CompositionPrincipes actifs
Certolizumab pégol, produit à partir de bactéries Escherichia coli génétiquement modifiées.
Excipients
Acétate de sodium (1.36 mg/ml), chlorure de sodium (7.31 mg/ml), eau pour préparation injectable q.s ad solutionem pour 1ml.
Quantité maximale de sodium par ml: 3.3 mg.
Indications/Possibilités d’emploiMaladie de Crohn
Cimzia est indiqué pour l'induction d'une réponse clinique, ainsi que pour le maintien d'une réponse clinique et d'une rémission chez les patients adultes présentant une maladie de Crohn active qui n'ont pas suffisamment répondu à un traitement conventionnel.
Polyarthrite rhumatoïde
Cimzia, en association au méthotrexate (MTX), est indiqué
·pour le traitement de la polyarthrite rhumatoïde (PR) active, modérée à sévère, de l'adulte, lorsque la réponse aux traitements de fond (disease-modifying anti-rheumatic drugs; DMARDs), y compris le MTX, est inadéquate,
·pour le traitement de la PR sévère, active et évolutive chez les adultes non précédemment traités par le MTX ou d'autres DMARDs.
Cimzia peut être administré en monothérapie en cas d'intolérance au MTX ou lorsque la poursuite du traitement par le MTX est inadaptée.
Il a été montré que Cimzia ralentit la progression des dommages structuraux articulaires mesurés par radiographie et améliore les capacités fonctionnelles, lorsqu'il est administré en association au MTX.
Arthrite psoriasique
Cimzia, en association au méthotrexate (MTX), est indiqué dans le traitement de l'arthrite psoriasique active de patients adultes lorsque la réponse au traitement de fond (DMARDs) est insuffisante.
Cimzia améliore la capacité fonctionnelle physique des patients atteints d'arthrite psoriasique.
Cimzia peut être administré en monothérapie en cas d'intolérance au méthotrexate ou lorsque la poursuite du traitement par le méthotrexate est inadaptée.
Spondylarthrite axiale
Cimzia est indiqué dans le traitement de la spondylarthrite axiale (axSpA) active sévère de patients adultes incluant des patients souffrant de spondylarthrite ankylosante active sévère (SA) (également appelée spondylarthrite axiale radiographique) et des patients présentant une spondylarthrite axiale active sévère sans signes radiographiques (également appelée spondylarthrite axiale non radiographique) qui n'ont pas répondu de manière adéquate au traitement conventionnel ou qui n'ont pas toléré les AINS (anti-inflammatoires non stéroïdiens). Les patients présentant une spondylarthrite axiale active sévère non radiographiques devraient montrer des signes objectifs d'inflammation à l'imagerie par résonance magnétique (IRM) et un taux élevé de CRP (protéine C réactive).
Psoriasis en plaques
Cimzia est indiqué pour le traitement du psoriasis en plaques modéré à sévère chez les patients adultes qui nécessitent un traitement systémique ou une photothérapie.
Posologie/Mode d’emploiLe traitement devrait être initié et contrôlé par des médecins spécialistes ayant l'expérience du diagnostic et du traitement des pathologies dans lesquelles Cimzia est indiqué.
Après une formation adaptée à la technique d'injection, les patients peuvent s'auto-injecter Cimzia, si leur médecin le juge approprié et si le suivi médical nécessaire est assuré. Les sites d'injection adaptés sont la cuisse ou l'abdomen.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Une dose de 200 mg est administrée en une injection sous-cutanée de 1 ml.
Une dose de 400 mg est administrée en deux injections sous-cutanées de 1 ml.
Instauration du traitement
Adultes (à partir de 18 ans):
La dose initiale de Cimzia recommandée chez les patients adultes est de 400 mg (administrés en 2 injections sous-cutanées de 200 mg chacune) au début (semaine 0) et aux semaines 2 et 4.
Traitement d'entretien
Adultes (à partir de 18 ans):
Maladie de Crohn
Cimzia est indiqué pour une utilisation par voie sous-cutanée chez l'adulte.
Après la dose initiale, la dose d'entretien de Cimzia recommandée chez les patients adultes présentant une maladie de Crohn est de 400 mg toutes les 4 semaines.
Polyarthrite rhumatoïde
Après la dose initiale, la dose d'entretien de Cimzia recommandée chez les patients adultes présentant une polyarthrite rhumatoïde est de 200 mg toutes les 2 semaines sous forme d'injection sous-cutanée. Dans les cas où une dose toutes les 2 semaines n'est pas réalisable, une dose de 400 mg toutes les 4 semaines s'est montrée sûre et efficace (cf. passage «Efficacité clinique»).
Lorsque cela est approprié, l'administration de MTX devra être poursuivie pendant le traitement par Cimzia.
Chez les patients non précédemment traités par méthotrexate, seule la posologie de 200 mg toutes les 2 semaines a été étudiée jusqu'à un an.
Arthrite psoriasique
Après la dose initiale, la dose d'entretien de Cimzia recommandée chez les patients adultes présentant une arthrite psoriasique est de 200 mg toutes les 2 semaines. En fonction de la réponse clinique du patient, on peut envisager de manière alternative une dose de 400 mg toutes les 4 semaines.
Spondylarthrite axiale
Après la dose initiale, la dose d'entretien de Cimzia recommandée chez les patients adultes présentant une spondylarthrite axiale est de 200 mg toutes les 2 semaines ou de 400 mg toutes les 4 semaines. Après au moins un an de traitement par Cimzia, chez les patients présentant une rémission persistante, une dose d'entretien réduite de 200 mg toutes les quatre semaines peut être envisagée (cf. passage «Efficacité clinique»).
Pour les indications susmentionnées (polyarthrite rhumatoïde, arthrite psoriasique et spondylarthrite axiale), les données disponibles suggèrent qu'une réponse clinique est habituellement obtenue au cours des 12 premières semaines de traitement. La poursuite du traitement devra être reconsidérée avec attention chez les patients pour lesquels aucun bénéfice thérapeutique n'a été observé à l'issue des 12 premières semaines de traitement.
Psoriasis en plaques
Après la dose initiale, la dose d'entretien de Cimzia chez les patients adultes présentant un psoriasis en plaques est de 200 mg toutes les 2 semaines. Une dose de 400 mg toutes les 2 semaines peut être envisagée chez les patients ayant une réponse insuffisante (voir «Efficacité clinique»).
Les données disponibles chez l'adulte ayant un psoriasis en plaques suggèrent qu'une réponse clinique est habituellement obtenue au cours des 16 premières semaines de traitement. La poursuite du traitement devra être reconsidérée avec attention chez les patients pour lesquels aucun bénéfice thérapeutique n'a été observé au cours des 16 premières semaines de traitement. Chez certains patients ayant obtenu une réponse initiale partielle, une amélioration ultérieure peut être observée en poursuivant le traitement au-delà de 16 semaines.
Durée du traitement
Maladie de Crohn:
La sécurité et l'efficacité de Cimzia n'ont jusqu'à présent pas été suffisamment vérifiées pour un traitement de plus de 6 mois.
Polyarthrite rhumatoïde chez les patients adultes sans traitement préalable par le MTX ou d'autres DMARD:
Chez ces patients, la sécurité et l'efficacité de Cimzia ont été démontrées sur une durée de traitement allant jusqu'à 1 an. Concernant le maintien d'une réponse au-delà d'un an, les données sont limitées (voir «Propriétés/Effets»).
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucune étude clinique spécifique portant sur l'évaluation des effets de troubles de la fonction hépatique sur la pharmacocinétique du certolizumab pégol n'a été menée. L'analyse pharmacocinétique portant sur la population n'a pas permis de tirer des conclusions concernant les conséquences de troubles de la fonction hépatique, car cette analyse ne comprenait qu'un faible nombre de patients présentant des troubles significatifs de la fonction hépatique.
Les données sont insuffisantes pour formuler des recommandations posologiques chez des patients présentant des troubles de la fonction hépatique.
Patients présentant des troubles de la fonction rénale
Aucune étude clinique spécifique portant sur l'évaluation des effets de troubles de la fonction rénale sur la pharmacocinétique du certolizumab pégol n'a été menée. L'analyse pharmacocinétique portant sur la population n'a toutefois montré aucun effet dépendant de la clairance de la créatinine; c.-à-d. que de légers troubles de la fonction rénale n'ont probablement aucune conséquence significative.
Les données sont insuffisantes pour recommander un dosage pour les patients souffrant de dysfonctionnement rénal.
Patients âgés (> 65 ans)
La prudence est de rigueur lors du traitement de sujets âgés. On ne dispose que de données limitées relatives à la sécurité et à l'efficacité chez des patients âgés atteints de la maladie de Crohn et de polyarthrite rhumatoïde; une analyse pharmacocinétique se rapportant à la population n'a toutefois pas mis en évidence d'effets liés à l'âge.
Aucun ajustement posologique n'est nécessaire chez les patients âgés.
Enfants et adolescents (0-17 ans)
L'utilisation de Cimzia dans la population pédiatrique n'est pas autorisée. Aucune recommandation posologique ne peut être donnée. Les données actuellement disponibles sont décrites dans les rubriques «Propriétés/Effets» et «Effets indésirables».
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients conformément à la composition.
Tuberculose évolutive ou autres infections sévères telles que sepsis, abcès ou infections opportunistes.
Insuffisance cardiaque modérée à sévère (de classe III/IV dans la classification NYHA).
Mises en garde et précautionsImmunosuppression
Étant donné que le TNF intervient dans l'apparition d'inflammations et module les immunoréactions cellulaires, il est possible que les agents anti-TNF tels que Cimzia aient un impact sur les réponses immunitaires aux infections et aux tumeurs malignes. En l'état actuel des connaissances, les effets d'un traitement par Cimzia sur le développement et l'évolution de tumeurs malignes, ainsi que d'infections actives et/ou chroniques ne sont pas totalement élucidés.
Les patients atteints de polyarthrite rhumatoïde peuvent ne pas présenter de symptômes caractéristiques d'une infection, dont la fièvre, en raison de leur maladie et des traitements médicamenteux concomitants. La détection précoce de toute infection est donc essentielle pour initier un traitement le plus rapidement possible.
Infections
Les patients doivent faire l'objet d'une surveillance attentive au regard des infections, comme une tuberculose, avant, pendant et après le traitement par Cimzia; il faut alors tenir compte de la longue demi-vie du produit. L'élimination de Cimzia pouvant durer jusqu'à 5 mois, la surveillance devra être poursuivie pendant toute cette période.
Chez les patients présentant des infections cliniquement importantes, y compris des infections chroniques ou localisées, aucun traitement par Cimzia ne doit être instauré avant que l'infection ne soit contrôlée.
Les patients chez qui une nouvelle infection apparaît pendant le traitement par Cimzia doivent faire l'objet d'une surveillance attentive.
Si une nouvelle infection grave apparaît chez un patient, il convient d'arrêter le traitement par Cimzia jusqu'à ce que les infections soient maîtrisées. Chez les patients ayant des antécédents d'infections récurrentes fréquentes ou d'infections opportunistes ou des maladies sous-jacentes les prédisposant aux infections, le médecin doit soigneusement évaluer l'utilisation de Cimzia.
Chez les patients qui ont été traités par des agents anti-TNF tels que Cimzia, des cas d'infections graves, de septicémie, de tuberculoses (également miliaires, disséminées et extrapulmonaires), de mycoses opportunistes, p.ex. histoplasmose, pneumocystose, zona, infections virales et/ou parasitaires, et quelques cas de décès, ont été rapportés.
Un nombre important de ces infections graves sont survenues chez des patients sous traitement concomitant par immunosuppresseurs susceptibles de les prédisposer à des infections en plus de leur maladie.
Tuberculose
Avant de débuter un traitement par Cimzia, une tuberculose active ou inactive (latente) doit être recherchée chez tous les patients. Cette recherche doit comprendre une anamnèse détaillée précisant les antécédents personnels de tuberculose, d'éventuels contacts antérieurs avec un patient présentant une tuberculose active et les traitements immunosuppresseurs anciens et/ou en cours.
Des tests appropriés, i.e. intradermoréaction ou d'autres tests biologiques de dépistage de la tuberculose et radiographie thoracique, devront être réalisés chez tous les patients. Il est rappelé aux médecins prescripteurs qu'une intradermoréaction ou d'autres tests biologiques de dépistage de la tuberculose peuvent s'avérer faussement négatifs, surtout chez un patient sévèrement malade ou immunodéprimé.
En cas de diagnostic d'une tuberculose latente, un traitement antituberculeux adapté doit être mis en œuvre avant de commencer le traitement par Cimzia, en tenant compte des recommandations nationales.
Un traitement antituberculeux doit également être envisagé avant l'initiation de Cimzia chez les patients ayant des antécédents de tuberculose latente ou active, pour lesquels l'administration d'un traitement antituberculeux approprié n'a pu être confirmée, ainsi que chez les patients à risque significatif de tuberculose malgré un test négatif pour une tuberculose latente. En cas de suspicion d'une tuberculose latente, des tests biologiques de dépistage de la tuberculose devront être envisagés avant de commencer le traitement par Cimzia, que le patient ait ou non été vacciné par le BCG.
En cas de diagnostic d'une tuberculose active, Cimzia ne doit pas être initié.
En cas de diagnostic ou de suspicion d'une tuberculose latente ou en présence de facteurs de risque considérables, il faut initier un traitement prophylactique de la tuberculose 1 mois avant de débuter le traitement par Cimzia. Le rapport bénéfice/risque du traitement par Cimzia devra être très soigneusement évalué. La décision d'entreprendre un traitement antituberculeux chez un patient individuel doit avoir lieu avec l'avis d'un médecin spécialisé ayant l'expérience de la tuberculose.
Malgré un traitement prophylactique préalable ou concomitant contre la tuberculose, des cas de tuberculose active sont apparus chez des patients qui étaient traités par des antagonistes du TNF, y compris Cimzia. Certains patients dont la tuberculose avait été traitée avec succès ont à nouveau développé une tuberculose pendant le traitement par des antagonistes du TNF, y compris Cimzia.
Tous les patients doivent être informés de la nécessité de consulter un médecin si des signes ou des symptômes évoquant une tuberculose (p. ex., toux persistante, asthénie/perte de poids, fébricule) apparaissent pendant ou après un traitement par Cimzia.
Réactivation du virus de l'hépatite B (VHB)
Une réactivation du virus de l'hépatite B s'est produite chez des patients porteurs chroniques de ce virus (c.-à-d. positifs pour l'antigène de surface) et traités par des antagonistes du TNF y compris Cimzia. Certains cas ont eu une issue fatale.
Une infection par VHB doit être recherchée avant d'initier un traitement par Cimzia. Pour les patients dont le test d'infection par le VHB est positif, il est recommandé de consulter un médecin spécialisé dans le traitement de l'hépatite B.
Aucune donnée adéquate n'est disponible concernant les patients porteurs du VHB traités par un anti-TNF, en association avec un traitement antiviral, en prévention d'une réactivation du VHB. Chez les porteurs du VHB nécessitant un traitement par anti-TNF, une surveillance étroite des signes cliniques et biologiques d'infection active par le VHB doit être mise en place et maintenue tout au long du traitement et pendant plusieurs mois après son arrêt, en particulier si le patient est également traité par des corticoïdes.
En cas de réactivation du VHB, Cimzia doit être interrompu et un traitement antiviral efficace ainsi qu'un traitement de soutien adapté doivent être initiés. La sécurité de la reprise du traitement par un anti-TNF après contrôle de la réactivation du VHB n'a pas été établie.
Par conséquent, les prescripteurs doivent envisager avec prudence la reprise du traitement par Cimzia dans cette situation et surveiller étroitement leurs patients.
Tumeurs malignes et troubles lymphoprolifératifs
Dans les études contrôlées menées avec Cimzia et d'autres agents anti-TNF, il a été observé plus de cas de lymphomes et d'autres tumeurs malignes parmi les patients ayant reçu ces agents que chez les patients du groupe témoin.
La fréquence était toutefois faible et le temps de suivi était plus court chez les patients sous placebo que chez les patients traités par des antagonistes du TNF.
Chez les patients souffrant de la maladie de Crohn ou d'une polyarthrite rhumatoïde, en particulier chez ceux chez qui la maladie est fortement active, nécessitant une exposition chronique aux immunosuppresseurs, il y a un risque plus important de développer des lymphomes que dans le reste de la population, et ce également en l'absence d'un traitement par anti-TNF. En l'état actuel des connaissances, tout risque de développer des lymphomes ou d'autres tumeurs malignes chez les patients traités avec un agent anti-TNF ne peut être écarté.
On ne dispose d'aucune étude relative aux patients présentant des antécédents de tumeurs malignes ou à la poursuite du traitement chez des patients qui ont développé une tumeur maligne lors du traitement par Cimzia. C'est pourquoi, chez ces patients tout traitement par Cimzia ne doit être envisagé qu'avec une prudence toute particulière.
Des cas de leucémie aiguë et chronique ont été rapportés en relation avec l'utilisation post-marketing des agents anti-TNF dans le traitement de la polyarthrite rhumatoïde et dans d'autres indications. Les patients souffrant de polyarthrite rhumatoïde peuvent présenter un risque plus élevé (jusqu'à 2 fois plus élevé) de développer une leucémie que le reste de la population, et ce même sans traitement par des agents anti-TNF.
Chez l'enfant et l'adolescent (≤18 ans) ayant été traités par des agents anti-TNF, des cas de tumeurs malignes ont été rapportés, dont certaines ont eu une issue fatale. Dans environ la moitié de ces cas, il s'agissait de lymphomes, lymphomes Hodgkiniens et non-Hodgkiniens inclus. Les autres cas comprenaient différentes autres tumeurs malignes, y compris des tumeurs malignes rares qui sont généralement en rapport avec l'immunosuppression et d'autres qui ne surviennent généralement ni chez l'enfant ni chez l'adolescent.
Les tumeurs malignes sont survenues après une durée médiane de 30 mois de traitement (fourchette de 1 à 84 mois). La plupart des patients ont simultanément reçu des immunosuppresseurs. Ces cas concernaient des notifications post-marketing issues de différentes sources, registres et notifications spontanées inclus.
Cimzia n'est pas indiqué chez l'enfant et l'adolescent.
Depuis la commercialisation, des cas de lymphome hépatosplénique à lymphocytes T (HSTCL) ont été rapportés chez des patients, majoritairement des adolescents et de jeunes hommes atteints d'une maladie intestinale inflammatoire chronique et ayant une thérapie concomitante d'aziathioprine ou de 6-mercaptopurine avec un antagoniste du TNF (cf. «Effets indésirables»). Cette forme rare de lymphome à lymphocytes T a une évolution très agressive et une issue généralement fatale.
