Propriétés/EffetsCode ATC
A10BK02
Mécanisme d'action
Le co-transporteur sodium-glucose de type 2 (SGLT2), exprimé dans les tubules rénaux proximaux, est responsable de la majorité de la réabsorption du glucose filtré depuis la lumière tubulaire. La canagliflozine est un inhibiteur du SGLT2, actif par voie orale. En inhibant le SGLT2, la canagliflozine diminue la réabsorption du glucose filtré et diminue le seuil rénal du glucose (RTG). Elle augmente ainsi l'excrétion urinaire du glucose (UGE: urinary glucose excretion), ce qui diminue les concentrations plasmatiques du glucose chez les patients atteints de diabète de type 2. Ce mécanisme n'est pas dépendant de l'insuline. L'augmentation de l'excrétion urinaire du glucose (UGE) résultant de l'inhibition du SGLT2 conduit par ailleurs à une diurèse osmotique, ainsi qu'à une diminution de la pression artérielle systolique par un effet diurétique. L'augmentation de l'excrétion urinaire du glucose (UGE) entraîne une perte de calories. L'UGE augmente le risque d'infections des voies urinaires et d'infections mycosiques génitales.
Les études n'ont pas mis en évidence de malabsorption du glucose sous canagliflozine.
La canagliflozine augmente l'apport de sodium au tube distal par un blocage de la réabsorption du sodium et du glucose SGLT2-dépendante, ce qui explique que le rétrocontrôle tubulo-glomérulaire augmente; cela est associé, dans les modèles diabétiques précliniques ainsi que dans les études cliniques, à une diminution de la pression intraglomérulaire et de l'hyperfiltration, et potentiellement à un effet néphro-protecteur.
Pharmacodynamique
Après l'administration orale de doses uniques et multiples de canagliflozine à des patients adultes atteints de diabète de type 2, des diminutions dose-dépendantes du RTG et des augmentations de l'excrétion urinaire du glucose ont été observées. Lors d'une valeur initiale de RTG d'environ 13 mmol/l, un abaissement maximal du RTG moyen sur 24 heures à environ 4 mmol/l à 5 mmol/l a été observé à la dose journalière de 300 mg chez les patients atteints de diabète de type 2 dans des études de phase 1, ce qui suggère que le risque d'hypoglycémie induite par le traitement est faible. Chez les sujets atteints de diabète de type 2 traités par 100 mg ou 300 mg de canagliflozine, la diminution du RTG a entraîné une augmentation de l'excrétion urinaire du glucose comprise entre 77 g/jour et 119 g/jour dans toutes les études de phase 1. L'excrétion urinaire du glucose observée correspond à une perte de 308 à 476 kcal/jour. Les diminutions du RTG et les augmentations de l'excrétion urinaire du glucose ont persisté pendant la période de traitement de 26 semaines chez des patients atteints de diabète de type 2. Après une légère augmentation (le plus souvent <400-500 ml) du volume urinaire quotidien, celui-ci a diminué à nouveau au cours des premiers jours du traitement. La canagliflozine a augmenté de façon transitoire l'excrétion urinaire d'acide urique (augmentation de 19% par rapport à la valeur initiale le premier jour et diminution à 6% le jour 2 et à 1% le jour 13). Ceci s'est accompagné d'une réduction persistante du taux sérique d'acide urique d'environ 20%.
Dans une étude à dose unique réalisée chez des patients adultes atteints de diabète de type 2, l'administration de 300 mg avant un repas mixte a retardé l'absorption intestinale du glucose et a diminué la glycémie postprandiale.
Électrophysiologie cardiaque
Dans une étude randomisée, en double aveugle, contrôlée contre placebo, avec un contrôle positif (moxifloxacine), 60 sujets adultes sains ont reçu une dose orale unique de 300 mg de canagliflozine, de 1200 mg de canagliflozine (quatre fois la dose maximale recommandée), de moxifloxacine ou de placebo. Aucune modification significative de l'intervalle QTc n'a été observée à la dose recommandée de 300 mg ni à la dose de 1200 mg. À la dose de 1200 mg, les concentrations plasmatiques maximales de canagliflozine ont été environ 1,4 fois supérieures aux concentrations maximales atteintes à l'état d'équilibre avec une dose de 300 mg une fois par jour.
Efficacité clinique
Efficacité glycémique et sécurité chez les patients adultes
Au total, 10 285 patients adultes atteints de diabète de type 2 ont participé à neuf études cliniques d'efficacité et de sécurité, contrôlées et en double aveugle, ayant évalué les effets d'Invokana sur le contrôle glycémique. La distribution ethnique était de 72% de Blancs, 16% d'Asiatiques, 4% de Noirs et 8% de membres d'autres groupes. 16% des patients étaient d'origine hispanique. Environ 58% des patients étaient de sexe masculin. L'âge moyen des patients était de 59,6 ans (fourchette de 21 ans à 96 ans). 3082 patients étaient âgés de 65 ans et plus et 510 patients de 75 ans et plus. 58% des patients présentaient un indice de masse corporelle (IMC) supérieur ou égal à 30 kg/m2. Pour le sous-groupe de patients présentant une insuffisance rénale modérée avec un DFGe initial compris entre 30 et <60 ml/min/1,73 m2, les données de 1087 patients, issues du programme de développement clinique, ont été regroupées et analysées.
