CompositionPrincipes actifs
Glycopyrronium sous forme de bromure de glycopyrronium.
Excipients
Lactose monohydraté, stéarate de magnésium.
Indications/Possibilités d’emploiSeebri Breezhaler est indiqué dans le traitement bronchodilatateur d'entretien administré une fois par jour pour atténuer les symptômes de patients atteints de bronchopneumopathie chronique obstructive (BPCO).
Posologie/Mode d’emploiPopulation générale
Posologie
La dose recommandée de Seebri Breezhaler est d'une inhalation du contenu d'une gélule de 50 microgrammes une fois par jour à l'aide de l'inhalateur Seebri Breezhaler. Cette dose journalière ne doit pas être dépassée.
Groupes de patients particuliers
Insuffisance rénale
Seebri Breezhaler peut être utilisé à la dose recommandée chez les patients avec insuffisance rénale légère à modérée. Chez les patients avec insuffisance rénale sévère et chez les patients dialysés en insuffisance rénale terminale, Seebri Breezhaler ne doit être utilisé que si le bénéfice attendu dépasse le risque potentiel (voir aussi «Mises en garde et précautions» ainsi que «Pharmacocinétique»).
Insuffisance hépatique
Aucune étude spécifique n'a été conduite chez des patients atteints d'un trouble de la fonction hépatique. Seebri Breezhaler est principalement éliminé par voie rénale.
Patients âgés
Seebri Breezhaler peut être administré à la dose recommandée chez les patients âgés de 75 ans ou plus.
Enfants et adolescents
Seebri Breezhaler ne doit pas être administré aux patients de moins de 18 ans.
Mode d'administration
Les gélules de Seebri Breezhaler ne doivent être utilisées qu'en inhalation orale et uniquement à l'aide de l'inhalateur Seebri Breezhaler. Les gélules de Seebri Breezhaler ne doivent pas être avalées (voir aussi «Surdosage»).
Il est recommandé d'inhaler Seebri Breezhaler une fois par jour, toujours à la même heure. En cas d'oubli d'une dose, la dose suivante doit être inhalée le plus rapidement possible. Les patients doivent être avertis de ne pas inhaler plus d'une dose par jour.
Les gélules de Seebri Breezhaler doivent toujours être conservées dans les plaquettes thermoformées, pour les maintenir à l'abri de l'humidité, et ne doivent en être retirées qu'IMMEDIATEMENT AVANT L'EMPLOI.
Lors de la prescription de Seebri Breezhaler, on instruira les patients quant à l'utilisation correcte de l'inhalateur. Il faut demander aux patients qui ne ressentent pas d'amélioration respiratoire s'ils avalent le médicament au lieu de l'inhaler.
Contre-indicationsHypersensibilité au principe actif, au lactose ou à un autre composant.
Mises en garde et précautionsN'est pas destiné au traitement de symptômes aigus.
Seebri Breezhaler est utilisé une fois par jour pour le traitement d'entretien à long terme et n'est pas indiqué dans le traitement des bronchospasmes aigus, autrement dit dans le traitement d'urgence.
Hypersensibilité
Des réactions d'hypersensibilité immédiate ont été rapportées après l'inhalation de Seebri Breezhaler. En cas d'apparition de signes de réaction allergique, en particulier d'angioœdème (y compris les difficultés respiratoires ou à la déglutition, le gonflement de la langue, des lèvres et du visage), d'urticaire ou d'éruption cutanée, le traitement par Seebri Breezhaler doit être immédiatement interrompu et un traitement alternatif doit être instauré.
Action anticholinergique
Comme les autres anticholinergiques, la prudence est de mise en cas d'administration de Seebri Breezhaler à des patients atteints de glaucome à angle fermé, de rétention urinaire ou de maladies cardiovasculaires sévères préexistantes.
On conseillera aux patients de veiller à ne pas laisser la poudre entrer en contact avec les yeux par des manipulations inappropriées et on leur expliquera quels sont les signes et les symptômes d'un glaucome aigu à angle fermé. Ils doivent être informés de la nécessité d'interrompre le traitement par Seebri Breezhaler et de s'adresser immédiatement à leur médecin s'ils présentent de tels signes ou symptômes.
Patients avec insuffisance rénale sévère
Chez les patients avec insuffisance rénale sévère (débit de filtration glomérulaire estimé à moins de 30 ml/min/1,73 m2), y compris les patients en insuffisance rénale terminale dialysés, Seebri Breezhaler ne doit être administré que si les bénéfices attendus dépassent les risques potentiels (voir «Pharmacocinétique»). Ces patients requièrent une surveillance étroite à la recherche d'effets indésirables éventuels.
Bronchospasmes paradoxaux
Comme avec les autres traitements inhalés, l'inhalation de Seebri Breezhaler peut donner lieu à des bronchospasmes paradoxaux potentiellement fatals. En cas de bronchospasme paradoxal, le traitement par Seebri Breezhaler sera immédiatement interrompu et un traitement alternatif sera instauré.
Patients présentant des antécédents de maladies cardiovasculaires
Les patients présentant une maladie cardiaque ischémique instable, une insuffisance ventriculaire gauche, des antécédents d'infarctus du myocarde, des arythmies (à l'exception de la fibrillation auriculaire chronique stable), des antécédents de syndrome du QT long ou d'allongement de l'intervalle QT selon la formule de Fridericia (> 450 ms pour les hommes ou > 470 ms pour les femmes) ont été exclus des études cliniques. Ainsi, on ne dispose que d'expériences limitées chez ces groupes de patients. Il convient de faire preuve de prudence lors de l'utilisation de Seebri Breezhaler chez ces groupes de patients.
