CompositionPrincipes actifs
Canagliflozine (sous forme d'hémihydrate de canagliflozine).
Excipients
Noyau du comprimé:
Lactose (39,3 mg par comprimé pelliculé de 100 mg et 117,8 mg par comprimé pelliculé de 300 mg), cellulose microcristalline, hydroxypropylcellulose, croscarmellose sodique (peut être produite à partir de capsules de graines de coton génétiquement modifiées; 0,84 mg de sodium par comprimé pelliculé de 100 mg et 2,52 mg de sodium par comprimé pelliculé de 300 mg), stéarate de magnésium.
Pelliculage:
Invokana 100 mg comprimés pelliculés: alcool polyvinylique, dioxyde de titane (E171), macrogol (3350), talc, oxyde de fer jaune (E172).
Invokana 300 mg comprimés pelliculés: alcool polyvinylique, dioxyde de titane (E171), macrogol (3350), talc.
Indications/Possibilités d’emploiInvokana est indiqué chez les adultes (à partir de 18 ans) atteints de diabète sucré de type 2 insuffisamment contrôlé, en complément d'un régime alimentaire et de l'exercice physique:
·en monothérapie;
·en association thérapeutique adjuvante avec d'autres médicaments hypoglycémiants (voir «Efficacité clinique» pour les résultats relatifs aux associations examinées dans le cadre d'études cliniques).
Invokana est indiqué pour la prévention d'événements cardiovasculaires chez les patients adultes atteints de diabète sucré de type 2 et d'une affection cardiovasculaire déjà manifeste (voir «Efficacité clinique»: résultats cardiovasculaires dans le programme CANVAS).
Invokana est indiqué pour réduire le risque de progression d'une néphropathie diabétique chez des patients adultes atteints de diabète sucré de type 2 et d'une albuminurie [ACR >300 mg/g] (voir «Posologie/Mode d'emploi» et «Efficacité clinique»).
Posologie/Mode d’emploiLa dose recommandée d'Invokana est de 100 mg une fois par jour. Si un contrôle glycémique plus étroit s'avère nécessaire, la dose peut être augmentée à 300 mg, sauf chez des patients présentant une limitation de la fonction rénale (DFGe <60 ml/min/1,73 m2) ou un risque d'effets indésirables liés à une diminution du volume intravasculaire (voir «Mises en garde et précautions»). Chez les patients présentant des signes de déplétion volémique, il est formellement recommandé de corriger cet état avant d'instaurer le traitement par Invokana (voir «Mises en garde et précautions»).
Tableau 1: Dose recommandée en fonction de la fonction rénale
L'effet hypoglycémiant d'Invokana diminue lorsque l'insuffisance rénale augmente. L'association avec d'autres principes actifs antihyperglycémiants doit donc être envisagée à temps (en particulier chez des patients présentant un DFGe <45).
|
DFGe (ml/min/1,73 m2)
ou ClCr (ml/min)
|
Dose quotidienne totale de canagliflozine
| |
≥60
|
Débuter le traitement par 100 mg une fois par jour avant le premier repas. En cas de diminution insuffisance de la glycémie et si la tolérance est bonne, la dose peut être augmentée à 300 mg.
| |
45 - <60
|
Limité à 100 mg une fois par jour avant le premier repas.
| |
30 - <45 *
| |
<30
|
Le traitement des patients présentant une albuminurie (ACR >300 mg/g) peut être poursuivi avec 100 mg une fois par jour avant le premier repas (il n'existe aucune donnée concernant l'instauration d'un traitement dans cette population).
Invokana doit être arrêté avant le début d'une dialyse ou avant une transplantation rénale.
|
*début du traitement uniquement chez les patients présentant une albuminurie (ACR >300 mg/g).
Lorsqu'Invokana est ajouté à l'insuline ou à un sécrétagogue de l'insuline (p.ex. sulfonylurée), une réduction de la dose d'insuline ou du sécrétagogue de l'insuline peut être envisagée pour diminuer le risque d'hypoglycémies (voir «Interactions» et «Effets indésirables»).
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée.
Invokana n'a pas été évalué chez les patients présentant une insuffisance hépatique sévère. L'utilisation chez ces patients n'est pas recommandée (voir «Pharmacocinétique»).
Patients âgés
La fonction rénale et le risque de déplétion volémique doivent être pris en considération (voir «Mises en garde et précautions»).
Enfants et adolescents
La sécurité et l'efficacité d'Invokana chez les enfants et les adolescents de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Prise retardée
En cas d'omission ou d'oubli d'une dose, le patient doit la prendre dès qu'il s'en aperçoit, le même jour. Il ne doit pas prendre deux doses le même jour, ni un autre jour.
Mode d'administration
Invokana doit être pris avant le premier repas de la journée. Les comprimés pelliculés doivent être avalés entiers.
Interruption provisoire en cas d'opérations
Le traitement par Invokana doit être interrompu lorsque c'est possible, au moins 3 jours avant toute opération ou intervention majeure associée à un jeûne prolongé. La prise d'Invokana peut reprendre lorsque le patient est stable cliniquement et s'alimente par voie orale (voir «Mises en garde et précautions»).
Contre-indicationsHypersensibilité à la canagliflozine ou à l'un des excipients.
Mises en garde et précautionsGénéralités
La sécurité et l'efficacité d'Invokana n'ont pas été évaluées chez les patients présentant un diabète de type 1. Invokana ne doit pas être utilisé chez ces patients.
Acidocétose diabétique
Invokana ne doit pas être utilisé pour le traitement d'une acidocétose diabétique (ACD).
Dans le cadre d'études cliniques réalisées chez des patients présentant un diabète sucré de type 2, des cas d'ACD associés à une hospitalisation, ont été rapportés pour 0,2% (17/11 078) des sujets traités par Invokana. Depuis la commercialisation, des cas graves d'ACD, dont certains ont mis la vie du patient en danger, ou des cas dont l'issue a été fatale ont été rapportés chez des patients présentant un diabète de type 2 traités par des inhibiteurs du SGLT2, y compris Invokana. La multiplication des cas où la glycémie n'était que modérément élevée (ACD euglycémique avec des valeurs de glycémie inférieures à 13,9 mmol/l [250 mg/dl]), ce qui est tout à fait atypique, a été frappante (voir «Effets indésirables»).
C'est la raison pour laquelle une ACD doit être envisagée lors du traitement par Invokana de patients atteints de diabète de type 2 qui présentent des symptômes tels que nausées, vomissements, manque d'appétit, douleurs abdominales, soif excessive, troubles respiratoires, confusion, haleine fruitée, épuisement ou fatigue inhabituels, même en l'absence d'hyperglycémie prononcée. Il convient de rechercher sans délai la présence de corps cétoniques chez les patients traités par Invokana qui présentent de tels symptômes, quelle que soit leur glycémie. L'acidocétose diabétique peut persister plus longtemps chez certains patients après l'arrêt d'Invokana, c'est-à-dire qu'elle peut persister au-delà de la durée attendue compte tenu de la demi-vie plasmatique de la canagliflozine. Une glycosurie prolongée a été observée en association avec une ACD persistante. L'excrétion urinaire du glucose se poursuit jusqu'à 3 jours après l'arrêt d'Invokana; cependant, des cas de persistance de l'ACD et de la glucosurie au-delà de 6 jours et parfois jusqu'à 2 semaines après l'arrêt d'inhibiteurs du SGLT2 ont été rapportés après commercialisation. Il convient également de noter que des facteurs supplémentaires (p.ex. carence en insuline) peuvent contribuer à une prolongation d'une ACD indépendamment de la canagliflozine.
En présence d'une ACD, le traitement par Invokana des patients présentant un diabète de type 2 doit immédiatement être arrêté. Le traitement par la canagliflozine doit être temporairement interrompu chez les patients présentant un diabète de type 2 qui sont hospitalisés à cause d'une intervention chirurgicale majeure ou d'une maladie aiguë grave, ou pour toutes les interventions associées à un jeûne prolongé. Il est recommandé de surveiller ces patients pour détecter la survenue éventuelle d'une ACD. Chez certains patients, l'ACD peut persister pendant une période encore plus longue.
Le traitement par Invokana peut être poursuivi lorsque l'état du patient s'est à nouveau stabilisé (voir «Posologie/Mode d'emploi»).
Le risque de survenue d'une ACD au cours d'un traitement par un inhibiteur du SGLT2 (p.ex. Invokana) pourrait être accru chez les patients suivant simultanément un régime très pauvre en glucides (lequel favorise encore plus l'activité métabolique conduisant à la formation de corps cétoniques), chez les patients très déshydratés, chez les patients ayant des antécédents d'ACD et chez les patients présentant une diminution significative de la fonction des cellules bêta. Invokana doit être utilisé avec prudence chez ces patients. La prudence est également de mise lors d'une réduction de la dose d'insuline chez les patients présentant un diabète de type 2 qui reçoivent Invokana en plus de l'insuline.
Amputation des membres inférieurs
Dans des études cliniques à long terme portant sur Invokana, menées chez des patients atteints de diabète de type 2 et d'une maladie cardiovasculaire (MCV) existante ou présentant au moins deux facteurs de risque de MCV, Invokana était associé à un risque accru d'amputation des membres inférieurs (6,3 contre 3,4 événements pour 1000 années-patients) par rapport au placebo, cette augmentation concernait surtout les orteils et le médio-pied (voir «Effets indésirables»). Au cours d'une étude clinique de longue durée menée chez des patients atteints de diabète de type 2 et d'une affection rénale chronique, aucune différence sur le plan du risque d'amputations des membres inférieurs n'a été observée par rapport au placebo chez des patients qui avaient été traités par 100 mg de canagliflozine. Le mécanisme sous-jacent n'étant pas établi, les facteurs de risque d'une amputation, outre les facteurs de risque généraux, sont inconnus.
Avant d'instaurer le traitement par Invokana, il faut tenir compte de facteurs présents dans l'anamnèse du patient qui augmentent, dans certains cas, le risque d'amputation. Par mesure de précaution, une surveillance attentive des patients présentant un risque accru d'amputation doit être envisagée, de même que le conseil aux patients concernant l'importance des soins préventifs réguliers des pieds et le maintien d'une hydratation suffisante. L'arrêt du traitement par Invokana doit également être envisagé chez les patients développant des effets pouvant entraîner une amputation tels qu'un ulcère cutané, une infection, une ostéomyélite ou une gangrène des membres inférieurs.
Utilisation chez les patients atteints d'insuffisance rénale
L'effet hypoglycémiant d'Invokana diminue avec la progression de l'altération rénale et devient inefficace en cas d'insuffisance modérée à sévère (DFGe <45 ml/min/1,73 m2). Les patients présentant une limitation modérée à sévère de la fonction rénale peuvent toutefois bénéficier de l'effet néphroprotecteur de la canagliflozine à une dose de 100 mg une fois par jour (voir «Posologie/Mode d'emploi»).
Chez les patients présentant un DFGe <60 ml/min/1,73 m2 ou une ClCr <60 ml/min, une incidence accrue d'effets indésirables liés à une diminution du volume intravasculaire a été rapportée (p.ex. vertiges positionnels, hypotension orthostatique ou hypotension en général). En outre, des augmentations plus fréquentes des taux de potassium et des augmentations plus importantes des taux sériques de créatinine et des taux d'azote uréique dans le sang (BUN) ont été rapportées chez ces patients (voir «Effets indésirables»).
C'est pourquoi la dose de canagliflozine doit être limitée à 100 mg par jour chez les patients présentant un DFGe <60 ml/min/1,73 m2 ou une ClCr <60 ml/min.
Néphropathie diabétique chez les patients atteints de diabète de type 2
Voir Tableau 1 pour les recommandations posologiques en fonction du débit de filtration glomérulaire estimé (DFGe).
Indépendamment du DFGe avant le traitement, les patients ayant reçu de la canagliflozine ont montré une diminution initiale du DFGe, qui s'est ensuite estompée avec le temps (voir «Effets indésirables» et «Propriétés pharmacocinétiques»).
Chez tous les patients prenant de la canagliflozine, la surveillance de la fonction rénale est recommandée comme suit:
·avant l'instauration du traitement par Invokana, puis au moins une fois par an (voir «Posologie/Mode d'emploi», «Effets indésirables», «Propriétés/Effets» et «Pharmacocinétique»),
·avant l'instauration d'un traitement médicamenteux concomitant susceptible d'altérer la fonction rénale, puis régulièrement ensuite,
·au moins 2 à 4 fois par an si la fonction rénale diminue au point de constituer une altération modérée à sévère.
Utilisation chez les patients présentant un risque d'effets indésirables liés à une déplétion volémique
En raison de son mécanisme d'action (augmentation de l'excrétion urinaire du glucose), Invokana induit une diurèse osmotique qui peut réduire le volume intravasculaire et diminuer la pression artérielle (voir «Propriétés/Effets»). Les patients les plus susceptibles de présenter des effets indésirables liés à une diminution du volume intravasculaire (p.ex. vertiges positionnels, hypotension orthostatique ou hypotension en général) ont été ceux traités par des diurétiques de l'anse, des inhibiteurs de l'ECA ou des antagonistes des récepteurs de l'angiotensine (ARA), les patients présentant une insuffisance rénale modérée ainsi que les patients âgés, p.ex. ≥75 ans (voir «Posologie/Mode d'emploi» et «Effets indésirables»).
Dans 8 études cliniques contrôlées, menées avec Invokana, des effets indésirables dose-dépendants (100 mg: 2,3%, 300 mg: 3,4%, traitements témoins: 1,5%) liés à une diminution du volume intravasculaire ont été observés et se sont produits le plus souvent au cours des trois premiers mois du traitement (voir «Effets indésirables»). En raison de la diminution du volume intravasculaire, des augmentations moyennes généralement faibles et dose-dépendantes de la créatinine sérique et parallèlement des réductions moyennes généralement faibles du DFGe ont été observées au cours des six premières semaines du traitement par Invokana. Des réductions plus importantes du DFGe (>30%) ont occasionnellement été observées chez les patients mentionnés ci-dessus, susceptibles de présenter des diminutions plus importantes du volume intravasculaire; celles-ci se sont le plus souvent à nouveau améliorées à la poursuite du traitement et n'ont que rarement nécessité l'interruption du traitement par Invokana (voir «Effets indésirables»).
Les patients doivent être informés de la nécessité de signaler tout symptôme d'une diminution du volume intravasculaire. Ces effets indésirables ont entraîné l'arrêt du traitement par Invokana dans de rares cas et ont fréquemment été traités par un ajustement du traitement antihypertenseur (y compris des diurétiques), tandis que le traitement par Invokana a été poursuivi. Chez les patients présentant une déplétion volémique, il est recommandé de corriger cet état avant d'instaurer le traitement par Invokana.
