CompositionPrincipes actifs
Dronedaronum hydrochloridum.
Excipients
Lactose monohydraté 41,65 mg, hypromellose (E464), amidon de maïs, crospovidone (E1202), poloxamère 407, silice colloïdale anhydre, stéarate de magnésium (E572), macrogol 6000, dioxide de titane (E171), cire de carnauba (E903).
Indications/Possibilités d’emploiMultaq est indiqué pour le maintien du rythme sinusal après une cardioversion réussie chez les patients cliniquement stables atteints de fibrillation auriculaire (FA) non permanente (fibrillation auriculaire paroxystique ou persistante) et pour diminuer la fréquence des hospitalisations d'origine cardiovasculaire dans ce groupe de patients.
En raison de son profil de sécurité, Multaq ne doit être prescrit qu'après avoir envisagé les alternatives thérapeutiques.
Multaq ne devrait pas être prescrit aux patients avec une dysfonction systolique ventriculaire gauche ou aux patients avec une insuffisance cardiaque ou des antécédents d'insuffisance cardiaque.
Posologie/Mode d’emploiPosologie usuelle
La posologie recommandée chez l'adulte est de 400 mg deux fois par jour, à raison d'un comprimé au repas du matin et un comprimé au repas du soir. Le comprimé ne doit pas être mâché ou broyé.
Le traitement par Multaq peut être commencé en ambulatoire.
Ajustement posologiques du fait d'effets indésirables / d'interactions
Tout traitement par un antiarythmique de classe I ou III (comme la flécaïnide, la propafénone, la quinidine, le disopyramide, le dofétilide, le sotalol, l'amiodarone) doit être arrêté avant de commencer le traitement par Multaq. La longue demi-vie de l'amiodarone et sa possible durée d'action prolongée après arrêt du traitement doivent être prises en considération lors du passage à Multaq (voir «Contre-indications»). Les données concernant le moment optimal du changement sont toutefois limitées.
Patients présentant des troubles de la fonction hépatique
Aucun ajustement posologique n'est requis chez les patients atteints d'insuffisance hépatique légère ou modérée (voir «Pharmacocinétique»). En revanche, Multaq est contreindiqué en cas d'insuffisance hépatique sévère, étant donné l'absence de données (voir «Contre-indications»).
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique n'est requis (voir «Pharmacocinétique»).
Patients âgés
Un grand nombre de patients âgés souffrant de FA (fibrillation auriculaire) ou de FLA (flutter auriculaire) ont été admis dans le programme clinique de Multaq (plus de 4500 patients âgés de 65 ans ou plus, dont plus de 2000 patients de 75 ans ou plus). L'efficacité et la sécurité étaient comparables chez les patients âgés sans co-morbidités cardiovasculaires associées et chez les patients plus jeunes. En présence de co-morbidités, la prudence est requise chez les patients de 75 ans ou plus (voir «Contre-indications» et «Mises en garde et précautions»).
Enfants et adolescents
On ne dispose pas d'expérience chez les enfants et les adolescents.
Contre-indications·Hypersensibilité au principe actif ou à l'un des excipients.
·Bloc atrioventriculaire (BAV) du deuxième ou troisième degré, bloc de branche complet, bloc distal, atteinte du nœud sinusal, troubles de conduction auriculaire, maladie du sinus (excepté en combinaison avec un stimulateur cardiaque en fonctionnement).
·Bradycardie < 50 bpm.
·Fibrillation auriculaire permanente.
·Conditions hémodynamiques instables.
·Insuffisance cardiaque, antécédents d'insuffisance cardiaque ou dysfonction systolique du ventricule gauche (voir «Mises en garde et précautions» et «Propriétés/Effets»).
·Pneumopathie interstitielle (y compris fibrose pulmonaire).
·Patients ayant déjà développé une atteinte hépatique ou pulmonaire sous traitement par amiodarone.
·Coadministration d'inhibiteurs puissants du CYP 3A4, tels que le kétoconazole, l'itraconazole, le voriconazole, le posaconazole, la télithromycine, la clarithromycine, la ciclosporine et le ritonavir (voir «Interactions»).
·Médicaments induisant des torsades de pointes, tels que les phénothiazines, le cisapride, le bépridil, les antidépresseurs tricycliques, la terfénadine et certains macrolides oraux tels que l'érythromycine, les antiarythmiques de classe I et III (voir «Interactions»).
·Intervalle QTc (Bazett) ≥500 msec.
·Insuffisance hépatique sévère.
·Grossesse (voir «Grossesse, allaitement»).
·Allaitement (voir «Grossesse, allaitement»).
Mises en garde et précautionsUne surveillance étroite et régulière des fonctions cardiaque, hépatique et pulmonaire doit être effectuée au cours du traitement par dronédarone (voir ci-dessous). En cas de récidive de fibrillation auriculaire, l'arrêt du traitement doit être envisagé. Le traitement doit être interrompu si une des contre-indications mentionnées ci-dessus se manifeste en cours de traitement. L'utilisation concomitante de médicaments à marge thérapeutique étroite comme la digoxine ou les anticoagulants requiert une surveillance particulière (clinique et contrôles étroits de l'INR et de la concentration de digoxine jusqu'à stabilisation).
Patients évoluant vers une FA permanente pendant le traitement
Une étude clinique chez des patients présentant une FA permanente (durée de la FA ≥6 mois ou de durée inconnue) et des facteurs de risque cardiovasculaires a été interrompue prématurément du fait d'une augmentation significative des décès d'origine cardiovasculaire, des accidents vasculaires cérébraux, des insuffisances cardiaques et des hospitalisations d'origine cardiovasculaire dans le groupe de patients sous dronédarone. Il est recommandé de réaliser un ECG périodiquement, au moins tous les 6 mois. Si des patients développent une FA permanente, le traitement par Multaq doit être arrêté.
Patients présentant une insuffisance cardiaque, des antécédents d'insuffisance cardiaque ou une dysfonction systolique ventriculaire gauche
L'utilisation de Multaq est contre-indiquée chez les patients présentant des conditions hémodynamiques instables, une insuffisance cardiaque, des antécédents d'insuffisance cardiaque, ou une fonction systolique ventriculaire gauche diminuée (voir «Contre-indications»). L'apparition de symptômes cliniques d'insuffisance cardiaque doit être étroitement surveillée. Des cas d'insuffisance cardiaque apparus ou aggravés au cours du traitement par Multaq ont été rapportés spontanément.
Les patients doivent être avertis qu'en cas de signes ou symptômes d'insuffisance cardiaque, tels qu'une prise de poids, un œdème ou une augmentation d'une dyspnée, il leur est conseillé de consulter. Si une insuffisance cardiaque se développe, le traitement par Multaq doit être interrompu.
La fonction systolique ventriculaire gauche devrait être régulièrement suivie au cours du traitement. En cas de développement d'une dysfonction systolique ventriculaire gauche, le traitement par Multaq doit être interrompu.
Atteinte hépatique
Depuis sa mise sur le marché en 2009, de rares cas d'atteinte hépatique sévère allant jusqu'à une insuffisance hépatique aiguë et nécessitant parfois une transplantation ont été rapportés sous traitement par dronédarone.