Excipients
Seebri Breezhaler contient le colorant azoïque jaune orangé S (E 110). Seebri Breezhaler doit être utilisé avec précaution chez les patients souffrant d'une hypersensibilité aux colorants azoïques, à l'acide acétylsalicylique et à d'autres inhibiteurs des prostaglandines.
Seebri Breezhaler contient du lactose et ne doit donc pas être utilisé chez les patients atteints d'un déficit sévère en lactase ou d'une galactosémie.
InteractionsInteractions pharmacodynamiques
L'utilisation simultanée de Seebri Breezhaler avec d'autres médicaments administrés par voie inhalée contenant des anticholinergiques n'a pas fait l'objet d'études et n'est par conséquent pas recommandée.
Seebri Breezhaler a été employé avec d'autres médicaments souvent utilisés dans le cadre du traitement de la BPCO. En l'occurrence, on n'a pas constaté de signes cliniques d'interactions médicamenteuses, même si aucune étude formelle n'a été menée sur les interactions. Les médicaments utilisés en même temps comprennent des bronchodilatateurs sympatomimétiques, des méthylxantines ainsi que des stéroïdes oraux et inhalés.
Chez les adultes sains, l'utilisation simultanée de Seebri Breezhaler et d'indacatérol inhalé par voie orale (un agoniste bêta2-adrénergique) n'a eu aucun effet sur la pharmacocinétique des deux substances à l'état d'équilibre de ces deux dernières.
Dans un essai clinique chez des sujets sains, la cimétidine, un inhibiteur du transport des cations organiques qui contribue probablement à l'élimination rénale du glycopyrronium, a augmenté l'exposition globale (AUC) au glycopyrronium de 22% et diminué sa clairance rénale de 23%.
Les essais in vitro suggèrent que Seebri Breezhaler n'inhibe pas et n'induit pas le métabolisme d'autres médicaments ou processus auxquels participent des transporteurs de médicaments. Les processus métaboliques impliquant plusieurs enzymes jouent un rôle secondaire dans l'élimination du glycopyrronium (voir «Pharmacocinétique»). Il est peu probable que l'inhibition ou l'induction du métabolisme du glycopyrronium entraîne une modification significative de l'exposition systémique au médicament.
Grossesse, allaitementFemmes en âge de procréer
Il n'y a pas de recommandations particulières chez les femmes en âge de procréer.
Grossesse
Il n'existe pas de données cliniques chez les patientes enceintes atteintes de BPCO. L'inhalation de Seebri Breezhaler n'a pas entraîné d'effets tératogènes chez le rat et le lapin (voir «Données précliniques»). Comme on ne dispose pas d'une expérience suffisante chez les femmes enceintes, Seebri Breezhaler ne doit être utilisé durant la grossesse qu'après une évaluation soigneuse du rapport bénéfices/risques.
Allaitement
On ignore si le bromure de glycopyrronium passe dans le lait maternel humain. L'administration de Seebri Breezhaler à des femmes qui allaitent ne doit donc être conseillée qu'après une évaluation soigneuse du rapport bénéfices/risques.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée sur l'influence de Seebri Breezhaler sur l'aptitude à la conduite et l'utilisation de machines.
Effets indésirablesLa sécurité et la tolérance de Seebri Breezhaler ont été évaluées chez 1353 patients avec BPCO à la dose recommandée de 50 µg une fois par jour. Parmi ces patients, 842 ont été traités durant au moins 26 semaines et 351 pendant au moins 52 semaines. Les grandes études cliniques sur l'efficacité et la sécurité n'incluaient pas de patients avec glaucome à angle fermé, hyperplasie symptomatique de la prostate, sténose du col vésical, insuffisance rénale modérée et maladies cardiovasculaires significatives (telles qu'infarctus du myocarde récent, troubles du rythme, insuffisance ventriculaire gauche).
Le profil de sécurité est caractérisé par des symptômes en rapport avec l'action anticholinergique, y compris une sécheresse buccale. En comparaison, les autres effets gastro-intestinaux et les signes de rétention urinaire étaient peu fréquents. Les effets indésirables relatifs à la tolérance locale comprenaient les irritations de la gorge, les rhinopharyngites, les rhinites et les sinusites. À la dose recommandée, Seebri Breezhaler n'exerce aucun effet sur la tension artérielle ni sur la fréquence cardiaque.
Les effets indésirables annoncés au cours des 6 premiers mois de deux essais pivot de phase III cumulés d'une durée respective de 6 et 12 mois, sont listés par classes de systèmes d'organes MedDRA. Les fréquences étaient définies de la manière suivante: très fréquents (≥1/10); fréquents (≥1/100 à < 1/10); occasionnels (≥1/1000 à < 1/100). À l'intérieur de chacun des groupes de fréquence, les réactions indésirables sont classées par ordre décroissant de sévérité.
Infections
Occasionnels: rhinite, cystite.
Troubles du métabolisme et de la nutrition
Occasionnels: hyperglycémie.
Troubles psychiatriques
Occasionnels: insomnies.
Troubles du système nerveux
Occasionnels: hypoesthésie.
Troubles cardiaques
Occasionnels: fibrillation auriculaire, palpitations.
Organes respiratoires
Occasionnels: obstruction des sinus, toux avec expectorations, irritation de la gorge, épistaxis.