Mélanome et carcinome à cellules de Merkel
Des mélanomes et des carcinomes à cellules de Merkel ont été rapportés chez des patients traités par des antagonistes du TNF, y compris Cimzia (voir «Effets indésirables»). Il est recommandé d'effectuer régulièrement des examens cutanés pour tous les patients, en particulier ceux présentant des facteurs de risque de cancer de la peau.
Bronchopneumopathie chronique obstructive (BPCO)
Lors d'une étude clinique exploratoire évaluant l'utilisation d'un autre anti-TNF, l'infliximab, chez des patients ayant une bronchopneumopathie chronique obstructive (BPCO) modérée à sévère, plus de cancers, majoritairement du poumon et de la tête et du cou, ont été rapportés chez les patients traités par infliximab que chez ceux du groupe contrôle. Tous les patients avaient des antécédents de tabagisme important. En conséquence, des précautions doivent être prises lors de l'utilisation d'un anti-TNF chez les patients souffrant de BPCO ainsi que chez les patients présentant un risque accru de cancer du fait d'un tabagisme important.
Insuffisance cardiaque
Dans une étude clinique relative à un autre antagoniste du TNF, on a observé une aggravation de l'insuffisance cardiaque et une mortalité accrue par insuffisance cardiaque congestive.
Des cas d'aggravation d'insuffisance cardiaque congestive ont également été observés chez des patients traités par Cimzia. Cimzia doit être utilisé avec précaution chez les patients atteints d'insuffisance cardiaque légère (classe I/II de la NYHA). Cimzia est contre-indiqué en cas d'insuffisance cardiaque modérée à sévère. Le traitement par Cimzia doit être arrêté chez les patients qui développent une nouvelle insuffisance cardiaque ou une aggravation de leur insuffisance cardiaque.
Seul un petit nombre d'événements cardiovasculaires ont été rapportés dans les études relatives à la maladie de Crohn. La prudence est de rigueur lors de l'utilisation de Cimzia chez des patients âgés et en présence de tels facteurs de risque prédisposant à des affections cardiovasculaires ou inflammatoires.
Atteintes hématologiques
Dans de rares cas, les agents anti-TNF ont été associés à des pancytopénies, comme une anémie aplasique. Des événements indésirables affectant le système hématologique, comme une cytopénie médicalement significative (entre autres, leucopénie, pancytopénie, thrombocytopénie), ont rarement été rapportés en association avec Cimzia. Chez les patients présentant des antécédents ou des manifestations récentes d'anomalies hématologiques significatives, la prudence est de rigueur concernant un traitement par Cimzia. Tous les patients doivent être informés de la nécessité de consulter immédiatement un médecin en présence de signes ou de symptômes évoquant une éventuelle affection sanguine ou une infection (p. ex., fièvre persistante, hématomes, hémorragies, pâleur). Chez les patients présentant des anomalies hématologiques significatives avérées, il faut envisager l'arrêt du traitement par Cimzia.
Atteintes neurologiques
Dans de rares cas, des agents anti-TNF ont été associés à une aggravation des symptômes cliniques et/ou des signes radiographiques d'atteintes démyélinisantes, sclérose en plaques incluse, et à des atteintes démyélinisantes périphériques, syndrome de Guillain-Barré inclus. Chez les patients présentant des antécédents ou des manifestations récentes d'atteintes démyélinisantes du système nerveux central ou périphérique, le médecin prescripteur qui envisage l'utilisation de Cimzia doit faire preuve de prudence. Chez les patients traités par Cimzia, on a rapporté de rares cas de troubles neurologiques, comme des convulsions, des névrites optiques, des neuropathies périphériques et myélite transverse.
Hypersensibilité
Dans de rares cas, l'administration de Cimzia a été associée aux symptômes suivants susceptibles de représenter une réaction d'hypersensibilité: angio-œdème, dyspnée, hypotension, éruptions cutanées, maladie sérique et urticaire. Quelques-unes de ces réactions d'hypersensibilité sont apparues dès la première administration de Cimzia. Lorsque de telles réactions apparaissent, l'administration de Cimzia doit être interrompue et un traitement approprié doit être instauré.
On ne dispose d'aucune donnée relative à l'utilisation de Cimzia chez des patients présentant une réaction d'hypersensibilité grave à un autre agent anti-TNF; chez ces patients, la prudence est de rigueur.
Hypersensibilité au latex
Le protecteur d'aiguille à l'intérieur du capuchon amovible de la seringue préremplie et du stylo pré-rempli Cimzia contient 7% d'un dérivé de caoutchouc naturel.
Le protecteur d'aiguille n'entre pas en contact direct avec le patient ou la personne réalisant l'injection. Néanmoins, un risque potentiel de réactions d'hypersensibilité ne peut pas être complètement exclu chez les personnes sensibles au latex.
Processus auto-immuns
Le traitement par Cimzia peut entraîner la formation d'auto-anticorps, ainsi que dans de rares cas, le développement d'un syndrome pseudolupique. Si des symptômes évocateurs d'un syndrome pseudolupique se développent chez un patient à la suite du traitement par Cimzia, le traitement doit être arrêté. Chez les patients ayant des antécédents de lupus, la prudence est de rigueur. On ne dispose d'aucune donnée relative à l'utilisation de Cimzia chez des patients ayant souffert d'un lupus ou d'autres maladies auto-immunes après l'administration d'un autre agent anti-TNF.
Vaccinations
Les patients traités par Cimzia peuvent être vaccinés excepté avec des vaccins vivants ou vivants atténués.
Aucune donnée n'est disponible sur la réponse à la vaccination ou sur la transmission secondaire d'infections par des vaccins vivants chez les patients traités par Cimzia. Il convient de ne pas administrer de vaccins vivants ou atténués en même temps que Cimzia.
Au cours d'un essai clinique contrôlé versus placebo chez des patients ayant une polyarthrite rhumatoïde, aucune différence n'a été observée quant à la réponse immunitaire entre les groupes Cimzia et placebo lors de l'administration simultanée du vaccin pneumococcique polyosidique ou du vaccin grippal et de Cimzia. Dans les deux groupes, un nombre similaire de patients a développé des anticorps protecteurs. Les patients traités par Cimzia et méthotrexate ont eu une réponse immunitaire humorale plus faible que les patients recevant Cimzia seul. La signification clinique de cette observation n'est pas connue.
Cimzia n'inhibe pas la réponse immunitaire humorale au vaccin pneumococcique polyosidique et au vaccin grippal.
Administration concomitante d'autres agents biologiques
Des infections sévères ont été observées dans des études cliniques au cours desquelles l'anakinra (un antagoniste de l'interleukine-1) ou l'abatacept et d'autres agents anti-TNF ont été administrés de façon concomitante, sans bénéfice clinique supplémentaire comparativement à l'anti-TNF administré seul. En raison de la nature des effets indésirables observés lors de l'association thérapeutique d'un anti-TNF et l'anakinra ou l'abatacept, des toxicités similaires peuvent résulter de l'association entre Cimzia et anakinra ou abatacept. On ne dispose d'aucune étude relative à l'administration concomitante de Cimzia et d'anakinra ou d'abatacept, c'est pourquoi cette association est déconseillée.
Chirurgie
L'expérience concernant la tolérance au cours d'interventions chirurgicales chez les patients traités par Cimzia est limitée. La demi-vie de quatorze jours du certolizumab pégol doit être prise en considération si une intervention chirurgicale est programmée. Un patient traité par Cimzia, nécessitant une intervention chirurgicale, doit être étroitement surveillé afin de détecter toute infection et des actions appropriées mises en œuvre.
Test de l'aPTT (ou TCA: temps de céphaline activé) in vitro
Une interférence avec certains tests de la coagulation a été détectée chez des patients traités par Cimzia. Cimzia peut entraîner des valeurs du TCA faussement élevées chez des patients sans anomalie de la coagulation. Aucune donnée ne prouve que le traitement par Cimzia ait un effet sur la coagulation in vivo. Au cours du traitement par Cimzia, l'interprétation de résultats anormaux des tests de la coagulation devra être prudente. Aucune interférence avec la détermination du temps de thrombine (TT) et du taux de prothrombine (TP) n'a été observée.
Les réactifs suivants n'ont pas montré une influence sur les résultats des tests de l'aPTT: Dade® Actin® FS activated PTT, Dade® Actin® FSL Activated PTT, HemosiL™ SynthAFax, HemosiL™ SynthASil, Platelin® LS (distribué aussi sous le nom de TriniCLOaPTT HS). D'autres tests de détermination du TCA pourraient également être affectés.
Obstruction de l'intestin grêle chez les patients atteints de la maladie de Crohn
L'absence de réponse au traitement de la maladie de Crohn pourrait être due à la présence d'une sténose fibreuse fixée nécessitant une intervention chirurgicale.
Patients âgés
Le traitement des patients de plus de 60 ans doit être envisagé avec prudence et une attention particulière portée au risque d'infections.
Fertilité chez l'homme
Lors d'une étude clinique évaluant les effets d'une dose unique de certolizumab pégol sur la qualité du sperme (quantité de sperme, numération et concentration de spermatozoïdes, motilité progressive, motilité globale en pourcentage, vitalité et morphologie), 20 hommes adultes volontaires sains ont été randomisés pour recevoir une dose unique de 400 mg de Cimzia par voie sous-cutanée ou un placebo. Pendant la période de suivi de 14 semaines, aucun effet du traitement par certolizumab pégol n'a été observé sur la qualité du sperme par rapport au placebo.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose (200 mg), c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsLe traitement concomitant avec le méthotrexate, les corticoïdes, les anti-inflammatoires non stéroïdiens (AINS) et les antalgiques n'a montré aucun effet sur la pharmacocinétique du certolizumab pégol lors d'une analyse pharmacocinétique de population menée chez des patients atteints de la maladie de Crohn et d'une polyarthrite rhumatoïde.
L'utilisation concomitante d'immunosuppresseurs (p. ex., le méthotrexate, la 6-mercaptopurine ou l'azathioprine) n'a entraîné aucun effet cliniquement important sur la pharmacocinétique du certolizumab pégol.
L'association de Cimzia et d'anakinra ou d'abatacept n'est pas recommandée (voir rubrique «Mises en garde et précautions»).
La pharmacocinétique du certolizumab pégol a également été étudiée dans le cadre d'une étude d'interactions pharmacocinétiques menée auprès de 16 patients souffrant de polyarthrite rhumatoïde, recevant des doses stables de méthotrexate (entre 5 et 17.5 mg par semaine).
L'administration concomitante de certolizumab pégol et de méthotrexate n'a entraîné aucun effet significatif sur la pharmacocinétique du méthotrexate, alors que la pharmacocinétique du certolizumab pégol était comparable à celle observée précédemment chez des sujets sains.
Grossesse, allaitementGrossesse
Les études animales utilisant un anti-TNFα de rat chez le rongeur n'ont révélé aucun signe évocateur d'une altération de la fertilité ou d'une fœtotoxicité. Les études d'évaluation au regard d'une toxicité sur la reproduction humaine sont insuffisantes (voir «Données précliniques»). Les études non-cliniques suggèrent que le taux de transfert placentaire d'un homologue Fab-fragment du certolizumab pegol (sans fragment Fc) est faible ou négligeable (voir «Données précliniques»).
Les données de plus de 1300 grossesses exposées à Cimzia avec des issues connues collectées de manière prospective n'ont pas mis en évidence d'effet tératogène de Cimzia. Ces données comprennent plus de 1000 grossesses au cours desquelles Cimzia a été utilisé au premier trimestre. Ces données doivent être interprétées avec prudence en raison de limitations méthodologiques (telles que des informations incomplètes, le manque de groupe témoin et trop peu de rapports).
Dans une étude clinique, 16 femmes ont été traitées pendant la grossesse avec certolizumab pegol (200 mg toutes les deux semaines ou 400 mg toutes les quatre semaines). Les concentrations plasmatiques de certolizumab pegol mesurées chez 14 nourrissons à la naissance étaient inférieures à la limite de détection (Below the Limit of Quantification - BLQ) pour 13 échantillons; pour un échantillon, une valeur de 0.042 µg/ml avec un ratio plasmatique nourrisson/mère de 0.09 % a été déterminée à la naissance. Aux semaines 4 et 8, toutes les concentrations plasmatiques chez les nourrissons étaient inférieures à la limite de détection.
Les données publiées indiquent que le risque d'issue défavorable sur la grossesse chez les femmes atteintes d'arthrite rhumatoïde, de spondylarthrite ankylosante ou de la maladie de Crohn est lié à l'activité de la maladie de la mère, car une maladie active augmente le risque d'issue défavorable de la grossesse, notamment de fausse-couche, de naissance prématurée (avant la 37ème semaine de grossesse), de faible poids à la naissance (<2500 g) et de petite taille mesurée à la l'âge gestationnel lors de la naissance.
Si un traitement par anti-TNF est clairement indiqué, Cimzia peut être envisagé lors de la planification d'une grossesse ou pendant une grossesse en cours.
En raison de son effet inhibiteur sur le TNFα, Cimzia, administré pendant la grossesse, pourrait affecter les réponses immunitaires normales du nouveau-né. Même si les taux de certolizumab pégol sont faibles chez le nourrisson, leur signification clinique n'est pas connue. Il est recommandé d'attendre au moins 5 mois après la dernière administration de Cimzia chez la mère au cours de la grossesse avant d'administrer des vaccins vivants ou atténués au nourrisson (par ex. vaccination BCG), à moins que le bénéfice l'emporte clairement sur le risque théorique d'une administration de vaccins vivants ou vivants atténués chez les nourrissons.
Femmes en âge de procréer
L'utilisation d'une contraception appropriée doit être envisagée chez les femmes en âge de procréer. Pour les femmes planifiant une grossesse, la poursuite de la contraception doit être envisagée pendant 5 mois après la dernière dose de Cimzia en raison de son taux d'élimination (voir la rubrique «Pharmacocinétique»). Toutefois, la nécessité de traitement chez les femmes doit également être prise en compte (voir ci-dessus).
Allaitement
Dans une étude clinique menée sur 17 femmes allaitantes traitées par Cimzia, un transfert minimal de certolizumab pégol du plasma dans le lait maternel a été observé. Le pourcentage de la dose maternelle de certolizumab pégol transféré au nourrisson dans les 24 heures a été estimé entre 0.04 % et 0.30 %. Comme le certolizumab pégol est une protéine éliminée dans le tractus gastroinestinal après administration orale, la biodisponibilité absolue attendue est très faible.
Les bénéfices de l'allaitement pour le nouveau-né en termes de développement et de santé doivent être mis en balance avec la nécessité clinique du traitement par Cimzia pour la mère et les effets secondaires possibles pour le nourrisson allaité liés à Cimzia ou à la maladie sous-jacente de la mère.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été conduite.
Effets indésirablesAu total, 1'350 sujets atteints de la maladie de Crohn et 4'049 patients atteints de polyarthrite rhumatoïde ont été traités par Cimzia; 426 sujets atteints de la maladie de Crohn et 1'137 sujets atteints de polyarthrite rhumatoïde ont reçu un placebo.
Les données ci-dessous sont basées sur les effets indésirables observés dans le cadre de ces études contrôlées et des études ouvertes de suivi.
Sur les 1'350 patients avec la maladie de Crohn, 498 ont été traités pendant 6 mois et 122 pendant 12 mois.
Dans les études contrôlées versus placebo, les patients traités par Cimzia ont eu une durée d'exposition environ 4 fois supérieure à celle des patients du groupe placebo. Cette différence d'exposition est principalement due aux sorties prématurées d'étude, plus fréquentes chez les patients ayant reçu le placebo. Par ailleurs, les études RA-I et RA-II prévoyaient une sortie obligatoire à la semaine 16 pour les non-répondeurs; la majorité de ces derniers avait reçu du placebo.
Dans les études contrôlées, on a observé des effets indésirables chez 40.4% des patients atteints de la maladie de Crohn et chez 34.0% des patients atteints de polyarthrite rhumatoïde du groupe Cimzia, ainsi que chez 38.7% des patients atteints de la maladie de Crohn et chez 24.9% des patients atteints de polyarthrite rhumatoïde du groupe placebo. Dans les études contrôlées, 8.2% des patients atteints de la maladie de Crohn et 4.4% des patients atteints de polyarthrite rhumatoïde du groupe Cimzia, ainsi que 6.4% des patients atteints de la maladie de Crohn et 2.7% des patients atteints de polyarthrite rhumatoïde du groupe placebo ont interrompu le traitement en raison d'effets indésirables.
Chez les patients atteints de la maladie de Crohn et de la polyarthrite rhumatoïde, les réactions indésirables les plus graves étaient des infections et des tumeurs malignes (voir «Mises en garde et précautions»).
Dans le cadre des études cliniques contrôlées relatives à Cimzia, les événements indésirables les plus fréquents chez les patients atteints de la maladie de Crohn étaient les céphalées (12% Cimzia, 17.1% placebo), les rhinopharyngites (8.9% Cimzia, 7.7% placebo) et une aggravation de la maladie de Crohn (8.9% Cimzia, 10.8% placebo).
Des études contrôlées contre placebo dans la polyarthrite rhumatoïde ont montré que les effets indésirables les plus fréquents étaient des infections et des infestations chez 14.4% des patients traités par Cimzia et 8.0% des patients recevant le placebo, des troubles de l'état de santé général et des problèmes au site d'injection chez 8.8% des patients traités par Cimzia et 7.4% des patients recevant le placebo, ainsi que des dysfonctionnements de la peau et du tissu cellulaire sous-cutané chez 7.0% des patients traités par Cimzia et 2.4% des patients recevant le placebo.
Chez les patients atteints de la maladie de Crohn, une aggravation de la maladie de Crohn (2.4% Cimzia, 2.1% placebo), des abcès péri-anaux (0.8% Cimzia, 0.3% placebo), un zona (0.6% Cimzia, 0% placebo) et des douleurs dans le bas-ventre (0.4% Cimzia, 0.3% placebo), étaient les causes les plus fréquentes d'arrêt du traitement par Cimzia.
Les événements indésirables les plus fréquents qui ont conduit à l'arrêt de la prise de Cimzia chez des patients souffrant de polyarthrite rhumatoïde (0.3% Cimzia, 0.6% placebo), étaient des infections tuberculeuses (0.3% Cimzia, 0% placebo), de la pneumonie, des éruptions cutanées, de l'urticaire et de la pyrexie (tous ces événements: 0.2% Cimzia, 0% placebo).
Patients pédiatriques atteints de la maladie de Crohn
La sécurité chez les patients pédiatriques n'a pas été établie. CIMZIA a été étudié pour le traitement de patients pédiatriques atteints d'une maladie de Crohn modérément à sévèrement active. L'étude a été interrompue prématurément en raison d'un nombre élevé de patients ayant abandonné le traitement - principalement en raison d'une aggravation de la maladie sous-jacente.