Chez les patients présentant des lésions hépatiques préexistantes, un traitement par dronédarone ne devrait être introduit qu'avec la plus grande précaution après une évaluation soigneuse des risques et des bénéfices. En présence d'une augmentation des ALT dépassant plus de 3 fois la limite supérieure de la norme, le traitement ne devrait pas être introduit.
Les patients doivent être informés et signaler immédiatement à leur médecin tout signe ou symptôme évoquant une atteinte hépatique (douleurs abdominales persistantes d'apparition récente, anorexie, nausées, vomissements, fièvre, malaise, fatigue, jaunisse, urines sombres ou démangeaisons).
Un contrôle de la fonction hépatique devrait être effectué avant le début du traitement par Multaq puis une semaine après le début du traitement, puis mensuellement pendant les 6 premiers mois, à 9 mois, 12 mois et périodiquement par la suite.
Si une atteinte hépatique est suspectée sur la base de signes cliniques ou biologiques (ALT > 3 fois la limite supérieure de la norme ou évolution à la hausse), le traitement devrait être interrompu. Des investigations complémentaires, une surveillance du patient et un suivi rapproché de la fonction hépatique sont nécessaires.
En présence d'une atteinte hépatique sous traitement par dronédarone et en l'absence d'une autre étiologie identifiée, le traitement par dronédarone ne devrait pas être réintroduit après normalisation de la fonction hépatique.
Prise en charge de l'élévation de la créatinine plasmatique
Il est recommandé de doser la créatinine plasmatique avant et 7 jours après l'instauration du traitement par dronédarone. Si on observe une élévation de la créatininémie, une nouvelle mesure doit être répétée après 7 jours. Si la créatinine est stable, cette valeur doit être utilisée comme nouvelle référence, ce phénomène étant attendu sous dronédarone. Si la créatine continue d'augmenter, des investigations complémentaires et l'arrêt du traitement devraient être envisagés. Un accroissement de la créatininémie ne contraint pas nécessairement à un arrêt du traitement par IECA (inhibiteurs de l'enzyme de conversion de l'angiotensine) ou ARA II (antagonistes des récepteurs de l'angiotensine II).
Une élévation de la créatinine plasmatique a été observée sous 400 mg de dronédarone deux fois par jour, aussi bien chez des sujets sains que chez des patients. Ce phénomène survient précocement après l'instauration du traitement et atteint un plateau au bout de 7 jours. Chez les patients qui présentent une FA, la hausse moyenne avoisine les 10 µmol/l. Les dosages reprennent leurs valeurs initiales dans la semaine qui suit l'arrêt du médicament. Dans une étude spécifique portant sur des sujets sains, le phénomène a semblé être lié à une inhibition de la sécrétion de créatinine au niveau tubulaire, sans effet sur la filtration glomérulaire ou le flux sanguin rénal. Le même mécanisme a également été décrit avec d'autres médicaments, tels que la cimétidine, le triméthoprime ou l'amiodarone. Une élévation de la créatinine plasmatique risque d'être mal interprétée et d'entraîner un arrêt injustifié des IECA ou des ARA II chez les patients qui nécessitent ce médicament.
Après l'autorisation de mise sur le marché, des cas faisant état d'une augmentation plus importante de la créatinine ont été rapportés, y compris des cas d'azotémie pré-rénale consécutive à une insuffisance cardiaque congestive, une hypoperfusion ou une hypovolémie. Dans la plupart des cas, cet effet était réversible après l'arrêt de la dronédarone. Un contrôle périodique de la fonction rénale doit être effectué et de nouvelles investigations doivent être envisagées si nécessaire.
Troubles électrolytiques
Étant donné que les antiarythmiques peuvent être inefficaces, voire arythmogènes en cas d'hypokaliémie, tout déficit de potassium ou de magnésium doit être corrigé avant l'instauration et au cours d'un traitement par dronédarone.
Allongement de l'intervalle QT
L'action pharmacologique de la dronédarone peut induire un allongement modéré (environ 10 msec) de l'intervalle QTc (Bazett), lié à la repolarisation prolongée. Ces modifications sont liées à l'effet thérapeutique de la dronédarone et ne traduisent pas une toxicité. Un suivi, y compris par ECG, est toutefois recommandé en cours de traitement. Si l'intervalle QTc (Bazett) est ≥500 msec, la dronédarone doit être arrêtée (voir «Contre-indications»).
Selon l'expérience clinique, la dronédarone présente un faible effet arythmogène. Une diminution des décès pour cause d'arythmie a été observée dans l'étude ATHENA (voir «Pharmacodynamique»).
Toutefois, des effets arythmogènes peuvent survenir dans des situations particulières, telles que l'utilisation concomitante de médicaments qui favorisent l'arythmie et/ou les troubles électrolytiques (voir «Mises en garde et précautions» et «Interactions»).
Atteintes pulmonaires
Des cas d'affections pulmonaires interstitielles incluant des pneumopathies et des fibroses pulmonaires ont été rapportés après la mise sur le marché. L'apparition d'une dyspnée ou d'une toux non productive peut être liée à une toxicité pulmonaire. Les patients devraient être soumis à une évaluation clinique soigneuse et le traitement arrêté en cas de toxicité pulmonaire confirmée.
InteractionsDigoxine: l'administration concomitante de dronédarone chez les patients recevant de la digoxine entraîne une augmentation de la digoxinémie plasmatique et accroît ainsi les signes et symptômes liés à une toxicité de la digoxine. Un suivi clinique, électrocardiographique et biologique est recommandé et la dose de digoxine doit être réduite de moitié. Un effet synergique sur la fréquence cardiaque et la conduction auriculo-ventriculaire est également possible.
L'administration concomitante de bêtabloquants ou d'antagonistes calciques ayant un effet ralentisseur sur le nœud sinusal et le nœud auriculo-ventriculaire doit être réalisée avec prudence. Ces médicaments doivent être initiés à faible dose et la titration doit être réalisée uniquement après contrôle ECG. Chez les patients recevant déjà un antagoniste calcique ou un bêtabloquant au moment de l'instauration d'un traitement par dronédarone, un ECG doit être réalisé, et leur dose doit être ajustée si nécessaire.
Les IMAO pourraient diminuer la clairance du métabolite actif de la dronédarone et doivent être en conséquence utilisés avec précaution.
Anticoagulation
Les patients devraient être correctement anti-coagulés selon les directives cliniques sur la FA. L'International Normalized Ratio (INR) devrait être étroitement surveillé après l'instauration d'un traitement par la dronédarone chez les patients sous antagonistes de la vitamine K (selon les indications de l'Information professionnelle correspondante).
Patients présentant une coronaropathie
La prudence est nécessaire chez les patients présentant une coronaropathie.