Chez les patients traités par Invokana, une surveillance étroite de l'état volémique, y compris de la fonction rénale et des électrolytes sériques, est recommandée en cas de maladies intercurrentes pouvant provoquer une déplétion volémique (p.ex. maladies gastro-intestinales). Chez les patients développant une déplétion volémique au cours du traitement par Invokana, une interruption temporaire du traitement peut être envisagée jusqu'à la correction de cet état. En cas d'interruption du traitement, une surveillance plus étroite de la glycémie doit être envisagée.
Chez les patients atteints d'une cardiopathie ischémique ou d'une maladie cérébrovasculaire connues, chez lesquels une diminution supplémentaire de la pression artérielle peut représenter un risque accru, un ajustement des mesures diététiques associées (p.ex. consommation de sel) et/ou du traitement antihypertenseur doit être envisagé avant d'instaurer le traitement par Invokana.
Patients âgés
Les patients âgés peuvent présenter un risque accru de déplétion volémique, sont plus fréquemment traités par des diurétiques et présentent souvent une insuffisance rénale. Chez les patients âgés de ≥75 ans, une incidence accrue d'effets indésirables liés à une diminution du volume intravasculaire (p. ex. vertiges positionnels, hypotension orthostatique ou hypotension en général) a été rapportée. Par ailleurs, des diminutions plus importantes du DFGe ont été rapportées chez ces patients (voir «Posologie/Mode d'emploi» et «Effets indésirables»).
Hyperkaliémie:
La probabilité de développement d'une hyperkaliémie au cours du traitement par Invokana est plus importante chez les patients présentant une insuffisance rénale modérée et prenant des médicaments influençant l'excrétion du potassium, par exemple des diurétiques épargneurs de potassium, ou des médicaments agissant sur le système rénine-angiotensine-aldostérone.
Le taux de potassium doit être contrôlé régulièrement après l'instauration du traitement par Invokana chez les patients présentant une insuffisance rénale et les patients prédisposés à une hyperkaliémie en raison de médicaments ou d'autres maladies.
Intolérance au lactose:
Les comprimés contiennent du lactose. Les patients atteints d'une intolérance héréditaire au galactose, d'un déficit en lactase de Lapp ou d'une malabsorption du glucose et du galactose ne doivent pas être traités par Invokana.
Hypoglycémie lors de l'association avec d'autres principes actifs antihyperglycémiants
L'insuline et les sécrétagogues de l'insuline (p.ex. sulfonylurées) peuvent provoquer des hypoglycémies. L'incidence des hypoglycémies est plus élevée lors de l'ajout d'Invokana à l'insuline ou à un sécrétagogue de l'insuline (p.ex. une sulfonylurée) que lors de l'association placebo plus insuline ou sécrétagogue de l'insuline.
C'est pourquoi une réduction de la dose d'insuline ou du sécrétagogue de l'insuline doit être envisagée pour diminuer le risque d'hypoglycémies (voir «Posologie/Mode d'emploi» et «Effets indésirables»).
Fasciite nécrosante périnéale (gangrène de Fournier)
Des données de la pharmacovigilance post-commercialisation ont révélé des cas de fasciite nécrosante du périnée (gangrène de Fournier), une infection nécrosante très rare, mais grave et potentiellement fatale, nécessitant une intervention chirurgicale en urgence, chez des patients atteints de diabète sucré qui avaient reçu des inhibiteurs du SGLT2, y compris Invokana. Cette affection a touché aussi bien les femmes que les hommes. Les issues sérieuses ont englobé des hospitalisations, plusieurs opérations et des cas de décès.
Les patients sous traitement par Invokana et présentant des douleurs ou une sensibilité à la pression, un érythème ou des œdèmes dans la région génitale ou périnéale, de la fièvre ou une sensation de malaise doivent être examinés à la recherche d'une fasciite nécrosante. En cas de suspicion correspondante, un traitement par des antibiotiques à large spectre doit être instauré immédiatement, accompagné le cas échéant d'un débridement chirurgical. La prise d'Invokana doit être interrompue et remplacée par un traitement alternatif adapté, avec une surveillance étroite de la glycémie.
Infections mycosiques génitales
En raison du mécanisme d'action de la glycosurie induite par le médicament, Invokana augmente le risque d'infections mycosiques génitales. Des cas de candidose vulvovaginale chez les femmes et de balanite ou balanoposthite chez les hommes ont été rapportés au cours d'études cliniques (voir «Effets indésirables»). La probabilité de développer une infection était plus élevée chez les hommes et les femmes ayant des antécédents de mycoses génitales. Les balanites ou les balanoposthites sont principalement survenues chez les hommes non circoncis. Un phimosis a été rapporté dans de rares cas et des circoncisions ont été occasionnellement pratiquées. La majorité des mycoses génitales ont été traitées par des médicaments antimycosiques topiques prescrits par un médecin ou utilisés en automédication, tout en poursuivant le traitement par Invokana.
Chez les hommes et les femmes présentant des infections mycosiques génitales récidivantes, l'intérêt de la poursuite du traitement par Invokana doit être discuté en évaluant individuellement le rapport bénéfice-risque.
Réactions d'hypersensibilité
Des réactions d'hypersensibilité considérées en partie comme sévères (p.ex. urticaire généralisée) ont été observées au cours du traitement par Invokana. Ces réactions sont généralement survenues dans les quelques heures ou jours suivant le début du traitement. Le traitement par Invokana doit être arrêté en cas d'apparition de telles réactions (voir «Contre-indications»).
Sodium
Invokana contient moins de 1 mmol (23 mg) de sodium par comprimé pelliculé, c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsInteractions pharmacocinétiques
Études in vitro
La canagliflozine est essentiellement métabolisée par glucuronoconjugaison médiée par l'UDP-glucuronosyltransférase 1A9 (UGT1A9) et 2B4 (UGT2B4).
Des études in vitro avec des microsomes hépatiques humains ont montré que la canagliflozine inhibe légèrement les isoenzymes 3A4, 2B6, 2C8 et 2C9 du CYP450. Toutefois, aucune interaction cliniquement significative n'a été observée dans une étude clinique. La canagliflozine n'a pas induit ou inhibé d'autres isoenzymes du CYP450. La canagliflozine ne devrait donc pas modifier la clairance métabolique de médicaments métabolisés par ces enzymes et administrés simultanément.
La canagliflozine est un substrat de la glycoprotéine-P (P-gp) et un inhibiteur faible de la P-gp.
La canagliflozine subit un métabolisme oxydatif minime (voir «Pharmacocinétique»). Il est donc improbable que d'autres médicaments aient des effets cliniquement significatifs sur la pharmacocinétique de la canagliflozine par l'intermédiaire du système du cytochrome P450.
Données in vivo
Effet d'autres médicaments sur la canagliflozine
Les effets d'autres médicaments sur la canagliflozine ont été évalués dans des études cliniques. La ciclosporine, l'hydrochlorothiazide, les contraceptifs oraux (éthinylestradiol et lévonorgestrel), la metformine et le probénécide n'ont pas eu d'effets cliniquement significatifs sur la pharmacocinétique de la canagliflozine.
Rifampicine: L'administration concomitante de rifampicine, un inducteur non sélectif de plusieurs enzymes UGT et transporteurs de médicaments, dont l'UGT1A9, l'UGT2B4, la P-gp et le MRP2, a diminué l'exposition à la canagliflozine. Cette diminution de l'exposition à la canagliflozine peut réduire l'efficacité. Lorsqu'un inducteur de ces UGT et systèmes de transport des médicaments (p.ex. rifampicine, phénytoïne, barbituriques, phénobarbital, ritonavir, carbamazépine, millepertuis (Hypericum perforatum)) doit être administré en même temps qu'Invokana, il convient de surveiller le taux d'HbA1c chez les patients traités par Invokana 100 mg une fois par jour et d'envisager une augmentation de la dose à 300 mg une fois par jour. D'autres traitements antihyperglycémiants doivent être envisagés chez les patients présentant un DFGe compris entre 45 ml/min/1,73 m2 et <60 ml/min/1,73 m2 ou une ClCr comprise entre 45 ml/min et <60 ml/min et recevant un traitement par un inducteur des enzymes UGT.
Tableau 2: Effets de médicaments concomitants sur la disponibilité systémique de la canagliflozine
|
Médicament concomitant
|
Dose du médicament concomitant1
|
Dose de canagliflozine1
|
Rapport des moyennes géométriques
(rapport avec/sans médicament concomitant)
Aucun effet=1,0
| |
AUC2
(IC à 90%)
|
Cmax
(IC à 90%)
| |
Ajustement de la dose éventuellement nécessaire (voir indications précédentes dans le texte):
| |
Rifampicine
|
600 mg une fois par jour pendant 8 jours
|
300 mg
|
0,49
(0,44; 0,54)
|
0,72
(0,61; 0,84)
| |
Aucun ajustement de la dose d'Invokana nécessaire pour:
| |
Ciclosporine
|
400 mg
|
300 mg une fois par jour pendant 8 jours
|
1,23
(1,19; 1,27)
|
1,01
(0,91; 1,11)
| |
Éthinylestradiol et lévonorgestrel
|
0,03 mg d'éthinylestradiol et 0,15 mg de lévonorgestrel
|
200 mg une fois par jour pendant 6 jours
|
0,91
(0,88; 0,94)
|
0,92
(0,84; 0,99)
| |
Hydrochloro-thiazide
|
25 mg une fois par jour pendant 35 jours
|
300 mg une fois par jour pendant 7 jours
|
1,12
(1,08; 1,17)
|
1,15
(1,06; 1,25)
| |
Metformine
|
2000 mg
|
300 mg une fois par jour pendant 8 jours
|
1,10
(1,05; 1,15)
|
1,05
(0,96; 1,16)
| |
Probénécide
|
500 mg deux fois par jour pendant 3 jours
|
300 mg une fois par jour pendant 17 jours
|
1,21
(1,16; 1,25)
|
1,13
(1,00; 1,28)
|
1 Dose unique, sauf indication contraire.
2 AUCinf pour les médicaments administrés en dose unique et AUC24h pour les médicaments administrés en plusieurs doses.
Effet de la canagliflozine sur d'autres médicaments
Dans les études d'interactions chez des sujets sains, la canagliflozine n'a pas eu d'effets cliniquement significatifs à l'état d'équilibre sur la pharmacocinétique de la metformine, des contraceptifs oraux (éthinylestradiol et lévonorgestrel), du glibenclamide, de la simvastatine, du paracétamol ou de l'hydrochlorothiazide.
Digoxine: L'association de 300 mg de canagliflozine une fois par jour pendant 7 jours à une dose unique de 0,5 mg de digoxine, suivie de 0,25 mg par jour pendant 6 jours, a entraîné une augmentation de 20% de l'AUC de la digoxine et de 36% de la Cmax de la digoxine, éventuellement en raison d'une interaction au niveau de la Pgp. Les patients prenant de la digoxine ou d'autres glucosides cardiotoniques (p.ex. digitoxine) doivent être surveillés de manière appropriée.
Lithium: l'utilisation concomitante d'un inhibiteur du SGLT2 avec du lithium peut diminuer la concentration sérique de lithium. Pendant l'initiation du traitement par la canagliflozine et lors de modifications de la posologie, la concentration sérique de lithium doit être surveillée plus fréquemment.
Tableau 3: Effet de la canagliflozine sur la disponibilité systémique de médicaments concomitants
|
Médicament concomitant
|
Dose du médicament concomitant1
|
Dose de canagliflozine1
|
Rapport des moyennes géométriques
(rapport avec/sans médicament concomitant)
Aucun effet = 1,0
| |
|
AUC2
(IC à 90%)
|
Cmax
(IC à 90%)
| |
Concernant la pertinence clinique, voir indications dans le texte:
| |
Digoxine
|
0,5 mg une fois par jour le premier jour, suivi de 0,25 mg une fois par jour pendant 6 jours
|
300 mg une fois par jour pendant 7 jours
|
Digoxine
|
1,20
(1,12; 1,28)
|
1,36
(1,21; 1,53)
| |
Aucun ajustement de la dose du médicament concomitant nécessaire pour:
| |
Éthinylestradiol et lévonorgestrel
|
0,03 mg d'éthinyl-estradiol et 0,15 mg de lévonorgestrel
|
200 mg une fois par jour pendant 6 jours
|
Éthinylestradiol
|
1,07
(0,99; 1,15)
|
1,22
(1,10; 1,35)
| |
Lévonorgestrel
|
1,06
(1,00; 1,13)
|
1,22
(1,11; 1,35)
| |
Glibenclamide
|
1,25 mg
|
200 mg une fois par jour pendant 6 jours
|
Glibenclamide
|
1,02
(0,98; 1,07)
|
0,93
(0,85; 1,01)
| |
3-cis-hydroxy-glibenclamide
|
1,01
(0,96; 1,07)
|
0,99
(0,91; 1,08)
| |
4-trans-hydroxy-glibenclamide
|
1,03
(0,97; 1,09)
|
0,96
(0,88; 1,04)
| |
Hydrochloro-thiazide
|
25 mg une fois par jour pendant 35 jours
|
300 mg une fois par jour pendant 7 jours
|
Hydrochlorothiazide
|
0,99
(0,95; 1,04)
|
0,94
(0,87; 1,01)
| |
Metformine
|
2000 mg
|
300 mg une fois par jour pendant 8 jours
|
Metformine
|
1,20
(1,08; 1,34)
|
1,06
(0,93; 1,20)
| |
Paracétamol
|
1000 mg
|
300 mg deux fois par jour pendant 25 jours
|
Paracétamol
|
1,063
(0,98; 1,14)
|
1,00
(0,92; 1,09)
| |
Simvastatine
|
40 mg
|
300 mg une fois par jour pendant 7 jours
|
Simvastatine
|
1,12
(0,94; 1,33)
|
1,09
(0,91; 1,31)
| |
Simvastatine acide
|
1,18
(1,03; 1,35)
|
1,26
(1,10; 1,45)
| |
Warfarine
|
30 mg
|
300 mg une fois par jour pendant 12 jours
|
(R)-warfarine
|
1,01
(0,96; 1,06)
|
1,03
(0,94; 1,13)
| |
(S)-warfarine
|
1,06
(1,00; 1,12)
|
1,01
(0,90; 1,13)
| |
INR
|
1,00
(0,98; 1,03)
|
1,05
(0,99; 1,12)
|
1 Dose unique, sauf indication contraire.
2 AUCinf pour les médicaments administrés en dose unique et AUC24h pour les médicaments administrés en plusieurs doses.
3 AUC0-12h.
Grossesse, allaitementGrossesse
Un diabète non contrôlé pendant la grossesse (gestationnel ou préexistant) est associé à un risque accru de malformations congénitales et de mortalité périnatale.
Il n'existe que des données limitées concernant l'emploi d'Invokana chez la femme enceinte. Des études expérimentales sur des animaux menées avec la canagliflozine administrée seule ainsi qu'avec la metformine ont montré des signes en faveur d'un effet toxique sur la reproduction (voir «Données précliniques»).