Dans une étude clinique contrôlée versus placebo et MTX (C-EARLY), Cimzia a été étudié en association avec le MTX versus placebo/MTX chez 879 patients atteints de PR, n'ayant pas reçu préalablement de traitement de fond (DMARDs), pendant une durée allant jusqu'à 104 semaines. Le profil de sécurité dans le groupe de patients PR traités par Cimzia n'ayant pas reçu préalablement de traitement par DMARDs concordait avec celui observé en cas de PR dans d'autres études.
Arthrite psoriasique
Cimzia a été étudié chez 409 patients ayant une arthrite psoriasique active dans une étude clinique contrôlée contre placebo (PsA001). Le profil de sécurité de Cimzia chez des patients ayant une arthrite psoriasique est cohérent avec le profil de sécurité observé dans la polyarthrite rhumatoïde et lors de l'utilisation antérieure de Cimzia.
Spondylarthrite axiale
Cimzia a été initialement étudié chez 325 patients ayant une spondylarthrite axiale (y compris spondylarthrite ankylosante et spondylarthrite axiale non radiographique) dans une étude clinique contrôlée versus placebo (AS001). Cimzia a également été étudié chez 317 patients ayant une spondylarthrite axiale non radiographique dans une étude contrôlée versus placebo pendant 52 semaines (AS0006).
Cimzia a également été étudié chez des patients atteints de spondyloarthrite axiale (incluant la spondylarthrite ankylosante et la spondyloarthrite axiale non radiographique) dans une étude clinique d'une durée maximale de 96 semaines, qui comprenait une phase d'induction de 48 semaines en ouvert (N = 736) suivie par une phase de 48 semaines contrôlée versus placebo (N = 313) pour les patients en rémission persistante (C-OPTIMISE).
Dans les trois études, le profil de sécurité de Cimzia chez ces patients a été cohérent avec le profil de sécurité observé dans la polyarthrite rhumatoïde et lors de l'utilisation antérieure de Cimzia.
Psoriasis en plaques
Cimzia a été étudié chez 1'112 patients ayant un psoriasis dans des études contrôlées et en ouvert avec une durée maximale de suivi de 3 ans. Le programme de phase III comportait une phase de 16 semaines contrôlée par placebo, avec une phase de 12 semaines contrôlée par traitement actif dans l'une des études sur 3 ans, suivie d'une période de 32 semaines avec dose en aveugle et d'une phase de traitement en ouvert de 96 semaines. Les profils de sécurité à long terme de Cimzia 400 mg toutes les 2 semaines et Cimzia 200 mg toutes les 2 semaines étaient généralement similaires et correspondaient aux observations antérieures avec Cimzia.
Au cours des essais cliniques de phases II et III contrôlés jusqu'à la semaine 16, la proportion de patients ayant présenté des événements indésirables graves était de 3.5% pour Cimzia et de 3.7% pour le placebo.
La proportion de patients ayant interrompu le traitement en raison d'événements indésirables dans les études cliniques contrôlées était de 1.5% chez les patients traités par Cimzia et de 1.4% chez les patients recevant le placebo.
Les effets indésirables les plus fréquents rapportés jusqu'à la semaine 16 ont été observés dans les classes de système d'organes «Infections et infestations parasitaires» chez 6.1% des patients traités par Cimzia et 7% des patients recevant le placebo, «Troubles généraux et anomalies au site d'administration» chez 4.1% des patients traités par Cimzia et 2.3% des patients recevant le placebo et «Affections de la peau et du tissu sous-cutané» chez 3.5% des patients traités par Cimzia et 2.8% des patients recevant le placebo.
Les effets médicamenteux indésirables (observés au cours des études cliniques et en post-commercialisation) survenant plus fréquemment chez les patients traités par Cimzia que chez ceux sous placebo sont présentés ci-dessous.
Les effets indésirables au moins possiblement liés à Cimzia sont listés ci-dessous par fréquence (c.-à-d. le nombre de patients chez qui cette réaction est attendue) et par classe de systèmes d'organes en utilisant la classification suivante:
«très fréquents» (≥1/10), «fréquents» (≥1/100 à <1/10), «occasionnels» (≥1/1000 à <1/100), «rares» (≥1/10'000 à <1/1000), «très rares» (<1/10'000).
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Infections et infestations parasitaires
Fréquent: infections bactériennes (incluant tuberculose (aussi miliaire, disséminée et extrapulmonaire) et abcès). Infections virales (incluant herpès, papillomavirus et influenza).
Occasionnel: infections fongiques (incluant pneumocystose, histoplasmose, candidose), sepsis (incluant défaillance multiviscérale).
Tumeurs bénignes, malignes et non précisées (incl. kystes et polypes)
Occasionnel: tumeurs organiques solides, tumeurs gastro-intestinales (incluant cancer du rectum, cancer de l'intestin grêle), tumeurs bénignes et kystes (incluant papillome cutané), malignomes hématologiques et lymphatiques (y compris les lymphomes et la leucémie).
Rare: lésions pré-cancéreuses (incluant leucoplasie orale, naevus mélanocytaire), carcinomes cutanés hors mélanomes (incluant carcinome baso-cellulaire), mélanome.
Inconnue: carcinome à cellules de Merkel, lymphome hépatosplénique à lymphocytes T.
Affections hématologiques et du système lymphatique
Fréquent: leucopénie (incluant lymphocytopénie, neutropénie), troubles éosinophiles.
Occasionnel: anémie, thrombocytopénie, leucocytose, lymphadénopathie (incluant lymphadénite), thrombocytose.
Rare: pancytopénie, anomalies morphologiques des globules blancs, splénomégalie, érythrocytose.
Affections du système immunitaire
Occasionnel: lupus érythémateux, vasculite, hypersensibilité médicamenteuse (réaction anaphylactique), nouvelle poussée ou aggravation de psoriasis (incluant psoriasis palmoplantaire pustuleux) et manifestations apparentées, manifestations allergiques (incluant allergies multiples), auto-anticorps positifs.
Rare: maladie sérique, sarcoïdose, œdème angioneurotique, panniculite (incluant érythème noueux), aggravation des symptômes de dermatomyosite *.
Affections endocriniennes
Rare: affections de la thyroïde.
Troubles du métabolisme et de la nutrition
Occasionnel: troubles électrolytiques (y compris: hypocalcémie, hypercalcémie, hypokaliémie, hyponatrémie), hypernatrémie), dyslipidémie, troubles de l'appétit (incluant anorexie), modifications du poids.
Rare: hypoalbuminémie, hypoprotéinémie, hyperuricémie, modifications des valeurs glycémiques, hémosidérose.
Affections psychiatriques
Occasionnel: anxiété et troubles de l'humeur (incluant symptômes associés).
Rare: tentatives de suicide, délires, altération mentale, hallucination, agression, troubles émotionnels.
Affections du système nerveux
Fréquent: troubles sensoriels (incluant paresthésie), céphalées (incluant migraine).
Occasionnel: tremblements, neuropathie périphérique, vertiges.
Rare: agueusie, amnésie, épilepsie (incluant grand mal), troubles du sommeil, troubles de la démyélinisation (incluant névrite des nerfs crâniens, troubles de la coordination, troubles extrapyramidaux, sclérose en plaques), névralgie du trijumeau, aphasie, aréflexie, troubles de la coordination et de l'équilibre, radiculopathie.
Affections oculaires
Occasionnel: troubles visuels (incluant baisse de la vision), troubles de la sécrétion lacrymale (incluant sécheresse oculaire, larmoiements), inflammation oculaire et palpébrale (conjonctivite), prurit oculaire.
Rare: névrite optique, glaucome, douleurs oculaires, hémorragie conjonctivale.
Affections de l'oreille et du labyrinthe
Occasionnel: acouphènes, vertiges, douleurs dans l'oreille.
Rare: perte de l'audition.
Affections cardiaques
Occasionnel: coronaropathies ischémiques (incluant infarctus du myocarde, angine de poitrine), arythmies (incluant fibrillations auriculaires), tachycardie, palpitations.
Rare: arrêt cardiaque soudain, cardiomyopathies (incluant insuffisance cardiaque, défaillance cardiaque), péricardite, bloc atrio-ventriculaire.
Affections vasculaires
Occasionnel: bouffées de chaleur, hypotension, hypertension, hémorragie ou saignement (toute localisation), hypercoagulation (incluant embolie pulmonaire, thrombophlébite), syncope (incluant perte de conscience), œdème (périphérique et facial), ecchymoses (incluant hématomes, pétéchies).
Rare: anomalie veineuse, artériosclérose, phlébite, choc, syndrome de Raynaud, AVC, apoplexie, livedo réticulaire, télangiectasie.
Affections respiratoires, thoraciques et médiastinales
Occasionnel: épanchement pleural (et symptômes associés) congestion et inflammation des voies respiratoires, asthme et symptômes associés, bronchopneumopathie chronique obstructive, dyspnée, toux.
Rare: pneumopathie interstitielle (incluant pneumonie), ulcère nasal
Affections gastro-intestinales
Occasionnel: nausées et vomissements.
Peu fréquent: signes et symptômes de la maladie de Crohn (incluant sténose), ulcérations et perforations gastro-intestinales, stomatite (aphteuse), ballonnements, dyspepsie, inflammation du tractus digestif (toute localisation), sécheresse oropharyngée.
Rare: ascites, occlusion intestinale, paresthésie orale, hypermotilité, hémorroïdes, fissures anales, douleurs à la déglutition.
Affections hépatobiliaires
Fréquent: hépatite (incluant élévation des enzymes hépatiques).
Occasionnel: taux élevés de la phosphatase alcaline dans le sang, taux élevés de bilirubine dans le sang, hépatopathies (incluant cirrhose), cholestase.
Rare: affections biliaires (incluant cholélithiase, cholécystite).
Affections de la peau et du tissu sous-cutané
Fréquent: éruption cutanée.
Occasionnel: alopécie, nouvelle poussée ou aggravation d'un psoriasis (y compris psoriasis palmoplantaire pustuleux) et affections apparentées, dermatite et eczéma (aussi généralisés), affection des glandes sudoripares, sécheresse cutanée, hyperhidrose, urticaire (également généralisée), acné, photosensibilité, ulcère cutané, anomalie des ongles et affections du lit unguéal.
Rare: exfoliation et desquamation cutanées, affections bulleuses, ulcère cutané, Pityriasis rosea, peau sensible à la douleur, décolorations de la peau, dermatose neutrophile fébrile aiguë, rosacée, anomalies de la texture des cheveux, syndrome de Steven Johnson*, érythème polymorphe*, réactions lichénoïdes.
Affections musculo-squelettiques, du tissu conjonctif et des os
Fréquent: douleurs de l'appareil moteur, signes et symptômes de polyarthrite rhumatoïde (y compris arthrite et affections des tendons).
Occasionnel: douleurs dans les extrémités, élévation de la créatine phosphokinase sérique, arthralgie, troubles musculaires (incluant spasmes musculaires, myalgie, augmentation du tonus musculaire).
Rare: faible masse osseuse, ostéochondrose, contracture de Dupuytren.
Affections des reins et des voies urinaires
Occasionnel: troubles de la fonction rénale, néphrolithiase, hématurie, symptômes vésicaux et urétraux, anomalies des valeurs urinaires (y compris chromaturie et odeur d'urine anormale).
Rare: néphropathie (incluant néphrite).
Affections des organes de reproduction et du sein
Occasionnel: troubles du cycle menstruel et des saignements utérins (incluant aménorrhée), douleurs pelviennes, affections mammaires.
Rare: avortement spontané, troubles de la fonction sexuelle (incluant baisse de la libido, azoospermie, balanite, écoulement vaginal.
Troubles généraux et anomalies au site d'administration
Fréquent: fièvre, douleurs (toute localisation), asthénie, prurit (toute localisation), réactions au site d'administration (incluant douleurs, ecchymoses, hématomes, rougeurs, œdème, irritations, colorations, nécroses, phlébites, ulcères), épuisement.
Occasionnel: frissons, malaise, syndrome pseudo-grippal, sueurs nocturnes, fistules (toute localisation), altération de la perception de la température (incluant sensation de froid).
Investigations
Occasionnel: allongement du temps de coagulation (voir «Mises en garde et précautions»).
Rare: hyperuricémie.
Lésions, intoxications et complications liées aux procédures
Occasionnel: fractures, lésions cutanées, retard à la cicatrisation.
Dans le cadre d'études cliniques, on a observé de rares cas de blessures et de suffocation.
* La relation de cause à effet avec Cimzia n'est pas prouvée, mais ces évènements sont connus sous le nom d'effet de classe des antagonistes du TNF.
Description de certains effets indésirables
Infections
Dans les études cliniques contrôlées relatives à la maladie de Crohn, l'incidence des infections était de 35.8% chez les patients traités par Cimzia et de 30.5% chez les patients sous placebo. Dans le cas de ces infections, il s'agissait en premier lieu de rhinopharyngite (8.9% Cimzia, 7.7% placebo), d'infections des voies urinaires (4.8% Cimzia, 5.2% placebo), d'infections des voies respiratoires supérieures (4.0% Cimzia, 2.3% placebo) et de grippe (3.1% Cimzia, 4.5% placebo). L'incidence des abcès gastrointestinaux était de 3.8% pour Cimzia et de 1.2% pour le placebo. Dans les études cliniques contrôlées contre placebo, le taux d'infections était de 1'053 par patient-année pour les patients traités par Cimzia et de 1'065 par patient-année pour les patients sous placebo.
Lors des études cliniques contrôlées versus placebo dans la polyarthrite rhumatoïde, l'incidence de nouveaux cas d'infections a été de 1.03 par patient-année pour tous les patients traités par Cimzia et de 0.92 par patient-année pour les patients recevant le placebo. Les infections étaient essentiellement des infections des voies respiratoires hautes, des infections à herpès virus, des infections urinaires et des infections des voies respiratoires basses.
Lors des études cliniques contrôlées versus placebo dans la polyarthrite rhumatoïde, il y a eu plus de cas d'infections graves dans les groupes de traitement par Cimzia (0.07 par patient-année; toutes doses confondues) que dans les groupes placebo (0.02 par patient-année).
Le taux d'infections graves était de 0.07 par patient-année dans le groupe posologique de 200 mg toutes les 2 semaines et de 0.05 par patient-année dans le groupe posologique de 400 mg toutes les 4 semaines.
Les infections graves les plus fréquentes incluaient des pneumonies et des tuberculoses. En outre, ces infections incluaient aussi des infections opportunistes invasives (p.ex. pneumocystose, nocardiose, histoplasmose et zona disséminé). Il n'y a pas de preuve d'une augmentation du risque d'infections en cas d'exposition prolongée dans le temps (voir rubrique «Mises en garde et précautions d'emploi»).
Lors des essais cliniques contrôlés versus placebo sur le psoriasis, le taux d'incidence des nouveaux cas d'infection a été de 1.37 par patient-année pour tous les patients traités par Cimzia et de 1.59 par patient-année pour les patients recevant le placebo. Les infections étaient en premier lieu des infections des voies respiratoires hautes et des infections virales (notamment des infections à herpès virus). L'incidence des infections graves était de 0.02 par patient-année chez les patients traités par Cimzia. Aucune infection grave n'a été rapportée chez les patients recevant le placebo. Il n'y a pas de preuve d'une augmentation du risque d'infections en cas d'exposition prolongée dans le temps.
Tumeurs malignes et carcinome à cellules de Merkel
Dans les études cliniques relatives à la maladie de Crohn, l'incidence globale de toutes les tumeurs malignes était de 0.5% chez les patients traités par Cimzia et de 0.5% chez les patients sous placebo. Le seul cas de lymphome est survenu chez un patient traité par placebo. Le taux d'incidence pour la première survenue de toutes les tumeurs malignes, lymphomes inclus (par 100 patients-années), dans la population de sécurité de la maladie de Crohn était de 1.97 pour le placebo (n=426) contre 0.88 pour Cimzia 400 mg (n=1'350) et 0.80 pour Cimzia à n'importe quelle dose (n=1'564). De plus, on a enregistré un cancer du poumon métastatique plus de 10 mois après la seconde et dernière dose de Cimzia.
Le taux d'incidence pour la première survenue (par 100 patients-années) de tous les événements de la classe de systèmes d'organes «tumeurs bénignes, malignes et non précisées (incl. kystes et polypes)» dans la population souffrant de polyarthrite rhumatoïde était de 2.63 pour le placebo (n=1'759) contre 2.77 pour Cimzia 200 mg (n=2'871) et 1.48 pour Cimzia 400 mg (n=347) et de 2.65 pour Cimzia à n'importe quelle dose (n=4'248).
Le carcinome mammaire et ovarien, le basaliome et le lymphome faisaient partie des tumeurs malignes observées dans les études contrôlées contre placebo et ouvertes relatives à la polyarthrite rhumatoïde. Le taux d'incidence des lymphomes a été de 0.05 pour 100 patients-années et le taux d'incidence des mélanomes de 0.08 pour 100 patients-années lors des études cliniques de Cimzia dans la polyarthrite rhumatoïde. Le nombre de cas indiqué est trop faible pour pouvoir identifier un effet du traitement (voir rubrique «Mises en garde et précautions»).
Un cas de lymphome est également survenu lors de l'étude clinique de phase III dans l'arthrite psoriasique.
En dehors des cancers de la peau non-mélanomes, 11 cancers, dont 1 cas de lymphome, ont été observés lors des études cliniques de Cimzia sur le psoriasis au cours desquelles 1'112 patients au total ont été traités, représentant 2'300 patients-année.
Chez les patients traités par des antagonistes du TNF y compris Cimzia, des cas de carcinome à cellules de Merkel ont été observés dans la phase Post-Marketing.
Des cas de lymphome hépatosplénique à lymphocytes T, une forme rare de lymphome à lymphocytes T avec progression très agressive de la maladie, voire fatale, ont été rapportés chez des patients qui avaient reçu un traitement par anti-TNF post-marketing. La majorité des cas signalés suite à l'utilisation d'antagonistes du TNF sont survenus chez des adolescents et des jeunes hommes atteints de la maladie de Crohn ou de colite ulcéreuse. Presque tous ces patients ont été traités, avant ou au moment du diagnostic, par de l'azathioprine immunosuppressive et/ou de la 6-mercaptopurine en combinaison avec un antagoniste TNF.
Insuffisance cardiaque congestive
Neuf cas d'insuffisance cardiaque congestive sous Cimzia (0.1 pour 100 patients-années) ont été observés dans les études cliniques contrôlées versus placebo et les études cliniques en ouvert menées avec des patients atteints de polyarthrite rhumatoïde.
Un cas d'insuffisance cardiaque congestive a été observé dans les études cliniques de Cimzia sur le psoriasis.
Anticorps anti-certolizumab pégol
Comme décrit ci-dessous, des anticorps, y compris des anticorps neutralisants, contre Cimzia ont été détectés dans des études cliniques. Les anticorps anti-Cimzia peuvent ainsi être associés à des concentrations plasmatiques plus faibles du principe actif.