Études contrôlées contre placebo
Invokana a été étudié en monothérapie, en bithérapie avec la metformine, en bithérapie avec une sulfonylurée, en trithérapie avec la metformine et une sulfonylurée, ainsi qu'en ajout à l'insuline (tableau 5). En résumé, le traitement par Invokana a entraîné des résultats cliniquement et statistiquement significatifs par rapport au placebo (p<0,001) en ce qui concerne le contrôle glycémique, y compris le taux d'HbA1c, la proportion de patients présentant un taux d'HbA1c <7%, les modifications de la glycémie à jeun (GAJ) par rapport à la valeur initiale et la glycémie postprandiale (GPP) (après 2 heures). En outre, des diminutions du poids corporel et de la pression artérielle systolique ont également été observées par rapport au placebo.
Tableau 5: Résultats d'efficacité des études cliniques contrôlées contre placeboa
|
Monothérapie (26 semaines)
| |
|
Invokana
|
Placebo
(N=192)
| |
100 mg
(N=195)
|
300 mg
(N=197)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,06
|
8,01
|
7,97
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-0,77
|
-1,03
|
0,14
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-0,91b
(-1,09; -0,73)
|
-1,16b
(-1,34; -0,99)
|
N/Ac
| |
Patients (%) ayant atteint un taux d'HbA1c<7%
|
44,5b
|
62,4b
|
20,6
| |
Glycémie à jeun (mmol/l)
| |
Valeur initiale (moyenne)
|
9,57
|
9,57
|
9,20
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,51
|
-1,94
|
0,46
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-1,97b
(-2,34; -1,60)
|
-2,41b
(-2,78; -2,03)
|
N/Ac
| |
Glycémie postprandiale à 2 heures (mmol/l)
| |
Valeur initiale (moyenne)
|
13,87
|
14,10
|
12,74
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-2,38
|
-3,27
|
0,29
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-2,67b
(-3,28; -2,05)
|
-3,55b
(-4,17; -2,94)
|
N/Ac
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
85,9
|
86,9
|
87,5
| |
% de variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-2,8
|
-3,9
|
-0,6
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-2,2b
(-2,9; -1,6)
|
-3,3b
(-4,0; -2,6)
|
N/Ac
|
|
Bithérapie avec la metformine (26 semaines)
| |
|
Invokana + metformine
|
Placebo + metformine
(N=183)
| |
100 mg
(N=368)
|
300 mg
(N=367)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
7,94
|
7,95
|
7,96
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-0,79
|
-0,94
|
-0,17
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-0,62b
(-0,76; -0,48)
|
-0,77b
(-0,91; -0,64)
|
N/Ac
| |
Patients (%) ayant atteint un taux d'HbA1c<7%
|
45,5
|
57,8
|
29,8
| |
Glycémie à jeun (mmol/l)
| |
Valeur initiale (moyenne)
|
9,36
|
9,59
|
9,12
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,52
|
-2,10
|
0,14
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-1,65b
(-1,99; -1,32)
|
-2,23b
(-2,57; -1,90)
|
N/Ac
| |
Glycémie postprandiale à 2 heures (mmol/l)
| |
Valeur initiale (moyenne)
|
14,30
|
14,54
|
13,81
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-2,66
|
-3,17
|
-0,55
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-2,12b
(-2,73; -1,51)
|
-2,62b
(-3,24; -2,01)
|
N/Ac
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
88,7
|
85,4
|
86,7
| |
% de variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-3,7
|
-4,2
|
-1,2
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-2,5b
(-3,1; -1,9)
|
-2,9b
(-3,5; -2,3)
|
N/Ac
|
|
Trithérapie avec la metformine et une sulfonylurée (26 semaines)
| |
|
Invokana + metformine et sulfonylurée
|
Placebo +
metformine et sulfonylurée
(N=156)
| |
100 mg
(N=157)
|
300 mg
(N=156)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,13
|
8,13
|
8,12
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-0,85
|
-1,06
|
-0,13
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-0,71b
(-0,90;-0,52)
|
-0,92b
(-1,11;-0,73)
|
N/Ac
| |
Patients (%) ayant atteint un taux d'HbA1c<7%
|
43,2b
|
56,6b
|
18,0
| |
Glycémie à jeun (mmol/l)
| |
Valeur initiale (moyenne)
|
9,60
|
9,34
|
9,42
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,01
