Propriétés/EffetsCode ATC
L04AB05
Le certolizumab pégol est un fragment Fab d'un anticorps recombinant humanisé. Le fragment Fab est produit à partir d'Escherichia coli, puis purifié et conjugué au polyéthylèneglycol (PEG).
Mécanisme d'action
Le certolizumab pégol se lie avec une grande affinité au TNF-α humain, avec une constante de dissociation (kd) de 90 pM. Le TNF-α est une cytokine pro-inflammatoire importante qui joue un rôle déterminant dans les processus inflammatoires. Le certolizumab pégol possède un effet neutralisant sélectif sur le TNF-α (IC90 de 4 ng/ml lors de l'inhibition du TNF-α humain dans le cadre d'un essai de cytotoxicité-fibrosarcome in vitro sur lignées cellulaires murines L929), mais ne neutralise pas la lymphotoxine α (TNF-β).
Le certolizumab pégol possède un effet neutralisant dose-dépendant démontrable sur le TNF humain associé à la membrane et soluble. L'incubation de monocytes avec du certolizumab pégol a entraîné une inhibition dose-dépendante de TNF-α induit par LPS, ainsi que la formation d'IL-1β dans des monocytes humains.
Le certolizumab pégol est dépourvu de région Fc («fragment crystallizable»), telle qu'elle existe normalement dans un anticorps complet; c'est la raison pour laquelle il y a absence de fixation au complément et de formation de cytotoxicité anticorps-dépendante, à médiation cellulaire. In vitro, cela n'entraîne pas d'apoptose dans les monocytes ou les lymphocytes humains du sang périphérique ou la dégranulation des neutrophiles.
Pharmacodynamique
Sans objet.
Efficacité clinique
Maladie de Crohn
L'efficacité et la sécurité de Cimzia ont été vérifiées dans le cadre de deux études en double aveugle, randomisées, contrôlées contre placebo, chez des patients âgés de ≥18 ans atteints de la maladie de Crohn active, de modérée à sévère, c'est-à-dire avec un Crohn's Disease Activity Index (CDAI) de 220 et 450 (y compris 220 et 450). Dans les deux études, Cimzia a été administré par voie sous-cutanée à une dose de 400 mg. L'utilisation concomitante de doses stables de produits contre la maladie de Crohn était autorisée.
Étude I
Dans le cas de l'étude I (PRECiSE 1), il s'agissait d'une étude randomisée, contrôlée contre placebo, menée auprès de 662 patients atteints de la maladie de Crohn active. Cimzia ou le placebo ont été administrés au cours des semaines 0, 2 et 4, puis toutes les 4 semaines jusqu'en semaine 24. La réponse clinique (response) était définie comme une diminution du CDAI d'au moins 100 points, une rémission clinique comme une valeur absolue du CDAI de 150 ou moins. Trois patients ont été exclus de l'analyse Intent-to-treat- (ITT-); deux des patients randomisés dans le groupe Cimzia n'ont pas reçu de doses; un patient randomisé dans le groupe placebo a été exclu.
Le tableau 1 présente les résultats concernant la réponse clinique (response). L'effet de Cimzia s'est installé en semaine 2. En semaine 6, le nombre de répondeurs était cliniquement significatif par rapport au placebo; c.-à-d. qu'une réponse clinique était atteinte. De plus, le nombre de répondeurs était cliniquement significatif tant en semaine 6 qu'en semaine 26, ce qui est le signe d'une réponse durable.
Tableau 1: PRECiSE 1 – Réponse clinique; totalité de la population à l'étude
|
Temps de mesure
|
Nombre (%) de répondeurs
IC 95%
| |
Placebo
(N=328)
|
Cimzia 400 mg
(N=331)
| |
Semaine 2
| |
N
|
326
|
328
| |
Responders
|
46 (14.1%)
|
82 (25.0%)§
| |
IC 95%
|
10.3%, 17.9%
|
20.3%, 29.7%
| |
Semaine 6 (induction)
| |
N
|
325
|
327
| |
Responder
|
87 (26.8%)
|
115 (35.2%)*
| |
IC 95%
|
22.0%, 31.6%
|
30.0%, 40.3%
| |
Semaine 26
| |
N
|
327
|
328
| |
Responders
|
87 (26.6%)
|
122 (37.2%)*
| |
IC 95%
|
21.8%, 31.4%
|
32.0%, 42.4%
| |
Semaines 6&26 (réponse durable)
| |
N
|
325
|
325
| |
Responder
|
52 (16.0%)
|
75 (23.1%)*
| |
IC 95%
|
12.0%, 20.0%
|
18.5%, 27.7%
|
§ valeur de p pas calculée
* valeur de p <0.05 test de régression logistique
L'utilisation d'immunosuppresseurs ou de corticostéroïdes avant le début de l'étude n'a eu aucun effet sur la réponse clinique de Cimzia. Cimzia était efficace en ce qui concerne l'induction et le maintien de la réponse dans la sous-population de patients préalablement traités par infliximab (semaine 6: 24.5% vs 20.0%; semaines 6 et 26: 15.5% vs 10.6% pour Cimzia ou placebo).
Cimzia était efficace en ce qui concerne l'induction et le maintien de la réponse dans la sous-population de patients n'ayant pas été préalablement traités par infliximab (semaine 6: 39.7% vs 29.2%; semaines 6 et 26: 26.3% vs 17.9% pour Cimzia ou placebo). Pendant l'étude, les effets de la maladie de Crohn sur les patients ont été évalués au moyen d'un questionnaire spécifique à la maladie, appelé le Inflammatory Bowel Disease Questionnaire (IBDQ), ainsi que d'un questionnaire (SF-36) relatif à l'état de santé général.
Le IBDQ (valeur globale ≥16 points par rapport aux valeurs initiales) a montré que, par rapport au groupe sous placebo, en semaines 6, 16 et 26 un nombre plus important de patients du groupe recevant 400 mg de Cimzia a présenté une amélioration cliniquement importante concernant le résultat thérapeutique.
Comparés au groupe sous placebo, en semaine 6 et en semaines 6 et 26, les patients ayant reçu 400 mg de Cimzia ont présenté une atténuation significative de la douleur (p ≤0.05) par rapport aux valeurs initiales, objectivée par la valeur relative aux douleurs physiques du questionnaire SF-36.
Étude II:
Dans le cas de l'étude II (PRECiSE 2), il s'agissait d'une étude randomisée portant sur l'arrêt du traitement et conduite auprès d'une population de 668 patients atteints de la maladie de Crohn active. Tous les patients inclus dans l'étude ont reçu 400 mg de Cimzia en semaines 0, 2 et 4; l'évaluation de la réponse clinique a eu lieu en semaine 6. La réponse clinique (response) était définie comme une diminution du CDAI d'au moins 100 points, une rémission clinique comme une valeur absolue du CDAI de 150 ou moins. Les répondeurs ont été randomisés pour recevoir un traitement d'entretien avec 400 mg de Cimzia ou un placebo (débutant en semaine 8) jusqu'en semaine 24.
Les patients n'ayant pas répondu en semaine 6 (non-responder) ont été exclus de l'étude. Trois patients ont été exclus de l'analyse ITT.
Au total, 668 patients atteints de la maladie de Crohn active ont reçu 400 mg de Cimzia comme traitement d'induction selon le principe du Open-label.
Dans ce groupe de patients, 428 (64.1%) ont répondu au traitement et ont été randomisés en semaine 6 pour recevoir un placebo (212; ITT 210) ou Cimzia 400 mg (216; ITT 215); ils ont reçu au moins une injection en double aveugle. L'effet s'est installé en semaine 2.
Parmi les répondeurs en semaine 6, 45.1% ont présenté une réponse clinique en semaine 2.
Le tableau 2 présente les résultats relatifs à la réponse clinique (response) ou à la rémission. En semaine 26, par rapport au groupe avec 3 doses de Cimzia+placebo, dans le groupe sous Cimzia un pourcentage significativement plus élevé de répondeurs de semaine 6 a présenté une réponse clinique ou une rémission clinique.
Tableau 2: PRECiSE 2 – réponse clinique (response) et rémission clinique dans la totalité de la population à l'étude
|
Temps de mesure
|
Réponse clinique (response)
Pourcentage (%)
|
Rémission clinique
Pourcentage (%)
| |
|
Cimzia 400 mg × 3
+ Placebo*
N=210
|
Cimzia 400 mg
N=215
|
Cimzia 400 mg × 3
+ Placebo*
N=210
|
Cimzia 400 mg
N=215
| |
Semaine 26
Réponse durable
| |
N
|
210
|
215
|
210
|
215
| |
Responder
|
76 (36.2%)
|
135 (62.8%)
|
60 (28.6%)
|
103 (47.9%)
| |
IC 95%
|
29.7%, 42.7%
|
56.3%, 69.3%
|
22.5%, 34.7%
|
41.2%, 54.6%
| |
Valeur de p
|
|
<0.001
|
|
<0.001
|
* Ce tableau se rapporte exclusivement à la partie en double aveugle de l'étude.
Figure 1: PRECiSE 2 – Évolution temporelle de la réponse clinique; totalité de la population ITT
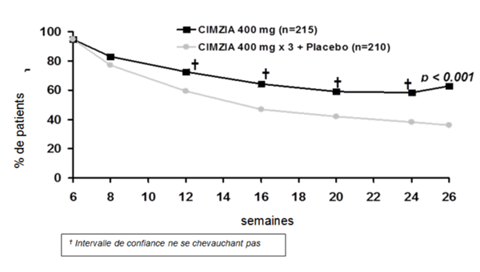
L'utilisation d'immunosuppresseurs ou de corticostéroïdes avant le début de l'étude n'a eu aucun effet sur la réponse clinique de Cimzia. Cimzia était efficace jusqu'en semaine 26 en ce qui concerne le maintien de la réponse, et ce tant dans la sous-population de patients préalablement traités par infliximab (44.2% vs 25.5% pour Cimzia ou placebo), que chez les patients n'ayant pas été préalablement traités par infliximab (68.7% ou 39.6%).