Troubles gastro-intestinaux
Fréquents: sécheresse buccale, gastro-entérite.
Occasionnels: dyspepsie, caries dentaires.
Troubles cutanés
Occasionnels: éruption cutanée.
Troubles musculosquelettiques
Occasionnels: douleurs des extrémités, douleurs thoraciques d'origine musculosquelettique.
Troubles rénaux et urinaires
Occasionnels: dysurie, uropathie obstructive.
Troubles généraux
Occasionnels: fatigue, asthénie.
Dans l'étude sur 12 mois, les effets supplémentaires suivants étaient plus fréquents sous Seebri Breezhaler que sous placebo: rhinopharyngite (9,0 vs 5,6%), vomissements (1,3 vs 0,7%), douleurs musculosquelettiques (1,1 vs 0,7%), douleurs de la nuque (1,3 vs 0,7%), diabète (0,8 vs 0%).
Effets indésirables médicamenteux issus de notifications spontanées et de la littérature médicale (fréquence inconnue)
Les effets secondaires médicamenteux suivants ont été signalés lors de l'expérience post-marketing avec Seebri Breezhaler: angioœdème, hypersensibilité, bronchospasmes paradoxaux, dysphonie, démangeaisons (fréquence inconnue).
Description de certains effets indésirables
L'effet indésirable anticholinergique le plus fréquent était la sécheresse buccale. La plupart des annonces concernant la sécheresse buccale était probablement en rapport avec le médicament et de degré léger. Aucun cas sévère n'a été rapporté. Un nombre plus élevé de néoplasies a été observé sous glycopyrronium que sous placebo. On ne connaît pas la cause de cette différence.
Des cas d'éruption cutanée en général légers ont été occasionnellement rapportés.
Groupes de patients particuliers
Chez les patients de plus de 75 ans, la fréquence des infections urinaires et des céphalées était plus élevée sous Seebri Breezhaler que sous placebo, atteignant respectivement 3,0 versus 1,5% et 2,3 versus 0%.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageA fortes doses, le glycopyrronium peut avoir des effets anticholinergiques qui peuvent nécessiter un traitement symptomatique.
L'inhalation orale répétée de Seebri Breezhaler à des doses totales de 100 et 200 µg une fois par jour durant 28 jours a été bien tolérée par des patients atteints de BPCO.
Une intoxication aiguë à la suite d'une prise orale involontaire de gélules de Seebri Breezhaler est peu probable en raison de sa faible biodisponibilité (environ 5%).
Après l'administration i.v. de 150 µg de bromure de glycopyrronium (correspondant à 120 µg de glycopyrronium) à des sujets sains, les concentrations plasmatiques maximales et l'exposition systémique totale étaient environ 50 fois et 6 fois supérieures à la concentration maximale et à l'exposition systémique totale qui ont été atteintes à l'état d'équilibre avec la dose de Seebri Breezhaler recommandée (50 µg une fois par jour). Ces doses étaient bien tolérées.
Propriétés/EffetsCode ATC
R03BB06
Mécanisme d'action
Seebri Breezhaler est un antagoniste des récepteurs muscariniques (anticholinergique) de longue durée d'action, administré par voie inhalée, indiqué dans le traitement bronchodilatateur d'entretien par voie inhalée de la BPCO. Les nerfs parasympathiques forment les voies nerveuses bronchoconstrictrices les plus importantes des voies aériennes et le tonus cholinergique est la principale composante réversible de l'obstruction des voies aériennes dans la BPCO. Seebri Breezhaler inhibe l'action bronchoconstrictrice de l'acétylcholine sur les cellules des muscles lisses des voies aériennes et produit ainsi une dilatation de ces dernières.
Des cinq sous-types de récepteurs muscariniques connus (M1-5), seuls les sous-types M1-3 ont une fonction physiologique bien définie dans le poumon humain. Le bromure de glycopyrronium est un antagoniste des récepteurs muscariniques ayant une forte affinité pour ces trois sous-types de récepteurs. Dans les essais de liaison compétitive, la substance a présenté une sélectivité 4 à 5 plus élevée pour les récepteurs M3 et M1 humains que pour le récepteur M2 humain. Comme cela a été démontré au cours des essais cliniques par l'observation des paramètres cinétiques de l'association/dissociation aux récepteurs et du délai d'action après l'inhalation, elle présente un délai d'action rapide.
On peut supposer, à partir de la demi-vie d'élimination terminale prolongée du glycopyrronium à la suite de l'inhalation de Seebri Breezhaler par rapport à la demi-vie après une injection i.v., que la longue durée d'action est partiellement due à la persistance des concentrations de principe actif dans les poumons (voir «Pharmacocinétique»).
Pharmacodynamique
Effets pharmacodynamiques primaires
Dans une série d'essais cliniques sur la pharmacocinétique et l'efficacité, Seebri Breezhaler a entraîné des améliorations tout à fait significatives de la fonction pulmonaire (mesurée par le volume expiratoire maximal par seconde, VEMS) durant 24 heures.
Les études pivot ont révélé un délai d'action rapide de l'ordre de 5 minutes après inhalation de Seebri Breezhaler avec une augmentation du VEMS de 0,091 l à 0,094 l versus valeur initiale. Dans l'étude sur 52 semaines, Seebri Breezhaler a induit une valeur de VEMS significativement plus élevée au jour 1 et à la semaine 26 que le tiotropium. Le VEMS était d'autre part numériquement supérieur durant les 4 premières heures après la prise de Seebri Breezhaler aux semaines 12 et 52 versus tiotropium.