Patients intolérants au galactose et au lactose
Compte tenu de la présence de lactose parmi les excipients, les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Femmes en âge de procréer
Les femmes préménopausées n'ayant pas subi d'hystérectomie ou d'ovariectomie doivent utiliser un moyen de contraception efficace pendant le traitement par Multaq. La dronédarone a eu des effets fœto-toxiques lors d'expérimentations animales à doses équivalentes aux posologies recommandées chez l'homme. Chez les femmes en âge de procréer, une méthode de contraception appropriée doit être proposée en tenant compte de leur condition médicale sous-jacente et des préférences liées à leur mode de vie (Voir «Grossesse, allaitement»).
Interactions
La dronédarone est principalement métabolisée par le CYP3A4 (voir «Pharmacocinétique»); il s'agit d'un inhibiteur modéré du CYP3A4 et léger du CYP2D6. Les inhibiteurs et inducteurs du CYP3A4 sont donc susceptibles d'interagir avec la dronédarone, et inversement, celle-ci peut interagir avec les substrats médicamenteux des CYP3A4 et CYP2D6. Elle peut aussi inhiber le transport par les P-glycoprotéines (PgP). La dronédarone et/ou ses métabolites ont aussi le potentiel d'inhiber l'OAT (transporteur d'anions organiques), l'OATP (polypeptide de transport d'anions organiques) et l'OCT (transporteur de cations organiques) in vitro.
La dronédarone n'exerce pas d'effet inhibiteur important sur CYP1A2, CYP2C9, CYP2C19, CYP2C8 et CYP2B6.
On peut également suspecter une interaction pharmacodynamique avec les bêtabloquants, les anticalciques et les digitaliques.
Au cours des essais cliniques, des patients traités par dronédarone ont reçu simultanément divers médicaments, tels que des bêtabloquants, des digitaliques, des anticalciques (y compris bradycardisants), des statines et des anticoagulants oraux.
Médicaments induisant des torsades de pointes
Les médicaments induisant des torsades de pointes tels que les phénothiazines, le cisapride, le bépridil, les antidépresseurs tricycliques, certains macrolides oraux, la terfénadine et les antiarythmiques de classe I et III, sont contre-indiqués pour éviter tout risque d'effet arythmogène.
Effet d'autres médicaments sur Multaq
Inhibiteurs puissants du CYP3A4
L'administration répétée de 200 mg de kétoconazole par jour a multiplié par 17 l'exposition à la dronédarone. L'utilisation concomitante de kétoconazole et d'autres inhibiteurs puissants du CYP3A4, tels que l'itraconazole, le voriconazole, le ritonavir, la télithromycine, la clarithromycine ou la ciclosporine est dès lors contre-indiquée (voir «Contre-indications»).
Inhibiteurs modérés ou faibles du CYP3A4: antagonistes calciques
Les anticalciques sont des substrats et/ou des inhibiteurs modérés du CYP3A4. Ceux qui présentent des propriétés bradycardisantes sont en outre susceptibles d'interagir avec Multaq au niveau pharmacodynamique.
L'administration répétée de diltiazem (240 mg deux fois par jour), de vérapamil (240 mg une fois par jour) et de nifédipine (20 mg deux fois par jour) a multiplié l'exposition à la dronédarone par un facteur 1,7; 1,4 et 1,2; respectivement. Les anticalciques voient également leur exposition s'accroître sous l'effet de la dronédarone (400 mg deux fois par jour) (facteur 1,4 et 1,5 pour le vérapamil et la nisoldipine, respectivement). Les essais cliniques n'ont pas révélé de problèmes de sécurité lors de la coadministration de la dronédarone et d'anticalciques à action bradycardisante.
De façon générale, étant donné l'interaction pharmacocinétique et le risque d'interaction pharmacodynamique, il convient toutefois de n'associer la dronédarone qu'avec précaution à des antagonistes calciques exerçant des effets dépresseurs sur le sinus et le nœud auriculoventriculaire, tels que le vérapamil et le diltiazem.
Érythromycine
D'autres inhibiteurs modérés du CYP3A4 sont aussi susceptibles d'augmenter l'exposition à la dronédarone. L'érythromycine, un macrolide oral, peut induire des torsades de pointe et est donc contre-indiqué (voir «Contre-indications»). Des doses répétées d'érythromycine (500 mg trois fois par jour pendant 10 jours) ont augmenté d'un facteur 3,8 l'exposition à la dronédarone à l'état d'équilibre.
Inducteurs du CYP3A4
La rifampicine (600 mg une fois par jour) a réduit de 80 % l'exposition à la dronédarone sans modifier notablement l'exposition à son métabolite actif. C'est pourquoi l'administration concomitante de rifampicine et d'autres inducteurs puissants du CYP3A4, tels que le phénobarbital, la carbamazépine, la phénytoïne et le millepertuis, n'est donc pas recommandée, parce qu'elle réduit l'exposition à la dronédarone.
Effet de Multaq sur d'autres médicaments
Interaction avec les médicaments métabolisés par le CYP3A4
·Statines substrats du CYP3A4 et/ou de la P-glycoprotéine
La dronédarone peut accroître l'exposition aux statines substrats du CYP3A4 et/ou de la P-gP. Son administration (400 mg deux fois par jour) a ainsi augmenté par un facteur respectivement de 4 et 2 l'exposition à la simvastatine et à la simvastatine acide. On s'attend à ce que la dronédarone puisse également élever l'exposition à la lovastatine, l'atorvastatine et la pravastatine dans des proportions similaires à celles observées pour la simvastatine acide. Une faible interaction de la dronédarone avec l'atorvastatine (facteur 1,7) a été observée. Lors d'essais cliniques, aucun problème d'innocuité n'a été constaté lors de l'administration simultanée de la dronédarone et de statines métabolisées par le CYP3A4.
Une faible interaction de la dronédarone avec les statines transportées par l'OATP telles que la rosuvastatine (facteur 1,4) a été observée.
Du fait de multiples mécanismes d'interaction sur les statines (CYP et transporteurs) et étant donné que les hautes doses de statines augmentent le risque de myopathie, il ne faut entreprendre qu'avec précaution l'utilisation concomitante des statines.
Des doses plus faibles d'initiation et d'entretien des statines devraient être envisagées en tenant compte des recommandations sur l'information destinée aux professionnels des statines et tout signe clinique de toxicité musculaire devrait être surveillé.
·Antagonistes calciques
L'interaction de la dronédarone sur les anticalciques est décrite ci-dessus.
·Sirolimus, tacrolimus
La dronédarone pourrait accroître les concentrations plasmatiques du tacrolimus et du sirolimus. Une surveillance de leurs concentrations plasmatiques et un ajustement adéquat des posologies est recommandé en cas de co-administration avec la dronédarone.
·Contraceptifs oraux
Aucune diminution des taux d'éthinylœstradiol et de lévonorgestrel n'a été observée chez des sujets sains recevant de la dronédarone (800 mg deux fois par jour) avec des contraceptifs oraux.