Les effets de la maladie de Crohn sur les patients ont été évalués pendant l'étude au moyen d'un questionnaire spécifique à la maladie, appelé le Inflammatory Bowel Disease Questionnaire (IBDQ), ainsi que d'un questionnaire (SF-36) relatif à l'état de santé général.
Le IBDQ (valeur globale ≥16 points par rapport aux valeurs initiales) a montré que, par rapport au groupe sous placebo, en semaines 16 et 26 un nombre plus important de patients du groupe recevant 400 mg de Cimzia a présenté une amélioration cliniquement importante concernant le résultat thérapeutique.
Comparés au groupe sous placebo, en semaine 26, les patients ayant reçu 400 mg de Cimzia ont présenté une atténuation significative de la douleur (p ≤0.05) par rapport aux valeurs initiales, objectivée par la valeur relative aux douleurs physiques du questionnaire SF-36.
Dans le questionnaire, les résultats relatifs au «rôle physique» étaient meilleurs pour Cimzia que pour le placebo; ils n'étaient toutefois pas statistiquement significatifs (p >0.05).
Cimzia a été étudié pour le traitement de patients pédiatriques atteints d'une maladie de Crohn modérément à sévèrement active (étude C87035 et son suivi à long terme CR0012). L'efficacité de Cimzia n'a pas été démontrée dans une étude de groupe parallèle, ouverte, randomisée et à doses multiples sur une période allant jusqu'à 62 semaines chez 99 sujets âgés de 6 à 17 ans (C87035). L'étude a été interrompue prématurément en raison d'un taux d'abandon élevé.
Polyarthrite rhumatoïde
Études RA-I, RA-II
L'efficacité et la tolérance de Cimzia ont été évaluées lors de 2 études cliniques randomisées, en double aveugle, contrôlées versus placebo, chez des patients âgés ≥18 ans ayant une polyarthrite rhumatoïde active diagnostiquée selon les critères de l'American College of Rheumatology (ACR), RA-I (RAPID 1) et RA-II (RAPID 2). Les patients avaient au moins 9 articulations gonflées et douloureuses et ils avaient une PR active depuis au moins 6 mois avant le début de l'étude. Au cours des deux études, Cimzia a été administré par voie sous-cutanée, en association avec le MTX par voie orale, préalablement administré pendant au moins 6 mois et à des doses stables d'au moins 10 mg par semaine pendant 2 mois. Il n'y a aucune expérience de l'administration de Cimzia en association avec des DMARDs autres que le MTX.
Étude C-EARLY
L'efficacité et la sécurité de Cimzia ont été étudiées dans une étude clinique randomisée, en double aveugle, contrôlée versus placebo, chez des patients adultes ayant une PR active modérée (3.5%) à sévère (96.5%) n'ayant pas été préalablement traités par DMARDs (C-EARLY). Dans l'étude C-EARLY, les patients avaient au moins 18 ans et avaient chacun au moins 4 articulations gonflées et douloureuses et un diagnostiqués depuis moins d'1 an (selon les critères de classification de l'ACR / European League Against Rheumatism [EULAR] de 2010). Le DAS28 de départ devait être >3.2 (activité de la maladie modérée à élevée) et le CRP était ≥10 mg/L ou l'ESR était ≥28 mm/h. En début d'étude, les patients avaient été diagnostiqués en moyenne 2.9 mois plus tôt et ils n'avaient pas reçu préalablement de traitement par DMARDs (y compris le MTX). 96.5% des patients de l'étude présentaient une PR sévère. Les patients ont reçu une dose initiale (semaines 0, 2 et 4) de 400 mg ou un placebo, puis une dose d'entretien de 200 mg de Cimzia ou un placebo toutes les 2 semaines pendant 52 semaines. Pour les deux bras Cimzia et placebo, le MTX a été initié à la semaine 0 (10 mg/semaine), augmenté jusqu'à la dose maximale tolérée jusqu'à la semaine 8 (au minimum 15 mg/semaine et au maximum 25 mg/semaine étaient autorisés) et maintenu tout au long de l'étude (après la semaine 8, la dose moyenne de MTX était de 22.3 mg/semaine dans le bras placebo et de 21.1 mg/semaine dans le bras Cimzia).
Dans le cadre d'une phase ultérieure, le maintien de la réponse a été étudié la 2ème année. Pour cela, les patients dans le groupe Cimzia 200 mg toutes les 2 semaines + MTX présentant une activité durablement faible de la maladie (DAS28-ESR < 3.2 aux semaines 40 et 52) ont été à nouveau randomisés à la semaine 52, dans le groupe Cimzia 200 mg toutes les 2 semaines + MTX, Cimzia 200 mg toutes les 4 semaines + MTX ou placebo + MTX (dans un rapport de 2:3:2). Les critères d'évaluation sont résumés dans le tableau 3.
Tableau 3: Description des essais cliniques
|
Numéro de l'étude
|
Nombre de patients
|
Posologie
|
Objectifs de l'étude
| |
RA-I
(52 semaines)
|
982
|
400 mg (0, 2, 4 semaines) avec le MTX
|
Évaluation du traitement des signes et symptômes et de l'inhibition des dommages structuraux.
| |
200 mg ou 400 mg toutes les 2 semaines avec le MTX
|
Co-critères principaux d'évaluation: ACR 20 à la semaine 24 et modification du mTSS à la semaine 52 par rapport au score initial.
| |
RA-II
(24 semaines)
|
619
|
400 mg (0, 2, 4 semaines) avec le MTX
|
Évaluation du traitement des signes et symptômes et de l'inhibition des dommages structuraux.
| |
200 mg ou 400 mg toutes les 2 semaines avec le MTX
|
Critère principal d'évaluation: ACR 20 à la semaine 24.
| |
C-EARLY
(jusqu'à la semaine 104)
|
879
|
400 mg (0, 2, 4 semaines) avec le MTX
200 mg toutes les 2 semaines avec le MTX
200 mg toutes les 4 semaines avec le MTX
(nouvelle randomisation après 52 semaines)
|
Évaluation en fonction du traitement des signes et symptômes et de l'inhibition de la progression des dommages structuraux chez les patients n'ayant pas reçu de traitement préalable par DMARDs
Critère principal d'évaluation à la semaine 52
pourcentage de participants en rémission persistante *
Critère principal d'évaluation à la semaine 104: maintien de la faible activité de la maladie **
|
mTSS: score total de Sharp modifié
* La rémission persistante à la semaine 52 est définie par un score DAS28-ESR <2.6 aux semaines 40 et 52.
** La faible activité de la maladie persistante à la semaine 104 est définie par un score DAS28-ESR <3.2 aux semaines 52 et 104, sans poussée de la maladie (= hausse du score DAS28-ESR >=0.6 et DAS28-ESR >3.2)
Réponse ACR
Les résultats des études cliniques RA-I et RA-II sont présentés dans le Tableau 4. Des réponses ACR 20 et ACR 50 supérieures de façon statistiquement significative par rapport au placebo ont été obtenues, respectivement, à partir de la semaine 1 et de la semaine 2, dans les deux études cliniques. Les réponses se sont maintenues jusqu'à la semaine 52 (RA-I) et 24 (RA-II). Sur les 783 patients initialement randomisés dans RA-I, 508 ont terminé les 52 semaines de la phase de traitement contrôlée versus placebo et sont entrés dans l'étude d'extension en ouvert. Parmi ceux-ci, 427 ont terminé les 2 années de l'étude d'extension en ouvert et ont donc eu une exposition à Cimzia de 148 semaines au total. La réponse ACR 20 à cette échéance était de 91%. La réduction du DAS28 (VS) par rapport à la valeur initiale a également été significativement plus importante (p <0.001) à la semaine 52 (RA-I) et à la semaine 24 (RA-II) par rapport au placebo, et s'est maintenue jusqu'à 2 ans dans l'étude d'extension en ouvert de l'étude RA-I.
Tableau 4: Réponse ACR dans les études cliniques RA-I et RA-II
|
|
Étude RA-I
Association avec le méthotrexate
(24 et 52 semaines)
|
Étude RA-II
Association avec le méthotrexate
(24 semaines)
| |
Réponse
|
Placebo +
MTX
N=199
|
Cimzia 200 mg
+ MTX toutes les
2 semaines
N=393
|
Placebo
+ MTX
N=127
|
Cimzia 200 mg
+ MTX toutes les
2 semaines
N=246
| |
ACR 20
| |
Semaine 24
|
14%
|
59%**
|
9%
|
57%**
| |
Semaine 52
|
13%
|
53%**
|
N/A
|
N/A
| |
ACR 50
| |
Semaine 24
|
8%
|
37%**
|
3%
|
33%**
| |
Semaine 52
|
8%
|
38%**
|
N/A
|
N/A
| |
ACR 70
| |
Semaine 24
|
3%
|
21%**
|
1%
|
16%*
| |
Semaine 52
|
4%
|
21%**
|
N/A
|
N/A
| |
Réponse clinique majeurea
|
1%
|
13%**
|
|
|
Cimzia versus placebo: * p ≤0.01, ** p <0.001
a La réponse clinique majeure est définie comme l'obtention d'une réponse ACR 70 lors de chaque évaluation sur une période continue de 6 mois.