L'action bronchodilatatrice de Seebri Breezhaler s'est maintenue durant 24 heures. Aucun indice suggérant une tachyphylaxie pour l'effet bronchodilatateur n'a été relevé après des doses répétées sur une période allant jusqu'à 52 semaines.
Effets pharmacodynamiques secondaires
Aucune modification de la fréquence cardiaque moyenne ou de l'intervalle QTc n'a été observée après l'inhalation de Seebri Breezhaler à des doses allant jusqu'à 176 microgrammes chez des patients avec BPCO. Dans une grande étude sur le QT chez 73 sujets sains, une dose inhalée unique de 352 microgrammes de glycopyrronium (8 fois la dose thérapeutique) n'a pas entraîné d'allongement de l'intervalle QTc et n'a induit qu'une discrète diminution de la fréquence cardiaque (effet maximal: -5,9 pulsations/min; effet moyen sur 24 heures: -2,8 pulsations/min) par rapport au placebo. L'effet de 150 microgrammes de bromure de glycopyrronium (soit 120 microgrammes de glycopyrronium) sur la fréquence cardiaque et l'intervalle QTc après injection intraveineuse a été évalué chez des sujets jeunes en bonne santé. Les concentrations maximales (Cmax) atteintes correspondaient à environ 50 fois l'exposition après l'inhalation de 44 microgrammes de glycopyrronium à l'état d'équilibre et n'étaient associées ni à une tachycardie, ni à des allongements de l'intervalle QTc. Une légère réduction de la fréquence cardiaque (différence moyenne sur 24 h: -2 pulsations/min versus placebo) a été observée; il s'agit d'un effet connu des anticholinergiques à faibles concentrations chez les sujets jeunes en bonne santé.
Au cours d'une étude «approfondie du QT/QTc» incluant 73 personnes en bonne santé, la dose inhalée de 352 microgrammes de Seebri Breezhaler (soit 8 fois la dose thérapeutique) n'a entraîné, par rapport au placebo, aucun allongement de l'intervalle QTc, mais a légèrement diminué la fréquence cardiaque (effet maximal 5,9 bpm; effet moyen sur 24 heures 2,8 bpm).
Efficacité clinique
Le programme de développement clinique de phase III de Seebri Breezhaler a compris deux études clés (une étude contrôlée par placebo sur 6 mois et une étude contrôlée par placebo et par substance active sur 12 mois), ayant inclus au total 1888 patients porteurs d'un diagnostic clinique de BPCO. Les patients étaient âgés de 40 ans ou plus et présentaient une anamnèse de tabagisme d'au moins 10 paquets-années, un VEMS post-bronchodilatateur < 80% et ≥30% de la valeur normale prédite, ainsi qu'un rapport VEMS/CVF inférieur à 70%. Les patients porteurs de cardiopathies et/ou avec des contre-indications aux anticholinergiques étaient exclus.
Fonction pulmonaire
Dans le cadre de ces essais cliniques, Seebri Breezhaler a entraîné, à la dose de 50 microgrammes une fois par jour, des améliorations cliniquement significatives de la fonction pulmonaire (mesurée par le volume expiratoire maximal par seconde, VEMS) durant 24 heures. Concernant le principal critère d'évaluation à 12 semaines (VEMS résiduel après 24 heures), Seebri Breezhaler a induit une amélioration de la bronchodilatation de 0,108 l et de 0,097 l versus placebo dans les études respectivement sur 6 mois et sur 12 mois (p < 0,001). Dans la seconde étude, l'amélioration versus placebo dans le groupe non aveugle sous 18 microgrammes de tiotropium une fois par jour était de 0,083 l (p < 0,001).
Dans les études pivot, l'effet de Seebri Breezhaler est apparu rapidement, dans les 5 minutes après l'inhalation, avec une augmentation du VEMS par rapport à la valeur initiale de l'ordre de 0,091 l à 0,094 l.
Les améliorations du VEMS résiduel moyen observées pour le principal critère d'évaluation (12 semaines) se sont maintenues pendant toute la durée de l'étude, aussi bien dans l'étude sur 6 mois que dans l'étude sur 12 mois. Le VEMS résiduel moyen a augmenté de 0,1008 l versus placebo à la semaine 12 de l'étude sur 6 mois et de 0,113 l à la semaine 26 de l'étude sur 6 mois; dans l'étude sur 12 mois, celui-ci a augmenté de 0,097 l à la semaine 12 et de 0,108 l à la semaine 52.
Ces données suggèrent que l'effet bronchodilatateur de Seebri Breezhaler sur 24 heures s'est maintenu tout au long d'une période d'un an à partir de la première dose.
Des mesures spirométriques ont été réalisées en série durant l'étude sur 6 mois au jour 1 (fig. 1-1), à la semaine 12 (fig. 1-2) et à la semaine 26. Dans l'étude sur 12 mois, des mesures spirométriques ont été réalisées en série au jour 1 (fig. 1-3), à la semaine 12 (fig. 1-4) et à la semaine 52.