Invokana ne doit pas être administré pendant la grossesse. En cas de survenue d'une grossesse, Invokana doit être arrêté.
Allaitement
On ignore si la canagliflozine et/ou ses métabolites sont excrétés dans le lait maternel. Des études expérimentales menées chez l'animal ont montré que la canagliflozine/ses métabolites passent dans le lait maternel (voir «Données précliniques»). Les données limitées dont on dispose n'excluent pas le risque théorique d'hypoglycémies chez l'enfant allaité. La décision d'arrêter Invokana ou d'arrêter l'allaitement doit être prise en tenant compte de l'utilité du médicament pour la mère et du risque potentiel pour l'enfant.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Les patients doivent cependant être avertis du risque d'hypoglycémie, lorsqu'Invokana est ajouté à l'insuline ou à un sécrétagogue de l'insuline. En outre, il convient d'attirer leur attention sur le risque accru d'effets indésirables liés à une diminution du volume intravasculaire, tels que vertiges positionnels (voir «Posologie/Mode d'emploi», «Mises en garde et précautions» et «Effets indésirables»).
Effets indésirablesRésumé du profil de sécurité
La sécurité de la canagliflozine a été évaluée chez 22 645 patients atteints de diabète de type 2, dont 13 278 patients traités par canagliflozine et 9367, par un comparateur, ayant reçu un médicament lors de 15 études cliniques contrôlées de phases 3 et 4 menées en double aveugle. Au total, 10 134 patients ont été traités dans le cadre de deux études cardiovasculaires dédiées, exposés en moyenne pendant 149 semaines (223 semaines dans CANVAS et 94 semaines dans CANVAS-R), et 8114 patients ont été traités lors de 12 études cliniques contrôlées de phases 3 et 4 menées en double aveugle sur une durée moyenne de traitement de 49 semaines. Dans une étude, au cours de laquelle les résultats thérapeutiques ont été examinés particulièrement sur le plan rénal, la durée moyenne d'exposition de la totalité des 4397 patients atteints de diabète de type 2 et d'une néphropathie diabétique était de 115 semaines.
L'évaluation primaire de la sécurité et de la tolérance a été réalisée au moyen d'une analyse regroupée (N=2313) des données regroupées de quatre études cliniques d'une durée de 26 semaines et contrôlées contre placebo. Les effets indésirables les plus fréquemment rapportés au cours du traitement ont été une hypoglycémie en association avec l'insuline ou des sulfonylurées, une candidose vulvovaginale, des infections des voies urinaires, une polyurie ou une pollakiurie. Les effets indésirables ayant entraîné un arrêt du traitement chez ≥0,5% de tous les patients traités par Invokana dans ces études ont été une candidose vulvovaginale (0,7% de toutes les femmes) et une balanite ou une balanoposthite (0,5% de tous les hommes).
Liste des effets indésirables
Les effets indésirables mentionnés ci-dessous sont survenus dans les études cliniques regroupées décrites plus haut, et contrôlées de placebo contre traitement actif. Les effets indésirables mentionnés ci-dessous sont classés par fréquence et par classes de systèmes d'organes (SOC). Les catégories de fréquence ont été définies selon la convention suivante: très fréquents (≥1/10), fréquents (≥1/100 et <1/10), occasionnels (≥1/1000 et <1/100), rares (≥1/10'000 et <1/1000), très rares (<1/10'000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Fréquence des effets indésirables (MedDRA) dans les études contrôlées de placebo contre traitement actifa et après la commercialisation
Infections et infestations
Très fréquents: candidose vulvovaginale**,f.
Fréquents: balanite ou balanoposthite**,g, infection des voies urinairese (pyélonéphrite et sepsis urinaire ont été rapportés après la mise sur le marché).
Rares: gangrène de Fournier (fasciite nécrosante du périnée)i.
Affections hématologiques et du système lymphatique
Fréquents: élévation du taux d'hématocrite.
Affections du système immunitaire
Rares: réaction anaphylactiquei.
Troubles du métabolisme et de la nutrition
Très fréquents: hypoglycémie en association avec l'insuline ou des sulfonylurées.
Fréquents: dyslipidémie.
Occasionnels: déshydratation*, élévation des taux sanguins de potassium, élévation des taux sanguins de phosphate.
Rares: acidocétose diabétiqueh.
Affections du système nerveux
Occasionnels: vertiges positionnels*, syncope*.
Affections vasculaires
Occasionnels: hypotension artérielle*, hypotension orthostatique*.
Affections gastro-intestinales
Fréquents: constipation, soifb, nausées.
Affections de la peau et du tissu sous-cutané
Occasionnels: éruption cutanéec, photosensibilité, urticaire.
Rares: angio-œdème.i
Affections musculo-squelettiques et du tissu conjonctif
Occasionnels: fractures osseuses.
Affections du rein et des voies urinaires
Fréquents: polyurie ou pollakiuried.
Occasionnels: élévation des taux sanguins de créatinine, élévation des taux sanguins d'urée, insuffisance rénale (principalement dans le contexte de déplétion volémique).
Actes médicaux et chirurgicaux
Occasionnels: amputations des membres inférieurs (principalement des orteils et du métatarse), en particulier chez les patients présentant un risque cardiovasculaire élevé.
* En rapport avec une diminution du volume intravasculaire; voir «Mises en garde et précautions».
** Voir «Mises en garde et précautions».
a Les profils des données de sécurité issues des études pivots individuelles (dont des études chez des patients atteints d'insuffisance rénale modérée, des patients âgés et des patients à risque cardiovasculaire et rénal accru) ont de manière générale correspondu aux effets indésirables énumérés ci-dessus.
b «Soif» comprend les termes soif, sécheresse buccale et polydipsie.
c «Éruption» comprend les termes éruption érythémateuse, éruption généralisée, éruption maculeuse, éruption maculo-papuleuse, éruption papuleuse, éruption prurigineuse, éruption pustuleuse et éruption vésiculeuse.
d «Polyurie ou pollakiurie» comprend les termes polyurie, pollakiurie, urgence mictionnelle, nycturie et augmentation de la quantité d'urine.
e «Infection des voies urinaires» comprend les termes infection des voies urinaires, cystite, infection rénale et urosepticémie. Il n'y a pas eu de déséquilibre entre Invokana 100 mg, Invokana 300 mg et le placebo en ce qui concerne les infections rénales et les urosepticémies.
f «Candidose vulvovaginale» comprend les termes candidose vulvovaginale, infection mycosique vulvovaginale, vulvovaginite, infection vaginale, vulvite et infection mycosique génitale.
g «Balanite ou balanoposthite» comprend les termes balanite, balanoposthite, balanite à Candida et infection mycosique génitale.
h Études cliniques de phase 3 et de phase 4, y compris des études non-CANVAS/non-CREDENCE, le programme CANVAS et les rapports spontanés
i Études cliniques de phases 3 et 4, y compris le programme CANVAS et CREDENCE
Description de certains effets indésirables
Acidocétose diabétique
Dans une étude de longue durée au cours de laquelle les résultats thérapeutiques ont été examinés particulièrement sur le plan rénal chez des patients atteints de diabète de type 2 et d'une néphropathie diabétique, le taux d'incidence de cas avérés d'acidocétose diabétique (ACD) était de 2,1 (0,5%, 12/2200) pour 1000 années-patients d'exposition à 100 mg de canagliflozine et de 0,3 (0,1%, 2/2197) pour 1000 années-patients d'exposition au placebo; parmi les 14 patients présentant une ACD, 8 patients (7 dans le groupe canagliflozine 100 mg et 1 dans le groupe placebo) avaient un DFGe situé entre 30 et 45 ml/min/1,73 m2 avant le traitement. (voir «Mises en garde et précautions»).
Amputation des membres inférieurs
Comme cela a été observé dans le programme CANVAS intégré, composé des deux études à long terme randomisées et contrôlées contre placebo CANVAS et CANVAS-R, incluant au total 10 134 patients, l'administration d'Invokana à des patients atteints de diabète de type 2 et d'une maladie cardiovasculaire existante ou présentant au moins deux facteurs de risque de maladie cardiovasculaire était associée à un risque environ 2 fois plus élevé d'amputation d'un membre inférieur. Sous Invokana, cela concernait 2,4% des patients contre 1,1% des patients sous placebo; les taux d'incidence correspondants pour 100 années-patients étaient de 0,63 et de 0,34 (hazard ratio HR=1,97 (IC à 95%: 1,41–2,75). Le déséquilibre est survenu dès les 26 premières semaines de traitement. Les patients des études CANVAS et CANVAS-R ont été suivis pendant une période moyenne de respectivement 5,7 et de 2,1 ans. Indépendamment du traitement par Invokana ou le placebo, le risque d'amputation était le plus élevé en début d'étude chez les patients qui avaient déjà des antécédents d'amputation, de maladie vasculaire périphérique et de neuropathie. Le risque d'amputation d'un membre inférieur n'était pas dose-dépendant. Au cours de l'étude CREDENCE, une étude de longue durée au cours de laquelle les résultats thérapeutiques ont été examinés particulièrement sur le plan rénal chez 4397 patients atteints de diabète de type 2 et d'une néphropathie diabétique, aucune différence sur le plan du risque d'amputations des membres inférieurs n'a été observée en lien avec l'utilisation de 100 mg de canagliflozine par rapport au placebo (12 événements pour 1000 années-patients contre 11 [HR: 1,11; IC à 95% 0,79, 1,56]) (voir «Mises en garde et précautions»).
Dans d'autres études portant sur Invokana en cas de diabète de type 2, ayant inclus une population générale totale de 8114 patients diabétiques, aucune différence n'a été observée concernant le risque d'amputation d'un membre inférieur par rapport aux groupes témoins (voir «Mises en garde et précautions»).
Effets indésirables liés à une diminution du volume intravasculaire
Dans l'analyse des données regroupées des quatre études d'une durée de 26 semaines, contrôlées contre placebo, l'incidence de tous les effets indésirables liés à une diminution du volume intravasculaire (p.ex. vertiges positionnels, hypotension orthostatique, déshydratation et syncope) a été de 1,2% pour Invokana 100 mg, de 1,3% pour Invokana 300 mg et de 1,1% pour le placebo. Dans les deux études contrôlées contre traitement actif, les incidences observées avec Invokana ont été similaires à celles observées avec les substances comparatives.
Dans une des études cardiovasculaires ciblées de longue durée (CANVAS), dans lesquelles les patients étaient généralement plus âgés, avec une prévalence accrue de comorbidités, le taux d'incidence des effets indésirables liés à une diminution du volume intravasculaire a été de 23,4 sous Invokana 100 mg, de 28,7 sous Invokana 300 mg et de 18,5 sous placebo, exprimés en événements pour 1000 patients-années d'exposition.
Pour évaluer les facteurs de risque relatifs à ces effets indésirables, une analyse plus vaste (n=12 441) de données regroupées de patients issus de 13 études contrôlées de phase 3 et de phase 4 a été réalisée en incluant les deux doses d'Invokana. Dans cette analyse regroupée, l'incidence de ces effets indésirables a été généralement plus élevée chez les patients traités par des diurétiques de l'anse, les patients présentant un DFGe initial compris entre 30 et <60 ml/min/1,73 m2 et les patients âgés de ≥75 ans. Chez les patients traités par des diurétiques de l'anse, les taux d'incidence ont été de 49,8% sous Invokana 100 mg et de 56,7% sous Invokana 300 mg, contre 41,5% événements pour 1000 années-patients d'exposition dans le groupe témoin. Chez les patients présentant un DFGe initial <60 ml/min/1,73 m2, les taux d'incidence ont été de 52,4 sous canagliflozine 100 mg et de 53,5 sous canagliflozine 300 mg, contre 31,1 événements pour 1000 années-patients d'exposition dans le groupe témoin. Chez les patients ≥75 ans, les taux d'incidence ont été de 52,7 sous canagliflozine 100 mg et de 60,8 sous canagliflozine 300 mg, contre 24,1 événements pour 1000 années-patients d'exposition dans le groupe témoin (voir «Posologie/Mode d'emploi» et «Mises en garde et précautions»).
Dans l'étude cardiovasculaire concernée et dans l'analyse de données groupées plus importante, ainsi qu'au cours d'une étude dans laquelle les résultats ont été analysés particulièrement sur le plan rénal, aucune augmentation du nombre d'interruptions liées aux effets indésirables et aux effets indésirables sévères associés à une déplétion volumique n'a été observée sous canagliflozine.
Hypoglycémie
Lors de l'utilisation d'Invokana en monothérapie ou en ajout à la metformine, l'incidence des hypoglycémies a été plutôt faible dans les groupes traités, d'environ 4% versus env. 2% sous placebo. Lors de l'administration d'Invokana en ajout à l'insuline, des hypoglycémies ont été rapportées chez respectivement 49,3%, 48,2% et 36,8% des patients sous Invokana 100 mg, Invokana 300 mg et placebo. Des hypoglycémies sévères sont survenues chez respectivement 1,8%, 2,7% et 2,5% des patients sous Invokana 100 mg, Invokana 300 mg et placebo. Lors de l'administration d'Invokana en ajout à des sulfonylurées, des épisodes d'hypoglycémie ont été observés chez respectivement 4,1%, 12,5% et 5,8% des patients sous Invokana 100 mg, Invokana 300 mg et placebo (voir «Posologie/Mode d'emploi» et «Interactions»).
Fasciite nécrosante périnéale (gangrène de Fournier)
La gangrène de Fournier a été identifiée comme effet indésirable spécifique à la classe des inhibiteurs du SGLT2 sur la base des rapports spontanés d'effets indésirables. Dans le cadre du programme de développement clinique de la canagliflozine de phases 3 et 4 (y compris le programme CANVAS et le programme CREDENCE), cet effet indésirable a été observé uniquement chez quatre patients (deux dans le bras sous canagliflozine et deux dans le bras témoin). Ces quatre cas de gangrène de Fournier ont tous été classés comme événements indésirables graves.
Sur la base de la fréquence observée au cours des études cliniques, cet effet indésirable a été classé comme «rare» (≥1/10 000 et <1/1000), [≥0,01% et <0,1%] (voir la liste des effets indésirables ci-dessus).
Infections mycosiques génitales
Une infection mycosique vulvovaginale (p.ex. vulvovaginite, candidose vulvovaginale) a été rapportée chez respectivement 10,4% et 11,4% des patientes traitées par Invokana 100 mg et Invokana 300 mg, contre 3,2% lors de l'administration du placebo. La plupart des cas de candidose vulvovaginale ont été rapportés durant les quatre premiers mois du traitement par la canagliflozine. 2,3% des patientes traitées par Invokana ont présenté plus d'une infection. Au total, 0,7% des patientes ont arrêté le traitement par Invokana en raison d'une candidose vulvovaginale (voir «Mises en garde et précautions»).