Les valeurs de p (test de Wald) sont citées pour la comparaison des traitements en utilisant une régression logistique avec le traitement et la région comme facteurs.
Pourcentage de réponse basé sur le nombre de sujets pour lesquels des données sont disponibles (n) concernant ce critère d'évaluation et cette échéance et qui peut différer de N.
Les critères principaux et secondaires majeurs de l'étude C-EARLY ont été atteints. Les principaux résultats de l'étude sont présentés dans le tableau 5.
Tableau 5: étude C-EARLY: pourcentage des patients en rémission persistante et en activité durablement faible de la maladie à la semaine 52
|
Réponse
|
Placebo + MTX
N=213
|
Cimzia 200 mg + MTX
N=655
| |
Rémission persistante*
(DAS28-ESR <2.6 aux semaines 40 et 52)
|
15.0%
|
28.9%**
| |
Activité durablement faible de la maladie
(DAS28-ESR ≤3.2 aux semaines 40 et 52)
|
28.6%
|
43.8%**
|
* Critère principal d'évaluation de l'étude C-EARLY (jusqu'à la semaine 52)
Population totale d'analyse, imputation des non-répondeurs pour les valeurs manquantes.
** Cimzia + MTX vs placebo + MTX: p<0,001
La valeur p a été estimée par un modèle de régression logistique selon les facteurs suivants: le traitement, la région et la durée depuis le diagnostic de PR à l'inclusion (≤4 mois vs >4 mois).
Une diminution plus importante du DAS28-ESR par rapport à l'inclusion a été observée chez les patients du groupe Cimzia+MTX par rapport au groupe placebo+MTX, dès la semaine 2 et jusqu'à la semaine 52 (p < 0.001 à chaque visite). Les taux de rémission (DAS28-ESR < 2.6), la faible activité de la maladie (DAS28-ESR ≤3.2), les réponses ACR50 et ACR70 à chaque visite ont démontré que le traitement par Cimzia+MTX a permis d'obtenir des réponses plus rapides et plus importantes que le traitement par placebo+MTX. Ces résultats ont été maintenus pendant les 52 semaines de traitement chez des patients non précédemment traités par DMARDs.
Chez les patients présentant une faible activité de la maladie à la semaine 52, une réponse numériquement plus fréquente a été obtenue dans le cadre de la poursuite de la thérapie combinée par Cimzia + MTX jusqu'à la semaine 104 que chez les patients qui avaient arrêté Cimzia (41/84, 48.8% contre 31/79, 39.2%; p=0.112).
Réponse radiographique
Dans l'étude RA-I, les dommages structuraux articulaires ont été évalués par radiographie et exprimés en termes de modification du mTSS et de ses composantes, le score d'érosion et le score de pincement articulaire (JSN de Joint Space Narrowing), à la semaine 52 par rapport aux scores initiaux. La progression des signes radiographiques a été significativement moindre chez les patients traités par Cimzia par rapport à ceux recevant le placebo, aux semaines 24 et 52 (voir tableau 6.). Dans le groupe placebo, 52% des patients n'ont pas présenté de progression radiographique (mTSS ≤0.0) à la semaine 52 versus 69% dans le groupe Cimzia 200 mg.
Tableau 6: Modifications sur 12 mois dans l'étude RA-I
|
|
Placebo + MTX
N=199
Moyenne (DS)
|
Cimzia 200 mg + MTX
N=393
Moyenne (DS)
|
Cimzia 200 mg + MTX –
Placebo + MTX
Différence moyenne
| |
mTSS
| |
Semaine 52
|
2.8 (7.8)
|
0.4 (5.7)
|
-2.4
| |
Score d'érosion
| |
Semaine 52
|
1.5 (4.3)
|
0.1 (2.5)
|
-1.4
| |
Score de pincement articulaire
| |
Semaine 52
|
1.4 (5.0)
|
0.4 (4.2)
|
-1.0
|
Les valeurs de p ont été <0.001 à la fois pour le mTSS et pour le score d'érosion et ≤0.01 pour le score de pincement articulaire. Une analyse ANCOVA a été réalisée sur la variation du score par rapport à la valeur initiale pour chaque mesure, avec la région et le traitement comme facteurs et la valeur initiale du score comme covariable.
Sur les 783 patients initialement randomisés dans RA-I, 508 ont terminé les 52 semaines de la phase de traitement contrôlée versus placebo et sont entrés dans la phase d'extension en ouvert. Le maintien de l'inhibition de la progression des dommages structuraux a été démontrée dans un sous-groupe de 449 de ces patients qui ont été traités pendant au moins 2 ans par Cimzia (RA-I et étude d'extension en ouvert) et avaient des données évaluables à l'échéance des 2 ans.
Dans l'étude C-EARLY, l'inhibition de la progression radiographique a été plus importante dans le bras Cimzia+MTX par rapport au bras placebo+MTX à la semaine 52 (voir tableau 7). A la semaine 52, 49,7 % des patients du groupe placebo+MTX n'ont présenté aucune progression radiographique (variation du mTSS ≤0,5) versus 70,3 % dans le groupe Cimzia+MTX (p < 0,001).
Tableau 7: Evolution radiologique à la semaine 52 dans l'étude C-EARLY
|
|
Placebo + MTX
N=163
Valeur moyenne (ET)
|
Cimzia 200 mg + MTX
N=528
Valeur moyenne (ET)
|
Cimzia 200 mg + MTX – Placebo + MTX
Différence*
| |
STSm
semaine 52
|
1.8 (4.3)
|
0.2 (3.2) **
|
-0.978 (-1.005, -0.500)
| |
Score d'érosion
semaine 52
|
1.1 (3.0)
|
0.1 (2.1) **
|
-0.500 (-0.508, -0.336)
| |
Score de pincement articulaire
semaine 52
|
0.7 (2.3)
|
0.1 (1.7) **
|
0.000 (0.000, 0.000)
|
Ensemble de données radiologiques avec extrapolation linéaire.
* Estimateur ponctuel de Hodges-Lehmann et intervalle de confiance asymptomatique (Moses) à 95%
** Cimzia + MTX vs placebo + MTX p<0.001. La valeur p a été estimée par analyse ANCOVA des rangs avec le traitement, la région et la durée depuis le diagnostic de PR (≤4 mois contre >4 mois) à l'inclusion comme facteurs et la valeur initiale du score comme co-variable.
Parmi les patients présentant une faible activité de la maladie à la semaine 52, une progression des dommages structuraux a été observée chez quelques patients seulement jusqu'à la semaine 104 (7/12, 10% des patients sous Cimzia 200 mg + MTX toutes les 2 semaines; 18/113 et 15/75, 20% des patients sous placebo + MTX). Les données ne permettent pas de tirer de conclusions comparables entre les posologies.
Capacité fonctionnelle et qualité de vie
Dans les études RA-I et RA-II, les patients traités par Cimzia ont rapporté des améliorations significatives, par rapport au placebo (p <0.001), de leurs capacités fonctionnelles évaluées par le questionnaire d'évaluation de l'état de santé – indice d'incapacité (HAQ-DI – Health Assessment Questionnaire – Disability Index) et de la fatigue évaluée par l'échelle d'évaluation de la fatigue (FAS – Fatigue Assessment Scale), à partir de la semaine 1 et jusqu'à la fin des études. Dans les deux études cliniques, les patients traités par Cimzia ont rapporté des améliorations significativement plus importantes du SF-36, scores résumés des composantes physiques et mentales (Physical and Mental Component Summaries) et du score de toutes les dimensions. L'amélioration des capacités fonctionnelles et de la qualité de vie liée à l'état de santé (HRQoL – health related quality of life) a été maintenue pendant 2 ans dans l'étude d'extension en ouvert de l'étude RA-I. Les patients traités par Cimzia ont rapporté des améliorations statistiquement significatives du Work Productivity Survey (questionnaire de productivité au travail) par rapport au placebo.
Dans l'étude C-EARLY, les patients traités par Cimzia + MTX ont rapporté une amélioration significative de leurs capacités fonctionnelles évaluées par le questionnaire HAQ-DI et de la douleur liée à l'arthrite évaluée par le PAAP (Patient Assessment of Arthritis Pain) à la semaine 52, par rapport aux patients traités par placebo + MTX (p<0.001 contre p<0.05). A la semaine 52, 48.1% des patients du groupe traité par Cimzia + MTX avaient atteint une capacité fonctionnelle normale (score HAQ-DI ≤0.05) contre 35.7% dans le groupe traité par placebo + MTX (p<0.01).
Spondylarthrite axiale (sous-populations avec spondylarthrite axiale non radiographique et spondylarthrite ankylosante)
AS001
L'efficacité et la sécurité du Cimzia ont été examinées dans le cadre d'une étude multicentrique randomisée, en double aveugle, contrôlée versus placebo (AS001). Celle-ci comprenait 325 patients ≥18 ans présentant une spondylarthrite axiale active de l'adulte depuis au moins 3 mois, définie selon les critères de classification pour la spondylarthrite de la société Assessment of Spondyloarthritis International Society (ASAS). La spondylarthrite axiale se caractérise par une spondylarthrite principalement avec une atteinte axiale et inclut le sous-groupe de maladies de la spondylite ankylosante, ainsi que le sous-groupe sans signe définitif de sacro-illite dans des radiographies conventionnelles sans produit de contraste, qualifié de spondylarthrite axiale non radiographique. La population participant à l'étude et atteinte de spondylarthrite axiale comprenait des sous-populations présentant aussi bien une spondylite ankylosante (SA) (également appelée spondylarthrite axiale radiographique) qu'une spondylarthrite axiale non radiographique. Les patients ont présenté une maladie active, laquelle a été définie par un indice d'activité de la spondylarthrite ankylosante de Bath (Bath Ankylosing Spondylitis Disease Activity Index - BASDAI) ≥4, des douleurs de la colonne vertébrale ≥4 sur une échelle d'évaluation numérique (NRS) de 0 à 10 et un taux de CRP élevé ou des signes actuels d'une sacro-illite via l'imagerie par résonance magnétique (IRM). Par ailleurs, les patients devaient avoir présenté une intolérance ou une réaction insuffisante face à au moins un AINS.