Les données des mesures spirométriques en série ont été utilisées pour calculer l'aire sous la courbe (AUC) standardisée du VEMS (en fonction du temps). Dans l'étude sur 6 mois, Seebri Breezhaler a induit une amélioration versus placebo de l'AUC du VEMS 0−24h de 0,133 l et de 0,199 l respectivement aux semaines 12 et 26 (p < 0,001). À la semaine 12 de l'étude sur 12 mois, Seebri Breezhaler a induit une amélioration versus placebo de l'AUC du VEMS 0−24h de 0,106 l (p < 0,001); pour le tiotropium, la différence du traitement par rapport au placebo était de 0,079 l (p = 0,014). À la semaine 52 de l'étude sur 12 mois, Seebri Breezhaler a entraîné une amélioration versus placebo de l'AUC du VEMS 0−24h de 0,106 l (p < 0,001); pour le tiotropium, la différence du traitement par rapport au placebo était de 0,040 l (p = 0,279).
L'importance de l'effet bronchodilatateur de Seebri Breezhaler dépend probablement du degré de réversibilité de la limitation du flux respiratoire au début de l'étude (celle-ci avait été évaluée par l'administration d'un antagoniste muscarinique bronchodilatateur à courte durée d'action): les analyses de sous-groupes secondaires correspondantes ont montré que les patients ayant le degré de réversibilité le plus faible au début de l'étude (< 5%) présentaient généralement aussi une réaction bronchodilatatrice plus faible que ceux dont le degré de réversibilité au même moment était plus élevé (≥5%). Chez les patients ayant le degré de réversibilité le plus faible au début de l'étude (< 5%), Seebri Breezhaler a entraîné, par rapport au placebo, une augmentation du VEMS résiduel de 0,072 l après 12 semaines (critère d'évaluation principal), et chez ceux ayant un degré de réversibilité plus élevé au même moment (≥5%), une augmentation du VEMS résiduel de 0,113 l (p < 0,05 dans chaque cas). Des résultats comparables ont été observés chez les patients sous tiotropium. Après 12 semaines de traitement par tiotropium, les patients ayant le degré de réversibilité le plus faible au début de l'étude (< 5%) ont présenté, par rapport au placebo, une augmentation du VEMS résiduel de 0,059 l, alors que ceux dont le degré de réversibilité était plus élevé au même moment (≥5%) ont présenté, par rapport au placebo, une augmentation du VEMS résiduel de 0,097 l.
Figure 1-1 Étude pivot sur six mois: données des spirométries en série (moyenne des moindres carrés du VEMS (l)) après la première dose
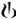
Figure 1-2 Étude pivot sur six mois: données des spirométries en série (moyenne des moindres carrés du VEMS (l)) à la semaine 12
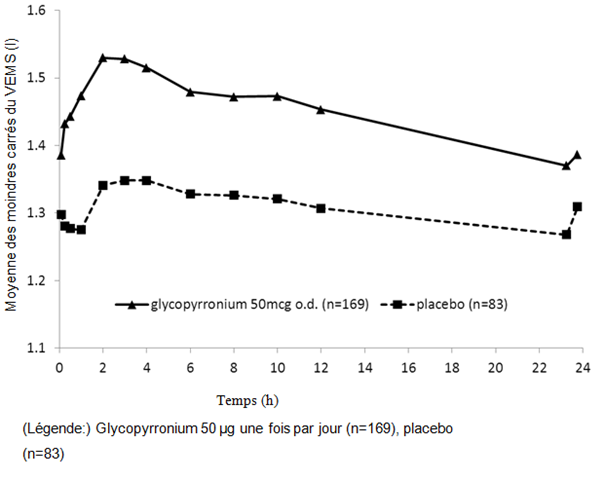
Figure 1-3 Étude pivot sur douze mois: données des spirométries en série (moyenne des moindres carrés du VEMS (l)) après la première dose
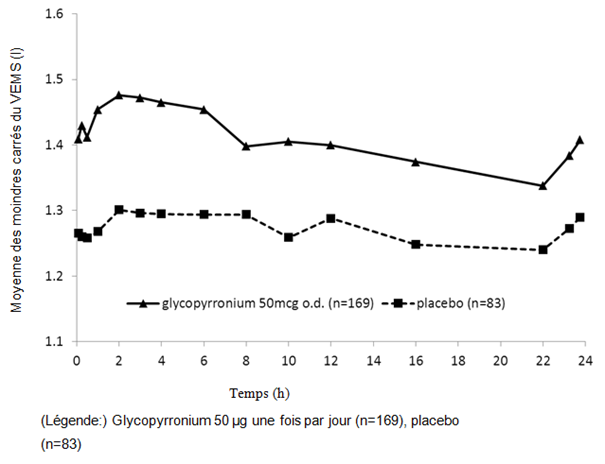
Figure 1-4 Étude pivot sur douze mois: données des spirométries en série (moyenne des moindres carrés du VEMS (l)) à la semaine 12
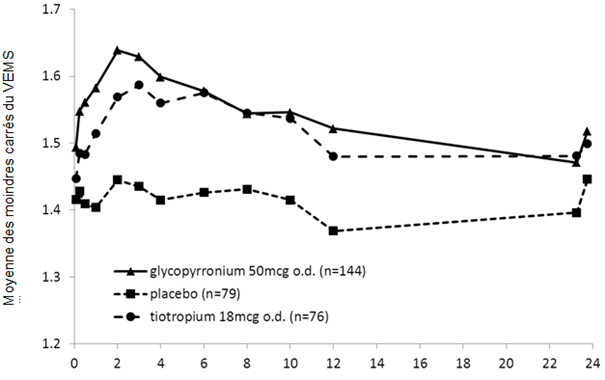
En plus de l'amélioration démontrée du VEMS, Seebri Breezhaler a aussi amélioré de manière consistante dans les deux essais pivot la capacité vitale forcée (CVF) et la capacité inspiratoire (CI). À la semaine 12, Seebri Breezhaler a induit dans les études sur 6 mois et sur 12 mois une augmentation respective de la CVF résiduelle moyenne versus placebo de 0,194 l et 0,183 l (p < 0,001). Seebri Breezhaler a amélioré la CI résiduelle à la semaine 12 des études sur 6 et 12 mois de 0,097 l et 0,129 l versus placebo (p < 0,001).