Interaction sur les médicaments métabolisés par le CYP2D6: bêtabloquants, antidépresseurs
·Bêtabloquants
La dronédarone peut augmenter l'exposition de bêtabloquants métabolisés par le CYP2D6. De plus, les bêtabloquants sont susceptibles d'interagir avec la dronédarone sur le plan pharmacodynamique. La dronédarone à raison de 800 mg par jour a multiplié l'exposition au métoprolol par 1,6 et au propranolol par 1,3 (c'est-à-dire bien moins que le facteur 6 qui distingue les métaboliseurs CYP2D6 lents et rapides). Lors d'essais cliniques, une bradycardie a été plus souvent observée lors de l'administration combinée de dronédarone et de bêtabloquants.
Étant donné l'interaction pharmacocinétique et une interaction pharmacodynamique possible, l'utilisation concomitante de bêtabloquants et de dronédarone exige des précautions.
·Antidépresseurs
La dronédarone étant un faible inhibiteur du CYP2D6 chez l'homme, on s'attend à une interaction limitée avec les médicaments antidépresseurs métabolisés par cette enzyme.
Interaction avec les substrats de la P-gP
·Digoxine
La dronédarone (400 mg deux fois par jour) a multiplié par 2,5 l'exposition à la digoxine en inhibant le transporteur P-gP. De plus, les digitaliques sont susceptibles d'interagir avec la dronédarone sur le plan pharmacodynamique. Lors d'essais cliniques, l'administration concomitante de la dronédarone et de digitaliques a provoqué une élévation des taux de digitaliques et/ou des troubles gastro-intestinaux.
Étant donné l'interaction pharmacocinétique et le risque d'interaction pharmacodynamique, l'utilisation simultanée de digoxine et de la dronédarone exige donc des précautions, et il faut surveiller étroitement les taux sériques de digoxine des patients, particulièrement au cours de la première semaine de co-administration. Un suivi clinique et un contrôle par ECG sont également recommandés, et la dose de digoxine devra être adaptée en conséquence.
·Dabigatran
La dronédarone est susceptible d'augmenter l'exposition au dabigatran en inhibant la P-gp. Par conséquent, il est recommandé de procéder à une évaluation des risques de survenue d'événements thromboemboliques et de saignements lors d'une co-administration. Une réduction de dose par rapport aux recommandations de l'information professionnelle doit être envisagée. L'exposition au dabigatran 150 mg une fois par jour est augmentée de 1,7 à 2 fois en cas de co-administration avec la dronédarone 400 mg deux fois par jour. Une étude de cohorte rétrospective aux Etats-Unis (USA) a montré que l'utilisation concomitante de dabigatran et de dronédarone chez les patients souffrant de fibrillation auriculaire non valvulaire (nv-VHF) n'a entraîné aucune augmentation des diagnostics de risque de saignement nécessitant une hospitalisation comparé à l'utilisation seule de dabigatran. Parmi les patients traités simultanément par dabigatran et dronédarone, un risque accru de saignements gastrointestinaux a été observé. Le traitement concomitant avec le dabigatran n'est pas recommandé.
·Autres substrats de la P-gP
La dronédarone inhibe la P-gP, des interactions peuvent dès lors survenir avec la doxorubicine, la fexofénadine et le talinolol.
Interaction avec les substrats du CYP3A4 et de la P-gp
La dronédarone est susceptible d'augmenter l'exposition aux inhibiteurs du facteur Xa en inhibant la P-gp ou le CYP3A4. Par conséquent, une évaluation des risques de survenue d'événements thromboemboliques et de saignements est nécessaire lors d'une co-administration. Une réduction du dosage doit être considérée selon les recommandations de l'information professionnelle concernée.
·Inhibiteurs du facteur Xa:
Rivaroxaban
La dronédarone est susceptible d'augmenter l'exposition au rivaroxaban et, par conséquent, une utilisation simultanée peut augmenter le risque de saignements.
Apixaban
La dronédarone peut augmenter l'exposition à l'apixaban. Cependant, en cas de co-administration avec des principes actifs qui ne sont pas des inhibiteurs puissants du CYP3A4 et de la P-gp, tels que la dronédarone, un ajustement posologique de l'apixaban n'est pas nécessaire, selon l'information professionnelle.
Edoxaban
La coadministration de dronédarone 400 mg deux fois par jour pendant 7 jours avec une dose unique d'édoxaban 60 mg au jour 5 a entraîné une augmentation de l'AUC et de la Cmax de 85 % et 46 % respectivement. La dose d'édoxaban doit être réduite conformément aux recommandations de l'information professionnelle de l'édoxaban.
Interaction avec warfarine et losartan (substrats du CYP2C9)
·Warfarine et autres antagonistes de la vitamine K
Des contrôles de l'INR sont recommandés en particulier pendant une à deux semaines au début d'un traitement par dronédarone.
Dans l'étude ATHENA, par rapport au groupe placebo, un nombre supérieur de patients sous traitement anticoagulant oral a connu une augmentation cliniquement significative (≥5) de l'INR, généralement 1 semaine après avoir commencé la dronédarone. Cependant, aucun risque excessif d'hémorragie n'a été observé dans le groupe dronédarone. Dans une étude d'interaction, la dronédarone (600 mg deux fois par jour) a multiplié par 1,2 les taux de S-warfarine, sans modifier R-warfarine et en n'augmentant l'International Normalized Ratio (INR) que d'un facteur 1,07.
Dans les déclarations spontanées de pharmacovigilance, des cas d'accroissement de l'INR avec ou sans événement hémorragique ont été rapportés chez des patients sous antagonistes oraux de la vitamine K commençant un traitement par dronédarone.
·Losartan et autres antagonistes des récepteurs de l'angiotensine II (ARA II)
Aucune interaction n'a été observée entre la dronédarone et losartan, elle n'est guère probable avec d'autres ARA II.
Interaction avec la théophylline (substrat du CYP1A2)
La dronédarone, 400 mg deux fois par jour, n'augmente pas l'exposition à la théophylline en état d'équilibre.
Interaction avec la metformine (substrat de l'OCT1 et l'OCT2)
La dronédarone n'a eu aucune influence pertinente du point de vue clinique sur la pharmacocinétique de la metformine. L'influence de la metformine sur la pharmacocinétique de la dronédarone n'a pas été étudiée car aucune interaction n'est attendue.
Interaction avec l'oméprazole (substrat du CYP2C19)
Aucune interaction n'a été observée entre la dronédarone et l'oméprazole, un substrat du CYP2C19.
Interaction avec le clopidogrel
Aucune interaction n'a été observée entre la dronédarone et le clopidogrel.
Autres informations
Le pantoprazole (40 mg une fois par jour), un médicament qui augmente le pH gastrique sans effet sur le cytochrome P450, n'a pas interagi de façon significative sur la pharmacocinétique de la dronédarone.
Jus de pamplemousse (inhibiteur du CYP3A4)
Des doses répétées de 300 ml de jus de pamplemousse, trois fois par jour, ont multiplié par 3 l'exposition à la dronédarone. Les patients doivent donc être avertis d'éviter les boissons contenant du jus de pamplemousse pendant qu'ils prennent la dronédarone.
Grossesse, allaitementOn ne dispose pas de données suffisantes concernant l'utilisation de la dronédarone chez la femme enceinte. Les expérimentations animales ont révélé une toxicité sur la reproduction (tératogénicité chez les rats) (voir «Données précliniques»).