Au total, 16% des patients avaient été précédemment exposés à un antagoniste de TNF. Les patients ont reçu une dose initiale de Cimzia de 400 mg dans les semaines 0, 2 et 4 (concernant les deux bras du traitement) ou un placebo, suivi(e) de soit 200 mg de Cimzia toutes les deux semaines soit de 400 mg de Cimzia toutes les quatre semaines, ou bien un placebo. 87.7% des patients ont reçu des AINS concomitants. Le critère principal d'évaluation de l'efficacité était le taux de réponse ASAS20 au cours de la douzième semaine. 153 patients ont participé à une sous-étude consacrée à l'imagerie.
Le taux de CRP est un marqueur sensible de l'inflammation et peut être élevé pour d'autres causes qu'une spondylarthrite axiale non radiographique. Pour cette raison, chez les patients présentant une spondylarthrite axiale active sévère sans signes radiographiques et sans preuve définitive de sacro-illite en radiographie, un taux élevé de CRP devrait être conforté par la démonstration d'une sacro-illite par IRM au niveau des articulations sacro-iliaques.
Réponse ASAS
Dans l'étude clinique AS001, 58% des patients traités sous 200 mg de Cimzia toutes les deux semaines et 64% des patients traités sous 400 mg de Cimzia toutes les quatre semaines ont obtenu une réponse ASAS20 au cours de la douzième semaine, contre 38% sous placebo (p <0.01).
Dans l'ensemble de la population, le pourcentage de répondeurs ASAS20 était cliniquement pertinent et significativement plus élevé à chaque examen dans les groupes de traitement recevant 200 mg de Cimzia toutes les deux semaines et 400 mg de Cimzia toutes les quatre semaines, que dans le groupe placebo (p ≤0.001 à chaque examen), après l'inclusion jusqu'à la semaine 24.
Des résultats comparables ont été obtenus dans les sous-populations de patients présentant une spondylarthrite ankylosante et de patients ayant une spondylarthrite axiale non radiographique (voir tableau 8).
Des améliorations significatives pour plusieurs composantes de l'activité de la spondylarthrite axiale [douleurs (douleurs d'ensemble de la colonne vertébrale), capacité fonctionnelle (indice fonctionnel de la spondylarthrite ankylosante de Bath - BASFI), inflammation (BASDAI valeur moyenne de Q5/6), douleurs nocturnes de la colonne vertébrale, fatigue, indice de mesure de la spondylarthrite ankylosante de Bath - BASMI] ont été en outre observées chez les patients traités par Cimzia par rapport aux patients ayant reçu le placebo.
Tableau 8: Réponse efficace à l'étude clinique AS001: réduction des signes et symptômes au sein des sous-populations souffrant de spondylite ankylosante et de spondylarthrite axiale non radiographique (pourcentage de patients)
|
Paramètre
|
Spondylarthrite ankylosante
|
Spondylarthrite axiale non
radiographique
| |
|
Placebo
N=57
|
Cimzia, tous schémas
d'administration(a)
N=121
|
Placebo
N=50
|
Cimzia, tous schémas
d'administration(a)
N=97
| |
ASAS20 (b,c)
| |
Semaine 12
|
37%
|
60%*
|
40%
|
61%*
| |
Semaine 24
|
33%
|
69%**
|
24%
|
68%**
| |
ASAS40 (c,d)
| |
Semaine 12
|
19%
|
45%**
|
16%
|
47%**
| |
Semaine 24
|
16%
|
53%**
|
14%
|
51%**
| |
ASAS 5/6 (c,d)
| |
Semaine 12
|
9%
|
42%**
|
8%
|
44%**
| |
Semaine 24
|
5%
|
40%**
|
4%
|
45%**
| |
Rémission partielle (c,d)
| |
Semaine 12
|
2%
|
20%**
|
6%
|
29%**
| |
Semaine 24
|
7%
|
28%**
|
10%
|
33%**
| |
BASDAI 50 (c,d)
| |
Semaine 12
|
11%
|
41%**
|
N/A
|
N/A
| |
Semaine 24
|
16%
|
49%**
|
N/A
|
N/A
|
(a) Cimzia - tous schémas d'administration = Données pour 200 mg de Cimzia toutes les deux semaines, suite à une dose initiale de 400 mg aux semaines 0, 2 et 4, et 400 mg de Cimzia toutes les quatre semaines, suite à une dose initiale de 400 mg aux semaines 0, 2 et 4
(b) Résultats du groupe randomisé
(c) Différence de traitement: Cimzia 200-Placebo, Cimzia 400-Placebo (correspond à un intervalle de confiance de 95% et la valeur de p) sont déterminés au moyen d'un test de Wald normal bilatéral en utilisant la méthode d'imputation des non-répondeurs (NRI).
(d) Population totale d'analyse
N/A = non disponible
*p ≤0.05, Cimzia vs placebo
**p <0.001, Cimzia vs placebo
Mobilité de la colonne vertébrale
La mobilité de la colonne vertébrale a été évaluée à l'inclusion, à la semaine 12 et à la semaine 24 au moyen du BASMI. Des différences cliniquement importantes et statistiquement significatives ont été observées à chaque visite après le début de l'étude chez les patients traités par Cimzia versus les patients ayant reçu le placebo.
La différence avec le placebo de la variation moyenne du BASMI linéaire par rapport à l'inclusion a été de -0.40 points (p <0.001) jusqu'à la semaine 12 et de -0.44 points (p <0.001) jusqu'à la semaine 24 chez les patients recevant du Cimzia. La différence avec le placebo a été plus importante (-0.60 et -0.59 points jusqu'à la semaine 12 respectivement semaine 24) dans la sous-population atteinte de spondylarthrite axiale non radiographique, que dans la sous-population atteinte de spondylite ankylosante (-0.21 et -0.32 points jusqu'à la semaine 12 respectivement semaine 24).
Inhibition de l'inflammation à l'imagerie par résonance magnétique (IRM)
Dans une sous-étude consacrée à l'imagerie, les signes d'inflammation ont été examinés à la semaine 12 à l'aide de l'IRM et exprimés comme variation par rapport à l'inclusion du score SPARCC (Spondyloarthritis Research Consortium of Canada - Consortium canadien de recherche sur la spondylarthrite) pour les articulations sacro-iliaques et du score de l'ASspiMRI a modifié (modification de Berlin) pour la colonne vertébrale. Une inhibition significative des signes d'inflammation dans les articulations sacro-iliaques ainsi que dans la colonne vertébrale a été observée dans le groupe de patients sous Cimzia (tout schéma d'administration), dans l'ensemble de la population de patients atteints de spondylarthrite axiale, ainsi que dans les sous-populations de patients atteints de spondylite ankylosante et de spondylarthrite axiale non radiographique. Cependant, cela n'a pas été le cas dans le groupe de patients sous placebo.
C-OPTIMISE
L'efficacité et la sécurité de la réduction de dose et de l'arrêt du traitement chez des patients en rémission persistante ont été évaluées chez des patients adultes (âgés de 18 à 45 ans) atteints d'une axSpA active précoce (durée des symptômes inférieure à 5 ans), présentant un score ASDAS ≥2,1 (et des critères d'inclusion de la maladie similaires à ceux de l'étude AS001) et une réponse insuffisante à au moins 2 AINS ou une intolérance ou contre-indication aux AINS. Les patients incluant les sous-populations SA et nr-axSpA ont été recrutés dans une phase d'induction en ouvert de 48 semaines (Partie A) au cours de laquelle ils ont tous reçu 3 doses de charge de Cimzia 400 mg aux Semaines 0, 2 et 4, suivies par Cimzia 200 mg toutes les 2 semaines de la Semaine 6 à la Semaine 46.
Les patients qui ont atteint une rémission persistante (définie comme le fait d'avoir une maladie inactive [ASDAS<1,3] sur une période d'au moins 12 semaines) et qui restaient en rémission à la Semaine 48, ont été randomisés dans la Partie B, et ont reçu Cimzia 200 mg toutes les 2 semaines (N = 104), Cimzia 200 mg toutes les 4 semaines (réduction de dose, N = 105), ou le placebo (arrêt du traitement, N = 104) pendant 48 semaines.
Le critère primaire d'efficacité correspondait au pourcentage de patients qui n'ont pas présenté de poussée au cours de la Partie B.
Les patients qui ont présenté une poussée au cours de la Partie B, c'est-à-dire ayant un score ASDAS ≥2,1 lors de 2 visites consécutives ou un score ASDAS > 3,5 lors de toute visite au cours de la Partie B, ont reçu un traitement de secours par Cimzia 200 mg toutes les 2 semaines pendant au moins 12 semaines (avec une dose de charge de Cimzia 400 mg à la Semaine 0, 2 et 4 chez les patients traités par placebo).