Efficacité sur les symptômes
La dose journalière unique de 50 µg de Seebri Breezhaler a diminué de manière significative la dyspnée estimée par le Transitional Dyspnea Index (TDI). Dans une analyse des données cumulées des deux études pivot sur 6 et 12 mois, le pourcentage de patients ayant répondu par une amélioration cliniquement significative du score TDI focal de ≥1 point à la semaine 26;était de 58,4% sous Seebri Breezhaler contre 46,4% chez les patients sous placebo et 53,4% chez les patients sous tiotropium. Les différences de taux de réponse étaient statistiquement significatives pour la comparaison Seebri Breezhaler versus placebo (< 0,001) et pour la comparaison tiotropium versus placebo (p = 0,009).
L'administration de Seebri Breezhaler 50 µg une fois par jour a également un effet significatif sur l'état de santé mesuré à l'aide du St. George's Respiratory Questionnaire (SGRQ). Une analyse des données cumulées des études pivot sur 6 et 12 mois a montré que le pourcentage des patients ayant répondu à Seebri Breezhaler par une amélioration cliniquement significative du score SGRQ global (≤ -4) à la semaine 26 était de 57,8% contre 47,6% chez les patients sous placebo et 61,0% chez ceux sous tiotropium. Les différences de taux de réponse étaient statistiquement significatives pour la comparaison Seebri Breezhaler versus placebo (< 0,001) et tiotropium versus placebo (p = 0,004).
Dans une analyse des données cumulées des études sur 6 et 12 mois, l'administration de Seebri Breezhaler 50 µg une fois par jour a significativement allongé le délai jusqu'à la première exacerbation modérée à sévère de la BPCO et diminué le nombre d'épisodes d'exacerbation modérée à sévère (les exacerbations étaient considérées comme modérées lorsqu'elles nécessitaient une corticothérapie systémique et/ou une antibiothérapie et comme sévères lorsqu'elles nécessitaient une hospitalisation). Le pourcentage de patients avec exacerbation de BPCO modérée à sévère de l'analyse cumulée d'une durée de 26 semaines était de 19,8% sous Seebri Breezhaler vs 27,2% sous placebo et le risque relatif estimé pour le délai jusqu'à des exacerbations modérées ou sévères était de 0,64 [IC à 95%: 0,520, 0,799; p < 0,001], ce qui correspond à une réduction du risque vs placebo de 36%. De la même façon, le risque relatif estimé pour le délai jusqu'à la première exacerbation sévère nécessitant une hospitalisation était de 0,39 [IC à 95%: 0,205, 0,728; p = 0,003]. Dans l'analyse cumulée d'une durée de 26 semaines, le taux d'exacerbations était significativement plus faible chez les patients sous Seebri Breezhaler que chez ceux sous placebo, le rapport des taux se situant vers 0,66 ([IC à 95%: 0,525, 0,841; p < 0,001]).
Par rapport au placebo, Seebri Breezhaler 50 µg une fois par jour a significativement réduit de 0,46 bouffées par jour (p = 0,005) le recours à la médication de secours pendant 26 semaines et de 0,37 bouffées par jour (p = 0,039) pendant 52 semaines durant les études sur 6 et 12 mois.
L'effet de Seebri Breezhaler en termes de réduction de l'hyperinflation dynamique et les améliorations de la tolérance de l'effort qui lui sont associées ont fait l'objet d'une étude randomisée, en double aveugle, contrôlée par placebo, chez 108 patients avec BPCO modérée à sévère. Seebri Breezhaler a atteint son effet maximal d'amélioration de la capacité inspiratoire à l'effort (0,23 l) et a entraîné une augmentation statistiquement significative de 43 secondes (une augmentation de 10%) de la durée d'effort après la première dose. Après 3 semaines de traitement, Seebri Breezhaler a amélioré la durée de l'effort de 89 secondes (une augmentation de 21%) et la capacité inspiratoire à l'effort a augmenté de 0,20 l.
Des mesures menées à l'aide de l'échelle de Borg ont montré que Seebri Breezhaler réduit la dyspnée et les douleurs des jambes. La diminution de la dyspnée de repos a également été mise en évidence à l'aide du Transitional Dyspnea Index (TDI).
PharmacocinétiqueAbsorption
Après l'inhalation orale à l'aide de l'inhalateur Seebri Breezhaler, le glycopyrronium a été rapidement absorbé et la concentration plasmatique maximale a été atteinte 5 minutes après l'administration.
La biodisponibilité absolue du glycopyrronium inhalé avec l'inhalateur Seebri Breezhaler a été estimée à environ 40%. Environ 90% de l'exposition systémique après l'inhalation repose sur une absorption pulmonaire et 10% sur une absorption gastro-intestinale. La biodisponibilité absolue du glycopyrronium pris par voie orale est estimée à environ 5%.