Pour une partie des patients ayant développés des anticorps anti-Cimzia, l'efficacité a été en conséquence plus faible.
Les données ci-dessous tiennent compte du pourcentage de patients dont les résultats de test ont été considérés comme positifs pour les anticorps anti-certolizumab pegol dosés par technique ELISA et ultérieurement par une méthode plus sensible, et dépendent fortement de la sensibilité et la spécificité du dosage.
L'incidence observée des anticorps (y compris des anticorps neutralisants) dans un dosage dépend essentiellement de plusieurs facteurs, notamment la sensibilité et la spécificité du dosage, la méthodologie du dosage, la manipulation des échantillons, l'heure du prélèvement des échantillons, les médicaments concomitants et la maladie sous-jacente. Pour ces raisons, une comparaison du taux des anticorps anti-Certolizumab pégol dans les études décrites ci-dessous avec le taux des anticorps dans d'autres études ou avec d'autres produits peut s'avérer trompeuse.
Maladie de Crohn
Le pourcentage total des patients ayant des anticorps anti-Cimzia détectables dans les études menées sur la maladie de Crohn était faible (7.9% des patients exposés en continu à Cimzia, une activité neutralisante in vitro ayant été détectée chez environ 6% de ces patients). Les patients traités simultanément par immunosuppresseurs ont présenté un taux de formation d'anticorps plus faible que les patients ne prenant pas d'immunosuppresseur dès leur admission dans l'étude (3.3% contre 11.1%).
Polyarthrite rhumatoïde
Le pourcentage total de patients ayant des anticorps anti-Cimzia détectables à au moins une occasion dans les études contrôlées versus placebo dans la polyarthrite rhumatoïde a été faible (9.6%; environ un tiers des patients anticorps positifs avait des anticorps ayant une activité neutralisante in vitro. Les patients traités simultanément par immunosuppresseurs (MTX) ont présenté un taux de formation d'anticorps plus faible que les patients ne prenant pas d'immunosuppresseurs à l'inclusion.
Arthrite psoriasique
Le pourcentage total de patients ayant des anticorps anti-Cimzia détectables à au moins une occasion avant la semaine 24 dans l'étude de phase III contrôlée versus placebo chez les patients ayant de l'arthrite psoriasique a été de 11.7%. Ce pourcentage est passé de 11.7% à 13.6% entre les semaines 24 et 48.
Psoriasis en plaques
Dans les études de phase III contrôlées versus placebo et versus comparateur actif, les pourcentages de patients ayant des anticorps anti-certolizumab pegol détectables au moins une occasion jusqu'à la semaine 48 étaient de 8,3% (22/265) et 19,2% (54/281) pour le certolizumab pegol 400 mg toutes les 2 semaines et le certolizumab pegol 200 mg toutes les 2 semaines, respectivement. Dans les études CIMPASI-1 et CIMPASI-2, 60 patients présentaient des anticorps, dont 27 étaient évaluables pour les anticorps neutralisants et testés positifs. La formation d'anticorps a été associée à une baisse de la concentration plasmatique en médicament et chez certains patients, à une efficacité réduite.
Spondylarthrite axiale
AS001
Le pourcentage total de patients ayant des anticorps anti-Certolizumab pegol détectables à au moins un dosage avant la semaine 24 était de 4.4% dans l'étude contrôlée versus placebo de phase III AS0001 chez les patients atteints de spondylarthrite axiale (sous-populations avec spondylarthrite ankylosante et spondylarthrite axiale non radiographique).
La formation d'anticorps était associée à une diminution de la concentration plasmatique du médicament.
Dans cette étude, le nombre de patients présentant des anticorps contre le certolizumab pegol était trop faible pour permettre une évaluation valable de l'influence de la formation d'anticorps sur l'efficacité.
Dans des études cliniques menées sur la maladie de Crohn et l'arthrite rhumatoïde, les patients ayant des anticorps détectables contre le certolizumab pégol ont présenté des concentrations plasmatiques inférieures de certolizumab pégol par rapport aux autres patients, et chez quelques patients souffrant d'arthrite rhumatoïde une efficacité moindre. Lors d'études menées sur la maladie de Crohn, il a été rapporté que les patients ayant des anticorps contre le certolizumab pégol (N=100) ont présentés les événements indésirables suivants au moins 3% plus fréquemment que les patients ne présentant pas ces anticorps (N=1'242): douleurs abdominales, douleurs articulaires, œdème périphérique, érythème noueux, érythème au point d'injection, douleurs au point d'injection, membres douloureux et infections des voies respiratoires inférieures.
Le nombre de patients ayant des anticorps anti-Cimzia détectables lors des études cliniques concernant l'arthrite psoriasique (PsA) et la spondylarthrite axiale (AxSpA) était trop faible pour évaluer de manière fiable l'impact sur l'efficacité.
AS0006 et C-OPTIMISE
Dans l'étude AS0006 (et ultérieurement dans l'étude C-OPTIMISE), un nouveau dosage, plus sensible et plus tolérant aux médicaments, a montré une incidence plus élevée chez les patients classés comme positifs à la détection d'anticorps.
L'incidence globale après 52 semaines de traitement de patients ayant des anticorps anti-Certolizumab pegol était de 97 % (248/255 patients) dans l'étude AS0006, et de 98,7 % (304/308 patients) dans l'étude C-OPTIMISE.
Seuls les titres d'anticorps les plus élevés étaient associés à des taux de plasmatiques réduits de certolizumab-pegol, cependant aucun impact sur l'efficacité clinique n'a été observé chez les patients présentant des titres d'anticorps élevés.
Environ 22 % (54/248) des patients qui étaient positifs aux anticorps anti-certolizumab pegol à un moment donné, ont présenté des anticorps classés comme neutralisants.
Des résultats similaires en relation avec les anticorps contre le certolizumab pegol ont été observés dans C-OPTIMISE. Les résultats de C-OPTIMISE indiquaient également qu'une réduction de la dose de certolizumab pegol à 200 mg toutes les 4 semaines ne changeait pas les résultats en termes d'immunogénicité.
Auto-anticorps
Dans le cadre d'études cliniques relatives à la maladie de Crohn, 3.7% des patients traités par Cimzia qui étaient AAN négatifs avant le début du traitement, ont développé des AAN positifs pendant l'étude, contre 1.7% des patients sous placebo. Les anti-dbADN correspondants étaient de 1.2% pour les patients traités par Cimzia contre 1.8% pour les patients sous placebo.
Parmi les sujets participant à l'étude d'homologation (étude pivot) dans la polyarthrite rhumatoïde qui étaient AAN négatifs à l'inclusion, 16.7% de ceux traités par Cimzia et 12.0% des patients sous placebo sont devenus AAN positifs. Parmi les sujets anticorps anti-ADNdb négatifs à l'inclusion, 2.2% de ceux traités par Cimzia et 1.0% des patients sous placebo, sont devenus anticorps anti-ADNdb positifs. Lors des études cliniques contrôlées versus placebo et des études d'extension en ouvert dans la polyarthrite rhumatoïde, des cas de syndrome lupique ont été rapportés peu fréquemment. De rares cas de maladies à médiation immunitaire ont été rapportés; leur imputabilité au traitement par Cimzia n'est pas connue. L'impact du traitement à long terme par Cimzia sur le développement de maladies auto-immunes n'est pas connu.
Réactions au site d'injection
Lors des études cliniques contrôlées versus placebo dans la polyarthrite rhumatoïde, 5.8% des patients traités par Cimzia ont présenté des réactions au site d'injection (érythème, prurit, hématome, douleur, gonflement ou ecchymoses) contre 4.8% des patients recevant le placebo. Des douleurs au site d'injection ont été observées chez 1.5% des patients traités par Cimzia, aucun cas n'ayant nécessité l'arrêt du médicament
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucun cas de surdosage n'a été rapporté.
La dose maximale tolérée de certolizumab pégol n'a pas été déterminée. Dans le cadre d'études cliniques, on n'a pas observé de toxicité dose-limitante. Dans ce cas, on a administré des doses multiples bien tolérées pouvant atteindre 800 mg (s.c.) ou 20 mg/kg (i.v.). Dans le cas d'un surdosage, il est recommandé de surveiller étroitement le patient à la recherche d'éventuelles réactions ou effets indésirables, ainsi que d'instaurer immédiatement un traitement symptomatique correspondant.
Propriétés/EffetsCode ATC
L04AB05
Le certolizumab pégol est un fragment Fab d'un anticorps recombinant humanisé. Le fragment Fab est produit à partir d'Escherichia coli, puis purifié et conjugué au polyéthylèneglycol (PEG).
Mécanisme d'action
Le certolizumab pégol se lie avec une grande affinité au TNF-α humain, avec une constante de dissociation (kd) de 90 pM. Le TNF-α est une cytokine pro-inflammatoire importante qui joue un rôle déterminant dans les processus inflammatoires. Le certolizumab pégol possède un effet neutralisant sélectif sur le TNF-α (IC90 de 4 ng/ml lors de l'inhibition du TNF-α humain dans le cadre d'un essai de cytotoxicité-fibrosarcome in vitro sur lignées cellulaires murines L929), mais ne neutralise pas la lymphotoxine α (TNF-β).
Le certolizumab pégol possède un effet neutralisant dose-dépendant démontrable sur le TNF humain associé à la membrane et soluble. L'incubation de monocytes avec du certolizumab pégol a entraîné une inhibition dose-dépendante de TNF-α induit par LPS, ainsi que la formation d'IL-1β dans des monocytes humains.
Le certolizumab pégol est dépourvu de région Fc («fragment crystallizable»), telle qu'elle existe normalement dans un anticorps complet; c'est la raison pour laquelle il y a absence de fixation au complément et de formation de cytotoxicité anticorps-dépendante, à médiation cellulaire. In vitro, cela n'entraîne pas d'apoptose dans les monocytes ou les lymphocytes humains du sang périphérique ou la dégranulation des neutrophiles.
Pharmacodynamique
Sans objet.
Efficacité clinique
Maladie de Crohn
L'efficacité et la sécurité de Cimzia ont été vérifiées dans le cadre de deux études en double aveugle, randomisées, contrôlées contre placebo, chez des patients âgés de ≥18 ans atteints de la maladie de Crohn active, de modérée à sévère, c'est-à-dire avec un Crohn's Disease Activity Index (CDAI) de 220 et 450 (y compris 220 et 450). Dans les deux études, Cimzia a été administré par voie sous-cutanée à une dose de 400 mg. L'utilisation concomitante de doses stables de produits contre la maladie de Crohn était autorisée.
Étude I
Dans le cas de l'étude I (PRECiSE 1), il s'agissait d'une étude randomisée, contrôlée contre placebo, menée auprès de 662 patients atteints de la maladie de Crohn active. Cimzia ou le placebo ont été administrés au cours des semaines 0, 2 et 4, puis toutes les 4 semaines jusqu'en semaine 24. La réponse clinique (response) était définie comme une diminution du CDAI d'au moins 100 points, une rémission clinique comme une valeur absolue du CDAI de 150 ou moins. Trois patients ont été exclus de l'analyse Intent-to-treat- (ITT-); deux des patients randomisés dans le groupe Cimzia n'ont pas reçu de doses; un patient randomisé dans le groupe placebo a été exclu.
Le tableau 1 présente les résultats concernant la réponse clinique (response). L'effet de Cimzia s'est installé en semaine 2. En semaine 6, le nombre de répondeurs était cliniquement significatif par rapport au placebo; c.-à-d. qu'une réponse clinique était atteinte. De plus, le nombre de répondeurs était cliniquement significatif tant en semaine 6 qu'en semaine 26, ce qui est le signe d'une réponse durable.
Tableau 1: PRECiSE 1 – Réponse clinique; totalité de la population à l'étude
|
Temps de mesure
|
Nombre (%) de répondeurs
IC 95%
| |
Placebo
(N=328)
|
Cimzia 400 mg
(N=331)
| |
Semaine 2
| |
N
|
326
|
328
| |
Responders
|
46 (14.1%)
|
82 (25.0%)§
| |
IC 95%
|
10.3%, 17.9%
|
20.3%, 29.7%
| |
Semaine 6 (induction)
| |
N
|
325
|
327
| |
Responder
|
87 (26.8%)
|
115 (35.2%)*
| |
IC 95%
|
22.0%, 31.6%
|
30.0%, 40.3%
| |
Semaine 26
| |
N
|
327
|
328
| |
Responders
|
87 (26.6%)
|
122 (37.2%)*
| |
IC 95%
|
21.8%, 31.4%
|
32.0%, 42.4%
| |
Semaines 6&26 (réponse durable)
| |
N
|
325
|
325
| |
Responder
|
52 (16.0%)
|
75 (23.1%)*
| |
IC 95%
|
12.0%, 20.0%
|
18.5%, 27.7%
|
§ valeur de p pas calculée
* valeur de p <0.05 test de régression logistique
L'utilisation d'immunosuppresseurs ou de corticostéroïdes avant le début de l'étude n'a eu aucun effet sur la réponse clinique de Cimzia. Cimzia était efficace en ce qui concerne l'induction et le maintien de la réponse dans la sous-population de patients préalablement traités par infliximab (semaine 6: 24.5% vs 20.0%; semaines 6 et 26: 15.5% vs 10.6% pour Cimzia ou placebo).
Cimzia était efficace en ce qui concerne l'induction et le maintien de la réponse dans la sous-population de patients n'ayant pas été préalablement traités par infliximab (semaine 6: 39.7% vs 29.2%; semaines 6 et 26: 26.3% vs 17.9% pour Cimzia ou placebo). Pendant l'étude, les effets de la maladie de Crohn sur les patients ont été évalués au moyen d'un questionnaire spécifique à la maladie, appelé le Inflammatory Bowel Disease Questionnaire (IBDQ), ainsi que d'un questionnaire (SF-36) relatif à l'état de santé général.
Le IBDQ (valeur globale ≥16 points par rapport aux valeurs initiales) a montré que, par rapport au groupe sous placebo, en semaines 6, 16 et 26 un nombre plus important de patients du groupe recevant 400 mg de Cimzia a présenté une amélioration cliniquement importante concernant le résultat thérapeutique.
Comparés au groupe sous placebo, en semaine 6 et en semaines 6 et 26, les patients ayant reçu 400 mg de Cimzia ont présenté une atténuation significative de la douleur (p ≤0.05) par rapport aux valeurs initiales, objectivée par la valeur relative aux douleurs physiques du questionnaire SF-36.
Étude II:
Dans le cas de l'étude II (PRECiSE 2), il s'agissait d'une étude randomisée portant sur l'arrêt du traitement et conduite auprès d'une population de 668 patients atteints de la maladie de Crohn active. Tous les patients inclus dans l'étude ont reçu 400 mg de Cimzia en semaines 0, 2 et 4; l'évaluation de la réponse clinique a eu lieu en semaine 6. La réponse clinique (response) était définie comme une diminution du CDAI d'au moins 100 points, une rémission clinique comme une valeur absolue du CDAI de 150 ou moins. Les répondeurs ont été randomisés pour recevoir un traitement d'entretien avec 400 mg de Cimzia ou un placebo (débutant en semaine 8) jusqu'en semaine 24.
Les patients n'ayant pas répondu en semaine 6 (non-responder) ont été exclus de l'étude. Trois patients ont été exclus de l'analyse ITT.
Au total, 668 patients atteints de la maladie de Crohn active ont reçu 400 mg de Cimzia comme traitement d'induction selon le principe du Open-label.
Dans ce groupe de patients, 428 (64.1%) ont répondu au traitement et ont été randomisés en semaine 6 pour recevoir un placebo (212; ITT 210) ou Cimzia 400 mg (216; ITT 215); ils ont reçu au moins une injection en double aveugle. L'effet s'est installé en semaine 2.
Parmi les répondeurs en semaine 6, 45.1% ont présenté une réponse clinique en semaine 2.
Le tableau 2 présente les résultats relatifs à la réponse clinique (response) ou à la rémission. En semaine 26, par rapport au groupe avec 3 doses de Cimzia+placebo, dans le groupe sous Cimzia un pourcentage significativement plus élevé de répondeurs de semaine 6 a présenté une réponse clinique ou une rémission clinique.
Tableau 2: PRECiSE 2 – réponse clinique (response) et rémission clinique dans la totalité de la population à l'étude
|
Temps de mesure
|
Réponse clinique (response)
Pourcentage (%)
|
Rémission clinique
Pourcentage (%)
| |
|
Cimzia 400 mg × 3
+ Placebo*
N=210
|
Cimzia 400 mg
N=215
|
Cimzia 400 mg × 3
+ Placebo*
N=210
|
Cimzia 400 mg
N=215
| |
Semaine 26
Réponse durable
| |
N
|
210
|
215
|
210
|
215
| |
Responder
|
76 (36.2%)
|
135 (62.8%)
|
60 (28.6%)
|
103 (47.9%)
| |
IC 95%
|
29.7%, 42.7%
|
56.3%, 69.3%
|
22.5%, 34.7%
|
41.2%, 54.6%
| |
Valeur de p
|
|
<0.001
|
|
<0.001
|
* Ce tableau se rapporte exclusivement à la partie en double aveugle de l'étude.
Figure 1: PRECiSE 2 – Évolution temporelle de la réponse clinique; totalité de la population ITT
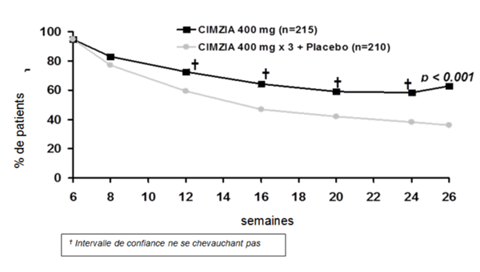
L'utilisation d'immunosuppresseurs ou de corticostéroïdes avant le début de l'étude n'a eu aucun effet sur la réponse clinique de Cimzia. Cimzia était efficace jusqu'en semaine 26 en ce qui concerne le maintien de la réponse, et ce tant dans la sous-population de patients préalablement traités par infliximab (44.2% vs 25.5% pour Cimzia ou placebo), que chez les patients n'ayant pas été préalablement traités par infliximab (68.7% ou 39.6%).
Les effets de la maladie de Crohn sur les patients ont été évalués pendant l'étude au moyen d'un questionnaire spécifique à la maladie, appelé le Inflammatory Bowel Disease Questionnaire (IBDQ), ainsi que d'un questionnaire (SF-36) relatif à l'état de santé général.
Le IBDQ (valeur globale ≥16 points par rapport aux valeurs initiales) a montré que, par rapport au groupe sous placebo, en semaines 16 et 26 un nombre plus important de patients du groupe recevant 400 mg de Cimzia a présenté une amélioration cliniquement importante concernant le résultat thérapeutique.
Comparés au groupe sous placebo, en semaine 26, les patients ayant reçu 400 mg de Cimzia ont présenté une atténuation significative de la douleur (p ≤0.05) par rapport aux valeurs initiales, objectivée par la valeur relative aux douleurs physiques du questionnaire SF-36.