Réponse clinique
Le pourcentage de patients qui ont atteint une rémission persistante à la Semaine 48 dans la Partie A s'élevait à 43,9 % pour la population globale d'axSpA, et était similaire dans les sous-populations nr-axSpA (45,3 %) et SA (42,8 %).
Parmi les patients qui ont été randomisés dans la Partie B (N = 313), une proportion supérieure statistiquement significative (p < 0,001, NRI) de patients n'a pas présenté de poussée en poursuivant le traitement par Cimzia 200 mg toutes les 2 semaines (83,7 %) ou Cimzia 200 mg toutes les 4 semaines (79,0 %) comparé au groupe en arrêt de traitement (20,2 %).
La différence dans le délai de survenue de poussée entre le groupe en arrêt de traitement et l'un ou l'autre des groupes de traitement par Cimzia, était statistiquement (p < 0,001 pour chaque comparaison) et cliniquement significative. Dans le groupe du placebo, les poussées ont commencé environ 8 semaines après l'arrêt de Cimzia, la majeure partie des poussées survenant dans les 24 heures suivant l'arrêt du traitement (Figure 2).
Figure 2: Courbe Kaplan-Meier du délai d'apparition d'une poussée
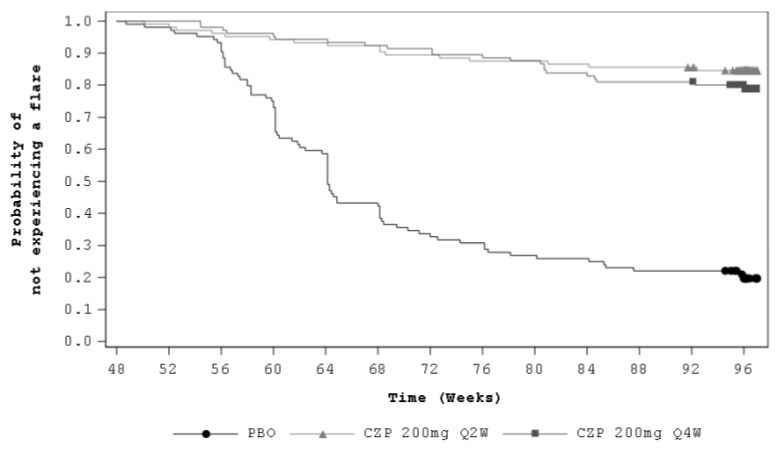
L'imputation des non-répondeurs (NRI) a été utilisée; les résultats concernent l'Ensemble randomisé.
Remarque: Le délai d'apparition d'une poussée a été défini comme la période commençant à la date de randomisation et se terminant à la date de la poussée. Pour les participants à l'étude qui n'ont pas eu de poussée, le délai d'apparition d'une poussée a été censuré à la date de la Visite de la Semaine 96.
Le diagramme de Kaplan-Meier a été tronqué à 97 semaines quand < 5 % des participants demeuraient dans l'étude.
Nouveau traitement des patients présentant une poussée
Dans la Partie B, 70 % (73/104) des patients traités par placebo, 14 % (15/105) des patients traités par Cimzia 200 mg toutes les 4 semaines et 6,7 % (7/104) des patients traités par Cimzia 200 mg toutes les 2 semaines ont présenté une poussée et ont ensuite été traités par Cimzia 200 mg toutes les 2 semaines.
Parmi les 73 patients ayant présenté une poussée dans le groupe affecté à l'arrêt du traitement, 71 patients ont suivi un traitement de secours par Cimzia pendant 12 semaines, et avaient des données ASDAS disponibles, dont 64 (90 %) présentaient un score ASDAS de maladie faible ou inactive (c'est-à-dire, tout ASDAS < 2,1) après 12 semaines de reprise du traitement en ouvert.
Le nombre de patients dans les deux groupes Cimzia présentant une poussée était trop faible pour permettre une évaluation valable des effets d'un nouveau traitement dans ce cas là.
D'après les résultats de C-OPTIMISE, une réduction de dose chez les patients en rémission persistante après une année de traitement par Cimzia peut être envisagée (voir «Posologie/Mode d'emploi»).
Spondylarthrite axiale non radiographique (nr-axSpA)
L'efficacité et la sécurité de Cimzia ont été étudiées dans une étude multicentrique randomisée, en double aveugle, contrôlée versus placebo de 52 semaines (AS0006) chez 317 patients ≥18 ans atteints de spondylarthrite axiale survenue à l'âge adulte et avec des douleurs dorsales depuis au moins 12 mois. Les patients devaient présenter des signes objectifs d'inflamations indiqués par des taux de protéine C-réactive (CRP) supérieur à la normale et/ou une sacro-iliite à l'imagerie par résonnance magnétique (IRM), indiquant une maladie inflammatoire [CRP positive (> ULN) et/ou IRM positive], mais sans signe radiographique définitif de lésions structurelles au niveau des articulations sacro-iliaques. Les patients avaient une maladie active définie par un score BASDAI ≥4, et des douleurs rachidiennes ≥4 sur une échelle NRS allant de 0 à 10. Les patients devaient présenter une intolérance ou une réponse inadéquate à au moins deux AINS. Les patients ont été traités par placebo ou par une dose initiale de 400 mg de Cimzia aux semaines 0, 2 et 4 puis par 200 mg de Cimzia toutes les 2 semaines. L'utilisation et l'ajustement de la dose du traitement standard (par ex. AINS, DMARD, corticostéroïdes et analgésiques) étaient autorisés à tout moment. De même, les patients pouvaient, à tout moment, passer du traitement de l'étude en double aveugle à un traitement ouvert avec Cimzia ou avec une autre substance. La principale variable d'efficacité était la réponse ASDAS-MI (Ankylosing Spondylitis Disease Activity Score major improvement) à la semaine 52. La réponse ASDAS-MI était définie comme une réduction de l'ASDAS (amélioration) ≥2,0 par rapport à la valeur de référence ou à l'atteinte de la plus petite valeur possible.
Dans le groupe Cimzia et dans le groupe placebo, 37 % et 41 % des patients, respectivement, présentaient une activité élevée (ASDAS ≥2,1; ≤3,5) et 62 % et 58 % des patients, respectivement, avaient une activité très élevée (ASDAS >3,5) de la maladie.
Réponse clinique
Dans l'étude AS006 en semaine 52, une proportion statistiquement significative des patients traités par Cimzia a obtenu une réponse ASDAS-MI par rapport aux patients ayant reçu le placebo. Les patients traités par Cimzia ont présenté des améliorations de plusieurs éléments de l'activité de la spondylarthrite axiale par rapport aux patients traités par placebo. Aussi bien en semaine 12 qu'en semaine 52, la réponse ASAS 40 était significativement supérieure par rapport au placebo. Les principaux résultats sont présentés dans les tableaux 9 et 10.
Tableau 9: réponse ASDAS-MI et ASAS-40 dans l'étude AS0006 (pourcentage des patients)
|
Paramètre
|
Placebo
N = 158
|
Cimziaa 200 mg toute les 2 semaines
N = 159
| |
ASDAS-MI
semaine 52
|
7%
|
47%*
| |
ASAS 40
semaine 12
semaine 52
|
11 %
16%
|
48 %*
57%*
|
a Cimzia administré toutes les 2 semaines après une dose initiale de 400 mg aux semaines 0, 2 et 4
* p<0,001
Tous les pourcentages représentent la proportion de patients ayant obtenu une réponse à l'ensemble de l'analyse.
Tableau 10: résultats BASDAI et BASFI dans l'étude AS0006
|
Paramètre
|
Placebo
N = 158
|
Cimzia(a) 200 mg toutes les 2 semaines
N = 159
| |
|
État initial
|
Semaine 12
|
Semaine 52
|
État initial
|
Semaine 12
|
Semaine 52
| |
BASDAI(b)
|
6,79 (1,28)
|
5,71 (2,09)
|
5,47 (2,30)
|
6,88 (1,40)
|
3,93 (2,21)
|
3,26 (2,47)
| |
BASFI(c)
|
5,44 (2,18)
|
4,95 (2,46)
|
4,71 (2,60)
|
5,41 (2,12)
|
3,20 (2,33)
|
2,68 (2,42)
|
(a) Cimzia administré toutes les 2 semaines après une dose initiale de 400 mg en semaine 0, 2 et 4
(b) BASDAI est l'indice „Bath Ankylosing Spondylitis Disease Activity NRS Index“, où 0 = nul et 10 = très grave
(c) BASFI est l'indice „Bath Ankylosing Spondylitis Functional NRS Index“, où 0 = facile et 0 = impossible
L'ensemble complet d'analyse avec indication de la dernière valeur (last observation carried forward, LOCF) a été utilisé.
Les signes d'inflammation ont été constatés à l'IRM à la semaine 12 et représentés sous la forme de modification de l'état initial du score SPARCC pour les articulations sacro-iliaques.
Une inhibition significative de l'inflammation des articulations sacro-iliaques a été constatée en semaine 12 et en semaine 52 chez les patients traités par Cimzia (-4,3 et -7,4 respectivement) par rapport aux patients traités par placebo (0,3 et 2,3 respectivement) par rapport à l'état initial.