|
-1,69
|
0,23
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-1,24b
(-1,75; -0,73)
|
-1,92b
(-2,43; -1,41)
|
N/Ac
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
93,5
|
93,5
|
90,8
| |
% de variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-2,1
|
-2,6
|
-0,7
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-1,4b
(-2,1;-0,7)
|
-2,0b
(-2,7;-1,3)
|
N/Ac
|
|
Ajout à l'insulined (18 semaines)
| |
|
Invokana + insuline
|
Placebo + insuline
(N=565)
| |
100 mg
(N=566)
|
300 mg
(N=587)
| |
HbA1c(%)
| |
Valeur initiale (moyenne)
|
8,33
|
8,27
|
8,20
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-0,63
|
-0,72
|
0,01
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-0,65b
(-0,73; -0,56)
|
-0,73b
(-0,82; -0,65)
|
N/Ac
| |
Patients (%) ayant atteint un taux d'HbA1c<7%
|
19,8
|
24,7
|
7,7
| |
Glycémie à jeun (mmol/l)
| |
Valeur initiale (moyenne)
|
9,43
|
9,33
|
9,38
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,03
|
-1,39
|
0,22
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 95%)
|
-1,25b
(-1,55; -0,96)
|
-1,61b
(-1,90;-1,31)
|
N/Ac
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
96,9
|
96,7
|
97,7
| |
% de variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,8
|
-2,3
|
0,1
| |
Différence par rapport au placebo (moyenne des moindres carrés) (IC à 97,5%)
|
-1,9b
(-2,2; -1,5)
|
-2,4b
(-2,8; -2,0)
|
N/Ac
|
a Population en intention de traiter en utilisant la dernière observation rapportée dans l'étude avant l'emploi d'un traitement glycémique de secours.
b p<0,001 par rapport au placebo.
c Non applicable.
d Invokana en ajout à l'insuline (avec ou sans autres médicaments antihyperglycémiants).
Traitement adjuvant: canagliflozine associée à la metformine et à un inhibiteur de la dipeptidyl peptidase-4
La canagliflozine a été étudiée en traitement adjuvant chez des patients adultes n'ayant pas obtenu un contrôle glycémique approprié sous traitement antérieur par la metformine et la sitagliptine et a été dosée conformément au schéma de titration (dose initiale de 100 mg; augmentation à 300 mg après 6 semaines chez les patients ayant un contrôle glycémique insuffisant, un DFGe approprié et ayant bien toléré la canagliflozine dosée à 100 mg).
Chez ces patients, par rapport au placebo, le contrôle glycémique s'est amélioré (voir tableau 6) après administration de canagliflozine. De plus, par rapport au placebo, la canagliflozine a entraîné une diminution du poids corporel et de la tension artérielle systolique.
Tableau 6: Résultats de l'étude clinique de 26 semaines contrôlée contre placebo portant sur la canagliflozine en association avec la metformine et la sitagliptine*
|
Paramètres d'efficacité
|
Placebo +
metformine et sitagliptine
(N=106)
|
Canagliflozine +
metformine et sitagliptine
(N=107)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,38
|
8,53
| |
Variation par rapport à la valeur initiale (moyenne ajustée)
|
-0,01
|
-0,91
| |
Différence par rapport au placebo (moyenne ajustée) (IC à 95%)†
|
|
-0,89‡
(-1,19; -0,59)
| |
Patients (%) ayant atteint un taux d'HbA1c <7%
|
12
|
32§
| |
Glycémie à jeun (mg/dl)
| |
Valeur initiale (moyenne)
|
180
|
186
| |
Variation par rapport à la valeur initiale (moyenne ajustée)
|
-3
|
-30
| |
Différence par rapport au placebo (moyenne ajustée) (IC à 95%)†
|
|
-27‡
(-40; -14)
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
89,9
|
93,8
| |
% de variation par rapport à la valeur initiale (moyenne ajustée)
|
-1,6
|
-3,4
| |
Différence par rapport au placebo (moyenne ajustée) (IC à 95%)†
|
|
-1,8‡
(-2,7; -0,9)
|
* Population en intention de traiter (Intent-to-treat-Population)
† La moyenne ajustée et l'IC ont été calculés avec un modèle mixte pour mesures répétées
‡ p<0,001
§ p<0,01
Études contrôlées contre traitement actif
Invokana a été comparé au glimépiride en bithérapie (avec la metformine) et à la sitagliptine en trithérapie (avec la metformine et une sulfonylurée) (tableau 7) chez des patients adultes.
