traitement d’urgence. Les 10 patients qui ont effectué la visite après 26 semaines avaient atteint un statut amélioré de MGFA post-intervention (MGFA PIS). Sept (70 %) patients avaient atteint une manifestation minimale de MAg réfractaire à la semaine 26.
Chez un patient (9,1 %), une détérioration clinique (crise myasthénique) a été observée au cours de l’évaluation primaire de la période de traitement, qui a nécessité un traitement d’urgence (EP) administré entre les visites d’étude de la semaine 22 et de la semaine 24. En conséquence, et sur décision du médecin, aucune évaluation de QMG, MG-ADL ou autre n’a été effectuée chez ce patient après la semaine 20 et il n’a pas été inclus dans la période de prolongation.
Au cours de l’évaluation primaire de la période de traitement chez les patients pédiatriques atteints de MAg réfractaire (étude ECU-MG-303), 1 patient sur 11 (9,1 %) a réduit la dose quotidienne d’anticholinestérase et 3 patients sur 11 (27,3 %) ont réduit leur dose quotidienne de corticoïde en raison d‘amélioration des symptômes de la MG.
Trouble du spectre de la neuromyélite optique
Soliris n’a pas été étudié chez les enfants et les adolescents atteints de NMOSD.
|