Arthrite psoriasique
L'efficacité et la sécurité du Cimzia ont été évaluées dans le cadre d'une étude clinique randomisée, multicentrique, en double aveugle, contrôlée versus placebo (PsA001), menée sur 409 patients d'âge ≥18 ans atteints d'arthrite psoriasique active de l'adulte depuis 6 mois au minimum, définie selon les critères de classification pour l'arthrite psoriasique (Classification Criteria for Psoriatic Arthritis, critères CASPAR). Les patients présentaient ≥3 articulations gonflées douloureuses, ainsi que soit une augmentation des marqueurs de la phase aiguë de l'inflammation soit une vitesse de sédimentation érythrocytaire (ESR – Erythrocyte sedimentation rate) ≥28 mm/heure. Les patients présentaient en outre des lésions cutanées psoriasiques actives ou des antécédents documentés de Psoriasis et ils avaient derrière eux au moins une tentative de traitement par DMARDs sans succès (médicaments antirhumatismaux modificateurs de la maladie). Le traitement antérieur par antagoniste du TNF était autorisé et environ 20% des patients avaient été précédemment traités par antagonistes du TNF. Les patients ont reçu soit 200 mg de Cimzia toutes les 2 semaines respectivement 400 mg de Cimzia toutes les 4 semaines ou d'un placebo toutes les 2 semaines, après des doses initiales de 400 mg de Cimzia aux semaines 0, 2 et 4 (dans les deux bras du traitement) ou d'un placebo. Les patients traités de manière concomitante par des AINS ou des DMARDs conventionnels représentaient respectivement 72.6% et 70.2% des patients. Les deux critères principaux d'évaluation étaient le pourcentage de patients ayant obtenu une réponse ACR 20 jusqu'à la semaine 12 et la modification du score total de Sharp modifié (modified Total Sharp Score, mTSS) à la semaine 24 par rapport à l'inclusion.
Réponse ACR
Les taux de réponses ACR 20, ACR 50 respectivement ACR 70 au traitement par Cimzia dans l'étude clinique PsA001 sont représentés dans le tableau 11. Les patients sous Cimzia ont présenté des taux de réponses ACR 20 statistiquement et significativement plus élevés d'un point de vue statistique aux semaines 12 et 24 (p <0,001) par rapport aux patients recevant le placebo. De plus, les patients recevant du Cimzia ont présenté des améliorations significatives aux semaines 12 et 24 en ce qui concerne les taux de réponses ACR 50 et ACR 70, ainsi que les composantes ACR individuelles (voir tableau 12), par rapport aux patients sous placebo. Les patients recevant 200 mg de Cimzia toutes les 2 semaines et les patients recevant 400 mg de Cimzia toutes les 4 semaines ont répondu de façon similaire au traitement.
Les patients recevant une comédication de méthotrexate ont présenté au cours de la semaine 24 des taux de réponses ACR 20, ACR 50 et ACR 70 plus importants (62%, 35% et 26%) que les patients sous monothérapie (53%, 32% et 20%).
Tableau 11: Réponse ACR dans l'étude clinique PsA001 (pourcentage de patients)
|
Réponse
|
Placebo
N=136
|
Cimzia(a) 200 mg
Q2W
N=138
|
Cimzia(b) 400 mg
Q4W
N=135
| |
ACR 20
| |
Semaine 12
|
24%
|
58%**
|
52%**
| |
Semaine 24
|
24%
|
64%**
|
56%**
| |
ACR 50
| |
Semaine 12
|
11%
|
36%**
|
33%**
| |
Semaine 24
|
13%
|
44%**
|
40%**
| |
ACR 70
| |
Semaine 12
|
3%
|
25%**
|
13%*
| |
Semaine 24
|
4%
|
28%**
|
24%**
|
(a) Cimzia toutes les 2 semaines, précédé de doses initiales de 400 mg aux semaines 0, 2 et 4
(b) Cimzia toutes les 4 semaines, précédé de doses initiales de 400 mg aux semaines 0, 2 et 4
* p <0.01, Cimzia versus placebo
** p <0.001, Cimzia versus placebo
Les résultats se sont basés sur la population randomisée. Différence de traitement: Cimzia 200 mg – placebo, Cimzia 400 mg – placebo (avec l'IC bilatéral à 95% et valeur de p) sont estimés avec des erreurs asymptotiques standard au moyen d'un test de Wald standard bilatéral en utilisant la méthode d'imputation des non-répondeurs (NRI – Non-Responder Imputation).
Tableau 12: Composantes des réponses ACR dans l'étude clinique PsA001
|
Paramètres
|
Placebo
N=136
|
Cimzia(a) 200 mg Q2W
N=138
|
Cimzia(b) 400 mg Q4W
N=135
| |
|
Valeurs initiales
|
Semaine 12
|
Semaine 24
|
Valeurs initiales
|
Semaine 12
|
Semaine 24
|
Valeurs initiales
|
Semaine 12
|
Semaine 24
| |
Nombre
d'articulations sensibles à la douleur
(0–68) (c)
|
19.9
|
16.5
|
17.0
|
21.5
|
11.2*
|
8.5*
|
19.6
|
11.2*
|
9.4*
| |
Nombre
d'articulations gonflées
(0-66) (c)
|
10.4
|
8.7
|
9.9
|
11.0
|
4.0*
|
3.1*
|
10.5
|
4.7*
|
3.0*
| |
Jugement médical global (c, d)
|
58.7
|
44.1
|
42.2
|
56.8
|
24.8*
|
19.6*
|
58.2
|
28.7*
|
21.1*
| |
Jugement global du patient (c, d)
|
57.0
|
50.2
|
49.0
|
60.2
|
32.6*
|
31.1*
|
60.2
|
39.6*
|
32.5*
| |
Douleur (c, e)
|
60.0
|
50.2
|
48.8
|
59.7
|
32.8*
|
31.1*
|
61.1
|
38.6*
|
32.7*
| |
Indice d'incapacité (HAQ) (c, f)
|
1.30
|
1.15
|
1.13
|
1.33
|
0.87*
|
0.81*
|
1.29
|
0.90*
|
0.86*
| |
CRP (mg/l)
|
18.56
|
14.75
|
14.66
|
15.36
|
5.67*
|
4.58*
|
13.71
|
6.34*
|
7.37*
|
(a) Cimzia toutes les 2 semaines, précédé de doses initiales de 400 mg aux semaines 0, 2 et 4
(b) Cimzia toutes les 4 semaines, précédé de doses initiales de 400 mg aux semaines 0, 2 et 4
(c) Approche LOCF (Last Observation Carried Forward – dernière observation reportée) pour des données manquantes, une interruption précoce de l'étude ou un adjuvant placebo
(d) Patient et Physician Global Assessment of Disease Activity (Jugement global de l'activité de la maladie par le patient respectivement le médecin), échelle VAS avec 0 = très bonne et 100 = très mauvaise
(e) The Patient Assessment of Arthritis Pain (L'évaluation de la douleur arthritique par le patient), échelle VAS avec 0 = aucune douleur et 100 = douleurs très fortes
(f) HAQ-DI (questionnaire d'évaluation de l'état de santé – indice d'incapacité), échelle à 4 points allant de 0 = aucune invalidité à 3 = grande invalidité
En ce qui concerne les valeurs représentées, il s'agit de données moyennes.
Les résultats se basent sur la population randomisée (imputation ou observation).
* p <0.001, Cimzia versus placebo
Le pourcentage de répondeurs ACR 20 était cliniquement pertinent et significativement plus élevé dans les groupes de traitement sous Cimzia 200 mg toutes les 2 semaines et Cimzia 400 mg toutes les 4 semaines, que dans le groupe de patients sous placebo (p ≤0.001 à toutes les visites), à toutes les visites après le début du traitement jusqu'à la semaine 24.
Réponse PASI
Les patients, ayant au minimum 3% à l'inclusion de la surface corporelle touchée par des lésions psoriasiques, ont été évalués au moyen de la réponse selon l'indice de surface et de gravité du psoriasis (Psoriasis Area and Severity Index – PASI), en ce qui concerne l'amélioration de leurs manifestations cutanées. Comme le montre le tableau 13, le pourcentage de patients était composé de ceux qui ont obtenu une réponse PASI 75 et une réponse PASI 90 à la semaine 24, dans les deux groupes posologiques de Cimzia (200 mg toutes les 2 semaines + 400 mg toutes les 4 semaines) (n=166) 61% respectivement 42%, par rapport à 15% respectivement 6% dans le groupe placebo (n=86) (p <0.001).
Tableau 13: Taux de réponse PASI dans l'étude clinique PsA001
|
Taux de réponse
|
Placebo
N=86
|
Cimzia(a) 200 mg Q2W
N=90
|
Cimzia(b) 400 mg Q4W
N=76
|
Cimzia (toutes les posologies)(c)
N=166
| |
|
Semaine 12
|
Semaine 24
|
Semaine 12
|
Semaine 24
|
Semaine 12
|
Semaine 24
|
Semaine 12
|
Semaine 24
| |
PASI 75
|
14%
|
15%
|
47%**
|
62%**
|
47%**
|
61%**
|
47%**
|
61%**
| |
PASI 90
|
5%
|
6%
|
22%**
|
47%**
|
20%*
|
36%**
|
21%**
|
42%**
|
(a) Cimzia toutes les 2 semaines, précédée de doses initiales de 400 mg aux semaines 0, 2 et 4
(b) Cimzia toutes les 4 semaines, précédée de doses initiales de 400 mg aux semaines 0, 2 et 4
(c) Cimzia (toutes les posologies) = données pour Cimzia 200 mg toutes les 2 semaines, précédée de doses initiales de 400 mg aux semaines 0, 2 et 4, plus données pour Cimzia 400 mg toutes les 4 semaines, précédée de doses initiales de 400 mg aux semaines 0, 2 et 4
* Valeur p <0.01, Cimzia vs placebo
** Valeur p <0.001, Cimzia vs placebo
Les résultats se sont basés sur la population randomisée.