Une balanite ou une balanoposthite à Candida a été rapportée chez respectivement 4,2% et 3,7% des hommes traités par Invokana 100 mg et Invokana 300 mg, contre 0,6% lors de l'administration du placebo. 0,9% des hommes traités par Invokana ont présenté plus d'une infection. Au total, 0,5% des hommes ont arrêté le traitement par Invokana en raison d'une balanite ou d'une balanoposthite à Candida. Dans une analyse regroupée de 10 études contrôlées, le taux d'incidence de phimosis a été de 5,6 événements pour 1000 années-patients d'exposition chez les hommes non circoncis. Dans cette analyse regroupée, le taux d'incidence de circoncisions a été de 3,8 événements pour 1000 années-patients d'exposition chez les hommes traités par la canagliflozine (voir «Mises en garde et précautions»).
Infections des voies urinaires
Les infections des voies urinaires ont été plus fréquentes lors de l'administration d'Invokana 100 mg ou 300 mg que sous placebo (5,9% et 4,3%, contre 4,0%). La plupart des infections étaient légères à modérées; la fréquence des effets indésirables sévères n'a pas été plus élevée. Les patients ont répondu au traitement habituel tout en poursuivant le traitement par la canagliflozine. Le traitement par la canagliflozine n'a été que rarement arrêté. L'incidence des infections répétées n'a pas été augmentée sous canagliflozine.
Fractures osseuses
Dans une étude cardiovasculaire (CANVAS) menée chez 4327 patients traités, présentant une maladie cardiovasculaire manifeste ou au moins deux facteurs de risque d'affection cardiovasculaire, les taux d'incidence de toutes les fractures osseuses relevées ont été de 15,9 pour 1000 années-patients d'exposition à la canagliflozine 100 mg, de 17,9 pour 1000 années-patients d'exposition à la canagliflozine 300 mg et de 10,9 pour 1000 années-patients d'exposition au placebo. Le hazard ratiocanagliflozine 100 mg/placebo correspondant a été de 1,45 [1,10; 1,92] IC à 95% et le HRcanagliflozine 300 mg/placebo de 1,64 [1,25; 2,15] IC à 95%. Le déséquilibre de ce taux de fractures est déjà survenu dans les 26 premières semaines de traitement. Le lien avec une augmentation de la fréquence des chutes n'a pas pu être confirmé avec certitude. Au cours de deux autres études de longue durée et d'études menées sur la population diabétique générale, aucune différence n'a été observée entre la canagliflozine et la substance de contrôle sur le plan du risque de fracture.
Dans une deuxième étude cardiovasculaire (CANVAS-R) menée chez 5807 patients traités, présentant une maladie cardiovasculaire manifeste ou au moins deux facteurs de risque d'affection cardiovasculaire, les taux d'incidence de toutes les fractures osseuses relevées ont été de 11,4 événements pour 1000 années-patients d'exposition à l'administration de canagliflozine et de 13,2 événements pour 1000 années-patients d'exposition au placebo.
Dans une étude de longue durée au cours de laquelle les résultats thérapeutiques de 4397 patients atteints de diabète de type 2 et d'une néphropathie diabétique ont été examinés particulièrement sur le plan rénal, les taux d'incidence de toutes les factures osseuses avérées étaient de 12,1 événements pour 1000 années-patients d'exposition, aussi bien en cas de prise de 100 mg de canagliflozine que dans le groupe placebo.
Dans d'autres études ayant inclus une population diabétique générale de 7729 patients et dans lesquelles l'incidence des fractures osseuses a été relevée, aucune différence du risque de fractures n'a été observée sous Invokana par rapport au groupe témoin. Les taux d'incidence de toutes les fractures osseuses relevées ont été de 11,8 événements pour 1000 années-patients d'exposition à l'administration de canagliflozine et de 10,8 événements pour 1000 années-patients d'exposition dans le groupe placebo. Après 104 semaines de traitement, la canagliflozine n'a pas présenté d'effet indésirable sur la densité minérale osseuse.
Photosensibilité
Des effets indésirables liés à une photosensibilité (incluant réactions de photosensibilité, photodermatose polymorphe et coup de soleil) sont survenus chez respectivement 0,1%, 0,2% et 0,2% des patients traités par les produits de comparaison, Invokana 100 mg et Invokana 300 mg.
Insuffisance rénale
Patients traités en raison d'un diabète de type 2 insuffisamment contrôlé
Invokana est associé à une augmentation dose-dépendante de la créatinine sérique et à une baisse concomitante du DFG estimé (tableau 4). Les modifications moyennes ont été plus importantes chez les patients présentant une insuffisance rénale modérée au début de l'étude (valeurs initiales).
Tableau 4: Modifications de la créatinine sérique et du DFGe en rapport avec Invokana au cours d'au total quatre études contrôlées contre placebo et d'une étude réalisée chez des patients présentant une insuffisance rénale modérée
|
|
Placebo
N=646
|
Invokana
100 mg
N=833
|
Invokana
300 mg
N=834
| |
Analyse regroupée de quatre études contrôlées
contre placebo
|
Valeurs initiales
|
Créatinine (µmol/l)
|
74,5
|
72,9
|
72,6
| |
DFGe (ml/min/1,73 m2)
|
87,0
|
88,3
|
88,8
| |
Modification à la semaine 6
|
Créatinine (µmol/l)
|
1,05
|
2,93
|
4,08
| |
DFGe (ml/min/1,73 m2)
|
-1,6
|
-3,8
|
-5,0
| |
Modification à la fin du traitement*
|
Créatinine (µmol/l)
|
0,91
|
1,96
|
2,99
| |
DFGe (ml/min/1,73 m2)
|
-1,6
|
-2,3
|
-3,4
|
|
|
Placebo
N=90
|
Invokana
100 mg
N=90
|
Invokana
300 mg
N=89
| |
Étude dans l'insuffisance rénale modérée
|
Valeurs initiales
|
Créatinine (µmol/l)
|
142,0
|
143,5
|
144,2
| |
DFGe (ml/min/1,73 m2)
|
40,1
|
39,7
|
38,5
| |
Modification à la semaine 3
|
Créatinine (µmol/l)
|
2,91
|
16,21
|
25,10
| |
DFGe (ml/min/1,73 m2)
|
-0,7
|
-4,6
|
-6,2
| |
Modification à la fin du traitement*
|
Créatinine (µmol/l)
|
6,04
|
14,04
|
16,22
| |
DFGe (ml/min/1,73 m2)
|
-1,5
|
-3,6
|
-4,0
|
* Semaine 26 dans la population mITT LOCF.
Dans l'analyse regroupée des quatre études contrôlées contre placebo menées chez des patients présentant une fonction rénale normale ou une insuffisance rénale légère au début de l'étude (valeurs initiales), la proportion de patients présentant au moins un événement d'une diminution significative de la fonction rénale (défini par un DFGe inférieur à 80 ml/min/1,73 m2 et une diminution du DFGe de 30% par rapport à la valeur initiale) a été de 2,1% dans le groupe placebo, de 2,0% sous Invokana 100 mg et de 4,1% sous Invokana 300 mg. À la fin du traitement, 0,5% des patients du groupe placebo, 0,7% des patients traités par Invokana 100 mg et 1,4% des patients traités par Invokana 300 mg présentaient une diminution significative de la fonction rénale.
Dans une étude menée chez des patients présentant une insuffisance rénale modérée, avec un DFGe initial compris entre 30 et <50 ml/min./1,73 m2 (DFGe initial moyen de 39 ml/min/1,73 m2) (voir «Propriétés/Effets, Populations spécifiques»), la proportion de patients présentant au moins un événement d'une diminution significative de la fonction rénale (défini par une diminution du DFGe de 30% par rapport à la valeur initiale) a été de 6,9% dans le groupe placebo, de 18% sous Invokana 100 mg et de 22,5% sous Invokana 300 mg. À la fin du traitement, 4,6% des patients du groupe placebo et 3,4% des patients traités par Invokana 100 mg ou 300 mg présentaient une diminution significative de la fonction rénale.
Dans une population regroupée de patients présentant une insuffisance rénale modérée (N=1087), avec un DFGe initial compris entre 30 et <60 ml/min/1,73 m2 (DFGe initial moyen de 48 ml/min/1,73 m2), l'incidence globale de ces événements a été plus faible que dans l'étude dédiée. Toutefois, une augmentation dose-dépendante des épisodes de diminution significative de la fonction rénale a été constatée par rapport au placebo.
L'utilisation d'Invokana a été associée à une fréquence accrue d'événements indésirables d'origine rénale (p.ex. augmentation de la créatinine sérique, diminution du débit de filtration glomérulaire, insuffisance rénale et défaillance rénale aigüe), notamment chez les patients présentant une insuffisance rénale modérée.
Dans l'analyse des données regroupées de patients présentant une insuffisance rénale modérée, la fréquence des événements indésirables rénaux a été de 3,7% dans le groupe placebo, de 8,9% sous Invokana 100 mg et de 9,3% sous Invokana 300 mg. Un arrêt du traitement en raison d'événements indésirables rénaux a eu lieu chez 1,0% des patients du groupe placebo, chez 1,2% des patients traités par Invokana 100 mg et chez 1,6% des patients traités par Invokana 300 mg (voir «Mises en garde et précautions»).
Patients atteints de diabète de type 2 traités en raison d'une néphropathie diabétique avec albuminurie (ACR >300 mg/g)
Dans une étude de longue durée dans laquelle les résultats thérapeutiques ont été particulièrement étudiés sur le plan rénal, menée chez des patients atteints de diabète de type 2 et d'une néphropathie chronique, les taux d'incidence de réactions indésirables en lien avec une déplétion volumique étaient similaires dans les sous-groupes de patients présentant un DFGe avant traitement de 45 à <60 ml/min/1,73 m2 [ClCr 45 à <60 ml/min]: 23 événements pour 1000 années-patients sous 100 mg de canagliflozine et 26 événements pour 1000 années-patients sous prise de placebo. Dans cette même étude, le taux d'incidence chez les patients présentant un DFGe avant traitement de 30 à <45 ml/min/1,73 m2 [ClCr 30 à <45 ml/min] était plus élevé en cas de prise de 100 mg de canagliflozine (49 événements pour 1000 années-patients) qu'en cas de prise de placebo (26 événements pour 1000 années-patients).
Dans une étude de longue durée dans laquelle les résultats thérapeutiques ont été particulièrement étudiés sur le plan rénal, aucune différence n'a été observée sur le plan des valeurs de potassium sérique, ni aucune augmentation des effets indésirables liés à l'hyperkaliémie ni aucune augmentation absolue (>6,5 mEq/l) ou relative (> à la limite supérieure de la norme et >15% de la valeur initiale) du potassium sérique en cas de prise de 100 mg de canagliflozine par rapport au placebo.
De manière générale, aucun déséquilibre n'a été observé entre les groupes de traitement sur le plan des valeurs anormales de phosphate, tant globalement qu'au sein de chaque catégorie de DFGe (45 à <60 ou 30 à <45 ml/min/1,73 m2 [ClCr 45 à <60 ou 30 à <45 ml/min]).
Chez les patients qui recevaient le placebo, une diminution progressive du DFGe est apparue, alors que les patients traités par 100 mg de canagliflozine, ont d'abord présenté une diminution du DFGe moyen qui s'est par la suite stabilisée (allant jusqu'à 3,5 ans). À la fin du traitement, le DFGe moyen du groupe placebo était inférieur de 1,6 ml/min/1,73 m2 au taux du groupe sous 100 mg de canagliflozine.
Examens de laboratoire
Les résultats des examens de laboratoire décrits ci-dessous proviennent de l'analyse des données regroupées des études cliniques d'une durée de 26 semaines, contrôlées contre placebo (sauf indication contraire).
Augmentation des taux sériques de potassium
Le pourcentage moyen d'augmentation des taux sériques de potassium par rapport aux valeurs initiales a été de 0,5% sous Invokana 100 mg et de 1,0% sous Invokana 300 mg, contre 0,6% sous placebo. Des épisodes d'augmentation des taux sériques de potassium (>5,4 mmol/l et augmentation de 15% par rapport au taux initial) ont été observés chez 4,4% des patients sous Invokana 100 mg, 7,0% des patients sous Invokana 300 mg et 4,8% des patients sous placebo. De manière générale, ces augmentations ont été légères (<6,5 mmol/l) et transitoires et n'ont pas nécessité de traitement spécifique.
Dans une étude réalisée chez des patients présentant une insuffisance rénale modérée (DFGe de 30 à <50 ml/min/1,73 m2) (voir «Propriétés/Effets, Populations spécifiques»), des augmentations dose-dépendantes et transitoires du potassium sérique ont déjà été observées précocement après l'instauration du traitement (c.-à-d. en l'espace de 3 semaines). Dans cette étude, des augmentations du potassium sérique supérieures à 5,4 mEq/l et de 15% par rapport à la valeur initiale sont survenues chez 16,1% des patients du groupe placebo, chez 12,4% des patients traités par Invokana 100 mg et chez 27,0% des patients traités par Invokana 300 mg. Des augmentations plus importantes (c.-à-d. d'au moins 6,5 mEq/l) ont été observées chez 1,1% des patients du groupe placebo et chez 2,2% des patients traités par Invokana 100 mg et Invokana 300 mg. Chez les patients présentant une insuffisance rénale modérée, les augmentations du potassium sérique ont été plus fréquentes lorsque le taux initial de potassium était déjà augmenté, ainsi qu'en cas d'utilisation de médicaments réduisant l'excrétion potassique, p.ex. de diurétiques épargneurs de potassium, d'inhibiteurs de l'ECA et d'antagonistes des récepteurs de l'angiotensine (voir «Mises en garde et précautions»).
Effets sur les lipides
Les modifications moyennes (modifications en pourcentage) du LDL-C par rapport aux taux initiaux ont été de +0,11 mmol/l (4,5%) sous Invokana 100 mg et de +0,21 mmol/l (8,0%) sous Invokana 300 mg comparativement au placebo. En ce qui concerne le cholestérol total, de plus faibles augmentations ont été observées par rapport au placebo, de 2,5% sous Invokana 100 mg et de 4,3% sous Invokana 300 mg. Les augmentations du cholestérol High-Density-Lipoprotein (HDL-C) ont été de 5,4% sous Invokana 100 mg et de 6,3% sous Invokana 300 mg par rapport au placebo. Les augmentations du non-HDL-C ont été de 0,05 mmol/l (1,5%) sous Invokana 100 mg et de 0,13 mmol/l (3,6%) sous Invokana 300 mg par rapport au placebo. Sous aucune des doses d'Invokana, le rapport LDL-C/HDL-C n'a présenté de modification par rapport au placebo. Les concentrations de particules d'apoB et de LDL-C (mesurées dans deux études) ainsi que de non-HDL-C ont plus faiblement augmenté par rapport aux variations du LDL-C.