Après des doses répétées à raison d'une inhalation par jour chez des patients avec BPCO, l'état d'équilibre pharmacocinétique (steady state) du glycopyrronium a été atteint en l'espace d'une semaine. Les valeurs moyennes des concentrations plasmatiques maximales et minimales de glycopyrronium à des doses de 50 µg une fois par jour se situaient respectivement autour de 166 pg/ml et 8 pg/ml. Sous des doses de 100 et 200 µg une fois par jour, l'exposition au glycopyrronium à l'état d'équilibre (AUC mesurée entre deux doses) était environ 1,4 à 1,7 fois plus élevée qu'après la première dose.
Distribution
Après injection i.v., le volume de distribution à l'état d'équilibre (Vss) du glycopyrronium était de 83 l et le volume de distribution dans la phase terminale (Vz) était de 376 l. Le volume de distribution apparent dans la phase terminale après inhalation (Vz/F) était de 7310 l, ce qui traduit l'élimination beaucoup plus lente de la substance après l'inhalation. La liaison in vitro du glycopyrronium aux protéines plasmatiques humaines était de 38% à 41% pour des concentrations de 1 à 10 ng/ml. Ces concentrations étaient au moins 6 fois plus élevées que la moyenne des pics de concentration atteints à l'état d'équilibre dans le plasma avec le schéma posologique de 50 µg une fois par jour.
Métabolisme
Les études du métabolisme in vitro ont mis en évidence des voies métaboliques comparables chez l'animal et chez l'homme pour le bromure de glycopyrronium. On n'a pas trouvé de métabolites spécifiquement humains. Une hydroxylation qui a conduit à la formation de différents métabolites mono- et bihydroxylés, a été observée, de même qu'une hydrolyse directe ayant entraîné la formation d'un dérivé de l'acide carbonique (M9).
Les études in vitro ont montré que plusieurs isoenzymes du CYP contribuent à la biotransformation oxydative du glycopyrronium. L'hydrolyse en M9 est probablement catalysée par des enzymes de la famille des cholinestérases.
Après inhalation, l'exposition systémique au M9 était en moyenne du même ordre de grandeur que l'exposition à la substance mère. Comme les essais in vitro n'ont pas trouvé de métabolisme pulmonaire et que la présence du M9 dans la circulation à la suite d'une dose i.v. était peu importante (environ 4% de la Cmax et de l'AUC de la substance mère), on admet que le M9 est formé par hydrolyse présystémique de la fraction avalée de la dose de bromure de glycopyrronium inhalée et/ou à la suite d'un métabolisme de premier passage. Après inhalation, comme après injection i.v., on n'a retrouvé que des quantités infimes de M9 dans les urines (soit ≤0,5% de la dose administrée). Des glucuronides et/ou des conjugués sulfatés de glycopyrronium ont été trouvés dans l'urine chez l'homme après inhalations répétées et correspondaient à environ 3% de la dose administrée.
Les essais d'inhibition in vitro suggèrent que le bromure de glycopyrronium n'a pas de propriété significative d'inhibition des CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 et CYP3A4/5, des transporteurs d'efflux MDR1, MRP2 et MXR, ni des transporteurs d'influx OATP1B1, OATP1B3, OAT1, OAT3, OCT1 ou OCT2.
Les essais d'induction enzymatique in vitro n'ont pas montré d'induction cliniquement significative des isoenzymes du cytochrome P450 testées, de l'UGT1A1, ni des transporteurs MDR1 et MRP2 par le bromure de glycopyrronium.
Élimination
Après l'injection i.v. de bromure de glycopyrronium marqué au [3H] chez l'homme, l'élimination urinaire moyenne de la radioactivité en 48 h correspondait à 85% de la dose. 5% de la dose ont en outre été retrouvés dans la bile. Le bilan des masses était donc presque équilibré.
L'élimination rénale de la substance mère est d'environ 60 à 70% de la clairance totale du glycopyrronium disponible au niveau systémique, tandis que les processus d'excrétion extrarénale représentent environ 30 à 40%. L'excrétion par voie biliaire contribue à l'élimination extrarénale, mais l'essentiel de la clairance extrarénale est le fait du métabolisme.
Après l'inhalation de doses monoquotidiennes uniques et répétées comprises entre 50 et 200 µg de glycopyrronium par des sujets sains et des patients avec BPCO, la clairance rénale moyenne du glycopyrronium se situait entre 17,4 et 24,4 l/h. Une sécrétion tubulaire active contribue à l'élimination rénale du glycopyrronium. Près de 20% de la dose ont été retrouvés dans l'urine sous forme de substance mère.
Les concentrations plasmatiques du glycopyrronium ont diminué de manière multiphasique. La demi-vie d'élimination terminale moyenne était beaucoup plus longue après inhalation (33 à 57 heures) qu'après injection intraveineuse (6,2 heures) et prise orale (2,8 heures). Le profil d'élimination suggère une absorption pulmonaire retardée et/ou un transfert de glycopyrronium dans la circulation systémique 24 h après l'inhalation et au-delà.
Linéarité
Chez les patients avec BPCO, l'exposition systémique et l'élimination urinaire totale du glycopyrronium à l'équilibre pharmacocinétique étaient augmentées à peu près proportionnellement aux doses sur une plage posologique allant de 50 µg à 200 µg.
Cinétique pour certains groupes de patients
Une analyse pharmacocinétique de population des données de patients avec BPCO a identifié le poids corporel et l'âge comme étant des facteurs contribuant à la variabilité interindividuelle de l'exposition systémique. Les données suggèrent que Seebri Breezhaler 50 µg une fois par jour peut aussi être donné sans risque supplémentaire aux patients BPCO âgés et dans tous les groupes de poids.
Le sexe, un tabagisme et la valeur initiale du VEMS n'ont eu aucune influence manifeste sur l'exposition systémique.