Dans le questionnaire, les résultats relatifs au «rôle physique» étaient meilleurs pour Cimzia que pour le placebo; ils n'étaient toutefois pas statistiquement significatifs (p >0.05).
Cimzia a été étudié pour le traitement de patients pédiatriques atteints d'une maladie de Crohn modérément à sévèrement active (étude C87035 et son suivi à long terme CR0012). L'efficacité de Cimzia n'a pas été démontrée dans une étude de groupe parallèle, ouverte, randomisée et à doses multiples sur une période allant jusqu'à 62 semaines chez 99 sujets âgés de 6 à 17 ans (C87035). L'étude a été interrompue prématurément en raison d'un taux d'abandon élevé.
Polyarthrite rhumatoïde
Études RA-I, RA-II
L'efficacité et la tolérance de Cimzia ont été évaluées lors de 2 études cliniques randomisées, en double aveugle, contrôlées versus placebo, chez des patients âgés ≥18 ans ayant une polyarthrite rhumatoïde active diagnostiquée selon les critères de l'American College of Rheumatology (ACR), RA-I (RAPID 1) et RA-II (RAPID 2). Les patients avaient au moins 9 articulations gonflées et douloureuses et ils avaient une PR active depuis au moins 6 mois avant le début de l'étude. Au cours des deux études, Cimzia a été administré par voie sous-cutanée, en association avec le MTX par voie orale, préalablement administré pendant au moins 6 mois et à des doses stables d'au moins 10 mg par semaine pendant 2 mois. Il n'y a aucune expérience de l'administration de Cimzia en association avec des DMARDs autres que le MTX.
Étude C-EARLY
L'efficacité et la sécurité de Cimzia ont été étudiées dans une étude clinique randomisée, en double aveugle, contrôlée versus placebo, chez des patients adultes ayant une PR active modérée (3.5%) à sévère (96.5%) n'ayant pas été préalablement traités par DMARDs (C-EARLY). Dans l'étude C-EARLY, les patients avaient au moins 18 ans et avaient chacun au moins 4 articulations gonflées et douloureuses et un diagnostiqués depuis moins d'1 an (selon les critères de classification de l'ACR / European League Against Rheumatism [EULAR] de 2010). Le DAS28 de départ devait être >3.2 (activité de la maladie modérée à élevée) et le CRP était ≥10 mg/L ou l'ESR était ≥28 mm/h. En début d'étude, les patients avaient été diagnostiqués en moyenne 2.9 mois plus tôt et ils n'avaient pas reçu préalablement de traitement par DMARDs (y compris le MTX). 96.5% des patients de l'étude présentaient une PR sévère. Les patients ont reçu une dose initiale (semaines 0, 2 et 4) de 400 mg ou un placebo, puis une dose d'entretien de 200 mg de Cimzia ou un placebo toutes les 2 semaines pendant 52 semaines. Pour les deux bras Cimzia et placebo, le MTX a été initié à la semaine 0 (10 mg/semaine), augmenté jusqu'à la dose maximale tolérée jusqu'à la semaine 8 (au minimum 15 mg/semaine et au maximum 25 mg/semaine étaient autorisés) et maintenu tout au long de l'étude (après la semaine 8, la dose moyenne de MTX était de 22.3 mg/semaine dans le bras placebo et de 21.1 mg/semaine dans le bras Cimzia).
Dans le cadre d'une phase ultérieure, le maintien de la réponse a été étudié la 2ème année. Pour cela, les patients dans le groupe Cimzia 200 mg toutes les 2 semaines + MTX présentant une activité durablement faible de la maladie (DAS28-ESR < 3.2 aux semaines 40 et 52) ont été à nouveau randomisés à la semaine 52, dans le groupe Cimzia 200 mg toutes les 2 semaines + MTX, Cimzia 200 mg toutes les 4 semaines + MTX ou placebo + MTX (dans un rapport de 2:3:2). Les critères d'évaluation sont résumés dans le tableau 3.
Tableau 3: Description des essais cliniques
|
Numéro de l'étude
|
Nombre de patients
|
Posologie
|
Objectifs de l'étude
| |
RA-I
(52 semaines)
|
982
|
400 mg (0, 2, 4 semaines) avec le MTX
|
Évaluation du traitement des signes et symptômes et de l'inhibition des dommages structuraux.
| |
200 mg ou 400 mg toutes les 2 semaines avec le MTX
|
Co-critères principaux d'évaluation: ACR 20 à la semaine 24 et modification du mTSS à la semaine 52 par rapport au score initial.
| |
RA-II
(24 semaines)
|
619
|
400 mg (0, 2, 4 semaines) avec le MTX
|
Évaluation du traitement des signes et symptômes et de l'inhibition des dommages structuraux.
| |
200 mg ou 400 mg toutes les 2 semaines avec le MTX
|
Critère principal d'évaluation: ACR 20 à la semaine 24.
| |
C-EARLY
(jusqu'à la semaine 104)
|
879
|
400 mg (0, 2, 4 semaines) avec le MTX
200 mg toutes les 2 semaines avec le MTX
200 mg toutes les 4 semaines avec le MTX
(nouvelle randomisation après 52 semaines)
|
Évaluation en fonction du traitement des signes et symptômes et de l'inhibition de la progression des dommages structuraux chez les patients n'ayant pas reçu de traitement préalable par DMARDs
Critère principal d'évaluation à la semaine 52
pourcentage de participants en rémission persistante *
Critère principal d'évaluation à la semaine 104: maintien de la faible activité de la maladie **
|
mTSS: score total de Sharp modifié
* La rémission persistante à la semaine 52 est définie par un score DAS28-ESR <2.6 aux semaines 40 et 52.
** La faible activité de la maladie persistante à la semaine 104 est définie par un score DAS28-ESR <3.2 aux semaines 52 et 104, sans poussée de la maladie (= hausse du score DAS28-ESR >=0.6 et DAS28-ESR >3.2)
Réponse ACR
Les résultats des études cliniques RA-I et RA-II sont présentés dans le Tableau 4. Des réponses ACR 20 et ACR 50 supérieures de façon statistiquement significative par rapport au placebo ont été obtenues, respectivement, à partir de la semaine 1 et de la semaine 2, dans les deux études cliniques. Les réponses se sont maintenues jusqu'à la semaine 52 (RA-I) et 24 (RA-II). Sur les 783 patients initialement randomisés dans RA-I, 508 ont terminé les 52 semaines de la phase de traitement contrôlée versus placebo et sont entrés dans l'étude d'extension en ouvert. Parmi ceux-ci, 427 ont terminé les 2 années de l'étude d'extension en ouvert et ont donc eu une exposition à Cimzia de 148 semaines au total. La réponse ACR 20 à cette échéance était de 91%. La réduction du DAS28 (VS) par rapport à la valeur initiale a également été significativement plus importante (p <0.001) à la semaine 52 (RA-I) et à la semaine 24 (RA-II) par rapport au placebo, et s'est maintenue jusqu'à 2 ans dans l'étude d'extension en ouvert de l'étude RA-I.
Tableau 4: Réponse ACR dans les études cliniques RA-I et RA-II
|
|
Étude RA-I
Association avec le méthotrexate
(24 et 52 semaines)
|
Étude RA-II
Association avec le méthotrexate
(24 semaines)
| |
Réponse
|
Placebo +
MTX
N=199
|
Cimzia 200 mg
+ MTX toutes les
2 semaines
N=393
|
Placebo
+ MTX
N=127
|
Cimzia 200 mg
+ MTX toutes les
2 semaines
N=246
| |
ACR 20
| |
Semaine 24
|
14%
|
59%**
|
9%
|
57%**
| |
Semaine 52
|
13%
|
53%**
|
N/A
|
N/A
| |
ACR 50
| |
Semaine 24
|
8%
|
37%**
|
3%
|
33%**
| |
Semaine 52
|
8%
|
38%**
|
N/A
|
N/A
| |
ACR 70
| |
Semaine 24
|
3%
|
21%**
|
1%
|
16%*
| |
Semaine 52
|
4%
|
21%**
|
N/A
|
N/A
| |
Réponse clinique majeurea
|
1%
|
13%**
|
|
|
Cimzia versus placebo: * p ≤0.01, ** p <0.001
a La réponse clinique majeure est définie comme l'obtention d'une réponse ACR 70 lors de chaque évaluation sur une période continue de 6 mois.
Les valeurs de p (test de Wald) sont citées pour la comparaison des traitements en utilisant une régression logistique avec le traitement et la région comme facteurs.
Pourcentage de réponse basé sur le nombre de sujets pour lesquels des données sont disponibles (n) concernant ce critère d'évaluation et cette échéance et qui peut différer de N.
Les critères principaux et secondaires majeurs de l'étude C-EARLY ont été atteints. Les principaux résultats de l'étude sont présentés dans le tableau 5.
Tableau 5: étude C-EARLY: pourcentage des patients en rémission persistante et en activité durablement faible de la maladie à la semaine 52
|
Réponse
|
Placebo + MTX
N=213
|
Cimzia 200 mg + MTX
N=655
| |
Rémission persistante*
(DAS28-ESR <2.6 aux semaines 40 et 52)
|
15.0%
|
28.9%**
| |
Activité durablement faible de la maladie
(DAS28-ESR ≤3.2 aux semaines 40 et 52)
|
28.6%
|
43.8%**
|
* Critère principal d'évaluation de l'étude C-EARLY (jusqu'à la semaine 52)
Population totale d'analyse, imputation des non-répondeurs pour les valeurs manquantes.
** Cimzia + MTX vs placebo + MTX: p<0,001
La valeur p a été estimée par un modèle de régression logistique selon les facteurs suivants: le traitement, la région et la durée depuis le diagnostic de PR à l'inclusion (≤4 mois vs >4 mois).
Une diminution plus importante du DAS28-ESR par rapport à l'inclusion a été observée chez les patients du groupe Cimzia+MTX par rapport au groupe placebo+MTX, dès la semaine 2 et jusqu'à la semaine 52 (p < 0.001 à chaque visite). Les taux de rémission (DAS28-ESR < 2.6), la faible activité de la maladie (DAS28-ESR ≤3.2), les réponses ACR50 et ACR70 à chaque visite ont démontré que le traitement par Cimzia+MTX a permis d'obtenir des réponses plus rapides et plus importantes que le traitement par placebo+MTX. Ces résultats ont été maintenus pendant les 52 semaines de traitement chez des patients non précédemment traités par DMARDs.
Chez les patients présentant une faible activité de la maladie à la semaine 52, une réponse numériquement plus fréquente a été obtenue dans le cadre de la poursuite de la thérapie combinée par Cimzia + MTX jusqu'à la semaine 104 que chez les patients qui avaient arrêté Cimzia (41/84, 48.8% contre 31/79, 39.2%; p=0.112).
Réponse radiographique
Dans l'étude RA-I, les dommages structuraux articulaires ont été évalués par radiographie et exprimés en termes de modification du mTSS et de ses composantes, le score d'érosion et le score de pincement articulaire (JSN de Joint Space Narrowing), à la semaine 52 par rapport aux scores initiaux. La progression des signes radiographiques a été significativement moindre chez les patients traités par Cimzia par rapport à ceux recevant le placebo, aux semaines 24 et 52 (voir tableau 6.). Dans le groupe placebo, 52% des patients n'ont pas présenté de progression radiographique (mTSS ≤0.0) à la semaine 52 versus 69% dans le groupe Cimzia 200 mg.
Tableau 6: Modifications sur 12 mois dans l'étude RA-I
|
|
Placebo + MTX
N=199
Moyenne (DS)
|
Cimzia 200 mg + MTX
N=393
Moyenne (DS)
|
Cimzia 200 mg + MTX –
Placebo + MTX
Différence moyenne
| |
mTSS
| |
Semaine 52
|
2.8 (7.8)
|
0.4 (5.7)
|
-2.4
| |
Score d'érosion
| |
Semaine 52
|
1.5 (4.3)
|
0.1 (2.5)
|
-1.4
| |
Score de pincement articulaire
| |
Semaine 52
|
1.4 (5.0)
|
0.4 (4.2)
|
-1.0
|
Les valeurs de p ont été <0.001 à la fois pour le mTSS et pour le score d'érosion et ≤0.01 pour le score de pincement articulaire. Une analyse ANCOVA a été réalisée sur la variation du score par rapport à la valeur initiale pour chaque mesure, avec la région et le traitement comme facteurs et la valeur initiale du score comme covariable.
Sur les 783 patients initialement randomisés dans RA-I, 508 ont terminé les 52 semaines de la phase de traitement contrôlée versus placebo et sont entrés dans la phase d'extension en ouvert. Le maintien de l'inhibition de la progression des dommages structuraux a été démontrée dans un sous-groupe de 449 de ces patients qui ont été traités pendant au moins 2 ans par Cimzia (RA-I et étude d'extension en ouvert) et avaient des données évaluables à l'échéance des 2 ans.
Dans l'étude C-EARLY, l'inhibition de la progression radiographique a été plus importante dans le bras Cimzia+MTX par rapport au bras placebo+MTX à la semaine 52 (voir tableau 7). A la semaine 52, 49,7 % des patients du groupe placebo+MTX n'ont présenté aucune progression radiographique (variation du mTSS ≤0,5) versus 70,3 % dans le groupe Cimzia+MTX (p < 0,001).
Tableau 7: Evolution radiologique à la semaine 52 dans l'étude C-EARLY
|
|
Placebo + MTX
N=163
Valeur moyenne (ET)
|
Cimzia 200 mg + MTX
N=528
Valeur moyenne (ET)
|
Cimzia 200 mg + MTX – Placebo + MTX
Différence*
| |
STSm
semaine 52
|
1.8 (4.3)
|
0.2 (3.2) **
|
-0.978 (-1.005, -0.500)
| |
Score d'érosion
semaine 52
|
1.1 (3.0)
|
0.1 (2.1) **
|
-0.500 (-0.508, -0.336)
| |
Score de pincement articulaire
semaine 52
|
0.7 (2.3)
|
0.1 (1.7) **
|
0.000 (0.000, 0.000)
|
Ensemble de données radiologiques avec extrapolation linéaire.
* Estimateur ponctuel de Hodges-Lehmann et intervalle de confiance asymptomatique (Moses) à 95%
** Cimzia + MTX vs placebo + MTX p<0.001. La valeur p a été estimée par analyse ANCOVA des rangs avec le traitement, la région et la durée depuis le diagnostic de PR (≤4 mois contre >4 mois) à l'inclusion comme facteurs et la valeur initiale du score comme co-variable.
Parmi les patients présentant une faible activité de la maladie à la semaine 52, une progression des dommages structuraux a été observée chez quelques patients seulement jusqu'à la semaine 104 (7/12, 10% des patients sous Cimzia 200 mg + MTX toutes les 2 semaines; 18/113 et 15/75, 20% des patients sous placebo + MTX). Les données ne permettent pas de tirer de conclusions comparables entre les posologies.
Capacité fonctionnelle et qualité de vie
Dans les études RA-I et RA-II, les patients traités par Cimzia ont rapporté des améliorations significatives, par rapport au placebo (p <0.001), de leurs capacités fonctionnelles évaluées par le questionnaire d'évaluation de l'état de santé – indice d'incapacité (HAQ-DI – Health Assessment Questionnaire – Disability Index) et de la fatigue évaluée par l'échelle d'évaluation de la fatigue (FAS – Fatigue Assessment Scale), à partir de la semaine 1 et jusqu'à la fin des études. Dans les deux études cliniques, les patients traités par Cimzia ont rapporté des améliorations significativement plus importantes du SF-36, scores résumés des composantes physiques et mentales (Physical and Mental Component Summaries) et du score de toutes les dimensions. L'amélioration des capacités fonctionnelles et de la qualité de vie liée à l'état de santé (HRQoL – health related quality of life) a été maintenue pendant 2 ans dans l'étude d'extension en ouvert de l'étude RA-I. Les patients traités par Cimzia ont rapporté des améliorations statistiquement significatives du Work Productivity Survey (questionnaire de productivité au travail) par rapport au placebo.
Dans l'étude C-EARLY, les patients traités par Cimzia + MTX ont rapporté une amélioration significative de leurs capacités fonctionnelles évaluées par le questionnaire HAQ-DI et de la douleur liée à l'arthrite évaluée par le PAAP (Patient Assessment of Arthritis Pain) à la semaine 52, par rapport aux patients traités par placebo + MTX (p<0.001 contre p<0.05). A la semaine 52, 48.1% des patients du groupe traité par Cimzia + MTX avaient atteint une capacité fonctionnelle normale (score HAQ-DI ≤0.05) contre 35.7% dans le groupe traité par placebo + MTX (p<0.01).
Spondylarthrite axiale (sous-populations avec spondylarthrite axiale non radiographique et spondylarthrite ankylosante)
AS001
L'efficacité et la sécurité du Cimzia ont été examinées dans le cadre d'une étude multicentrique randomisée, en double aveugle, contrôlée versus placebo (AS001). Celle-ci comprenait 325 patients ≥18 ans présentant une spondylarthrite axiale active de l'adulte depuis au moins 3 mois, définie selon les critères de classification pour la spondylarthrite de la société Assessment of Spondyloarthritis International Society (ASAS). La spondylarthrite axiale se caractérise par une spondylarthrite principalement avec une atteinte axiale et inclut le sous-groupe de maladies de la spondylite ankylosante, ainsi que le sous-groupe sans signe définitif de sacro-illite dans des radiographies conventionnelles sans produit de contraste, qualifié de spondylarthrite axiale non radiographique. La population participant à l'étude et atteinte de spondylarthrite axiale comprenait des sous-populations présentant aussi bien une spondylite ankylosante (SA) (également appelée spondylarthrite axiale radiographique) qu'une spondylarthrite axiale non radiographique. Les patients ont présenté une maladie active, laquelle a été définie par un indice d'activité de la spondylarthrite ankylosante de Bath (Bath Ankylosing Spondylitis Disease Activity Index - BASDAI) ≥4, des douleurs de la colonne vertébrale ≥4 sur une échelle d'évaluation numérique (NRS) de 0 à 10 et un taux de CRP élevé ou des signes actuels d'une sacro-illite via l'imagerie par résonance magnétique (IRM). Par ailleurs, les patients devaient avoir présenté une intolérance ou une réaction insuffisante face à au moins un AINS.
Au total, 16% des patients avaient été précédemment exposés à un antagoniste de TNF. Les patients ont reçu une dose initiale de Cimzia de 400 mg dans les semaines 0, 2 et 4 (concernant les deux bras du traitement) ou un placebo, suivi(e) de soit 200 mg de Cimzia toutes les deux semaines soit de 400 mg de Cimzia toutes les quatre semaines, ou bien un placebo. 87.7% des patients ont reçu des AINS concomitants. Le critère principal d'évaluation de l'efficacité était le taux de réponse ASAS20 au cours de la douzième semaine. 153 patients ont participé à une sous-étude consacrée à l'imagerie.