Avant de commencer le traitement par Multaq, il convient de s'assurer que les femmes en âge de procréer ne sont pas enceintes.
La dronédarone est dès lors contre-indiquée chez la femme enceinte (voir «Contre-indications»).
Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant un traitement par Multaq et pendant 7 jours après l'administration de la dernière dose.
On ignore si la dronédarone est excrétée dans le lait humain.
Des études effectuées sur des animaux ont révélé une excrétion de la dronédarone et de ses métabolites dans le lait. Les femmes ne doivent pas allaiter quand elles prennent Multaq et pendant 7 jours (environ 5 demi-vies) après la dernière dose (voir «Contre-indications»).
La dronédarone ne semble pas entraîner d'atteinte à la fertilité lors d'expérimentations animales.
Effet sur l’aptitude à la conduite et l’utilisation de machinesLes effets sur l'aptitude à conduire des véhicules et à utiliser des machines n'ont pas été étudiés.
Effets indésirablesLe profil d'innocuité de la dronédarone, administrée à raison de 400 mg deux fois par jour, à des patients atteints de FA ou de FLA se fonde sur 5 études contrôlées contre placebo: ATHENA, EURIDIS, ADONIS, ERATO et DAFNE. Un total de 6285 patients ont été randomisés et traités, dont 3282 sous dronédarone, 400 mg deux fois par jour, et 2875 sous placebo.
Le temps d'exposition moyen durant ces études était de 13 mois. Dans ATHENA, le suivi maximum était de 30 mois.
L'évaluation de facteurs intrinsèques, tels que le sexe ou l'âge, sur l'incidence des divers effets indésirables sous traitement, n'a pas laissé penser qu'un sous-groupe particulier était plus exposé.
Dans les études cliniques, un arrêt prématuré motivé par des réactions défavorables est survenu dans 11,8 % des cas sous dronédarone contre 7,7 % sous placebo. Les motifs les plus fréquemment évoqués pour arrêter le traitement par Multaq étaient des troubles gastro-intestinaux (3,2 % contre 1,8 % sous placebo).
Les effets indésirables le plus souvent observés sous dronédarone, 400 mg deux fois par jour, dans les 5 études consistaient en diarrhée, nausée et vomissement, fatigue et asthénie.
Les effets indésirables associés à la prise de dronédarone, 400 mg deux fois par jour, par des patients atteints de FA ou de FLA, sont présentés par systèmes d'organe et par ordre décroissant de fréquence.
Les effets indésirables relevant de la classe d'organe appelée «investigations» sont décrits séparément.
Les fréquences sont définies comme suit: très fréquent (≥1/10), fréquent (≥1/100 à < 1/10); occasionnel (≥1/1000 à < 1/100); rare (≥1/10 000 à < 1/1000); très rare (< 1/10 000).
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Affections cardiaques
Très fréquent: insuffisance cardiaque. Dans les 5 études contrôlées versus placebo, une insuffisance cardiaque a été rapportée à des taux comparables dans le groupe dronédarone et dans le groupe placebo (très fréquent, 11,2 % contre 10,9 %). Ce taux doit être considéré dans le contexte d'une incidence sous-jacente élevée d'insuffisance cardiaque chez les patients souffrant de FA.
Des cas d'insuffisance cardiaque ont été également rapportés après commercialisation (fréquence indéterminée): voir «Mises en garde et précautions».
Fréquent: bradycardie.
Rare: flutter auriculaire avec conduction auriculo-ventriculaire de type 1:1.
Affections respiratoires, thoraciques et médiastinales
Occasionnel: affections pulmonaires interstitielles comprenant pneumopathies interstitielles et fibroses pulmonaires. Dans les 5 études contrôlées par placebo, 0,6 % des patients dans le groupe dronédarone ont présenté des évènements pulmonaires contre 0,8 % dans le groupe placebo. Des cas de maladie pulmonaire interstitielle comprenant pneumopathies interstitielles et fibroses pulmonaires ont été rapportés depuis la mise sur le marché de Multaq (fréquence inconnue). Un certain nombre de patients avaient été préalablement exposés à l'amiodarone.
Affections du système nerveux
Fréquent: dysgueusie.
Rare: agueusie.
Affections gastro-intestinales
Fréquent: diarrhée, vomissements, nausées, douleurs abdominales, dyspepsie.
Affections hépatobiliaires (voir «Mises en garde et précautions»)
Fréquent: anomalies des tests de la fonction hépatique.
Rare: atteinte hépatocellulaire, y compris insuffisance hépatique aiguë mettant en jeu le pronostic vital.
Affections vasculaires
Rare: vascularite, y compris vascularite leucocytoclasique.
Affections de la peau et du tissu sous-cutané
Fréquent: éruptions (y compris généralisées, maculaires, maculo-papuleuses), prurit.
Occasionnel: érythèmes (y compris érythème et éruption cutanée érythémateuse), eczéma, réaction de photosensibilité, dermite allergique, dermatite.
Troubles généraux et anomalies au site d'administration
Fréquent: fatigue, asthénie.
Affections du système immunitaire
Rare: réactions anaphylactiques, y compris œdème de Quincke.
En outre, les données de laboratoire et les paramètres d'ECG suivants ont été très fréquemment (> 1/10) signalés sous dronédarone, 400 mg deux fois par jour:
·Taux sanguin de créatinine augmenté de ≥10 % cinq jours après l'instauration du traitement.
·Allongement de l'intervalle QTc (Bazett) (> 450 msec chez les hommes, > 470 msec chez les femmes).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageOn ignore si la dronédarone et/ou ses métabolites peuvent être extraits par dialyse (hémodialyse, dialyse péritonéale ou hémofiltration).
On ne dispose d'aucun antidote spécifique. En cas de surdosage, un traitement de soutien doit être instauré, qui visera le soulagement des symptômes.
Propriétés/EffetsCode ATC
C01BD07
Mécanisme d'action
Le mécanisme d'action précis de la dronédarone chez l'homme n'est pas connu. La dronédarone possède les propriétés anti-arrhythmiques des quatre classes de Vaughan-Williams, mais la contribution sur l'effet clinique de chacune de ces activités n'est pas connue. Chez les animaux, la dronédarone prévient la fibrillation auriculaire ou normalise le rythme sinusal selon le modèle utilisé. Elle prévient également la tachycardie et la fibrillation ventriculaires dans plusieurs modèles animaux. Ces effets résultent très vraisemblablement de ses propriétés électrophysiologiques, qui relèvent de chacune des quatre classes de Vaughan-Williams. La dronédarone bloque de multiples canaux en inhibant les flux de potassium (y compris IK(Ach), IKur, IKr, IKs) et en prolongeant ainsi le potentiel d'action cardiaque et les périodes réfractaires (Classe III). Elle inhibe également les flux de sodium (Classe I) et de calcium (Classe IV). Enfin, elle entrave les activités adrénergiques de façon non compétitive (Classe II).