Troubles de la fonction rénale
L'exposition systémique au bromure de glycopyrronium est influencée par les troubles de la fonction rénale. Une augmentation légère à modérée de l'exposition systémique totale (AUClast) allant jusqu'à 1,4 fois la valeur de base a été observée chez les patients avec insuffisance rénale légère à modérée. Chez les patients en insuffisance rénale sévère ou terminale, l'augmentation observée était d'un facteur allant jusqu'à 2,2. Une analyse pharmacocinétique de population a montré que Seebri Breezhaler peut être administré à la dose recommandée chez les patients atteints d'une BPCO et d'une insuffisance rénale légère à modérée (débit de filtration glomérulaire estimé DFGe ≥30 ml/min/1,73 m2).
Troubles de la fonction hépatique
Aucun essai clinique n'a été réalisé chez des patients présentant des troubles de la fonction hépatique. Le glycopyrronium est essentiellement éliminé de la circulation par voie rénale (voir «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l'efficacité de Seebri Breezhaler n'ont pas fait l'objet d'études chez les enfants et les adolescents de moins de 18 ans. Seebri Breezhaler ne doit pas être utilisé chez les patients pédiatriques.
Patients âgés
Le mécanisme d'élimination et les résultats des analyses pharmacocinétiques de population suggèrent qu'aucune adaptation posologique n'est nécessaire chez les patients âgés.
Origine ethnique
On n'a pas observé de différences significatives, du point de vue de l'exposition systémique totale (AUC), entre des sujets japonais et caucasiens après l'inhalation de bromure de glycopyrronium. On ne dispose pas de données pharmacocinétiques suffisantes pour d'autres groupes ethniques ou races.
Données précliniquesLes données noncliniques, basées sur des études conventionnelles de pharmacologie de sécurité, de toxicité en cas d'administration répétée, de génotoxicité, de potentiel carcinogène, de toxicité de reproduction et de toxicité du développement n'indiquent pas de risques particuliers chez l'homme.
Les effets observés lors des études de toxicité après inhalations de doses répétées ont pu être attribués à des exacerbations des effets pharmacologiques attendus du bromure de glycopyrronium ou à une légère réaction d'irritation locale. Ces dernières comprenaient des augmentations légères à modérées de la fréquence cardiaque chez le chien et plusieurs modifications réversibles associées à une réduction de la sécrétion des glandes salivaires et lacrymales, ainsi que des glandes de Harder et du pharynx chez le rat et le chien. Des opacifications du cristallin observées dans les essais chroniques chez le rat ont également été décrites sous d'autres antagonistes muscariniques et sont considérées comme des altérations spécifiques de l'espèce et de portée limitée pour les patients dans le contexte thérapeutique. Les observations faites au niveau des voies respiratoires du rat comprennent des modifications dégénératives/régénératives et des inflammations des fosses nasales et du larynx, correspondant à de légères irritations locales. De minimes lésions épithéliales des poumons au niveau bronchioloalvéolaire ont aussi été relevées chez le rat et sont considérées comme un léger phénomène d'hormèse. Toutes ces observations ont été faites lors d'expositions dépassant suffisamment les expositions maximales chez l'homme et n'ont donc que peu de signification pour l'utilisation clinique.
Les études de génotoxicité n'ont pas révélé de potentiel mutagène ni clastogène pour le bromure de glycopyrronium. Les études de carcinogénicité chez des souris transgéniques avec administration orale et chez des rats avec administration par inhalation n'ont pas montré d'indices de carcinogénicité à une exposition systémique (AUC) environ 53 fois supérieure chez des souris et environ 75 fois supérieure chez des rats à la dose maximale recommandée de 50 µg une fois par jour chez l'homme.
Les données publiées sur le bromure de glycopyrronium ne révèlent pas de problèmes de toxicité de reproduction. L'administration par voie inhalée de Seebri Breezhaler n'a pas entraîné d'effet tératogène chez le rat ni chez le lapin. Les études de reproduction chez le rat et d'autres données issues de l'expérimentation animale n'indiquent pas d'effets délétères sur la fertilité masculine ou féminine, ni sur le développement pré- et postnatal.
Le bromure de glycopyrronium et ses métabolites n'ont pas passé la barrière placentaire des souris, des lapines et des chiennes portantes de manière significative. Le bromure de glycopyrronium (y compris ses métabolites) a été excrété dans le lait de rates allaitantes et a atteint dans le lait des concentrations jusqu'à 10 fois supérieures à celles mesurées dans le plasma maternel.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver hors de portée des enfants.
Conserver dans l'emballage d'origine, à l'abri de l'humidité et ne pas conserver au-dessus de 25 °C.
Remarques concernant la manipulation
Pour les informations concernant l'administration et le mode d'emploi du produit, voir «Posologie/Mode d'emploi». Des instructions détaillées pour l'utilisation figurent dans l'information destinée aux patients. On utilise l'inhalateur du nouvel emballage avec chaque nouvelle prescription d'Seebri Breezhaler.
Numéro d’autorisation62580 (Swissmedic)
PrésentationGélules contenant 50 microgrammes de poudre pour inhalation:
Emballage de 30 gélules, 1 inhalateur [B]
Emballage multiple de 3 emballages contenant chacun 30 gélules et un inhalateur [B]
Titulaire de l’autorisationNovartis Pharma Schweiz AG, Risch, Domicile: 6343 Rotkreuz
Mise à jour de l’informationDécembre 2020
|