Augmentation des taux d'hémoglobine
Des augmentations minimes des concentrations d'hémoglobine ont été observées sous Invokana 100 mg et 300 mg par rapport aux valeurs initiales (+3,5% ou +4,7 g/l et +3,8% ou 5,1 g/l), tandis qu'une légère diminution a été observée sous placebo (-1,1% ou -0,18 g/l). Conformément à cela, s'agissant du nombre d'érythrocytes et de l'hématocrite, des augmentations minimes des pourcentages moyens de variations par rapport aux valeurs initiales ont été observées.
À la fin du traitement, 4,0% des patients sous Invokana 100 mg, 2,7% des patients sous Invokana 300 mg et 0,8% des patients sous placebo présentaient des taux d'hémoglobine supérieurs à la limite de la normale.
Augmentations du magnésium sérique
Des augmentations dose-dépendantes du magnésium sérique ont été observées peu de temps (en l'espace de 6 semaines) après l'instauration d'un traitement par Invokana et ont persisté au cours du traitement. Dans l'analyse regroupée des quatre études contrôlées contre placebo, la modification moyenne du taux sérique de magnésium a été de +8,1% sous Invokana 100 mg et de +9,3% sous Invokana 300 mg, contre -0,6% dans le groupe placebo. Dans une étude menée chez des patients présentant une insuffisance rénale modérée (voir «Propriétés/Effets, Populations spécifiques»), les concentrations sériques de magnésium ont augmenté de 0,2% dans le groupe placebo, de 9,2% sous Invokana 100 mg et de 14,8% sous Invokana 300 mg.
Augmentations du phosphate sérique
Des augmentations dose-dépendantes du taux sérique de phosphate ont été observées au cours du traitement par Invokana. Dans l'analyse regroupée des quatre études contrôlées contre placebo, le pourcentage moyen d'augmentation du taux sérique de phosphate a été de 3,6% sous Invokana 100 mg et de 5,1% sous Invokana 300 mg, contre 1,5% dans le groupe placebo. Dans une étude menée chez des patients présentant une insuffisance rénale modérée (voir «Propriétés/Effets, Populations spécifiques»), les concentrations sériques de phosphate ont augmenté de 1,2% dans le groupe placebo, de 5,0% sous Invokana 100 mg et de 9,3% sous Invokana 300 mg.
Diminution du débit de filtration glomérulaire
Au cours d'études de longue durée sur l'issue cardiovasculaire, une diminution du DFGe est survenue au début du traitement par la canagliflozine. Celle-ci était réversible à l'arrêt du traitement par la canagliflozine. Lors de la poursuite du traitement par la canagliflozine, le DFGe a de nouveau augmenté progressivement au cours des études (jusqu'à 6,5 ans) et a atteint un DFGe moyen >70 ml/min/1,73 m2 à la fin de l'étude. En revanche, une diminution progressive du DFGe a été observée chez les patients du groupe contrôle sous placebo.
Effets indésirables dans des populations spécifiques
Patients âgés
Le profil de sécurité observé chez les patients âgés a correspondu de manière générale à celui observé chez les patients plus jeunes. Les patients âgés de ≥75 ans ont cependant présenté une incidence accrue d'effets indésirables liés à une diminution du volume intravasculaire (tels que vertiges positionnels, hypotension artérielle, hypotension orthostatique). Les taux d'incidence de ces événements ont été respectivement de 52,7, de 60,8 et de 24,1 événements pour 1000 années-patients d'exposition à la canagliflozine 100 mg, à la canagliflozine 300 mg et au placebo. Une diminution du DFGe de - 3,41 ml/min/1,73 m2 sous canagliflozine 100 mg, de -4,67 ml/min/1,73 m2 sous canagliflozine 300 mg et de -4,15 ml/min/1,73 m2 sous placebo a été observée (voir «Posologie/Mode d'emploi» et «Mises en garde et précautions»).
Patients insuffisants rénaux
Les patients présentant un DFGe initial <60 ml/min/1,73 m2 ou une ClCr <60 ml/min ont eu une incidence accrue d'effets indésirables liés à une diminution du volume intravasculaire (p.ex. vertiges positionnels, hypotension orthostatique, hypotension). Les taux d'incidence de ces événements ont été respectivement de 53, 51 et 31 événements pour 1000 années-patients d'exposition à la canagliflozine 100 mg, à la canagliflozine 300 mg et au placebo (voir «Posologie/Mode d'emploi» et «Mises en garde et précautions»).
Les taux d'incidence globale des augmentations des taux sériques de potassium ont été plus élevés chez les patients présentant une insuffisance rénale modérée. Les taux d'incidence de ces événements ont été respectivement de 49, de 61 et de 54 événements pour 1000 années-patients d'exposition à la canagliflozine 100 mg, à la canagliflozine 300 mg et au placebo. Ces augmentations ont été le plus souvent transitoires et n'ont pas nécessité de traitement spécifique.
Chez les patients présentant une insuffisance rénale modérée, des augmentations des taux sériques de créatinine de 9,2 µmol/l et des taux sanguins d'azote uréique d'environ 1,0 mmol/l ont été observées avec les deux doses de canagliflozine. Les taux d'incidence d'une plus forte diminution du DFGe (>30%) à un moment quelconque du traitement ont été de 73 sous canagliflozine 100 mg et de 81 sous canagliflozine 300 mg, contre 65 événements pour 1000 années-patients d'exposition sous placebo. Lors de la dernière mesure réalisée après le début de l'étude, les taux d'incidence de telles diminutions ont été de 33 événements sous canagliflozine 100 mg, de 27 événements sous canagliflozine 300 mg et de 37 événements pour 1000 années-patients d'exposition sous placebo (voir «Mises en garde et précautions»).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSignes et symptômes
L'administration à des sujets sains de doses uniques d'Invokana allant jusqu'à 1600 mg et l'administration à des patients atteints de diabète de type 2 de 300 mg d'Invokana deux fois par jour pendant 12 semaines ont été généralement bien tolérées.
Traitement
En cas de surdosage, il convient de recourir aux mesures de soutien habituelles, p.ex. élimination de substances non absorbées du tractus gastro-intestinal, surveillance clinique et traitement de soutien en fonction de l'état clinique du patient. La canagliflozine a été très faiblement éliminée de l'organisme pendant une hémodialyse de quatre heures. Il est improbable que la canagliflozine soit dialysable par dialyse péritonéale.
Propriétés/EffetsCode ATC
A10BK02
Mécanisme d'action
Le co-transporteur sodium-glucose de type 2 (SGLT2), exprimé dans les tubules rénaux proximaux, est responsable de la majorité de la réabsorption du glucose filtré depuis la lumière tubulaire. La canagliflozine est un inhibiteur du SGLT2, actif par voie orale. En inhibant le SGLT2, la canagliflozine diminue la réabsorption du glucose filtré et diminue le seuil rénal du glucose (RTG). Elle augmente ainsi l'excrétion urinaire du glucose (UGE: urinary glucose excretion), ce qui diminue les concentrations plasmatiques du glucose chez les patients atteints de diabète de type 2. Ce mécanisme n'est pas dépendant de l'insuline. L'augmentation de l'excrétion urinaire du glucose (UGE) résultant de l'inhibition du SGLT2 conduit par ailleurs à une diurèse osmotique, ainsi qu'à une diminution de la pression artérielle systolique par un effet diurétique. L'augmentation de l'excrétion urinaire du glucose (UGE) entraîne une perte de calories. L'UGE augmente le risque d'infections des voies urinaires et d'infections mycosiques génitales.
Les études n'ont pas mis en évidence de malabsorption du glucose sous canagliflozine.
La canagliflozine augmente l'apport de sodium au tube distal par un blocage de la réabsorption du sodium et du glucose SGLT2-dépendante, ce qui explique que le rétrocontrôle tubulo-glomérulaire augmente; cela est associé, dans les modèles diabétiques précliniques ainsi que dans les études cliniques, à une diminution de la pression intraglomérulaire et de l'hyperfiltration, et potentiellement à un effet néphro-protecteur.
Pharmacodynamique
Après l'administration orale de doses uniques et multiples de canagliflozine à des patients atteints de diabète de type 2, des diminutions dose-dépendantes du RTG et des augmentations de l'excrétion urinaire du glucose ont été observées. Lors d'une valeur initiale de RTG d'environ 13 mmol/l, un abaissement maximal du RTG moyen sur 24 heures à environ 4 mmol/l à 5 mmol/l a été observé à la dose journalière de 300 mg chez les patients atteints de diabète de type 2 dans des études de phase 1, ce qui suggère que le risque d'hypoglycémie induite par le traitement est faible. Chez les sujets atteints de diabète de type 2 traités par 100 mg ou 300 mg de canagliflozine, la diminution du RTG a entraîné une augmentation de l'excrétion urinaire du glucose comprise entre 77 g/jour et 119 g/jour dans toutes les études de phase 1. L'excrétion urinaire du glucose observée correspond à une perte de 308 à 476 kcal/jour. Les diminutions du RTG et les augmentations de l'excrétion urinaire du glucose ont persisté pendant la période de traitement de 26 semaines chez des patients atteints de diabète de type 2. Après une légère augmentation (le plus souvent <400-500 ml) du volume urinaire quotidien, celui-ci a diminué à nouveau au cours des premiers jours du traitement. La canagliflozine a augmenté de façon transitoire l'excrétion urinaire d'acide urique (augmentation de 19% par rapport à la valeur initiale le premier jour et diminution à 6% le jour 2 et à 1% le jour 13). Ceci s'est accompagné d'une réduction persistante du taux sérique d'acide urique d'environ 20%.
Dans une étude à dose unique réalisée chez des patients atteints de diabète de type 2, l'administration de 300 mg avant un repas mixte a retardé l'absorption intestinale du glucose et a diminué la glycémie postprandiale.
Électrophysiologie cardiaque
Dans une étude randomisée, en double aveugle, contrôlée contre placebo, avec un contrôle positif (moxifloxacine), 60 sujets sains ont reçu une dose orale unique de 300 mg de canagliflozine, de 1200 mg de canagliflozine (quatre fois la dose maximale recommandée), de moxifloxacine ou de placebo. Aucune modification significative de l'intervalle QTc n'a été observée à la dose recommandée de 300 mg ni à la dose de 1200 mg. À la dose de 1200 mg, les concentrations plasmatiques maximales de canagliflozine ont été environ 1,4 fois supérieures aux concentrations maximales atteintes à l'état d'équilibre avec une dose de 300 mg une fois par jour.
Efficacité clinique
Efficacité glycémique et sécurité
Au total, 10 285 patients atteints de diabète de type 2 ont participé à neuf études cliniques d'efficacité et de sécurité, contrôlées et en double aveugle, ayant évalué les effets d'Invokana sur le contrôle glycémique. La distribution ethnique était de 72% de Blancs, 16% d'Asiatiques, 4% de Noirs et 8% de membres d'autres groupes. 16% des patients étaient d'origine hispanique. Environ 58% des patients étaient de sexe masculin. L'âge moyen des patients était de 59,6 ans (fourchette de 21 ans à 96 ans). 3082 patients étaient âgés de 65 ans et plus et 510 patients de 75 ans et plus. 58% des patients présentaient un indice de masse corporelle (IMC) supérieur ou égal à 30 kg/m2. Pour le sous-groupe de patients présentant une insuffisance rénale modérée avec un DFGe initial compris entre 30 et <60 ml/min/1,73 m2, les données de 1087 patients, issues du programme de développement clinique, ont été regroupées et analysées.
Études contrôlées contre placebo
Invokana a été étudié en monothérapie, en bithérapie avec la metformine, en bithérapie avec une sulfonylurée, en trithérapie avec la metformine et une sulfonylurée, ainsi qu'en ajout à l'insuline (tableau 5). En résumé, le traitement par Invokana a entraîné des résultats cliniquement et statistiquement significatifs par rapport au placebo (p<0,001) en ce qui concerne le contrôle glycémique, y compris le taux d'HbA1c, la proportion de patients présentant un taux d'HbA1c <7%, les modifications de la glycémie à jeun (GAJ) par rapport à la valeur initiale et la glycémie postprandiale (GPP) (après 2 heures). En outre, des diminutions du poids corporel et de la pression artérielle systolique ont également été observées par rapport au placebo.
Tableau 5: Résultats d'efficacité des études cliniques contrôlées contre placeboa
|
Monothérapie (26 semaines)
| |
|
Invokana
|
Placebo
(N=192)
| |
100 mg
(N=195)
|
300 mg
(N=197)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,06
|
8,01
|
7,97
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-0,77
|
-1,03
|
0,14
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-0,91b
(-1,09; -0,73)
|
-1,16b
(-1,34; -0,99)
|
N/Ac
| |
Patients (%) ayant atteint un taux d'HbA1c<7%
|
44,5b
|
62,4b
|
20,6
| |
Glycémie à jeun (mmol/l)
| |
Valeur initiale (moyenne)
|
9,57
|
9,57
|
9,20
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,51
|
-1,94
|
0,46
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-1,97b
(-2,34; -1,60)
|
-2,41b
(-2,78; -2,03)
|
N/Ac
| |
Glycémie postprandiale à 2 heures (mmol/l)
| |
Valeur initiale (moyenne)
|
13,87
|
14,10
|
12,74
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-2,38
|
-3,27
|
0,29
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-2,67b
(-3,28; -2,05)
|
-3,55b
(-4,17; -2,94)
|
N/Ac
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
85,9
|
86,9
|
87,5
| |
% de variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-2,8
|
-3,9
|
-0,6
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-2,2b
(-2,9; -1,6)
|
-3,3b
(-4,0; -2,6)
|
N/Ac
|
|
Bithérapie avec la metformine (26 semaines)
| |
|
Invokana + metformine
|
Placebo + metformine
(N=183)
| |
100 mg
(N=368)
|
300 mg
(N=367)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
7,94
|
7,95
|
7,96
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-0,79
|
-0,94
|
-0,17
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-0,62b
(-0,76; -0,48)
|
-0,77b
(-0,91; -0,64)
|
N/Ac
| |
Patients (%) ayant atteint un taux d'HbA1c<7%
|
45,5
|
57,8
|
29,8
| |
Glycémie à jeun (mmol/l)
| |
Valeur initiale (moyenne)
|
9,36
|
9,59
|
9,12
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,52
|
-2,10
|
0,14
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-1,65b
(-1,99; -1,32)
|
-2,23b
(-2,57; -1,90)
|
N/Ac
| |
Glycémie postprandiale à 2 heures (mmol/l)
| |
Valeur initiale (moyenne)
|
14,30
|
14,54
|
13,81
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-2,66
|
-3,17
|
-0,55
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-2,12b
(-2,73; -1,51)
|
-2,62b
(-3,24; -2,01)
|
N/Ac
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
88,7
|
85,4
|
86,7
| |
% de variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-3,7
|
-4,2
|
-1,2
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-2,5b
(-3,1; -1,9)
|
-2,9b
(-3,5; -2,3)
|
N/Ac
|
|
Trithérapie avec la metformine et une sulfonylurée (26 semaines)
| |
|
Invokana + metformine et sulfonylurée
|
Placebo +
metformine et sulfonylurée
(N=156)
| |
100 mg
(N=157)
|
300 mg
(N=156)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,13
|
8,13
|
8,12
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-0,85
|
-1,06
|
-0,13
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-0,71b
(-0,90;-0,52)
|
-0,92b
(-1,11;-0,73)
|
N/Ac
| |
Patients (%) ayant atteint un taux d'HbA1c<7%
|
43,2b
|
56,6b
|
18,0
| |
Glycémie à jeun (mmol/l)
| |
Valeur initiale (moyenne)
|
9,60
|
9,34
|
9,42
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,01
|
-1,69
|
0,23
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-1,24b
(-1,75; -0,73)
|
-1,92b
(-2,43; -1,41)
|
N/Ac
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
93,5
|
93,5
|
90,8
| |
% de variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-2,1
|
-2,6
|
-0,7
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-1,4b
(-2,1;-0,7)
|
-2,0b
(-2,7;-1,3)
|
N/Ac
|
|
Ajout à l'insulined (18 semaines)
| |
|
Invokana + insuline
|
Placebo + insuline
(N=565)
| |
100 mg
(N=566)
|
300 mg
(N=587)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,33
|
8,27
|
8,20
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-0,63
|
-0,72
|
0,01
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-0,65b
(-0,73; -0,56)
|
-0,73b
(-0,82; -0,65)
|
N/Ac
| |
Patients (%) ayant atteint un taux d'HbA1c<7%
|
19,8
|
24,7
|
7,7
| |
Glycémie à jeun (mmol/l)
| |
Valeur initiale (moyenne)
|
9,43
|
9,33
|
9,38
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,03
|
-1,39
|
0,22
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-1,25b
(-1,55; -0,96)
|
-1,61b
(-1,90;-1,31)
|
N/Ac
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
96,9
|
96,7
|
97,7
| |
% de variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,8
|
-2,3
|
0,1
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 97,5%)
|
-1.9b
(-2,2; -1,5)
|
-2.4b
(-2,8; -2,0)
|
N/Ac
|
a Population en intention de traiter en utilisant la dernière observation rapportée dans l'étude avant l'emploi d'un traitement glycémique de secours.