Pharmacodynamique
Dans les modèles animaux, la dronédarone ralentit la fréquence cardiaque, allonge le phénomène de Wenckebach, ainsi que les intervalles AH, PQ, QT, sans modifier notablement les intervalles QTc, HV et QRS ou en les allongeant faiblement. Elle prolonge les périodes réfractaires de l'oreillette, du nœud auriculo-ventriculaire et du ventricule avec un degré minime de «reverse-use dependency».
La dronédarone diminue la pression artérielle et la contractilité myocardique (dP/dt max) sans modifier la fraction d'éjection ventriculaire gauche, et réduit la consommation en oxygène du myocarde.
La dronédarone présente des propriétés vasodilatatrices, plus prononcées au niveau des artères coronaires (par activation de la voie de l'oxyde nitrique) qu'à celui des artères périphériques.
La dronédarone exerce des effets anti-adrénergiques indirects; elle réduit la réaction alpha-adrénergique de la tension artérielle à l'épinéphrine, ainsi que les réactions bêta 1 et bêta 2 à l'isoprotérénol.
Efficacité clinique
Réduction du risque d'hospitalisation d'origine cardiovasculaire ou de décès
L'efficacité de la dronédarone dans la réduction du risque d'hospitalisation d'origine cardiovasculaire ou de décès de toute cause a été démontrée par ATHENA, une étude multicentrique, multinationale, à double insu, randomisée et contrôlée par placebo chez des patients atteints de FA ou de FLA ou présentant des antécédents correspondants ainsi que d'autres facteurs de risque.
Les patients comptaient au moins un facteur de risque (tels que: âge, hypertension, diabète, antécédent d'AVC, diamètre de l'oreillette gauche ≥50 mm ou FEVG < 0,40), avec des épisodes de FA/FLA et de rythme sinusal documentés au cours des 6 mois précédents. Les patients pouvaient être en FA/FLA ou en rythme sinusal après conversion spontanée ou à la suite d'une autre procédure.
Quatre mille six cent vingt-huit (4628) patients ont été randomisés pour recevoir, pendant des périodes allant jusqu'à 30 mois maximum (suivi médian: 22 mois) soit de la dronédarone, 400 mg deux fois par jour (2301 patients), soit un placebo (2327 patients), en plus d'un traitement classique comprenant des bêtabloquants (71 %), des IECA ou ARA II (69 %), des digitaliques (14 %), des antagonistes calciques (14 %), des statines (39 %), des anticoagulants oraux (60 %), une thérapie antiplaquettaire chronique (5 %) et/ou des diurétiques (54 %).
Le critère d'évaluation primaire de l'étude était le délai de première hospitalisation d'origine cardiovasculaire ou de décès de toute cause.
Les critères d'évaluation secondaires étaient le délai de décès de toute cause, le délai de première hospitalisation d'origine cardiovasculaire, ou le délai de décès pour cause cardiovasculaire.
Le délai de mort subite a également été évalué.
Les patients étaient âgés de 23 à 97 ans, et 42 % d'entre eux avaient plus de 75 ans. Quarante-sept pour cent (47 %) des patients étaient de sexe féminin, et la majorité était d'origine caucasienne (89 %).
La plupart des sujets souffraient d'hypertension (86 %) et d'une maladie cardiaque structurelle (60 %) (y compris maladie coronaire: 30 %; insuffisance cardiaque congestive (ICC): 30 %; dysfonction ventriculaire gauche avec fraction d'éjection < 45 %: 12 %). Vingt-cinq pour cent (25 %) présentaient une FA à l'inclusion dans l'étude.
La dronédarone a réduit l'incidence des hospitalisations d'origine cardiovasculaire ou de décès de toute cause de 24,2 % par rapport au placebo (p = 2 x 10-8).
La figure 1 représente les courbes d'incidence cumulée des événements. Ces courbes se sont séparées précocement, pour continuer à diverger pendant les 30 mois de la période de suivi.
Figure 1 – Courbes d'incidence cumulée selon Kaplan-Meier, allant de la randomisation à la première hospitalisation d'origine cardiovasculaire ou au décès de toute cause
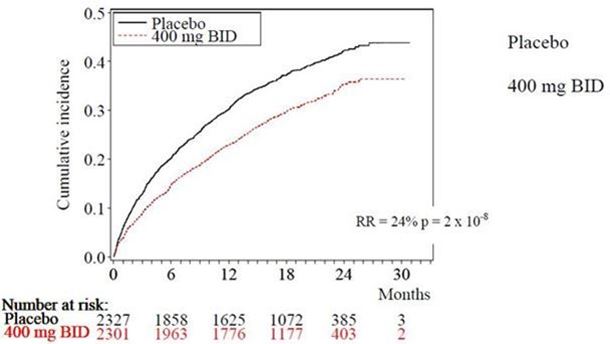
La diminution des hospitalisations d'origine cardiovasculaire ou des décès de toute cause a été du même ordre dans tous les sous-groupes, indépendamment des caractéristiques de départ ou des médicaments utilisés (IECA ou ARA II; bêtabloquants, digitaliques, statines, antagonistes calciques, diurétiques) (voir figure 2).
Figure 2 – Estimations du risque relatif (dronédarone 400 mg deux fois par jour, par rapport au placebo) avec 95 % d'intervalle de confiance, en fonction du choix des caractéristiques à l'inclusion - première hospitalisation d'origine cardiovasculaire ou décès de toute cause

a Déterminé à partir d'un modèle de régression de Cox.
b Valeur de p d'une interaction entre les caractéristiques de départ et le traitement, calculée à partir d'un modèle de régression de Cox.
c Anticalciques dotés d'effets bradycardisants limités aux diltiazem, vérapamil et bépridil.
Des résultats similaires ont été observés sur le plan de l'incidence des hospitalisations d'origine cardiovasculaire, avec une réduction du risque de 25,5 % (p = 9 x 10-9).
Bien qu'une réduction des hospitalisations pour FA ait été prépondérante, le risque de première hospitalisation d'origine cardiovasculaire autre que FA/FLA était significativement diminué de 14,5 % dans le groupe dronédarone par comparaison au placebo (p = 0,0162).
On observait également dans le groupe dronédarone un nombre inférieur d'hospitalisations pour aggravation d'une ICC [3,4 % contre 4,0 % (placebo)], infarctus du myocarde ou angor instable [2,1 % contre 2,6 % (placebo)] ou d'un AIT/AVC [1,2 % contre 1,5 % (placebo)].
Les taux d'hospitalisations pour hémorragie majeure [0,9 % contre 1 % (placebo)], syncope [0,9 % contre 1 % (placebo)] ou arythmie ventriculaire (y compris extrasystoles, tachycardie, fibrillation et autres arythmies ventriculaires) [0,4 % contre 0,3 % (placebo)] étaient similaires dans les deux groupes.
En outre, la durée totale d'hospitalisation sous dronédarone était inférieure à celle sous placebo [9995 nuitées contre 13 986 (placebo)], avec une forte réduction (47 %) du nombre de nuits d'hospitalisation d'origine cardiovasculaire en unités de soins intensifs / unité de soins cardiologiques.