Le taux de CRP est un marqueur sensible de l'inflammation et peut être élevé pour d'autres causes qu'une spondylarthrite axiale non radiographique. Pour cette raison, chez les patients présentant une spondylarthrite axiale active sévère sans signes radiographiques et sans preuve définitive de sacro-illite en radiographie, un taux élevé de CRP devrait être conforté par la démonstration d'une sacro-illite par IRM au niveau des articulations sacro-iliaques.
Réponse ASAS
Dans l'étude clinique AS001, 58% des patients traités sous 200 mg de Cimzia toutes les deux semaines et 64% des patients traités sous 400 mg de Cimzia toutes les quatre semaines ont obtenu une réponse ASAS20 au cours de la douzième semaine, contre 38% sous placebo (p <0.01).
Dans l'ensemble de la population, le pourcentage de répondeurs ASAS20 était cliniquement pertinent et significativement plus élevé à chaque examen dans les groupes de traitement recevant 200 mg de Cimzia toutes les deux semaines et 400 mg de Cimzia toutes les quatre semaines, que dans le groupe placebo (p ≤0.001 à chaque examen), après l'inclusion jusqu'à la semaine 24.
Des résultats comparables ont été obtenus dans les sous-populations de patients présentant une spondylarthrite ankylosante et de patients ayant une spondylarthrite axiale non radiographique (voir tableau 8).
Des améliorations significatives pour plusieurs composantes de l'activité de la spondylarthrite axiale [douleurs (douleurs d'ensemble de la colonne vertébrale), capacité fonctionnelle (indice fonctionnel de la spondylarthrite ankylosante de Bath - BASFI), inflammation (BASDAI valeur moyenne de Q5/6), douleurs nocturnes de la colonne vertébrale, fatigue, indice de mesure de la spondylarthrite ankylosante de Bath - BASMI] ont été en outre observées chez les patients traités par Cimzia par rapport aux patients ayant reçu le placebo.
Tableau 8: Réponse efficace à l'étude clinique AS001: réduction des signes et symptômes au sein des sous-populations souffrant de spondylite ankylosante et de spondylarthrite axiale non radiographique (pourcentage de patients)
|
Paramètre
|
Spondylarthrite ankylosante
|
Spondylarthrite axiale non
radiographique
| |
|
Placebo
N=57
|
Cimzia, tous schémas
d'administration(a)
N=121
|
Placebo
N=50
|
Cimzia, tous schémas
d'administration(a)
N=97
| |
ASAS20 (b,c)
| |
Semaine 12
|
37%
|
60%*
|
40%
|
61%*
| |
Semaine 24
|
33%
|
69%**
|
24%
|
68%**
| |
ASAS40 (c,d)
| |
Semaine 12
|
19%
|
45%**
|
16%
|
47%**
| |
Semaine 24
|
16%
|
53%**
|
14%
|
51%**
| |
ASAS 5/6 (c,d)
| |
Semaine 12
|
9%
|
42%**
|
8%
|
44%**
| |
Semaine 24
|
5%
|
40%**
|
4%
|
45%**
| |
Rémission partielle (c,d)
| |
Semaine 12
|
2%
|
20%**
|
6%
|
29%**
| |
Semaine 24
|
7%
|
28%**
|
10%
|
33%**
| |
BASDAI 50 (c,d)
| |
Semaine 12
|
11%
|
41%**
|
N/A
|
N/A
| |
Semaine 24
|
16%
|
49%**
|
N/A
|
N/A
|
(a) Cimzia - tous schémas d'administration = Données pour 200 mg de Cimzia toutes les deux semaines, suite à une dose initiale de 400 mg aux semaines 0, 2 et 4, et 400 mg de Cimzia toutes les quatre semaines, suite à une dose initiale de 400 mg aux semaines 0, 2 et 4
(b) Résultats du groupe randomisé
(c) Différence de traitement: Cimzia 200-Placebo, Cimzia 400-Placebo (correspond à un intervalle de confiance de 95% et la valeur de p) sont déterminés au moyen d'un test de Wald normal bilatéral en utilisant la méthode d'imputation des non-répondeurs (NRI).
(d) Population totale d'analyse
N/A = non disponible
*p ≤0.05, Cimzia vs placebo
**p <0.001, Cimzia vs placebo
Mobilité de la colonne vertébrale
La mobilité de la colonne vertébrale a été évaluée à l'inclusion, à la semaine 12 et à la semaine 24 au moyen du BASMI. Des différences cliniquement importantes et statistiquement significatives ont été observées à chaque visite après le début de l'étude chez les patients traités par Cimzia versus les patients ayant reçu le placebo.
La différence avec le placebo de la variation moyenne du BASMI linéaire par rapport à l'inclusion a été de -0.40 points (p <0.001) jusqu'à la semaine 12 et de -0.44 points (p <0.001) jusqu'à la semaine 24 chez les patients recevant du Cimzia. La différence avec le placebo a été plus importante (-0.60 et -0.59 points jusqu'à la semaine 12 respectivement semaine 24) dans la sous-population atteinte de spondylarthrite axiale non radiographique, que dans la sous-population atteinte de spondylite ankylosante (-0.21 et -0.32 points jusqu'à la semaine 12 respectivement semaine 24).
Inhibition de l'inflammation à l'imagerie par résonance magnétique (IRM)
Dans une sous-étude consacrée à l'imagerie, les signes d'inflammation ont été examinés à la semaine 12 à l'aide de l'IRM et exprimés comme variation par rapport à l'inclusion du score SPARCC (Spondyloarthritis Research Consortium of Canada - Consortium canadien de recherche sur la spondylarthrite) pour les articulations sacro-iliaques et du score de l'ASspiMRI a modifié (modification de Berlin) pour la colonne vertébrale. Une inhibition significative des signes d'inflammation dans les articulations sacro-iliaques ainsi que dans la colonne vertébrale a été observée dans le groupe de patients sous Cimzia (tout schéma d'administration), dans l'ensemble de la population de patients atteints de spondylarthrite axiale, ainsi que dans les sous-populations de patients atteints de spondylite ankylosante et de spondylarthrite axiale non radiographique. Cependant, cela n'a pas été le cas dans le groupe de patients sous placebo.
C-OPTIMISE
L'efficacité et la sécurité de la réduction de dose et de l'arrêt du traitement chez des patients en rémission persistante ont été évaluées chez des patients adultes (âgés de 18 à 45 ans) atteints d'une axSpA active précoce (durée des symptômes inférieure à 5 ans), présentant un score ASDAS ≥2,1 (et des critères d'inclusion de la maladie similaires à ceux de l'étude AS001) et une réponse insuffisante à au moins 2 AINS ou une intolérance ou contre-indication aux AINS. Les patients incluant les sous-populations SA et nr-axSpA ont été recrutés dans une phase d'induction en ouvert de 48 semaines (Partie A) au cours de laquelle ils ont tous reçu 3 doses de charge de Cimzia 400 mg aux Semaines 0, 2 et 4, suivies par Cimzia 200 mg toutes les 2 semaines de la Semaine 6 à la Semaine 46.
Les patients qui ont atteint une rémission persistante (définie comme le fait d'avoir une maladie inactive [ASDAS<1,3] sur une période d'au moins 12 semaines) et qui restaient en rémission à la Semaine 48, ont été randomisés dans la Partie B, et ont reçu Cimzia 200 mg toutes les 2 semaines (N = 104), Cimzia 200 mg toutes les 4 semaines (réduction de dose, N = 105), ou le placebo (arrêt du traitement, N = 104) pendant 48 semaines.
Le critère primaire d'efficacité correspondait au pourcentage de patients qui n'ont pas présenté de poussée au cours de la Partie B.
Les patients qui ont présenté une poussée au cours de la Partie B, c'est-à-dire ayant un score ASDAS ≥2,1 lors de 2 visites consécutives ou un score ASDAS > 3,5 lors de toute visite au cours de la Partie B, ont reçu un traitement de secours par Cimzia 200 mg toutes les 2 semaines pendant au moins 12 semaines (avec une dose de charge de Cimzia 400 mg à la Semaine 0, 2 et 4 chez les patients traités par placebo).
Réponse clinique
Le pourcentage de patients qui ont atteint une rémission persistante à la Semaine 48 dans la Partie A s'élevait à 43,9 % pour la population globale d'axSpA, et était similaire dans les sous-populations nr-axSpA (45,3 %) et SA (42,8 %).
Parmi les patients qui ont été randomisés dans la Partie B (N = 313), une proportion supérieure statistiquement significative (p < 0,001, NRI) de patients n'a pas présenté de poussée en poursuivant le traitement par Cimzia 200 mg toutes les 2 semaines (83,7 %) ou Cimzia 200 mg toutes les 4 semaines (79,0 %) comparé au groupe en arrêt de traitement (20,2 %).
La différence dans le délai de survenue de poussée entre le groupe en arrêt de traitement et l'un ou l'autre des groupes de traitement par Cimzia, était statistiquement (p < 0,001 pour chaque comparaison) et cliniquement significative. Dans le groupe du placebo, les poussées ont commencé environ 8 semaines après l'arrêt de Cimzia, la majeure partie des poussées survenant dans les 24 heures suivant l'arrêt du traitement (Figure 2).
Figure 2: Courbe Kaplan-Meier du délai d'apparition d'une poussée
L'imputation des non-répondeurs (NRI) a été utilisée; les résultats concernent l'Ensemble randomisé.
Remarque: Le délai d'apparition d'une poussée a été défini comme la période commençant à la date de randomisation et se terminant à la date de la poussée. Pour les participants à l'étude qui n'ont pas eu de poussée, le délai d'apparition d'une poussée a été censuré à la date de la Visite de la Semaine 96.
Le diagramme de Kaplan-Meier a été tronqué à 97 semaines quand < 5 % des participants demeuraient dans l'étude.
Nouveau traitement des patients présentant une poussée
Dans la Partie B, 70 % (73/104) des patients traités par placebo, 14 % (15/105) des patients traités par Cimzia 200 mg toutes les 4 semaines et 6,7 % (7/104) des patients traités par Cimzia 200 mg toutes les 2 semaines ont présenté une poussée et ont ensuite été traités par Cimzia 200 mg toutes les 2 semaines.
Parmi les 73 patients ayant présenté une poussée dans le groupe affecté à l'arrêt du traitement, 71 patients ont suivi un traitement de secours par Cimzia pendant 12 semaines, et avaient des données ASDAS disponibles, dont 64 (90 %) présentaient un score ASDAS de maladie faible ou inactive (c'est-à-dire, tout ASDAS < 2,1) après 12 semaines de reprise du traitement en ouvert.
Le nombre de patients dans les deux groupes Cimzia présentant une poussée était trop faible pour permettre une évaluation valable des effets d'un nouveau traitement dans ce cas là.
D'après les résultats de C-OPTIMISE, une réduction de dose chez les patients en rémission persistante après une année de traitement par Cimzia peut être envisagée (voir «Posologie/Mode d'emploi»).
Spondylarthrite axiale non radiographique (nr-axSpA)
L'efficacité et la sécurité de Cimzia ont été étudiées dans une étude multicentrique randomisée, en double aveugle, contrôlée versus placebo de 52 semaines (AS0006) chez 317 patients ≥18 ans atteints de spondylarthrite axiale survenue à l'âge adulte et avec des douleurs dorsales depuis au moins 12 mois. Les patients devaient présenter des signes objectifs d'inflamations indiqués par des taux de protéine C-réactive (CRP) supérieur à la normale et/ou une sacro-iliite à l'imagerie par résonnance magnétique (IRM), indiquant une maladie inflammatoire [CRP positive (> ULN) et/ou IRM positive], mais sans signe radiographique définitif de lésions structurelles au niveau des articulations sacro-iliaques. Les patients avaient une maladie active définie par un score BASDAI ≥4, et des douleurs rachidiennes ≥4 sur une échelle NRS allant de 0 à 10. Les patients devaient présenter une intolérance ou une réponse inadéquate à au moins deux AINS. Les patients ont été traités par placebo ou par une dose initiale de 400 mg de Cimzia aux semaines 0, 2 et 4 puis par 200 mg de Cimzia toutes les 2 semaines. L'utilisation et l'ajustement de la dose du traitement standard (par ex. AINS, DMARD, corticostéroïdes et analgésiques) étaient autorisés à tout moment. De même, les patients pouvaient, à tout moment, passer du traitement de l'étude en double aveugle à un traitement ouvert avec Cimzia ou avec une autre substance. La principale variable d'efficacité était la réponse ASDAS-MI (Ankylosing Spondylitis Disease Activity Score major improvement) à la semaine 52. La réponse ASDAS-MI était définie comme une réduction de l'ASDAS (amélioration) ≥2,0 par rapport à la valeur de référence ou à l'atteinte de la plus petite valeur possible.
Dans le groupe Cimzia et dans le groupe placebo, 37 % et 41 % des patients, respectivement, présentaient une activité élevée (ASDAS ≥2,1; ≤3,5) et 62 % et 58 % des patients, respectivement, avaient une activité très élevée (ASDAS >3,5) de la maladie.
Réponse clinique
Dans l'étude AS006 en semaine 52, une proportion statistiquement significative des patients traités par Cimzia a obtenu une réponse ASDAS-MI par rapport aux patients ayant reçu le placebo. Les patients traités par Cimzia ont présenté des améliorations de plusieurs éléments de l'activité de la spondylarthrite axiale par rapport aux patients traités par placebo. Aussi bien en semaine 12 qu'en semaine 52, la réponse ASAS 40 était significativement supérieure par rapport au placebo. Les principaux résultats sont présentés dans les tableaux 9 et 10.
Tableau 9: réponse ASDAS-MI et ASAS-40 dans l'étude AS0006 (pourcentage des patients)
|
Paramètre
|
Placebo
N = 158
|
Cimziaa 200 mg toute les 2 semaines
N = 159
| |
ASDAS-MI
semaine 52
|
7%
|
47%*
| |
ASAS 40
semaine 12
semaine 52
|
11 %
16%
|
48 %*
57%*
|
a Cimzia administré toutes les 2 semaines après une dose initiale de 400 mg aux semaines 0, 2 et 4
* p<0,001
Tous les pourcentages représentent la proportion de patients ayant obtenu une réponse à l'ensemble de l'analyse.
Tableau 10: résultats BASDAI et BASFI dans l'étude AS0006
|
Paramètre
|
Placebo
N = 158
|
Cimzia(a) 200 mg toutes les 2 semaines
N = 159
| |
|
État initial
|
Semaine 12
|
Semaine 52
|
État initial
|
Semaine 12
|
Semaine 52
| |
BASDAI(b)
|
6,79 (1,28)
|
5,71 (2,09)
|
5,47 (2,30)
|
6,88 (1,40)
|
3,93 (2,21)
|
3,26 (2,47)
| |
BASFI(c)
|
5,44 (2,18)
|
4,95 (2,46)
|
4,71 (2,60)
|
5,41 (2,12)
|
3,20 (2,33)
|
2,68 (2,42)
|
(a) Cimzia administré toutes les 2 semaines après une dose initiale de 400 mg en semaine 0, 2 et 4
(b) BASDAI est l'indice „Bath Ankylosing Spondylitis Disease Activity NRS Index“, où 0 = nul et 10 = très grave
(c) BASFI est l'indice „Bath Ankylosing Spondylitis Functional NRS Index“, où 0 = facile et 0 = impossible
L'ensemble complet d'analyse avec indication de la dernière valeur (last observation carried forward, LOCF) a été utilisé.
Les signes d'inflammation ont été constatés à l'IRM à la semaine 12 et représentés sous la forme de modification de l'état initial du score SPARCC pour les articulations sacro-iliaques.
Une inhibition significative de l'inflammation des articulations sacro-iliaques a été constatée en semaine 12 et en semaine 52 chez les patients traités par Cimzia (-4,3 et -7,4 respectivement) par rapport aux patients traités par placebo (0,3 et 2,3 respectivement) par rapport à l'état initial.
Arthrite psoriasique
L'efficacité et la sécurité du Cimzia ont été évaluées dans le cadre d'une étude clinique randomisée, multicentrique, en double aveugle, contrôlée versus placebo (PsA001), menée sur 409 patients d'âge ≥18 ans atteints d'arthrite psoriasique active de l'adulte depuis 6 mois au minimum, définie selon les critères de classification pour l'arthrite psoriasique (Classification Criteria for Psoriatic Arthritis, critères CASPAR). Les patients présentaient ≥3 articulations gonflées douloureuses, ainsi que soit une augmentation des marqueurs de la phase aiguë de l'inflammation soit une vitesse de sédimentation érythrocytaire (ESR – Erythrocyte sedimentation rate) ≥28 mm/heure. Les patients présentaient en outre des lésions cutanées psoriasiques actives ou des antécédents documentés de Psoriasis et ils avaient derrière eux au moins une tentative de traitement par DMARDs sans succès (médicaments antirhumatismaux modificateurs de la maladie). Le traitement antérieur par antagoniste du TNF était autorisé et environ 20% des patients avaient été précédemment traités par antagonistes du TNF. Les patients ont reçu soit 200 mg de Cimzia toutes les 2 semaines respectivement 400 mg de Cimzia toutes les 4 semaines ou d'un placebo toutes les 2 semaines, après des doses initiales de 400 mg de Cimzia aux semaines 0, 2 et 4 (dans les deux bras du traitement) ou d'un placebo. Les patients traités de manière concomitante par des AINS ou des DMARDs conventionnels représentaient respectivement 72.6% et 70.2% des patients. Les deux critères principaux d'évaluation étaient le pourcentage de patients ayant obtenu une réponse ACR 20 jusqu'à la semaine 12 et la modification du score total de Sharp modifié (modified Total Sharp Score, mTSS) à la semaine 24 par rapport à l'inclusion.
Réponse ACR
Les taux de réponses ACR 20, ACR 50 respectivement ACR 70 au traitement par Cimzia dans l'étude clinique PsA001 sont représentés dans le tableau 11. Les patients sous Cimzia ont présenté des taux de réponses ACR 20 statistiquement et significativement plus élevés d'un point de vue statistique aux semaines 12 et 24 (p <0,001) par rapport aux patients recevant le placebo. De plus, les patients recevant du Cimzia ont présenté des améliorations significatives aux semaines 12 et 24 en ce qui concerne les taux de réponses ACR 50 et ACR 70, ainsi que les composantes ACR individuelles (voir tableau 12), par rapport aux patients sous placebo. Les patients recevant 200 mg de Cimzia toutes les 2 semaines et les patients recevant 400 mg de Cimzia toutes les 4 semaines ont répondu de façon similaire au traitement.
Les patients recevant une comédication de méthotrexate ont présenté au cours de la semaine 24 des taux de réponses ACR 20, ACR 50 et ACR 70 plus importants (62%, 35% et 26%) que les patients sous monothérapie (53%, 32% et 20%).