b p<0,001 par rapport au placebo.
c Non applicable.
d Invokana en ajout à l'insuline (avec ou sans autres médicaments antihyperglycémiants).
Traitement adjuvant: canagliflozine associée à la metformine et à un inhibiteur de la dipeptidyl peptidase-4
La canagliflozine a été étudiée en traitement adjuvant chez des patients n'ayant pas obtenu un contrôle glycémique approprié sous traitement antérieur par la metformine et la sitagliptine et a été dosée conformément au schéma de titration (dose initiale de 100 mg; augmentation à 300 mg après 6 semaines chez les patients ayant un contrôle glycémique insuffisant, un DFGe approprié et ayant bien toléré la canagliflozine dosée à 100 mg).
Chez ces patients, par rapport au placebo, le contrôle glycémique s'est amélioré (voir tableau 6) après administration de canagliflozine. De plus, par rapport au placebo, la canagliflozine a entraîné une diminution du poids corporel et de la tension artérielle systolique.
Tableau 6: Résultats de l'étude clinique de 26 semaines contrôlée contre placebo portant sur la canagliflozine en association avec la metformine et la sitagliptine*
|
Paramètres d'efficacité
|
Placebo +
metformine et sitagliptine
(N=106)
|
Canagliflozine +
metformine et sitagliptine
(N=107)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,38
|
8,53
| |
Variation par rapport à la valeur initiale (moyenne ajustée)
|
-0,01
|
-0,91
| |
Différence par rapport au placebo (moyenne ajustée) (IC à 95%)†
|
|
-0,89‡
(-1,19; -0,59)
| |
Patients (%) ayant atteint un taux d'HbA1c <7%
|
12
|
32§
| |
Glycémie à jeun (mg/dl)
| |
Valeur initiale (moyenne)
|
180
|
186
| |
Variation par rapport à la valeur initiale (moyenne ajustée)
|
-3
|
-30
| |
Différence par rapport au placebo (moyenne ajustée) (IC à 95%)†
|
|
-27‡
(-40; -14)
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
89,9
|
93,8
| |
% de variation par rapport à la valeur initiale (moyenne ajustée)
|
-1,6
|
-3,4
| |
Différence par rapport au placebo (moyenne ajustée) (IC à 95%)†
|
|
-1,8‡
(-2,7; -0,9)
|
* Population en intention de traiter (Intent-to-treat-Population)
† La moyenne ajustée et l'IC ont été calculés avec un modèle mixte pour mesures répétées
‡ p<0,001
§ p<0,01
Études contrôlées contre traitement actif
Invokana a été comparé au glimépiride en bithérapie (avec la metformine) et à la sitagliptine en trithérapie (avec la metformine et une sulfonylurée) (tableau 7).
Tableau 7: Résultats d'efficacité des études contrôlées contre traitement actifa
|
Comparaison au glimépiride en bithérapie avec la metformine (52 semaines)
| |
|
Invokana + metformine
|
Glimépiride (titré) +
metformine
(N=482)
| |
100 mg
(N=483)
|
300 mg
(N=485)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
7,78
|
7,79
|
7,83
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-0,82
|
-0,93
|
-0,81
| |
Différence par rapport au glimépiride (moyenne des moindres carrés) (IC à 95%)
|
-0,01b
(−0,11;0,09)
|
-0,12b
(−0,22; −0,02)
|
N/Ac
| |
Patients (%) ayant atteint un taux d'HbA1c<7%
|
53,6
|
60,1
|
55,8
| |
Glycémie à jeun (mmol/l)
| |
Valeur initiale (moyenne)
|
9,18
|
9,09
|
9,20
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,35
|
-1,52
|
-1,02
| |
Différence par rapport au glimépiride (moyenne des moindres carrés) (IC à 95%)
|
-0,33
(−0,56; -0,11)
|
-0,51
(−0,73; −0,28)
|
N/Ac
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
86,8
|
86,6
|
86,6
| |
% de variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-4,2
|
-4,7
|
1,0
| |
Différence par rapport au glimépiride (moyenne des moindres carrés) (IC à 95%)
|
-5,2d
(−5,7; −4,7)
|
-5,7d
(−6,2; −5,1)
|
N/Ac
|
|
Comparaison à la sitagliptine en trithérapie avec la metformine et une sulfonylurée (52 semaines)
| |
|
Invokana 300 mg +
metformine et sulfonylurée
(N=377)
|
Sitagliptine 100 mg +
metformine et sulfonylurée
(N=378)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,12
|
8,13
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,03
|
-0,66
| |
Différence par rapport à la sitagliptine (moyenne des moindres carrés) (IC à 95%)
|
-0,37e (-0,50; -0,25)
|
N/Ac
| |
Patients (%) ayant atteint un taux d'HbA1c<7%
|
47,6
|
35,3
| |
Glycémie à jeun (mmol/l)
| |
Valeur initiale (moyenne)
|
9,42
|
9,09
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,66
|
-0,32
| |
Différence par rapport à la sitagliptine (moyenne des moindres carrés) (IC à 95%)
|
-1,34 (-1,66; -1,01)
|
N/Ac
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
87,6
|
89,6
| |
% de variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-2,5
|
0,3
| |
Différence par rapport à la sitagliptine (moyenne des moindres carrés) (IC à 95%)
|
-2,8d (-3,3; -2,2)
|
N/Ac
|
a Population en intention de traiter en utilisant la LOCF dans l'étude avant l'emploi d'un traitement glycémique de secours.
b Invokana+metformine est considéré comme non inférieur à glimépiride+metformine, car la limite supérieure de l'intervalle de confiance est inférieure au seuil de non-infériorité prédéfini (<0,3%).
c Non applicable.
d p<0,01.
e Invokana+metformine+sulfonylurée est considéré comme non inférieur à sitagliptine+metformine+sulfonylurée, car la limite supérieure de l'intervalle de confiance est inférieure au seuil de non-infériorité prédéfini (<0,3%).
Canagliflozine en association avec la metformine en traitement initial
La canagliflozine a été étudiée en association avec la metformine chez des patients atteints de diabète sucré de type 2 non préalablement traités chez qui des mesures alimentaires et l'exercice physique n'ont, à eux seuls, pas permis d'obtenir un contrôle glycémique approprié. L'association thérapeutique initiale (canagliflozine à 100 mg et metformine XR ou canagliflozine à 300 mg et metformine XR) était supérieure en termes d'amélioration du taux d'HbA1c par rapport à la canagliflozine en monothérapie (à 100 mg ou 300 mg) et à la metformine en monothérapie (voir tableau 8).
Tableau 8: Résultats de l'étude clinique de 26 semaines contrôlée contre principe actif portant sur la canagliflozine en association avec la metformine en traitement initial*
|
Paramètres d'efficacité
|
Metformine XR
(N=237)
|
Canagliflozine à 100 mg
(N=237)
|
Canagliflozine à 300 mg
(N=238)
|
Canagliflozine à 100 mg +
metformine XR
(N=237)
|
Canagliflozine à 300 mg +
metformine XR
(N=237)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,81
|
8,78
|
8,77
|
8,83
|
8,90
| |
Variation par rapport à la valeur initiale (moyenne ajustée)
|
-1,30
|
-1,37
|
-1,42
|
-1,77
|
-1,78
| |
Différence par rapport à 100 mg de canagliflozine (moyenne ajustée) (IC à 95%)†
|
|
|
|
-0,40‡
(-0,59, -0,21)
|
| |
Différence par rapport à 300 mg de canagliflozine (moyenne ajustée) (IC à 95%)†
|
|
|
|
|
-0,36‡
(-0,56, -0,17)
| |
Différence par rapport à la metformine XR (moyenne ajustée) (IC à 95%)†
|
|
-0,06‡
(-0,26, 0,13)
|
-0,11‡
(-0,31, 0,08)
|
-0,46‡
(-0,66, -0,27)
|
-0,48‡
(-0,67, -0,28)
| |
Patients (%) ayant atteint un taux d'HbA1c <7%
|
43
|
39
|
43
|
50§§
|
57§§
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
92,1
|
90,3
|
93,0
|
88,3
|
91,5
| |
% de variation par rapport à la valeur initiale (moyenne ajustée)
|
-2,1
|
-3,0
|
-3,9
|
-3,5
|
-4,2
| |
Différence par rapport à la metformine XR (moyenne ajustée) (IC à 95%)†
|
|
-0,9§§
(-1,6, -0,2)
|
-1,8§
(-2,6, -1,1)
|
-1,4‡
(-2,1, -0,6)
|
-2,1‡
(-2,9, -1,4)
|
* Population en intention de traiter
† Moyenne ajustée des moindres carrés (moyenne LS) pour les covariables incluant la valeur initiale et le facteur de stratification
‡ p=0,001 ajusté
§ p<0,01 ajusté
§§ p<0,05 ajusté
Populations spécifiques
Dans trois études sur des populations spécifiques (patients âgés, patients présentant un DFGe compris entre 30 et <50 ml/min/1,73 m2 et patients présentant une affection cardiovasculaire ou un risque accru d'une telle affection), Invokana a été ajouté au traitement antidiabétique actuel des patients (régime, monothérapie, association thérapeutique).
Patients âgés
Une étude en double aveugle, contrôlée contre placebo, d'une durée de 26 semaines, a été menée chez au total 714 patients âgés de ≥55 à ≤80 ans (dont 227 patients âgés de 65 à <75 ans et 46 patients âgés de 75 à <85 ans) et présentant un contrôle glycémique insuffisant avec le traitement antidiabétique actuel (régime et activité physique seuls ou associés à des principes actifs antihyperglycémiants oraux ou parentéraux). Des modifications statistiquement significatives (p<0,001) du taux d'HbA1c par rapport à la valeur initiale, de -0,57% sous Invokana 100 mg et de -0,70% sous Invokana 300 mg, ont été observées comparativement au placebo. Par ailleurs, une diminution statistiquement significative de la GAJ a été observée et un pourcentage plus élevé de patients a atteint un taux d'HbA1c <7,0%, comparativement au placebo (voir «Posologie/Mode d'emploi» et «Effets indésirables»).
Patients présentant un DFGe compris entre 45 et <60 ml/min/1,73 m2
Dans une analyse regroupée des données de patients (N=722) présentant un DFGe initial compris entre 45 ml/min/1,73 m2 et <60 ml/min/1,73 m2, la canagliflozine à la dose de 100 mg a entraîné une réduction du taux d'HbA1c de -0,47% par rapport au placebo. Les patients présentant un DFGe initial compris entre 45 ml/min/1,73 m2 et <60 ml/min/1,73 m2 ont présenté une diminution moyenne du poids corporel de -1,8% par rapport au placebo sous 100 mg de canagliflozine.
La majorité des patients présentant un DFGe initial compris entre 45 et <60 ml/min/1,73 m2 a été traité par l'insuline et/ou des sulfonylurées (85% [614/722]). Dans cette analyse des données regroupées, 27,3% des patients sous placebo, 39,8% des patients sous Invokana 100 mg et 41,8% des patients sous Invokana 300 mg ont eu ≥1 hypoglycémie. Le traitement concomitant par l'insuline et/ou des sulfonylurées explique cette incidence accrue d'hypoglycémies (voir «Effets indésirables»).
Pression artérielle
Dans les études contrôlées contre placebo, les diminutions moyennes de la pression artérielle systolique par rapport au placebo ont été les suivantes: -3,9 mmHg sous Invokana 100 mg, -5,3 mmHg sous Invokana 300 mg et -0,1 mmHg sous placebo. L'effet sur la pression diastolique a été moins important dans la même population, avec des modifications moyennes de -2,1 mmHg sous Invokana 100 mg, de -2,5 mmHg sous Invokana 300 mg et de -0,3 mmHg sous placebo. Aucune modification notable de la fréquence cardiaque n'a été observée.
Résultats cardiovasculaires dans le programme CANVAS
L'effet de la canagliflozine sur le risque cardiovasculaire chez les adultes atteints de diabète de type 2 et présentant une maladie cardiovasculaire (CV) manifeste ou un risque CV (deux risques CV ou plus) a été évalué au cours du programme CANVAS (étude CANVAS et étude CANVAS-R). Il s'agissait d'études multicentriques, multinationales, randomisées en double aveugle avec des groupes parallèles et avec des critères d'inclusion et d'exclusion ainsi que des collectifs de patients similaires. Ces études ont comparé le risque de survenue d'un événement indésirable cardiovasculaire majeur (ECM, défini comme un décès consécutif à un événement cardiovasculaire, un infarctus du myocarde non fatal ou un accident vasculaire cérébral non fatal) entre la canagliflozine et un placebo dans le contexte d'un traitement standard du diabète et d'une maladie cardiovasculaire athérosclérotique.