Différence de traitement: Cimzia 200 mg – placebo, Cimzia 400 mg – placebo (avec intervalle de confiance de 95% correspondant et la valeur p) sont estimés avec des erreurs asymptotiques standard au moyen d'un test de Wald standard bilatéral. Remarque: la méthode NRI (Non-Responder-Imputation) a été utilisée.
Les patients qui ont présenté une enthésite ont été évalués du point de vue de l'amélioration moyenne de l'indice de Leeds pour l'enthésite (Leeds Enthesitis Index – LEI). Les patients sous 200 mg de Cimzia toutes les 2 semaines ou 400 mg toutes les 4 semaines ont présenté une amélioration plus importante de l'enthésite (-1.8; -1.7) que les patients recevant le placebo (-0.9) dans la semaine 12 (p <0.001 respectivement p <0.01) et la semaine 24 (200 mg toutes les 2 semaines: -2.0; 400 mg toutes les 4 semaines: 1.8; placebo: -1.1) (p <0.001 respectivement p <0.01).
Les mêmes schémas de posologie ont également montré une réduction significative de la dactylite (modification moyenne du score initial (-30.40; -45.46) par rapport aux patients traités par placebo (-16.79) dans la semaine 12.
Réponse radiologique
Dans l'étude clinique PsA001, la progression des dommages articulaires a été évaluée par radiographie entre la période d'inclusion et la semaine 24 et exprimée en termes de modification du mTSS et de ses composantes dans le score d'érosion (SE) et le score de pincement articulaire (JSN, de Joint Space Narrowing). Le score mTSS-Score a été modifié en ajoutant les articulations interphalangiennes distales pour arthrite psoriasique. L'effet du traitement suivant a été observé avec une imputation post hoc: le traitement par Cimzia par rapport au traitement par placebo à la semaine 24 a inhibé la progression radiologique de la modification du mTSS par rapport à la valeur initiale (la valeur moyenne LS [± SE] du score était de 0.28 [± 0.07] dans le groupe de patients sous placebo par rapport aux 0.06 [± 0.06] dans l'ensemble du groupe Cimzia; p=0.007). Une différence statistiquement significative par rapport au placebo a été observée dans le groupe de patients recevant 200 mg de CZP Q2W (p=0.004), mais pas dans le groupe de patients recevant 400 mg de CZP Q4W (p=0.072).
Réaction en termes de capacité fonctionnelle et de critères d'évaluation liés à la santé
Dans l'étude clinique PsA001, les patients traités par Cimzia à partir de la semaine 2 ont rapporté de nettes améliorations de leurs capacités fonctionnelles physiques (déterminées au moyen du questionnaire d'évaluation de l'état de santé – indice d'incapacité (HAQ – DI Disability Index)») «Health Assessment Questionnaire – Disability Index)») ainsi que de la douleur (déterminée au moyen de l'évaluation par le patient de la douleur arthritique «Patient Assessment of Arthritis Pain [PAAP]») (voir tableau 12). La différence a été significative pour la PAAP après la semaine 1. Les patients recevant du Cimzia ont rapporté une amélioration de la fatigue évaluée au moyen de l'échelle d'évaluation de la fatigue («FASCA – Fatigue Assessment Scale») de la semaine 2 à la semaine 24, par rapport au placebo
Les patients recevant du Cimzia ont rapporté des améliorations significatives de leur qualité de vie liée à leur santé dans le questionnaire relatif à l'indice de qualité de vie spécifique du RhPso (PsAQoL) à partir de la semaine 2, ainsi que dans tous les «scores» résumés des composantes physiques et mentales du questionnaire SF 36 de la semaine 4 à la semaine 24.
Les patients traités par Cimzia ont connu des améliorations des limites de productivité dues à l'arthrite psoriasique sur leur lieu de travail et à leur domicile sur la base du «Work Productivity Survey» (questionnaire de productivité au travail) par rapport aux patients recevant le placebo de la semaine 4 à la semaine 24.
Psoriasis en plaques
L'efficacité et la sécurité du Cimzia ont été testées dans deux études contrôlées versus placebo (COMPASI-1 et CIMPASI-2) et une étude contrôlée versus placebo et versus un comparateur actif (CIMPACT) chez des patients âgés de ≥18 ans atteints de psoriasis en plaques chronique modéré à sévère sur une période d'au moins 6 mois. Les patients présentaient un score PASI (Psoriasis Area and Severity Index-Score) ≥12, une atteinte ≥10 % de la surface corporelle (BSA, Body Surface Area), un score PGA ≥3 (évaluation globale du médecin, Physician Global Assessment) et nécessitaient un traitement systémique et/ou une photothérapie et/ou une chimiothérapie. Les patients en échec «primaire» à tout traitement biologique antérieur (défini par l'absence de réponse au cours des 12 premières semaines de traitement) ont été exclus des études de phase III (CIMPASI-1, CIMPASI-2 et CIMPACT). L'efficacité et la sécurité du Cimzia ont été évaluées par rapport à étanercept dans l'étude CIMPACT.
Dans les études CIMPASI-1 et CIMPASI-2, les co-critères primaires d'efficacité étaient le taux de répondeurs PASI 75 et le pourcentage de patients présentant un score PGA «blanchi» ou «quasiment blanchi» (avec une diminution d'au moins 2 points par rapport à l'inclusion) à la semaine 16. Dans l'étude CIMPACT, le critère primaire d'efficacité était le taux de répondeurs PASI 75 à la semaine 12. Les taux de répondeurs PASI 75 et PGA à la semaine 16 étaient des critères secondaires. Le taux de répondeurs PASI 90 à la semaine 16 était un critère secondaire dans les 3 études.
234 patients et 227 patients ont été évalués dans les études CIMPASI-1 et CIMPASI-2, respectivement. Dans les deux études, les patients ont été randomisés pour recevoir un placebo ou Cimzia 200 mg toutes les 2 semaines (après une dose initiale de Cimzia de 400 mg aux semaines 0, 2 et 4) ou Cimzia 400 mg toutes les 2 semaines. À la semaine 16, les patients randomisés dans les bras Cimzia et répondeurs PASI 50 ont continué à recevoir Cimzia jusqu'à la semaine 48 à la même dose randomisée. Les patients qui avaient été initialement randomisés dans le bras placebo et qui avaient obtenu une réponse PASI 50, mais pas une réponse PASI 75 à la Semaine 16 ont reçu Cimzia 200 mg toutes les 2 semaines (avec une dose initiale de Cimzia de 400 mg aux semaines 16, 18, et 20). Les patients qui présentaient une réponse insuffisante à la semaine 16 (non-répondeurs PASI 50) étaient éligibles à recevoir Cimzia 400 mg toutes les 2 semaines pendant 128 semaines maximum.
559 patients ont été évalués dans l'étude CIMPACT. Les patients ont été randomisés pour recevoir un placebo ou Cimzia 200 mg toutes les 2 semaines (après une dose initiale de Cimzia de 400 mg aux semaines 0, 2 et 4) ou Cimzia 400 mg toutes les 2 semaines jusqu'à la semaine 16, ou étanercept 50 mg deux fois par semaine jusqu'à la semaine 12. Les patients qui avaient été initialement randomisés dans le bras Cimzia et répondeurs PASI 75 à la semaine 16 ont été de nouveau randomisés en fonction de leur schéma posologique initial. Les patients sous Cimzia 200 mg toutes les 2 semaines ont été de nouveau randomisés dans le bras Cimzia 200 mg toutes les 2 semaines, le bras Cimzia 400 mg toutes les 4 semaines ou le bras placebo. Les patients sous Cimzia 400 mg toutes les 2 semaines ont été de nouveau randomisés dans le bras Cimzia 400 mg toutes les 2 semaines, le bras Cimzia 200 mg toutes les 2 semaines, ou dans le bras placebo. L'étude était en double aveugle contrôlée versus placebo jusqu'à la semaine 48. Tous les sujets non-répondeurs PASI 75 à la semaine 16 ont intégré un bras d'échappement et ont reçu 400 mg de Cimzia en ouvert toutes les 2 semaines pendant 128 semaines maximum.
Lors des trois études, une phase de traitement en ouvert de 96 semaines succédait à une phase d'entretien en aveugle de 48 semaines pour les patients répondeurs PASI 50 à la semaine 48. Tous les patients sous traitement en aveugle ont commencé la période en ouvert avec Cimzia 200 mg toutes les 2 semaines. Au cours de la phase de traitement en ouvert, les ajustements de dose entre Cimzia 200 mg toutes les 2 semaines et Cimzia 400 mg toutes les 2 semaines étaient autorisés en l'absence d'une réponse PASI 50 ou à l'appréciation du médecin investigateur. La durée maximale des études était en tout de 144 semaines.
Parmi les 850 patients randomisés pour recevoir le placebo ou Cimzia dans ces études contrôlées versus placebo, 29% étaient naïfs de traitement systémique antérieur pour le traitement d'un psoriasis. 47% avaient reçu une photothérapie ou une chimiothérapie antérieure et 30% avaient reçu un traitement biologique antérieur pour le traitement d'un psoriasis. Parmi ces 850 patients, 14% avaient reçu au moins un anti-TNF, 13% avaient reçu un anti-IL-17 et 5% avaient reçu un anti-IL-12/23. 18% des patients ont rapporté des antécédents de rhumatisme psoriasique à l'inclusion. Le score PASI moyen à l'inclusion était de 20 et variait entre 12 et 69. Le score PGA à l'inclusion était modéré (70%) à sévère (30%). La BSA moyenne à l'inclusion était de 25% et variait entre 10% et 96%.