Tableau 11: Réponse ACR dans l'étude clinique PsA001 (pourcentage de patients)
|
Réponse
|
Placebo
N=136
|
Cimzia(a) 200 mg
Q2W
N=138
|
Cimzia(b) 400 mg
Q4W
N=135
| |
ACR 20
| |
Semaine 12
|
24%
|
58%**
|
52%**
| |
Semaine 24
|
24%
|
64%**
|
56%**
| |
ACR 50
| |
Semaine 12
|
11%
|
36%**
|
33%**
| |
Semaine 24
|
13%
|
44%**
|
40%**
| |
ACR 70
| |
Semaine 12
|
3%
|
25%**
|
13%*
| |
Semaine 24
|
4%
|
28%**
|
24%**
|
(a) Cimzia toutes les 2 semaines, précédé de doses initiales de 400 mg aux semaines 0, 2 et 4
(b) Cimzia toutes les 4 semaines, précédé de doses initiales de 400 mg aux semaines 0, 2 et 4
* p <0.01, Cimzia versus placebo
** p <0.001, Cimzia versus placebo
Les résultats se sont basés sur la population randomisée. Différence de traitement: Cimzia 200 mg – placebo, Cimzia 400 mg – placebo (avec l'IC bilatéral à 95% et valeur de p) sont estimés avec des erreurs asymptotiques standard au moyen d'un test de Wald standard bilatéral en utilisant la méthode d'imputation des non-répondeurs (NRI – Non-Responder Imputation).
Tableau 12: Composantes des réponses ACR dans l'étude clinique PsA001
|
Paramètres
|
Placebo
N=136
|
Cimzia(a) 200 mg Q2W
N=138
|
Cimzia(b) 400 mg Q4W
N=135
| |
|
Valeurs initiales
|
Semaine 12
|
Semaine 24
|
Valeurs initiales
|
Semaine 12
|
Semaine 24
|
Valeurs initiales
|
Semaine 12
|
Semaine 24
| |
Nombre
d'articulations sensibles à la douleur
(0–68) (c)
|
19.9
|
16.5
|
17.0
|
21.5
|
11.2*
|
8.5*
|
19.6
|
11.2*
|
9.4*
| |
Nombre
d'articulations gonflées
(0-66) (c)
|
10.4
|
8.7
|
9.9
|
11.0
|
4.0*
|
3.1*
|
10.5
|
4.7*
|
3.0*
| |
Jugement médical global (c, d)
|
58.7
|
44.1
|
42.2
|
56.8
|
24.8*
|
19.6*
|
58.2
|
28.7*
|
21.1*
| |
Jugement global du patient (c, d)
|
57.0
|
50.2
|
49.0
|
60.2
|
32.6*
|
31.1*
|
60.2
|
39.6*
|
32.5*
| |
Douleur (c, e)
|
60.0
|
50.2
|
48.8
|
59.7
|
32.8*
|
31.1*
|
61.1
|
38.6*
|
32.7*
| |
Indice d'incapacité (HAQ) (c, f)
|
1.30
|
1.15
|
1.13
|
1.33
|
0.87*
|
0.81*
|
1.29
|
0.90*
|
0.86*
| |
CRP (mg/l)
|
18.56
|
14.75
|
14.66
|
15.36
|
5.67*
|
4.58*
|
13.71
|
6.34*
|
7.37*
|
(a) Cimzia toutes les 2 semaines, précédé de doses initiales de 400 mg aux semaines 0, 2 et 4
(b) Cimzia toutes les 4 semaines, précédé de doses initiales de 400 mg aux semaines 0, 2 et 4
(c) Approche LOCF (Last Observation Carried Forward – dernière observation reportée) pour des données manquantes, une interruption précoce de l'étude ou un adjuvant placebo
(d) Patient et Physician Global Assessment of Disease Activity (Jugement global de l'activité de la maladie par le patient respectivement le médecin), échelle VAS avec 0 = très bonne et 100 = très mauvaise
(e) The Patient Assessment of Arthritis Pain (L'évaluation de la douleur arthritique par le patient), échelle VAS avec 0 = aucune douleur et 100 = douleurs très fortes
(f) HAQ-DI (questionnaire d'évaluation de l'état de santé – indice d'incapacité), échelle à 4 points allant de 0 = aucune invalidité à 3 = grande invalidité
En ce qui concerne les valeurs représentées, il s'agit de données moyennes.
Les résultats se basent sur la population randomisée (imputation ou observation).
* p <0.001, Cimzia versus placebo
Le pourcentage de répondeurs ACR 20 était cliniquement pertinent et significativement plus élevé dans les groupes de traitement sous Cimzia 200 mg toutes les 2 semaines et Cimzia 400 mg toutes les 4 semaines, que dans le groupe de patients sous placebo (p ≤0.001 à toutes les visites), à toutes les visites après le début du traitement jusqu'à la semaine 24.
Réponse PASI
Les patients, ayant au minimum 3% à l'inclusion de la surface corporelle touchée par des lésions psoriasiques, ont été évalués au moyen de la réponse selon l'indice de surface et de gravité du psoriasis (Psoriasis Area and Severity Index – PASI), en ce qui concerne l'amélioration de leurs manifestations cutanées. Comme le montre le tableau 13, le pourcentage de patients était composé de ceux qui ont obtenu une réponse PASI 75 et une réponse PASI 90 à la semaine 24, dans les deux groupes posologiques de Cimzia (200 mg toutes les 2 semaines + 400 mg toutes les 4 semaines) (n=166) 61% respectivement 42%, par rapport à 15% respectivement 6% dans le groupe placebo (n=86) (p <0.001).
Tableau 13: Taux de réponse PASI dans l'étude clinique PsA001
|
Taux de réponse
|
Placebo
N=86
|
Cimzia(a) 200 mg Q2W
N=90
|
Cimzia(b) 400 mg Q4W
N=76
|
Cimzia (toutes les posologies)(c)
N=166
| |
|
Semaine 12
|
Semaine 24
|
Semaine 12
|
Semaine 24
|
Semaine 12
|
Semaine 24
|
Semaine 12
|
Semaine 24
| |
PASI 75
|
14%
|
15%
|
47%**
|
62%**
|
47%**
|
61%**
|
47%**
|
61%**
| |
PASI 90
|
5%
|
6%
|
22%**
|
47%**
|
20%*
|
36%**
|
21%**
|
42%**
|
(a) Cimzia toutes les 2 semaines, précédée de doses initiales de 400 mg aux semaines 0, 2 et 4
(b) Cimzia toutes les 4 semaines, précédée de doses initiales de 400 mg aux semaines 0, 2 et 4
(c) Cimzia (toutes les posologies) = données pour Cimzia 200 mg toutes les 2 semaines, précédée de doses initiales de 400 mg aux semaines 0, 2 et 4, plus données pour Cimzia 400 mg toutes les 4 semaines, précédée de doses initiales de 400 mg aux semaines 0, 2 et 4
* Valeur p <0.01, Cimzia vs placebo
** Valeur p <0.001, Cimzia vs placebo
Les résultats se sont basés sur la population randomisée.
Différence de traitement: Cimzia 200 mg – placebo, Cimzia 400 mg – placebo (avec intervalle de confiance de 95% correspondant et la valeur p) sont estimés avec des erreurs asymptotiques standard au moyen d'un test de Wald standard bilatéral. Remarque: la méthode NRI (Non-Responder-Imputation) a été utilisée.
Les patients qui ont présenté une enthésite ont été évalués du point de vue de l'amélioration moyenne de l'indice de Leeds pour l'enthésite (Leeds Enthesitis Index – LEI). Les patients sous 200 mg de Cimzia toutes les 2 semaines ou 400 mg toutes les 4 semaines ont présenté une amélioration plus importante de l'enthésite (-1.8; -1.7) que les patients recevant le placebo (-0.9) dans la semaine 12 (p <0.001 respectivement p <0.01) et la semaine 24 (200 mg toutes les 2 semaines: -2.0; 400 mg toutes les 4 semaines: 1.8; placebo: -1.1) (p <0.001 respectivement p <0.01).
Les mêmes schémas de posologie ont également montré une réduction significative de la dactylite (modification moyenne du score initial (-30.40; -45.46) par rapport aux patients traités par placebo (-16.79) dans la semaine 12.
Réponse radiologique
Dans l'étude clinique PsA001, la progression des dommages articulaires a été évaluée par radiographie entre la période d'inclusion et la semaine 24 et exprimée en termes de modification du mTSS et de ses composantes dans le score d'érosion (SE) et le score de pincement articulaire (JSN, de Joint Space Narrowing). Le score mTSS-Score a été modifié en ajoutant les articulations interphalangiennes distales pour arthrite psoriasique. L'effet du traitement suivant a été observé avec une imputation post hoc: le traitement par Cimzia par rapport au traitement par placebo à la semaine 24 a inhibé la progression radiologique de la modification du mTSS par rapport à la valeur initiale (la valeur moyenne LS [± SE] du score était de 0.28 [± 0.07] dans le groupe de patients sous placebo par rapport aux 0.06 [± 0.06] dans l'ensemble du groupe Cimzia; p=0.007). Une différence statistiquement significative par rapport au placebo a été observée dans le groupe de patients recevant 200 mg de CZP Q2W (p=0.004), mais pas dans le groupe de patients recevant 400 mg de CZP Q4W (p=0.072).
Réaction en termes de capacité fonctionnelle et de critères d'évaluation liés à la santé
Dans l'étude clinique PsA001, les patients traités par Cimzia à partir de la semaine 2 ont rapporté de nettes améliorations de leurs capacités fonctionnelles physiques (déterminées au moyen du questionnaire d'évaluation de l'état de santé – indice d'incapacité (HAQ – DI Disability Index)») «Health Assessment Questionnaire – Disability Index)») ainsi que de la douleur (déterminée au moyen de l'évaluation par le patient de la douleur arthritique «Patient Assessment of Arthritis Pain [PAAP]») (voir tableau 12). La différence a été significative pour la PAAP après la semaine 1. Les patients recevant du Cimzia ont rapporté une amélioration de la fatigue évaluée au moyen de l'échelle d'évaluation de la fatigue («FASCA – Fatigue Assessment Scale») de la semaine 2 à la semaine 24, par rapport au placebo
Les patients recevant du Cimzia ont rapporté des améliorations significatives de leur qualité de vie liée à leur santé dans le questionnaire relatif à l'indice de qualité de vie spécifique du RhPso (PsAQoL) à partir de la semaine 2, ainsi que dans tous les «scores» résumés des composantes physiques et mentales du questionnaire SF 36 de la semaine 4 à la semaine 24.
Les patients traités par Cimzia ont connu des améliorations des limites de productivité dues à l'arthrite psoriasique sur leur lieu de travail et à leur domicile sur la base du «Work Productivity Survey» (questionnaire de productivité au travail) par rapport aux patients recevant le placebo de la semaine 4 à la semaine 24.
Psoriasis en plaques
L'efficacité et la sécurité du Cimzia ont été testées dans deux études contrôlées versus placebo (COMPASI-1 et CIMPASI-2) et une étude contrôlée versus placebo et versus un comparateur actif (CIMPACT) chez des patients âgés de ≥18 ans atteints de psoriasis en plaques chronique modéré à sévère sur une période d'au moins 6 mois. Les patients présentaient un score PASI (Psoriasis Area and Severity Index-Score) ≥12, une atteinte ≥10 % de la surface corporelle (BSA, Body Surface Area), un score PGA ≥3 (évaluation globale du médecin, Physician Global Assessment) et nécessitaient un traitement systémique et/ou une photothérapie et/ou une chimiothérapie. Les patients en échec «primaire» à tout traitement biologique antérieur (défini par l'absence de réponse au cours des 12 premières semaines de traitement) ont été exclus des études de phase III (CIMPASI-1, CIMPASI-2 et CIMPACT). L'efficacité et la sécurité du Cimzia ont été évaluées par rapport à étanercept dans l'étude CIMPACT.
Dans les études CIMPASI-1 et CIMPASI-2, les co-critères primaires d'efficacité étaient le taux de répondeurs PASI 75 et le pourcentage de patients présentant un score PGA «blanchi» ou «quasiment blanchi» (avec une diminution d'au moins 2 points par rapport à l'inclusion) à la semaine 16. Dans l'étude CIMPACT, le critère primaire d'efficacité était le taux de répondeurs PASI 75 à la semaine 12. Les taux de répondeurs PASI 75 et PGA à la semaine 16 étaient des critères secondaires. Le taux de répondeurs PASI 90 à la semaine 16 était un critère secondaire dans les 3 études.
234 patients et 227 patients ont été évalués dans les études CIMPASI-1 et CIMPASI-2, respectivement. Dans les deux études, les patients ont été randomisés pour recevoir un placebo ou Cimzia 200 mg toutes les 2 semaines (après une dose initiale de Cimzia de 400 mg aux semaines 0, 2 et 4) ou Cimzia 400 mg toutes les 2 semaines. À la semaine 16, les patients randomisés dans les bras Cimzia et répondeurs PASI 50 ont continué à recevoir Cimzia jusqu'à la semaine 48 à la même dose randomisée. Les patients qui avaient été initialement randomisés dans le bras placebo et qui avaient obtenu une réponse PASI 50, mais pas une réponse PASI 75 à la Semaine 16 ont reçu Cimzia 200 mg toutes les 2 semaines (avec une dose initiale de Cimzia de 400 mg aux semaines 16, 18, et 20). Les patients qui présentaient une réponse insuffisante à la semaine 16 (non-répondeurs PASI 50) étaient éligibles à recevoir Cimzia 400 mg toutes les 2 semaines pendant 128 semaines maximum.
559 patients ont été évalués dans l'étude CIMPACT. Les patients ont été randomisés pour recevoir un placebo ou Cimzia 200 mg toutes les 2 semaines (après une dose initiale de Cimzia de 400 mg aux semaines 0, 2 et 4) ou Cimzia 400 mg toutes les 2 semaines jusqu'à la semaine 16, ou étanercept 50 mg deux fois par semaine jusqu'à la semaine 12. Les patients qui avaient été initialement randomisés dans le bras Cimzia et répondeurs PASI 75 à la semaine 16 ont été de nouveau randomisés en fonction de leur schéma posologique initial. Les patients sous Cimzia 200 mg toutes les 2 semaines ont été de nouveau randomisés dans le bras Cimzia 200 mg toutes les 2 semaines, le bras Cimzia 400 mg toutes les 4 semaines ou le bras placebo. Les patients sous Cimzia 400 mg toutes les 2 semaines ont été de nouveau randomisés dans le bras Cimzia 400 mg toutes les 2 semaines, le bras Cimzia 200 mg toutes les 2 semaines, ou dans le bras placebo. L'étude était en double aveugle contrôlée versus placebo jusqu'à la semaine 48. Tous les sujets non-répondeurs PASI 75 à la semaine 16 ont intégré un bras d'échappement et ont reçu 400 mg de Cimzia en ouvert toutes les 2 semaines pendant 128 semaines maximum.
Lors des trois études, une phase de traitement en ouvert de 96 semaines succédait à une phase d'entretien en aveugle de 48 semaines pour les patients répondeurs PASI 50 à la semaine 48. Tous les patients sous traitement en aveugle ont commencé la période en ouvert avec Cimzia 200 mg toutes les 2 semaines. Au cours de la phase de traitement en ouvert, les ajustements de dose entre Cimzia 200 mg toutes les 2 semaines et Cimzia 400 mg toutes les 2 semaines étaient autorisés en l'absence d'une réponse PASI 50 ou à l'appréciation du médecin investigateur. La durée maximale des études était en tout de 144 semaines.
Parmi les 850 patients randomisés pour recevoir le placebo ou Cimzia dans ces études contrôlées versus placebo, 29% étaient naïfs de traitement systémique antérieur pour le traitement d'un psoriasis. 47% avaient reçu une photothérapie ou une chimiothérapie antérieure et 30% avaient reçu un traitement biologique antérieur pour le traitement d'un psoriasis. Parmi ces 850 patients, 14% avaient reçu au moins un anti-TNF, 13% avaient reçu un anti-IL-17 et 5% avaient reçu un anti-IL-12/23. 18% des patients ont rapporté des antécédents de rhumatisme psoriasique à l'inclusion. Le score PASI moyen à l'inclusion était de 20 et variait entre 12 et 69. Le score PGA à l'inclusion était modéré (70%) à sévère (30%). La BSA moyenne à l'inclusion était de 25% et variait entre 10% et 96%.
Les principaux résultats des études CIMPASI-1 et CIMPASI-2 sont reportés dans le tableau 14.
Tableau 14: Réponse clinique dans les études CIMPASI-1 et CIMPASI-2 à la semaine 16 et à la semaine 48
|
|
Semaine 16
|
Semaine 48
| |
CIMPASI-1
| |
|
Placebo
N=51
|
Cimzia 200 mg toutes les 2 semainesa)
N=95
|
Cimzia 400 mg toutes les 2 semaines
N=88
|
Cimzia 200 mg toutes les 2 semaines
N=95
|
Cimzia 400 mg toutes les 2 semaines
N=88
| |
PGA blanchi ou quasiment blanchib)
|
4.2%
|
47.0%*
|
57.9%*
|
52.7%
|
69.5%
| |
PASI 75
|
6.5%
|
66.5%*
|
75.8%*
|
67.2%
|
87.1%
| |
PASI 90
|
0.4%
|
35.8%*
|
43.6%*
|
42.8%
|
60.2%
| |
CIMPASI-2
| |
|
Placebo
N=49
|
Cimzia 200 mg toutes les 2 semaines a)
N=91
|
Cimzia 400 mg toutes les 2 semaines
N=87
|
Cimzia 200 mg toutes les 2 semaines
N=91
|
Cimzia 400 mg toutes les 2 semaines
N=87
| |
PGA blanchi ou quasiment blanchib)
|
2.0%
|
66.8%*
|
71.6%*
|
72.6%
|
66.6%
| |
PASI 75
|
11.6%
|
81.4%*
|
82.6%*
|
78.7%
|
81.3%
| |
PASI 90
|
4.5%
|
52.6%*
|
55.4%*
|
59.6%
|
62.0%
|
a) Cimzia 200 mg, administré toutes les 2 semaines, après une dose initiale de 400 mg aux semaines 0, 2, 4.
b) Echelle PGA à 5 catégories. L'atteinte du critère «blanchi» (0) ou «quasiment blanchi» (1) était définie par l'absence de signes de psoriasis ou une coloration des lésions normale à rose, l'absence d'épaississement de la plaque et une desquamation absente ou minime.
* p<0.0001, Cimzia vs. placebo
Les taux de répondeurs et les valeurs p pour les scores PASI et PGA ont été calculés à l'aide d'un modèle de régression logistique dans lequel les données manquantes ont été imputées à l'aide d'une imputation multiple avec la méthode MCMC. Les patients ayant échappé ou été sortis de l'étude (sur la base d'une non atteinte de réponse PASI 50) ont été considérés comme des non-répondeurs à la semaine 48.