Dans l'étude CANVAS les patients ont été randomisés par un procédé aléatoire selon un ratio 1:1:1 et assignés à un traitement par canagliflozine 100 mg, canagliflozine 300 mg ou placebo pertinent. Dans l'étude CANVAS-R les patients ont été randomisés par un procédé aléatoire selon un ratio 1:1 et assignés à un traitement par canagliflozine 100 mg ou placebo pertinent. À la discrétion du médecin investigateur, une augmentation de la dose à 300 mg a été autorisée après la semaine 13, en fonction de la tolérance et des besoins glycémiques. Pour garantir aux sujets de recevoir un traitement contre le diabète et l'athérosclérose conforme au standard de prise en charge, un ajustement des traitements concomitants correspondants a été autorisé selon l'appréciation de l'investigateur.
Au total 10 134 patients (4327 dans l'étude CANVAS et 5807 dans l'étude CANVAS-R; au total 4344 sujets ont été attribués aléatoirement au groupe placebo et 5790 au traitement par canagliflozine) ont été traités sur une période de 149 semaines (valeur moyenne; la durée du traitement a été de 223 semaines dans l'étude CANVAS et de 94 semaines dans l'étude CANVAS-R). Le statut vital a été saisi chez 99,6% des participants dans les deux études. Environ 78% des sujets étaient caucasiens, 13% étaient asiatiques et 3% étaient noirs. L'âge moyen était de 63 ans et environ 64% étaient de sexe masculin.
Au moment de la sélection, tous les patients de l'étude présentaient un diabète sucré de type 2 insuffisamment contrôlé (HbA1c ≥7,0% à ≤10,5%). La valeur moyenne de l'HbA1c au début de l'étude était de 8,2% et la durée moyenne du diabète était de 13,5 ans. Environ 31%, 21% et respectivement 18% des sujets présentaient des antécédents de neuropathie, de rétinopathie et de néphropathie. 80% des patients présentaient une fonction rénale normale ou légèrement insuffisante au début de l'étude et 20% présentaient une insuffisance rénale modérée (DFGe moyen: 77 ml/min/1,73 m2). Au début de l'étude les patients suivaient un traitement par un antidiabétique au moins, incluant la metformine (77%), l'insuline (50%) et une sulfonylurée (43%).
66 pour-cent des sujets présentaient des antécédents de maladie cardiovasculaire manifeste, dont 56% une maladie coronarienne, 19% une affection cérébrovasculaire et 21% une maladie vasculaire périphérique; 14% présentaient des antécédents d'insuffisance cardiaque. Au début de l'étude la pression artérielle systolique moyenne était de 137 mmHg et la pression artérielle diastolique moyenne de 78 mmHg, le taux moyen de LDL s'élevait à 89 mg/dl, le taux moyen de HDL à 46 mg/dl et le rapport albuminurie/créatininurie (ACR) à 115 mg/g. Au début de l'étude près de 80% des patients étaient sous traitement par des inhibiteurs du système rénine-angiotensine, 54% sous traitement par des bêtabloquants, 13% sous traitement par des diurétiques de l'anse, 36% sous traitement par d'autres diurétiques, 75% sous traitement par des statines et 74% sous traitement par des antiagrégants plaquettaires (principalement de l'acide acétylsalicylique).
Le critère d'évaluation principal du programme CANVAS a été la durée de la période écoulée jusqu'à la première apparition d'un ECM. Les HR pour l'EMC et l'IC à 95% chez les patients traités par la canagliflozine, comparés aux patients du groupe placebo, ont été évalués à l'aide d'un modèle de régression de Cox constant, avec une stratification selon l'étude et une maladie cardiovasculaire manifeste.
La canagliflozine a entraîné une réduction significative du risque de première survenue du critère d'évaluation principal combiné ECM (HR: 0,86; IC à 95% 0,75, 0,97) auquel chaque composante ECM a contribué. Les résultats pour les deux dosages recommandés de canagliflozine (100 mg et 300 mg) étaient cohérents avec les résultats obtenus pour les données regroupées. L'efficacité de la canagliflozine concernant les ECM était différente selon que les patients présentaient déjà au début de l'étude une maladie cardiovasculaire manifeste (HR: 0,82; IC à 95% 0,72, 0,95) ou seulement des facteurs de risque cardiovasculaire (HR: 0,98; IC à 95% 0,74, 1,30).
2011 patients présentaient un DFGe situé entre 30 et <60 ml/min/1,73 m2. Les résultats portant sur l'ECM dans ce sous-groupe ont été en corrélation avec les résultats globaux.
Tableau 9: Effet du traitement pour le critère d'évaluation principal et ses composantes
|
|
Placebo
N=4347
Taux d'événements pour 1000 années-patients
|
Canagliflozine
N=5795
Taux d'événements pour 1000 années-patients
|
Hazard Ratio
(IC à 95%)
| |
Combinaison de décès consécutif à un événement cardiovasculaire, d'infarctus du myocarde non fatal, ou d'accident vasculaire cérébral non fatal (durée de l'intervalle jusqu'à la première apparition; groupe d'analyse en intention de traiter)1
|
31,48
|
26,93
|
0,86 (0,75-0,97)
| |
Décès consécutif à un événement cardiovasculaire
|
12,84
|
11,60
|
0,87 (0,72-1,06)
| |
Infarctus du myocarde non fatal
|
11,61
|
9,74
|
0,85 (0,69-1,05)
| |
Accident vasculaire cérébral non fatal
|
8,39
|
7,12
|
0,90 (0,71-1,15)
|
1 Valeur de p pour la supériorité (bilatérale) = 0,0158
Résultats selon les sous-groupes présence ou absence d'une maladie cardiovasculaire manifeste:
L'analyse primaire de l'efficacité s'est basée sur la population globale. Une analyse par sous-groupes prédéfinie pour les patients avec ou sans maladie cardiovasculaire manifeste antérieure n'a certes révélé aucun indice net d'hétérogénéité de l'efficacité entre ces deux groupes (valeur de p d'interaction = 0,1803); toutefois, une réduction cliniquement pertinente du risque cardiovasculaire n'a été observée que chez les patients présentant une maladie cardiovasculaire manifeste antérieure (HR: 0,82; IC à 95% 0,72, 0,95) et non chez les patients sans maladie manifeste antérieure (HR: 0,98; IC à 95%: 0,74, 1,30).
Basé sur l'estimateur de Kaplan-Meier de la première apparition d'un ECM, une diminution de l'ECM dans le groupe traité par la canagliflozine a déjà été observée à la semaine 26 et s'est poursuivie pendant toute l'étude restante.
Mortalité globale:
Dans le groupe combiné de canagliflozine, le HR pour la mortalité globale a été de 0,87 (0,74; 1,01), comparé au placebo.
Insuffisance cardiaque traitée à l'hôpital
En comparaison avec le placebo, la canagliflozine a diminué le risque d'insuffisance cardiaque nécessitant une hospitalisation (HR: 0,67; IC à 95% [0,52; 0,87]).
Critères d'évaluation rénaux dans le programme CANVAS
Dans le programme CANVAS, le HR pour le temps jusqu'à la survenue de la première néphropathie confirmée (doublement de la créatinine sérique, nécessité d'une thérapie de remplacement rénal, décès d'origine rénale) était de 0,53 (IC à 95%: 0,33; 0,84) pour la canagliflozine (1,5 événement pour 1000 années-patients) par rapport au placebo (2,8 événements pour 1000 années-patients). De plus, la canagliflozine a réduit la progression de l'albuminurie de 25,8% contre 29,2% sous placebo (HR: 0,73; IC à 95%: 0,67, 0,79), chez des patients présentant une normo- ou microalbuminurie avant le début du traitement. Bien que le traitement par des inhibiteurs de l'ECA et/ou un ARA ne représentait pas un critère d'inclusion dans le programme CANVAS, 80% des participants prenaient un inhibiteur de l'ECA et/ou un ARA au moment de la randomisation.
Résultats rénaux de l'étude CREDENCE
L'effet de 100 mg de canagliflozine sur les résultats rénaux observés chez des adultes atteints de diabète sucré de type 2 et d'une néphropathie diabétique (ND) définie par un taux de filtration glomérulaire estimé (DFGe) de 30 à <90 ml/min/1,73 m2 ou une ClCr de 30 à <90 ml/min et une albuminurie (>300 à 5000 mg/g de créatinine) a été estimé au cours de l'étude CREDENCE («Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation Trial»). Il s'agissait d'une étude en groupes parallèles contrôlés par placebo, multicentrique, internationale, randomisée (1:1), en double aveugle, guidée par les événements. Au total, 4401 participants ont été randomisés afin de former l'ensemble des données d'analyse en ITT (Intent-To-Treat), utilisé pour les analyses d'efficacité principales, alors que 4397 participants traités étaient disponibles pour l'évaluation de la sécurité. Au cours de l'étude CREDENCE, le risque de survenue d'une néphropathie diabétique (définie comme critère d'évaluation composite par l'insuffisance rénale terminale, le doublement de la créatinine sérique et le décès d'origine rénale ou cardiovasculaire) a été examiné et comparé lors de la prise de 100 mg de canagliflozine ou d'un placebo sur fond de traitement standard d'une néphropathie diabétique (notamment inhibiteur de l'enzyme de conversion de l'angiotensine [inhibiteur de l'ECA] et antagoniste du récepteur de l'angiotensine [ARA]).
Au cours de l'étude CREDENCE les sujets ont été traités par 100 mg de canagliflozine ou le placebo, stratifiés d'après le DFGe au moment du dépistage (30 à <45, 45 à <60, 60 à <90 ml/min/1,73 m2) ou la ClCr (30 à <45, 45<60, 60 à <90 ml/min) jusqu'au commencement de la dialyse ou jusqu'à la transplantation rénale.
Au total 4397 sujets ont été traités, la durée moyenne d'exposition était de 115 semaines. Le statut vital a été saisi chez 99,9% des sujets de l'étude. L'âge moyen était de 63 ans, 66% étaient de sexe masculin.
La valeur initiale moyenne de l'HbA1c était de 8,3%, la valeur initiale médiane du quotient albumine/créatinine dans l'urine était de 927 mg/g. Les principes actifs antihyperglycémiants les plus fréquemment utilisés en début d'étude étaient l'insuline (65,5%), les dérivés biguanides (57,8%) et les sulfonylurées (28,8%). Presque tous les sujets (99,9%) recevaient un inhibiteur de l'ECA ou un ARA au moment de la randomisation. Environ 92% des sujets recevaient un traitement cardiovasculaire au début de l'étude (en dehors des inhibiteurs de l'ECA et des ARA), environ 60% étaient traités par un principe actif antithrombotique (y compris l'acide acétylsalicylique) et 69% par des statines.
Au début de l'étude, le DFGe moyen était de 56,2 ml/min/1,73 m2 (environ 60% de la population de l'étude présentait un DFGe <60 ml/min/1,73 m2). En moyenne, les participants étaient diabétiques depuis environ 16 ans. Le pourcentage de patients présentant des antécédents d'affection cardiovasculaire était de 50,4%; un antécédent d'insuffisance cardiaque était présent chez 14,8% des patients. En plus de la néphropathie diabétique, environ 64% de la population souffrait d'au moins une autre complication microvasculaire.
Pour le critère d'évaluation principal et pour les critères d'évaluation secondaires, les HR et l'IC à 95% correspondant ont été évalués à l'aide d'un modèle stratifié de régression aléatoire proportionnelle de Cox (avec le traitement comme variable explicative et stratifiés d'après le DFGe au moment du dépistage [30 à <45, 45 à <60, 60 à <90 ml/min/1,73 m2] ou la ClCr [30 à <45, 45 à <60, 60 à <90 ml/min]).
Le critère d'évaluation principal composite de l'étude CREDENCE était le délai jusqu'à/au a) la première survenue d'une insuffisance rénale terminale (IRT; définie par la chute du DFGe en dessous de 15 ml/min/1,73 m2 ou de la ClCr sous 15 ml/min) ou le début de la dialyse chronique ou une transplantation rénale, b) doublement de la créatinine sérique, c) décès d'origine rénale, ou d) décès d'origine cardiovasculaire.
La canagliflozine à 100 mg a diminué le risque de première survenue du critère d'évaluation principal composite (IRT, doublement de la créatinine sérique et décès d'origine rénale ou cardiovasculaire), de manière significative [p <0,0001; HR: 0,70; IC à 95%: 0,59, 0,82] (voir Figure 1). L'effet du traitement était comparable dans tous les sous-groupes, dont les trois sous-groupes définis par le DFGe et chez les sujets présentant ou non des antécédents d'affection cardiovasculaire.
Sur la base de la courbe de Kaplan-Meier du délai jusqu'à la première survenue du critère d'évaluation principal composite, l'effet sur le traitement de 100 mg de canagliflozine était reconnaissable à partir de la semaine 52 et se poursuivait jusqu'à la fin de l'étude.
Les effets de 100 mg de canagliflozine sur les critères d'évaluation secondaires sont résumés dans la Figure 1.
Figure 1: Effet du traitement sur les critères d'évaluation principaux et secondaires et leurs composantes
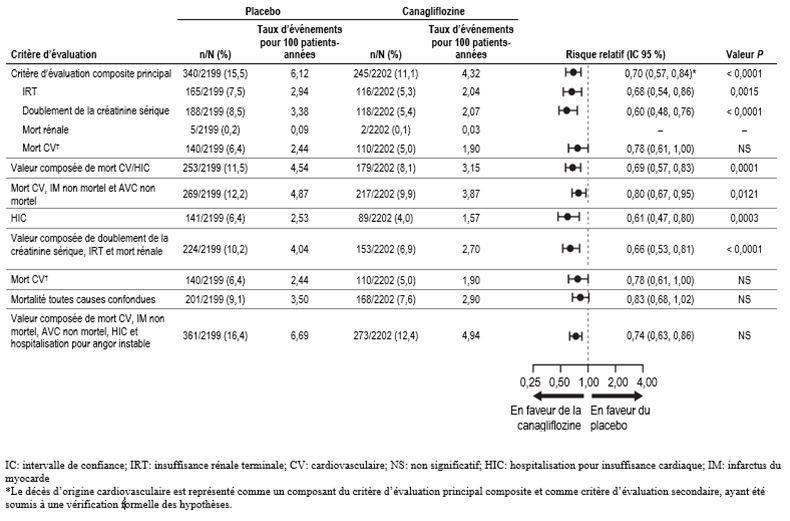
Comme le représente la Figure 2, le DFGe a baissé linéairement et progressivement chez les patients traités par placebo; en revanche, dans le groupe canagliflozine, on a observé une réduction abrupte à la semaine 3, qui s'estompait par la suite; la réduction moyenne calculée par la méthode des moindres carrés était inférieure après la semaine 52 dans le groupe canagliflozine par rapport au groupe placebo, l'effet du traitement se poursuivait jusqu'à la fin du traitement.