Les principaux résultats des études CIMPASI-1 et CIMPASI-2 sont reportés dans le tableau 14.
Tableau 14: Réponse clinique dans les études CIMPASI-1 et CIMPASI-2 à la semaine 16 et à la semaine 48
|
|
Semaine 16
|
Semaine 48
| |
CIMPASI-1
| |
|
Placebo
N=51
|
Cimzia 200 mg toutes les 2 semainesa)
N=95
|
Cimzia 400 mg toutes les 2 semaines
N=88
|
Cimzia 200 mg toutes les 2 semaines
N=95
|
Cimzia 400 mg toutes les 2 semaines
N=88
| |
PGA blanchi ou quasiment blanchib)
|
4.2%
|
47.0%*
|
57.9%*
|
52.7%
|
69.5%
| |
PASI 75
|
6.5%
|
66.5%*
|
75.8%*
|
67.2%
|
87.1%
| |
PASI 90
|
0.4%
|
35.8%*
|
43.6%*
|
42.8%
|
60.2%
| |
CIMPASI-2
| |
|
Placebo
N=49
|
Cimzia 200 mg toutes les 2 semaines a)
N=91
|
Cimzia 400 mg toutes les 2 semaines
N=87
|
Cimzia 200 mg toutes les 2 semaines
N=91
|
Cimzia 400 mg toutes les 2 semaines
N=87
| |
PGA blanchi ou quasiment blanchib)
|
2.0%
|
66.8%*
|
71.6%*
|
72.6%
|
66.6%
| |
PASI 75
|
11.6%
|
81.4%*
|
82.6%*
|
78.7%
|
81.3%
| |
PASI 90
|
4.5%
|
52.6%*
|
55.4%*
|
59.6%
|
62.0%
|
a) Cimzia 200 mg, administré toutes les 2 semaines, après une dose initiale de 400 mg aux semaines 0, 2, 4.
b) Echelle PGA à 5 catégories. L'atteinte du critère «blanchi» (0) ou «quasiment blanchi» (1) était définie par l'absence de signes de psoriasis ou une coloration des lésions normale à rose, l'absence d'épaississement de la plaque et une desquamation absente ou minime.
* p<0.0001, Cimzia vs. placebo
Les taux de répondeurs et les valeurs p pour les scores PASI et PGA ont été calculés à l'aide d'un modèle de régression logistique dans lequel les données manquantes ont été imputées à l'aide d'une imputation multiple avec la méthode MCMC. Les patients ayant échappé ou été sortis de l'étude (sur la base d'une non atteinte de réponse PASI 50) ont été considérés comme des non-répondeurs à la semaine 48.
Les résultats sont issus de l'ensemble randomisé.
Les principaux résultats de l'étude CIMPACT sont reportés dans le tableau 15.
Tableau 15: Réponse clinique dans l'étude CIMPACT à la semaine 12 et à la semaine 16
|
|
Semaine 12
|
Semaine 16
| |
|
Placebo
N=57
|
Cimzia 200 mg toutes les 2 semainesa)
N=165
|
Cimzia 400 mg toutes les 2 semaines
N=167
|
Placebo
N=57
|
Cimzia 200 mg toutes les 2 semaines
N=165
|
Cimzia 400 mg
toutes les 2 semainesN=167
| |
PASI 75
|
5%
|
61.3%*
|
66.7%*
|
3.8%
|
68.2%*
|
74.7%*
| |
PASI 90
|
0.2%
|
31.2%*
|
34.0%*
|
0.3%
|
39.8%*
|
49.1%*
| |
PGA blanchi ou quasiment blanchib)
|
1.9%
|
39.8%**
|
50.3%*
|
3.4%
|
48.3%*
|
58.4%*
|
a) Cimzia 200 mg, administré toutes les 2 semaines, après une dose initiale de 400 mg aux semaines 0, 2, 4.
b) Echelle PGA à 5 catégories. L'atteinte du critère «blanchi» (0) ou «quasiment blanchi» (1) était définie par l'absence de signes de psoriasis ou une coloration des lésions normale à rose, l'absence d'épaississement de la plaque et une desquamation absente ou minime.
* p<0.0001, Cimzia vs. placebo
** p<0.0001, Cimzia vs. placebo
Taux de répondeurs et valeurs p basés sur un modèle de régression logistique. Les données manquantes ont été imputées à l'aide d'une imputation multiple basée sur la méthode MCMC.
Les résultats sont issus de l'ensemble randomisé.
Dans l'étude CIMPACT, Cimzia 400 mg toutes les 2 semaines versus l'étanercept 50 mg deux fois par semaine, au bout de 12 semaines, a démontré une supériorité concernant le taux de répondeurs PASI 75 (66.7% contre 53.3%; p < 0.05). Cimzia 200 mg toutes les 2 semaines versus l'étanercept 50 mg deux fois par semaine, a démontré une non- infériorité concernant le taux de répondeurs PASI 75, avec une marge de non-infériorité préalablement définie de 10 % (61.3%; différence entre l'étanercept et Cimzia 200 mg toutes les 2 semaines 8,0% [IC à 95% –2.9; 18.9]).
Dans les trois études, le taux de répondeurs PASI 75 et le taux de patients présentant un score PGA «blanchi» ou «quasiment blanchi» du bras Cimzia était significativement supérieur à celui du bras placebo à partir de la semaine 4.
Les deux doses de Cimzia ont démontré une efficacité par rapport au placebo, quels que soient l'âge, le sexe, le poids corporel, l'IMC, l'antériorité du psoriasis, les antécédents de traitements systémiques et les antécédents de traitements biologiques.
Maintien de la réponse
Dans une analyse intégrée des études CIMPASI-1 et CIMPASI-2, parmi les patients qui étaient répondeurs PASI 75 à la semaine 16 et qui avaient reçu Cimzia 400 mg toutes les 2 semaines (134 patients sur les 175 randomisés) ou Cimzia 200 mg une semaine sur 2 (132 patients sur les 186 randomisés), les taux de maintien de la réponse à la semaine 48 étaient de 98.0% et de 87.5%, respectivement. Parmi les patients qui présentaient un score PGA blanchi ou quasiment blanchi à la semaine 16 et qui avaient reçu Cimzia 400 mg toutes les 2 semaines (103 patients sur 175) ou Cimzia 200 mg toutes les 2 semaines (95 patients sur 186), les taux de maintien de la réponse à la semaine 48 étaient de 85.9 % et de 84.3 % respectivement.
Ces taux de réponse étaient calculés sur un modèle de régression logistique dans lequel les données manquantes ont été imputées à l'aide d'une imputation multiple (méthode MCMC) sur 48 et 144 semaines. Les résultats à 48 semaines de la phase de traitement en ouvert pendant 96 semaines chez les répondeurs PASI 75 indiquaient que la réponse se maintient majoritairement sous Cimzia 200 mg toutes les 2 semaines.
Dans l'étude CIMPACT, parmi les répondeurs PASI 75 à la semaine 16 qui avaient reçu Cimzia 400 mg toutes les 2 semaines et qui avaient été de nouveau randomisés dans le bras Cimzia 400 mg toutes les 2 semaines, ou le bras Cimzia 200 mg toutes les 2 semaines, ou le bras placebo, le pourcentage de répondeurs PASI 75 à la semaine 48 était supérieur dans les groupes Cimzia à celui du groupe placebo (98.0%, 80.0%, et 36.0%, respectivement). Parmi les répondeurs PASI 75 à la semaine 16 qui avaient reçu Cimzia 200 mg toutes les 2 semaines et qui avaient été de nouveau randomisés dans le bras Cimzia 400 mg toutes les 4 semaines, ou le bras Cimzia 200 mg toutes les 2 semaines, ou le bras placebo, le pourcentage de répondeurs PASI 75 à la semaine 48 était également supérieur dans les groupes Cimzia à celui du groupe placebo (88.6%, 79.5%, et 45.5%, respectivement). L'imputation des non-répondeurs a été utilisée pour les données manquantes. Les résultats de la période suivante de traitement en ouvert sur 96 semaines chez les répondeurs PASI 75 ont démontré que la réponse se maintient majoritairement sous Cimzia 200 mg toutes les 2 semaines.
Qualité de vie / résultats du point de vue des patients
Des améliorations statistiquement significatives à la semaine 16 (CIMPASI-1 et CIMPASI-2) du DLQI (indice dermatologique de qualité de vie, Dermatology Life Quality Index) versus placebo ont été démontrées par rapport à l'inclusion. Les diminutions moyennes (améliorations) du DLQI par rapport à l'inclusion variaient de -8.9 à -11.1 avec Cimzia 200 mg toutes les 2 semaines, de -9.6 à -10,0 avec Cimzia 400 mg toutes les 2 semaines, versus -2.9 à -3.3 pour le placebo à la semaine 16.
De plus, à la semaine 16, le traitement par Cimzia était associé à une plus grande proportion de patients ayant obtenu un score DLQI de 0 ou 1 (Cimzia 400 mg toutes les 2 semaines, 45.5% et 50.6% respectivement; Cimzia 200 mg toutes les 2 semaines, 47.4% et 46.2% respectivement, versus placebo, 5.9% et 8.2% respectivement).
Les patients traités par Cimzia ont rapporté des améliorations plus importantes par rapport au placebo dans l'échelle HADS-D (échelle d'anxiété et de dépression à l'hôpital - dimension dépressive, Hospital Anxiety and Depression Scale).
|