Les résultats sont issus de l'ensemble randomisé.
Les principaux résultats de l'étude CIMPACT sont reportés dans le tableau 15.
Tableau 15: Réponse clinique dans l'étude CIMPACT à la semaine 12 et à la semaine 16
|
|
Semaine 12
|
Semaine 16
| |
|
Placebo
N=57
|
Cimzia 200 mg toutes les 2 semainesa)
N=165
|
Cimzia 400 mg toutes les 2 semaines
N=167
|
Placebo
N=57
|
Cimzia 200 mg toutes les 2 semaines
N=165
|
Cimzia 400 mg
toutes les 2 semainesN=167
| |
PASI 75
|
5%
|
61.3%*
|
66.7%*
|
3.8%
|
68.2%*
|
74.7%*
| |
PASI 90
|
0.2%
|
31.2%*
|
34.0%*
|
0.3%
|
39.8%*
|
49.1%*
| |
PGA blanchi ou quasiment blanchib)
|
1.9%
|
39.8%**
|
50.3%*
|
3.4%
|
48.3%*
|
58.4%*
|
a) Cimzia 200 mg, administré toutes les 2 semaines, après une dose initiale de 400 mg aux semaines 0, 2, 4.
b) Echelle PGA à 5 catégories. L'atteinte du critère «blanchi» (0) ou «quasiment blanchi» (1) était définie par l'absence de signes de psoriasis ou une coloration des lésions normale à rose, l'absence d'épaississement de la plaque et une desquamation absente ou minime.
* p<0.0001, Cimzia vs. placebo
** p<0.0001, Cimzia vs. placebo
Taux de répondeurs et valeurs p basés sur un modèle de régression logistique. Les données manquantes ont été imputées à l'aide d'une imputation multiple basée sur la méthode MCMC.
Les résultats sont issus de l'ensemble randomisé.
Dans l'étude CIMPACT, Cimzia 400 mg toutes les 2 semaines versus l'étanercept 50 mg deux fois par semaine, au bout de 12 semaines, a démontré une supériorité concernant le taux de répondeurs PASI 75 (66.7% contre 53.3%; p < 0.05). Cimzia 200 mg toutes les 2 semaines versus l'étanercept 50 mg deux fois par semaine, a démontré une non- infériorité concernant le taux de répondeurs PASI 75, avec une marge de non-infériorité préalablement définie de 10 % (61.3%; différence entre l'étanercept et Cimzia 200 mg toutes les 2 semaines 8,0% [IC à 95% –2.9; 18.9]).
Dans les trois études, le taux de répondeurs PASI 75 et le taux de patients présentant un score PGA «blanchi» ou «quasiment blanchi» du bras Cimzia était significativement supérieur à celui du bras placebo à partir de la semaine 4.
Les deux doses de Cimzia ont démontré une efficacité par rapport au placebo, quels que soient l'âge, le sexe, le poids corporel, l'IMC, l'antériorité du psoriasis, les antécédents de traitements systémiques et les antécédents de traitements biologiques.
Maintien de la réponse
Dans une analyse intégrée des études CIMPASI-1 et CIMPASI-2, parmi les patients qui étaient répondeurs PASI 75 à la semaine 16 et qui avaient reçu Cimzia 400 mg toutes les 2 semaines (134 patients sur les 175 randomisés) ou Cimzia 200 mg une semaine sur 2 (132 patients sur les 186 randomisés), les taux de maintien de la réponse à la semaine 48 étaient de 98.0% et de 87.5%, respectivement. Parmi les patients qui présentaient un score PGA blanchi ou quasiment blanchi à la semaine 16 et qui avaient reçu Cimzia 400 mg toutes les 2 semaines (103 patients sur 175) ou Cimzia 200 mg toutes les 2 semaines (95 patients sur 186), les taux de maintien de la réponse à la semaine 48 étaient de 85.9 % et de 84.3 % respectivement.
Ces taux de réponse étaient calculés sur un modèle de régression logistique dans lequel les données manquantes ont été imputées à l'aide d'une imputation multiple (méthode MCMC) sur 48 et 144 semaines. Les résultats à 48 semaines de la phase de traitement en ouvert pendant 96 semaines chez les répondeurs PASI 75 indiquaient que la réponse se maintient majoritairement sous Cimzia 200 mg toutes les 2 semaines.
Dans l'étude CIMPACT, parmi les répondeurs PASI 75 à la semaine 16 qui avaient reçu Cimzia 400 mg toutes les 2 semaines et qui avaient été de nouveau randomisés dans le bras Cimzia 400 mg toutes les 2 semaines, ou le bras Cimzia 200 mg toutes les 2 semaines, ou le bras placebo, le pourcentage de répondeurs PASI 75 à la semaine 48 était supérieur dans les groupes Cimzia à celui du groupe placebo (98.0%, 80.0%, et 36.0%, respectivement). Parmi les répondeurs PASI 75 à la semaine 16 qui avaient reçu Cimzia 200 mg toutes les 2 semaines et qui avaient été de nouveau randomisés dans le bras Cimzia 400 mg toutes les 4 semaines, ou le bras Cimzia 200 mg toutes les 2 semaines, ou le bras placebo, le pourcentage de répondeurs PASI 75 à la semaine 48 était également supérieur dans les groupes Cimzia à celui du groupe placebo (88.6%, 79.5%, et 45.5%, respectivement). L'imputation des non-répondeurs a été utilisée pour les données manquantes. Les résultats de la période suivante de traitement en ouvert sur 96 semaines chez les répondeurs PASI 75 ont démontré que la réponse se maintient majoritairement sous Cimzia 200 mg toutes les 2 semaines.
Qualité de vie / résultats du point de vue des patients
Des améliorations statistiquement significatives à la semaine 16 (CIMPASI-1 et CIMPASI-2) du DLQI (indice dermatologique de qualité de vie, Dermatology Life Quality Index) versus placebo ont été démontrées par rapport à l'inclusion. Les diminutions moyennes (améliorations) du DLQI par rapport à l'inclusion variaient de -8.9 à -11.1 avec Cimzia 200 mg toutes les 2 semaines, de -9.6 à -10,0 avec Cimzia 400 mg toutes les 2 semaines, versus -2.9 à -3.3 pour le placebo à la semaine 16.
De plus, à la semaine 16, le traitement par Cimzia était associé à une plus grande proportion de patients ayant obtenu un score DLQI de 0 ou 1 (Cimzia 400 mg toutes les 2 semaines, 45.5% et 50.6% respectivement; Cimzia 200 mg toutes les 2 semaines, 47.4% et 46.2% respectivement, versus placebo, 5.9% et 8.2% respectivement).
Les patients traités par Cimzia ont rapporté des améliorations plus importantes par rapport au placebo dans l'échelle HADS-D (échelle d'anxiété et de dépression à l'hôpital - dimension dépressive, Hospital Anxiety and Depression Scale).
PharmacocinétiqueLes concentrations plasmatiques de certolizumab pégol ont généralement été proportionnelles à la dose.
La pharmacocinétique observée chez les patients ayant une maladie de Crohn, une polyarthrite rhumatoïde et un psoriasis a été cohérente avec celle observée chez les volontaires sains.
L'évaluation de la pharmacocinétique de certolizumab pégol a eu lieu dans le cadre d'une analyse pharmacocinétique croisée entre différentes études se rapportant à la population en se fondant sur les données de 1'580 sujets, dont 1'268 patients atteints de la maladie de Crohn et de 234 patients atteints de polyarthrite rhumatoïde ainsi que de 78 volontaires sains. L'analyse pharmacocinétique se rapportant à la population a montré que ni l'âge, ni le sexe, ni la clairance de la créatinine, ni la numération leucocytaire, n'influençaient la pharmacocinétique du certolizumab pégol. En raison du faible nombre de patients présentant des troubles importants de la fonction hépatique, l'analyse pharmacocinétique se rapportant à la population n'a pas permis de tirer des conclusions relatives à l'effet des troubles de la fonction hépatique.
Absorption
Lors d'une administration sous-cutanée, des pics de concentration de certolizumab pégol ont été atteints de 54 à 171 heures après l'injection. Lors d'une administration sous-cutanée, la biodisponibilité (F) du certolizumab pégol est d'env. 80% (dispersion: 76%-88%) par rapport à une administration intraveineuse.
Distribution
Dans le cadre de l'analyse pharmacocinétique se rapportant à la population, s'appuyant en majeure partie sur des données issues de patients atteints de la maladie de Crohn, le volume de distribution central (Vc) a été estimé à 4.0 l, avec une variabilité interindividuelle (% CV) de 16.9%. Le volume de distribution à l'état d'équilibre était de 6.4 l. Le volume apparent de distribution (Vd/F) a été estimé à 8,01 l lors d'une analyse pharmacocinétique de population chez des patients ayant une polyarthrite rhumatoïde et à 4.71 l lors d'une analyse pharmacocinétique basée on de patients atteints de psoriasis en plaques.
La distribution comparative du certolizumab pégol dans les tissus a été déterminée chez des rats après administration intraveineuse d'une dose unique. Chez le rat, la distribution tissulaire de 125I-CDP870 était comparable à celle d'un anticorps IgG complètement humanisé sur une période de 48 heures. Dans une étude ex vivo supplémentaire, on a examiné une réactivité croisée potentielle du certolizumab pégol avec des cryosections de tissus humains normaux. Le certolizumab pégol n'a montré aucune réactivité avec une sélection standard déterminée de tissus humains normaux.
Métabolisme
Par pégylation (liaison covalente de polymères PEG à des peptides), l'élimination de ces unités hors de la circulation sanguine est ralentie par différents mécanismes, comme une clairance rénale moins importante, une diminution de la protéolyse et une immunogénicité réduite. Dans des études cliniques, on a enregistré des anticorps anti-Certolizumab pegol chez 7.9% (voir la rubrique «Effets indésirables»).
Dans le cas du certolizumab pégol il s'agit d'un fragment Fab d'un anticorps qui est conjugué au PEG afin d'allonger la demi-vie d'élimination plasmatique terminale du Fab de manière à ce qu'elle soit comparable à celle d'un produit à base d'anticorps complets.
Élimination
La demi-vie d'élimination terminale (t½) était d'environ 14 jours pour toutes les doses étudiées. Lors d'une administration intraveineuse à des sujets sains, la clairance se situait entre 9.21 ml/h et 14.38 ml/h. Après administration sous-cutanée à des sujets sains, la clairance se situait entre 10.58 ml/h et 12.13 ml/h; dans le cadre de l'analyse pharmacocinétique se rapportant à la population de patients avec la maladie de Crohn, la clairance de la population globale a été estimée à 17.25 ml/h, avec une variabilité interindividuelle de 38.3% (CV) et une «inter-occasion variability» de 16.4%. Après administration sous-cutanée, la clairance a été estimée à 21.0 ml/h lors d'une analyse pharmacocinétique de population atteinte de polyarthrite rhumatoïde, avec une variabilité interindividuelle de 30.8% (CV) et une variabilité intra-individuelle de 22.0%. Après administration sous-cutanée chez les patients atteints de psoriasis, la clairance était de 14 ml/h avec une variabilité inter-individuelle de 22.2 % (CV). L'élimination par voie rénale s'est révélée être la voie d'élimination du composant PEG déconjugué du certolizumab pégol.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Aucune étude clinique spécifique portant sur l'évaluation des effets de troubles de la fonction hépatique sur la pharmacocinétique du certolizumab pégol n'a été menée. L'analyse pharmacocinétique portant sur la population n'a pas permis de tirer des conclusions concernant les conséquences de troubles de la fonction hépatique car cette analyse ne comprenait qu'un faible nombre de patients présentant des troubles significatifs de la fonction hépatique.
Troubles de la fonction rénale
Aucune étude clinique spécifique portant sur l'évaluation des effets de troubles de la fonction rénale sur la pharmacocinétique du certolizumab pégol ou de sa fraction PEG n'a été menée. L'analyse pharmacocinétique portant sur la population de patients ayant une insuffisance rénale légère n'a toutefois montré aucun effet dépendant de la clairance de la créatinine; c.-à-d. que de légers troubles de la fonction rénale n'ont probablement aucune conséquence significative.
Les données sont insuffisantes pour recommander des posologies dans l'insuffisance rénale modérée ou sévère. La pharmacocinétique de la fraction PEG (polyéthylène glycol) du certolizumab pégol devrait dépendre de la fonction rénale mais n'a pas été évaluée chez l'insuffisant rénal.
Patients âgés
Maladie de Crohn
Aucune étude clinique particulière n'a été conduite chez les sujets âgés; une analyse pharmacocinétique se rapportant à la population n'a toutefois pas mis en évidence d'effets liés à l'âge chez 1'580 patients.
Polyarthrite rhumatoïde
Aucune étude clinique spécifique n'a été réalisée chez le sujet âgé. Aucun effet de l'âge n'a toutefois été observé lors d'une analyse pharmacocinétique de population réalisée chez des patients ayant une polyarthrite rhumatoïde, parmi lesquels 78 (13.2% de la population) étaient âgés de 65 ans et plus, et dont le plus âgé avait 83 ans.
Psoriasis en plaques
Aucun effet de l'âge n'a été observé lors d'une analyse pharmacocinétique de patients adultes atteints de psoriasis en plaques.
Enfants et adolescents
On ne dispose d'aucune étude relative à l'utilisation de certolizumab pégol chez l'enfant.
Sexe
Aucune étude clinique particulière relative à l'influence du sexe sur la pharmacocinétique du certolizumab pégol n'a été conduite; une analyse pharmacocinétique se rapportant à la population n'a toutefois pas mis en évidence d'effets liés au sexe chez 1'580 patients atteints de la maladie de Crohn et chez 1'768 patients atteints de polyarthrite rhumatoïde.
Race/Ethnie
L'analyse pharmacocinétique se rapportant à la population a entraîné une augmentation d'env. 15% de la clairance chez les patients caucasiens par rapport aux patients non-caucasiens; ce résultat n'est pas cliniquement significatif. Les paramètres pharmacocinétiques de sujets japonais étaient comparables à ceux de sujets caucasiens après administration sous-cutanée en trois niveaux posologiques dans le cadre d'une étude de biodisponibilité.
Données précliniquesLes études pivots de sécurité préclinique ont été conduites chez le singe Cynomolgus adultes (y compris des études de 13 et 26 semaines avec administration sous-cutanée répétée) et chez des jeunes singes Cynomologus (étude de 52 semaines avec administration sous-cutanée répétée). Les examens histopathologiques ont montré une vacuolisation cellulaire, principalement dans les macrophages, dans un certain nombre de sites (ganglions lymphatiques, sites d'injection, rate, surrénales, utérus, col utérin, plexus choroïde cérébral et cellules épithéliales des plexus choroïdes). Il est vraisemblable que cette observation soit due à la capture cellulaire de la fraction PEG.
Dans toutes les études, les modifications ont été en partie réversibles après les phases d'entretien de 13, 26 ou 52 semaines.
Des études fonctionnelles in vitro de macrophages humains vacuolisés ont indiqué que toutes les fonctions étudiées étaient conservées. Les études in vivo conduites chez le singe Cynomolgus adultes n'ont révélé aucun signe morphologique de dégénérescence ou de troubles fonctionnels.
Les études chez le rat ont indiqué que >90% du PEG administré étaient éliminés dans les 3 mois suivant l'administration d'une dose unique, l'urine étant la principale voie d'excrétion.
Aucun effet indésirable n'a été observé sur le bien-être maternel ou la fertilité des femelles, les indices de développement embryo- fœtal, péri- et post-natal chez le rat, en utilisant un fragment Fab' de rongeur anti-TNFα de rat PEGylé (cTN3 PF), après suppression prolongée du TNFα.
Des effets sur la motilité et la densité des spermatozoïdes ont été observés chez des rats mâles traités au cTN3PF, sans conséquence apparente sur la fertilité.
Les études de distribution ont démontré que le passage transplacentaire ainsi que le passage dans le lait maternel du cTN3 PF sont négligeables. Dans des études animales, on a enregistré un passage du cTN3 PF dans le lait maternel de rates avec un rapport lait-plasma d'environ 10%. Le certolizumab pégol ne se lie pas au récepteur Fc néonatal humain (FcRn). Les données issues d'un modèle humain de transfert placentaire en circuit fermé ex-vivo suggèrent un transfert faible ou négligeable dans le compartiment fœtal. De plus, des expériences de transcytose médiée par le FcRn dans des cellules transfectées avec le FcRn humain ont mis en évidence un transfert négligeable.
Mutagénicité
Aucune mutagénicité ou clastogénicité n'a été démontrée dans les études précliniques.
Carcinogénicité
Aucune étude sur la cancérogénicité de Cimzia n'a été réalisée.
Toxicité sur la reproduction
Dans des études animales menées chez des rats recevant jusqu'à 100 mg/kg d'un fragment Fab pégylé d'un anticorps anti-murin de rongeur (cTN3 PF) semblable au certolizumab pégol, aucun signe de toxicité maternelle ou de tératogénicité n'a été observé. Dans une étude sur le développement embryofoetal des malformations spontanées des reins sont survenues à la dose de 20 mg/kg, qui ont été observées chez des témoins historiques, mais qui n'étaient pas dose-dépendantes.
Remarques particulièresIncompatibilités
Cimzia ne doit pas être mélangé avec un autre médicament.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8°C).
Ne pas congeler.
Conserver le récipient dans son carton pour le protéger de la lumière.
Tenir hors de portée des enfants.
Les seringues et stylos pré-remplis Cimzia peuvent être conservés à température ambiante (jusqu'à 25 °C) pendant une seule période de 10 jours à l'abri de la lumière. Les seringues et stylos pré-remplis doivent être jetés s'ils ne sont pas utilisés dans ces 10 jours, même s'ils ont été placés à nouveau dans le réfrigérateur.
Remarques concernant la manipulation
Ce médicament est à usage unique. Étant donné que l'injection de Cimzia ne devrait seulement se faire que si le liquide a atteint la température ambiante, il est par conséquent nécessaire de sortir la seringue pré-remplie de Cimzia du réfrigérateur 30 minutes avant l'utilisation, respectivement, de sortir le stylo pré-rempli de Cimzia du réfrigérateur 30 à 45 minutes avant l'utilisation. Ne réchauffez en aucun cas la seringue, respectivement le stylo d'une autre manière.
Numéro d’autorisation60096, 66590 (Swissmedic).
PrésentationCimzia: emballage de 2 seringues pré-remplies et 2 tampons imbibés d'alcool [B].
Cimzia AutoClicks: emballage de 2 stylos pré-remplis et 2 tampons imbibés d'alcool [B].
Titulaire de l’autorisationUCB-Pharma SA, Bulle.
Mise à jour de l’informationDécembre 2024
|