Le nombre de décès était moindre dans le groupe dronédarone 400 mg deux fois par jour [n = 116 contre 139 (placebo), réduction du risque 15,6 %, p = 0,176], avec une nette diminution de 30,2 % du risque de décès d'origine cardiovasculaire [p = 0,025; 2,8 % contre 4,0 % (placebo)]. La raison principale de ce phénomène résidait en une réduction de 59,5 % du risque de mort subite d'origine cardiaque [p = 0,0031; 0,6 % contre 1,5 % (placebo)] et une réduction de 38,3 % du risque de décès par accident vasculaire cérébral [p = 0,2021; 0,5 % contre 0,8 % (placebo)].
Les courbes d'incidence cumulée selon Kaplan-Meier (voir figures 3 et 4) couvrant la période allant de la randomisation au décès cardiovasculaire indiquent un effet précoce et soutenu dans le temps de la dronédarone sur la mortalité.
Figure 3 - Courbes d'incidence cumulée selon Kaplan-Meier de la randomisation au décès cardiovasculaire durant l'étude

Figure 4 - Courbes d'incidence cumulée selon Kaplan-Meier des morts cardiaques subites durant l'étude

Maintien du rythme sinusal
Dans EURIDIS et ADONIS, un total de 1237 personnes avec antécédent de FA ou de FLA ont été randomisés en ambulatoire pour recevoir soit la dronédarone 400 mg deux fois par jour (n = 828) ou soit un placebo (n = 409) en plus de traitements conventionnels (comprenant anticoagulants oraux, bêtabloquants, IECA ou ARA II, agents antiplaquettaires, diurétiques, statines, digitaliques et anticalciques). Ces patients avaient présenté au moins un épisode de FA ou FLA prouvé par ECG au cours des 3 mois précédents, étaient en rythme sinusal depuis au moins une heure et ont été suivis pendant 12 mois.
Ils étaient âgés de 20 à 88 ans, en majorité d'origine caucasienne (97 %) et de sexe masculin (69 %). Les comorbidités les plus fréquentes étaient l'hypertension (56,8 %) et une maladie cardiaque structurelle (41,5 %), y compris une maladie coronarienne (21,8 %).
Les données tant cumulées que séparées d'EURIDIS et ADONIS ont révélé que la dronédarone allongeait de façon équilibrée le délai de première récidive de FA ou FLA (critère d'évaluation primaire). Par comparaison au placebo, la dronédarone réduisait de 25,5 % le risque de première récidive de FA ou FLA au cours des 12 mois de l'étude (p = 0,00007). Le délai médian entre la randomisation et cette première récidive de FA ou FLA dans le groupe dronédarone était de 116 jours, c'est-à-dire 2,2 fois supérieur à celui observé dans le groupe placebo (53 jours). La majorité (60 %) des premières rechutes étaient symptomatiques. La dronédarone allongeait également le délai de première récidive symptomatique de FA ou FLA dans les deux études (p = 0,0003). Sous dronédarone 400 mg deux fois par jour, la proportion de patients n'ayant pas présenté de première récidive symptomatique de FA ou FLA atteignait 62,3 % au bout d'un an.
Dans le cadre de DAFNE, où la dronédarone était instaurée avant la cardioversion, le délai médian de récidive de FA, diagnostiquée par surveillance ECG à 12 dérivations transtéléphoniques (TTEM), était de 60 jours sous dronédarone 400 mg deux fois par jour contre 5 jours dans le groupe placebo. La dronédarone 400 mg deux fois par jour abaissait de 55 % (p = 0,001) le risque de première récidive de FA par rapport aux résultats observés sous placebo au cours des 6 mois de l'étude.
Etude ANDROMEDA
Des patients hospitalisés peu auparavant pour une insuffisance cardiaque symptomatique et une dysfonction systolique grave du ventricule gauche (indice de mobilité de la paroi ≤1,2) ont été randomisés pour recevoir soit Multaq, 400 mg deux fois par jour, soit un placebo adéquat, avec un critère d'évaluation principal composé de la mortalité de toutes causes et des hospitalisations pour insuffisance cardiaque. Après le recrutement de 627 des 1000 patients prévus (310 et 317 dans les groupes dronédarone et placebo, respectivement) et un suivi médian de 63 jours, l'étude a été arrêtée en raison d'un excès de mortalité dans le groupe dronédarone.
Vingt-cinq (25) patients du groupe dronédarone (8,1 %) contre 12 dans le groupe placebo (3,8 %) sont décédés, (RR 2,13; IC 95 %: 1,07 à 4,25; p = 0,027). La principale cause de décès a consisté en une aggravation de l'insuffisance cardiaque. Un nombre supérieur d'hospitalisations pour raisons cardiovasculaires a également été constaté dans le groupe dronédarone (71 contre 51 sous placebo) (voir «Contre-indications»).
Les populations recrutées dans les études ANDROMEDA et ATHENA étaient significativement différentes. Les patients admis dans ANDROMEDA souffraient d'une insuffisance cardiaque relativement grave et avaient été hospitalisés ou envoyés à une consultation spécialisée pour une aggravation de leurs symptômes d'insuffisance cardiaque, notamment une dyspnée. L'état clinique de ces patients pouvait s'être amélioré au moment de l'admission, et c'est l'évolution de leur décompensation qui les caractérisait. Les patients admis dans ANDROMEDA appartenaient principalement aux classes NYHA II (40 %) et III (57 %), et seuls 38 % comptaient des antécédents de fibrillation ou de flutter auriculaire (FA/FLA) (25 % présentaient une FA au moment de la randomisation). En revanche, l'étude ATHENA comptait 71 % de patients sans insuffisance cardiaque, 25 % d'individus appartenant aux classes NYHA I ou II et seulement 4 % de sujets de classe III. Tous les patients présentaient un antécédent de FA/FAL.
Patients présentant une fibrillation auriculaire permanente
L'étude PALLAS était une étude randomisée, contrôlée contre placebo, destinée à investiguer le bénéfice clinique de la dronédarone (400 mg, 2 fois par jour) ajoutée au traitement habituel chez des patients présentant une fibrillation auriculaire permanente et des facteurs de risque cardiovasculaires (environ 68 % des patients présentaient une insuffisance cardiaque, 41 % une maladie coronarienne, 28 % des antécédents d'accident vasculaire cérébral ou d'accident ischémique transitoire, 21 % une fraction d'éjection systolique ventriculaire gauche ≤40 % et 18 % des patients de ≥75 ans avait un diabète et une hypertension). L'étude a dû être interrompue prématurément après randomisation de 3236 patients en raison de l'augmentation significative des insuffisances cardiaques (HR 2,16: 1,57 – 2,98), des accidents vasculaires cérébraux (HR 2,32: 1,11 – 4,88) et des décès d'origine cardiovasculaire (HR 2,11: 1,00 – 4,49) dans le groupe traité par dronédarone. (Voir «Contre-indications» et «Mises en garde et Précautions»).