Figure 2: Variation moyenne des moindres carrés du DFGe par rapport aux valeurs initiales au fil du temps (analyses des patients sous traitement)
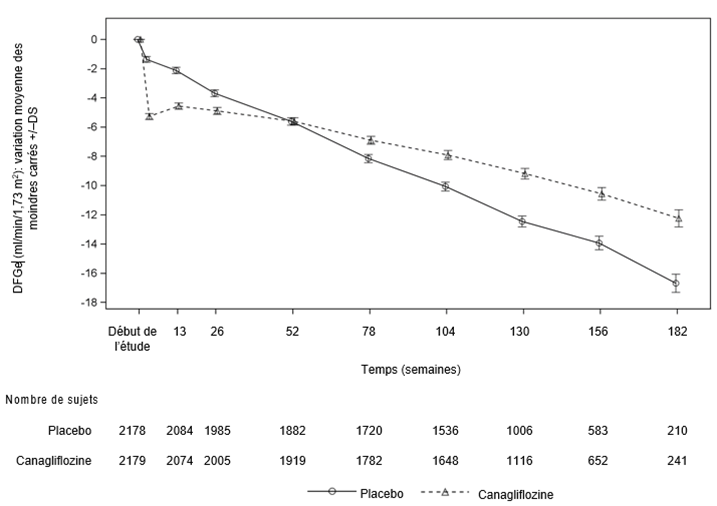
Au cours de l'étude CREDENCE, le taux d'incidence des effets indésirables rénaux du groupe 100 mg de canagliflozine était inférieur à celui du groupe placebo (57 pour 1000 années-patients dans le groupe canagliflozine et 79 pour 1000 années-patients dans le groupe placebo).
PharmacocinétiqueLa pharmacocinétique de la canagliflozine est globalement comparable chez les sujets sains et chez les patients atteints de diabète de type 2.
Absorption
La biodisponibilité absolue moyenne de la canagliflozine administrée par voie orale est d'environ 65%. L'administration concomitante de canagliflozine et d'un repas riche en graisses n'a pas eu d'effets sur la pharmacocinétique de la canagliflozine; Invokana peut donc être pris au cours ou en dehors des repas. Toutefois, en raison de la diminution potentielle des excursions glycémiques postprandiales liées au retard d'absorption intestinale du glucose, il est recommandé de prendre Invokana de préférence avant le premier repas de la journée (voir «Posologie/Mode d'emploi»).
Après administration orale d'une dose unique de 100 mg ou de 300 mg à des sujets sains, la canagliflozine a été rapidement absorbée et les concentrations plasmatiques maximales (Tmax médian) ont été mesurées 1 à 2 heures après la prise. L'AUC moyenne a été respectivement de 6,8 µg.h/ml et de 22,9 µg.h/ml et la concentration maximale (Cmax) respectivement de 1,1 µg.h/ml et 2,8 µg.h/ml après des doses de 100 mg et 300 mg. L'augmentation de la Cmax plasmatique et de l'AUC de la canagliflozine a été proportionnelle à la dose entre 50 mg et 300 mg. L'état d'équilibre a été atteint après 4 à 5 jours, après administration d'une dose de 100 mg et 300 mg de canagliflozine une fois par jour. La canagliflozine ne présente pas un profil pharmacocinétique temps-dépendant et s'est accumulée dans le plasma, atteignant des concentrations jusqu'à 36% plus élevées après des doses multiples de 100 mg et 300 mg.
Distribution
Après une perfusion intraveineuse unique chez des sujets sains, le volume moyen de distribution de la canagliflozine à l'état d'équilibre a été de 83,5 l, ce qui indique que la distribution tissulaire est importante. La canagliflozine est fortement liée aux protéines plasmatiques (99%), principalement à l'albumine. La liaison aux protéines est indépendante des concentrations plasmatiques de canagliflozine. La liaison aux protéines plasmatiques n'est pas modifiée de façon significative chez les patients atteints d'insuffisance rénale ou hépatique. Le cycle entéro-hépatique de la canagliflozine est peu important.
Métabolisme
La O-glucurono-conjugaison est la principale voie d'élimination métabolique de la canagliflozine, laquelle est transformée essentiellement par l'UGT1A9 et l'UGT2B4 en deux métabolites O-glucuronides inactifs. Des augmentations de l'AUC de la canagliflozine de 26% ont été observées chez des sujets porteurs de l'allèle UGT1A9*3 et de 18% chez des sujets porteurs de l'allèle UGT2B4*2. Ces augmentations de l'exposition à la canagliflozine ne sont pas considérées comme cliniquement significatives. Le métabolisme (oxydatif) de la canagliflozine, médié par le CYP3A4, est minime (environ 7%) chez l'être humain.
Élimination
Après administration orale d'une dose unique de [C14]canagliflozine à des sujets sains, au total environ 61% de la dose radioactive ont été éliminés dans les fèces, respectivement 41,5%, 7,0% et 3,2% sous forme de canagliflozine, de métabolite hydroxylé et de métabolite O-glucuronide. Environ 33% de la dose radioactive administrée ont été éliminés dans l'urine, essentiellement sous forme de métabolites O-glucuronides (30,5%). Moins de 1% de la dose a été éliminé dans l'urine sous forme de canagliflozine inchangée. La clairance rénale pour les doses de 100 mg et 300 mg a varié entre 1,30 et 1,55 ml/min.
La canagliflozine est un médicament à faible clairance, c.-à-d. avec une clairance systémique moyenne d'environ 192 ml/min après administration intraveineuse à des sujets sains.
La demi-vie terminale apparente (t½) a été de 10,6 heures pour la dose de 100 mg et de 13,1 heures pour la dose de 300 mg.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Après administration d'une dose unique de 300 mg de canagliflozine, les rapports des moyennes géométriques de la Cmax et de l'AUC∞ de la canagliflozine ont été respectivement de 107% et 110% chez les patients présentant une insuffisance hépatique légère (classe A de Child-Pugh) par rapport aux valeurs observées chez les patients ayant une fonction hépatique normale et respectivement de 96% et 111% chez les patients présentant une insuffisance hépatique modérée (classe B de Child-Pugh) par rapport aux valeurs observées chez les patients ayant une fonction hépatique normale.
Ces différences ne sont pas considérées comme cliniquement significatives. Aucun ajustement de la dose n'est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée. Il n'existe aucune expérience clinique de l'utilisation chez les patients de classe C de Child-Pugh (insuffisance hépatique sévère); Invokana n'est donc pas recommandé pour ce groupe de patients.
Troubles de la fonction rénale
La pharmacocinétique de la canagliflozine à la dose de 200 mg a été évaluée dans une étude à dose unique ouverte, menée chez des sujets présentant différents degrés d'insuffisance rénale [classifiée selon le DFG estimé à l'aide de la formule Modification-of-Diet-in-Renal-Disease (MDRD)] par rapport à des sujets sains. L'étude a inclus 3 sujets présentant une fonction rénale normale (DFGe ≥90 ml/min/1,73 m2), 10 sujets présentant une insuffisance rénale légère (DFGe compris entre 60 et <90 ml/min/1,73 m2), 9 sujets présentant une insuffisance rénale modérée (DFGe compris entre 30 et <60 ml/min/1,73 m2) et 10 sujets présentant une insuffisance rénale sévère (DFGe compris entre 15 et <30 ml/min/1,73 m2) ainsi que 8 sujets en insuffisance rénale terminale (ESRD) sous hémodialyse.
La Cmax de la canagliflozine était augmentée de 13% chez les sujets atteints d'insuffisance rénale légère, de 29% chez les sujets atteints d'insuffisance rénale modérée et de 29% chez les sujets atteints d'insuffisance rénale sévère, mais n'était pas augmentée chez les patients hémodialysés. Par rapport aux sujet sains, l'AUC plasmatique de la canagliflozine a augmenté de 17%, 63% et 50% chez les sujets présentant une insuffisance rénale légère, modérée et sévère, mais a été semblable chez les sujets atteints d'ESRD et chez les sujets sains (voir «Posologie/Mode d'emploi», «Mises en garde et précautions» et «Effets indésirables»).
La canagliflozine a été éliminée de façon négligeable par hémodialyse.
Patients âgés
D'après une analyse pharmacocinétique de population, l'âge n'a pas eu d'effet cliniquement significatif sur la pharmacocinétique de la canagliflozine (voir «Posologie/Mode d'emploi», «Mises en garde et précautions» et «Effets indésirables»).
Enfants et adolescents
Une étude de phase 1 a évalué la pharmacocinétique et la pharmacodynamique de la canagliflozine chez des enfants et des adolescents atteints d'un diabète sucré de type 2 âgés d'au moins 10 ans et de moins de 18 ans. La réponse pharmacocinétique et pharmacodynamique observée a correspondu à celle des adultes.
Autres populations
Aucun ajustement de la dose en fonction du sexe, de l'origine ethnique ou de l'indice de masse corporelle (IMC) n'est nécessaire. D'après une analyse pharmacocinétique de population, ces caractéristiques n'ont eu aucun effet cliniquement significatif sur la pharmacocinétique de la canagliflozine.
Données précliniquesLes données précliniques issues des études conventionnelles de pharmacologie de sécurité, de toxicité en administration répétée, de génotoxicité et de toxicité de reproduction n'ont révélé aucun risque particulier pour l'être humain. Dans les études de toxicité sur le développement, des troubles minimes du développement des reins et de la maturation fœtale (formation osseuse) n'ont pas pu être exclus. Une étude réalisée chez des rats juvéniles ayant reçu de la canagliflozine du jour 21 postnatal au jour 90 n'a pas montré d'augmentation de la sensibilité par rapport aux effets observés chez les rats adultes.
Carcinogénicité
La canagliflozine n'a entraîné aucune augmentation de l'incidence des tumeurs chez des souris CDI mâles et femelles, dans une étude de deux ans réalisée avec des doses de 10, 30 et 100 mg/kg. La dose la plus élevée, de 100 mg/kg, correspondait à jusqu'à 14 fois l'exposition (sur la base de l'AUC) à la dose clinique de 300 mg. Une incidence accrue de tumeurs testiculaires à cellules de Leydig a été observée chez des rats Sprague-Dawley mâles à toutes les doses testées de canagliflozine de 10, 30 et 100 mg/kg; la dose la plus faible, de 10 mg/kg, correspondait à environ 1,5 fois l'exposition (sur la base de l'AUC) à la dose clinique de 300 mg. Chez des rats Sprague-Dawley mâles et femelles, les doses plus élevées de canagliflozine (100 mg/kg) ont entraîné une augmentation de l'incidence des phéochromocytomes et des tumeurs tubulaires rénales; sur la base de l'exposition (selon l'AUC), la No-Observable-Effect-Level (NOEL) de 30 mg/kg/jour pour les phéochromocytomes et les tumeurs tubulaires rénales correspond à environ 4,5 fois l'exposition à la dose clinique journalière de 300 mg. D'après des études mécanistiques précliniques et cliniques, les tumeurs à cellules de Leydig, les tumeurs tubulaires rénales et les phéochromocytomes sont spécifiques au rat. La cause des tumeurs tubulaires rénales et des phéochromocytomes induits par la canagliflozine chez le rat semble être une malabsorption des glucides secondaire à l'effet inhibiteur de la canagliflozine sur le SGLT1 intestinal dans l'intestin du rat; les études mécanistiques cliniques n'ont pas mis en évidence de malabsorption des glucides chez l'être humain à des doses de canagliflozine jusqu'à deux fois supérieures à la dose maximale recommandée en clinique. Les tumeurs à cellules de Leydig sont associées à une augmentation de l'hormone lutéinisante (LH), ce qui est un mécanisme connu de formation des tumeurs à cellules de Leydig chez le rat. Dans une étude clinique de 12 semaines, aucune augmentation du taux basal de LH n'a été observée chez les hommes traités par la canagliflozine.
Mutagénicité
La canagliflozine n'a pas été mutagène dans le test d'Ames avec ou sans activation métabolique. La canagliflozine a été mutagène dans un test du lymphome de souris in vitro, mais seulement après activation métabolique. La canagliflozine n'a été ni mutagène ni clastogène chez le rat dans un test du micronoyau in vivo après administration orale ainsi que dans un test des comètes in vivo après administration orale.
Toxicité de reproduction
Dans une étude sur le développement prénatal et postnatal, la canagliflozine, administrée à des rats femelles du jour 6 de la gestation au jour 20 de la lactation, à des doses toxiques pour les mères >30 mg/kg/jour (correspondant à ≥5,9 fois l'exposition humaine à la canagliflozine à la dose maximale recommandée chez l'être humain [DMRH]), a entraîné une diminution du poids corporel chez la progéniture mâle et femelle. La toxicité maternelle s'est limitée à une diminution de la prise de poids corporel.
Dans des études chez le rat, la canagliflozine n'a eu aucun effet négatif sur le développement embryonnaire précoce, le comportement d'accouplement et la fertilité jusqu'aux doses maximales de 100 mg/kg (jusqu'à 19 fois l'exposition (sur la base de l'AUC) à la dose clinique de 300 mg).
Les données pharmacodynamiques/toxicologiques issues d'études expérimentales menées chez l'animal ont mis en évidence l'excrétion de la canagliflozine/de ses métabolites dans le lait maternel ainsi que des effets pharmacologiques chez la progéniture allaitée des rates et chez les rats juvéniles ayant reçu de la canagliflozine.
Remarques particulièresInfluence sur les méthodes de diagnostic
Interférences du médicament avec les analyses de laboratoire
Tests de 1,5-AG
Les augmentations de l'excrétion urinaire du glucose induites par la canagliflozine peuvent diminuer la rétro-résorption de 1,5-anhydroglucitol (1,5-AG) et rendre la mesure du 1,5-AG peu fiable pour évaluer le contrôle de la glycémie. Par conséquent, les tests de 1,5-AG ne doivent pas être utilisés par les patients sous canagliflozine pour évaluer le contrôle de la glycémie. Pour toute information plus détaillée, il est conseillé de contacter le fabricant du test 1,5-AG.
Test de glucose urinaire
En raison du mécanisme d'action, les patients qui prennent Invokana auront un test de glycosurie positif.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques concernant le stockage
Conserver à 15-30°C.
Conserver dans l'emballage d'origine.
Conserver hors de portée des enfants.
Numéro d’autorisation62956 (Swissmedic).
PrésentationInvokana comprimés pelliculés à 100 mg: 30, 100 comprimés pelliculés (B)
Invokana comprimés pelliculés à 300 mg: 30, 100 comprimés pelliculés (B)
Invokana est disponible dans des plaquettes thermoformées unidoses perforées en PVC/aluminium contenant chacune 10 comprimés pelliculés.
Titulaire de l’autorisationJanssen-Cilag AG, Zug, ZG.
Mise à jour de l’informationMai 2024.
|