PharmacocinétiqueAbsorption
Après son administration orale lors d'un repas, la dronédarone est bien absorbée (au moins 70 %). Toutefois, du fait d'un métabolisme de premier passage présystémique, la biodisponibilité absolue de ce médicament (pris avec de la nourriture) est de 15 %. La consommation concomitante d'aliments multiplie la biodisponibilité du produit par un facteur 2 à 4. Après une administration orale lors d'un repas, les concentrations plasmatiques maximum en dronédarone et en son principal métabolite actif circulant (métabolite N-débutylé) sont atteintes en 3 à 6 heures. Après la prise répétée de 400 mg, deux fois par jour, l'état d'équilibre s'observe dans les 4 à 8 jours du traitement, et le taux moyen d'accumulation de la dronédarone oscille entre 2,6 et 4,5. La Cmax moyenne de la dronédarone atteint 84 à 147 ng/ml à l'état d'équilibre, et l'exposition au principal métabolite N-débutylé est similaire à celle observée pour la substance mère. La pharmacocinétique de la dronédarone et de son métabolite N-débutylé dévie modérément de la règle de la proportionnalité à la dose: un doublement de la dose entraîne une hausse de la Cmax et de l'AUC par un facteur d'environ 2,5 à 3,0.
Distribution
La liaison de la dronédarone et son métabolite N-débutylé aux protéines plasmatiques est > 98 % in vitro et n'est pas saturable. Les deux composés se lient principalement à l'albumine. Après une administration intraveineuse (IV), le volume de distribution à l'état d'équilibre (Véq) oscille entre 1200 et 1400 l.
Métabolisme
La dronédarone est fortement métabolisée, principalement par le CYP3A4 (voir «Interactions»). La principale voie métabolique comprend une N-débutylation qui conduit à la formation du principal métabolite circulant et actif, suivie d'une d'oxydation; une déamination oxydative qui conduit à la formation d'un métabolite inactif, l'acide propionique, suivie d'une oxydation, et une oxydation directe. Les oxydases monoamines contribuent partiellement au métabolisme du métabolite actif de la dronédarone. Le métabolite N-débutylé présente une activité pharmacodynamique mais est 3 à 10 fois moins puissant que la dronédarone.
Élimination
Après administration orale d'une dose marquée, environ 6 % de la quantité ingérée est excrétée dans l'urine, principalement sous la forme de métabolites (la forme inchangée n'est pas excrétée dans l'urine), et 84 % sont excrétés dans les fèces, principalement sous la forme de métabolites. Après une administration IV, la clairance plasmatique de la dronédarone oscille entre 130 et 150 l/h. La demi-vie d'élimination terminale du médicament avoisine les 25 à 30 heures, et celle de son métabolite N-débutylé, les 20 à 25 heures. La dronédarone et son métabolite sont complètement éliminés du plasma des patients dans les 2 semaines qui suivent la fin d'un traitement par 400 mg deux fois par jour.
Cinétique pour certains groupes de patients
La pharmacocinétique de la dronédarone chez les patients atteints de fibrillation auriculaire est semblable à celle observée chez les sujets en bonne santé. Les principales sources de variabilité de l'exposition à la dronédarone (âge, sexe, poids corporel, traitement concomitant par des inhibiteurs faibles ou modérés du CYP3A4) restent d'amplitude modeste (moins d'un facteur 2).
Sexe
L'exposition à la dronédarone est en moyenne 30 % plus élevée chez les femmes que chez les hommes.
Patients âgés
Sur le nombre total de participants aux études cliniques de la dronédarone, 73 % étaient âgés de 65 ans ou plus, et 34 % avaient 75 ans ou plus. L'exposition à la dronédarone est 23 % plus élevée chez les sujets de 65 ans ou plus que chez les moins de 65 ans.
Insuffisance hépatique
Chez les sujets qui présentent une insuffisance hépatique modérée, les expositions à la dronédarone totales et non liées sont multipliées par 1,3 et 2, respectivement. En revanche, l'exposition au métabolite actif est diminuée d'un facteur 1,6 à 1,9 (voir «Posologie/Mode d'emploi»).
L'effet de l'insuffisance hépatique sévère sur la pharmacocinétique de la dronédarone n'a pas été évalué (voir «Contre-indications»).
Insuffisance rénale
Des patients insuffisants rénaux figuraient dans les études cliniques. De façon cohérente avec la très faible excrétion rénale de la dronédarone, aucune modification pharmacocinétique n'a été observée chez ces personnes, en particulier chez celles dont l'insuffisance rénale était sévère (voir «Posologie/Mode d'emploi»).
Données précliniquesLa dronédarone n'a pas présenté d'effet génotoxique lors d'un test du micronucleus pratiqué in vivo sur des souris, ainsi que de quatre tests effectués in vitro:
le test d'Ames avec ou sans activation métabolique, un test de réparation de l'ADN réalisé sur des hépatocytes de rat, une recherche de mutation génétique sur des fibroblastes de hamsters et enfin, une étude cytogénétique portant sur des lymphocytes humains.
Dans des études de carcinogénicité par voie orale d'une durée de 2 ans, la posologie la plus élevée de la dronédarone administrée pendant 24 mois était de 70 mg/kg/jour chez le rat et de 300 mg/kg/jour chez la souris.
Les observations ont consisté en une élévation de l'incidence des tumeurs de la glande mammaire chez les souris femelles, des sarcomes histiocytaires chez la souris et des hémangiomes au niveau des ganglions lymphatiques mésentériques du rat; tous seulement sous la plus forte posologie testée (correspondant à une exposition 5 à 10 fois plus élevée que celle constatée à dose thérapeutique chez l'être humain). Les hémangiomes ne sont pas des modifications précancéreuses et ils ne se transforment pas en hémangiosarcomes malins, que ce soit chez les animaux ou chez l'être humain. Aucune de ces observations n'a été considérée comme pertinente pour l'être humain.
Dans des études de toxicité chronique, une phospholipidose légère et réversible (accumulation de macrophages spumeux) a été observée dans les ganglions lymphatiques mésentériques, principalement chez le rat. Cet effet est considéré comme spécifique à cette espèce et non pertinent pour l'être humain.
Une très légère élévation de créatinine plasmatique a été observée chez le rat, à partir de la dose de 10 mg/kg/jour de dronédarone dans une étude de toxicité en doses répétées pendant 6 mois; cet effet n'est pas le signe d'une atteinte rénale, puisqu'aucune modification histologique rénale n'a été observée.
Des effets phototoxiques ont été observés chez des cobayes traités par dronédarone à fortes doses.
L'administration de doses élevées de dronédarone a entraîné d'importants effets sur le développement embryo-fœtal du rat, tels qu'une multiplication des pertes après nidation, une réduction des poids fœtaux et placentaires et des malformations externes, viscérales et squelettiques.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
A conserver dans l'emballage d'origine à 15-30 °C.
Numéro d’autorisation59292 (Swissmedic).
PrésentationMultaq comprimés blancs oblongs pelliculés à 400 mg: 60 (B)
Titulaire de l’autorisationsanofi-aventis (suisse) sa, 1214 Vernier/GE
Mise à jour de l’informationSeptembre 2024
|