Tableau 7: Résultats d'efficacité des études contrôlées contre traitement actifa
|
Comparaison au glimépiride en bithérapie avec la metformine (52 semaines)
| |
|
Invokana + metformine
|
Glimépiride (titré) +
metformine
(N=482)
| |
100 mg
(N=483)
|
300 mg
(N=485)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
7,78
|
7,79
|
7,83
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-0,82
|
-0,93
|
-0,81
| |
Différence par rapport au glimépiride (moyenne des moindres carrés) (IC à 95%)
|
-0,01b
(−0,11;0,09)
|
-0,12b
(−0,22; −0,02)
|
N/Ac
| |
Patients (%) ayant atteint un taux d'HbA1c<7%
|
53,6
|
60,1
|
55,8
| |
Glycémie à jeun (mmol/l)
| |
Valeur initiale (moyenne)
|
9,18
|
9,09
|
9,20
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,35
|
-1,52
|
-1,02
| |
Différence par rapport au glimépiride (moyenne des moindres carrés) (IC à 95%)
|
-0,33
(−0,56; -0,11)
|
-0,51
(−0,73; −0,28)
|
N/Ac
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
86,8
|
86,6
|
86,6
| |
% de variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-4,2
|
-4,7
|
1,0
| |
Différence par rapport au glimépiride (moyenne des moindres carrés) (IC à 95%)
|
-5,2d
(−5,7; −4,7)
|
-5,7d
(−6,2; −5,1)
|
N/Ac
|
|
Comparaison à la sitagliptine en trithérapie avec la metformine et une sulfonylurée (52 semaines)
| |
|
Invokana 300 mg +
metformine et sulfonylurée
(N=377)
|
Sitagliptine 100 mg +
metformine et sulfonylurée
(N=378)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,12
|
8,13
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,03
|
-0,66
| |
Différence par rapport à la sitagliptine (moyenne des moindres carrés) (IC à 95%)
|
-0,37e (-0,50; -0,25)
|
N/Ac
| |
Patients (%) ayant atteint un taux d'HbA1c<7%
|
47,6
|
35,3
| |
Glycémie à jeun (mmol/l)
| |
Valeur initiale (moyenne)
|
9,42
|
9,09
| |
Variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-1,66
|
-0,32
| |
Différence par rapport à la sitagliptine (moyenne des moindres carrés) (IC à 95%)
|
-1,34 (-1,66; -1,01)
|
N/Ac
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
87,6
|
89,6
| |
% de variation par rapport à la valeur initiale (moyenne des moindres carrés)
|
-2,5
|
0,3
| |
Différence par rapport à la sitagliptine (moyenne des moindres carrés) (IC à 95%)
|
-2,8d (-3,3; -2,2)
|
N/Ac
|
a Population en intention de traiter en utilisant la LOCF dans l'étude avant l'emploi d'un traitement glycémique de secours.
b Invokana+metformine est considéré comme non inférieur à glimépiride+metformine, car la limite supérieure de l'intervalle de confiance est inférieure au seuil de non-infériorité prédéfini (<0,3%).
c Non applicable.
d p<0,01.
e Invokana+metformine+sulfonylurée est considéré comme non inférieur à sitagliptine+metformine+sulfonylurée, car la limite supérieure de l'intervalle de confiance est inférieure au seuil de non-infériorité prédéfini (<0,3%).
Canagliflozine en association avec la metformine en traitement initial
La canagliflozine a été étudiée en association avec la metformine chez des patients adultes atteints de diabète sucré de type 2 non préalablement traités chez qui des mesures alimentaires et l'exercice physique n'ont, à eux seuls, pas permis d'obtenir un contrôle glycémique approprié. L'association thérapeutique initiale (canagliflozine à 100 mg et metformine XR ou canagliflozine à 300 mg et metformine XR) était supérieure en termes d'amélioration du taux d'HbA1c par rapport à la canagliflozine en monothérapie (à 100 mg ou 300 mg) et à la metformine en monothérapie (voir tableau 8).
Tableau 8: Résultats de l'étude clinique de 26 semaines contrôlée contre principe actif portant sur la canagliflozine en association avec la metformine en traitement initial*
|
Paramètres d'efficacité
|
Metformine XR
(N=237)
|
Canagliflozine à 100 mg
(N=237)
|
Canagliflozine à 300 mg
(N=238)
|
Canagliflozine à 100 mg +
metformine XR
(N=237)
|
Canagliflozine à 300 mg +
metformine XR
(N=237)
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,81
|
8,78
|
8,77
|
8,83
|
8,90
| |
Variation par rapport à la valeur initiale (moyenne ajustée)
|
-1,30
|
-1,37
|
-1,42
|
-1,77
|
-1,78
| |
Différence par rapport à 100 mg de canagliflozine (moyenne ajustée) (IC à 95%)†
|
|
|
|
-0,40‡
(-0,59, -0,21)
|
| |
Différence par rapport à 300 mg de canagliflozine (moyenne ajustée) (IC à 95%)†
|
|
|
|
|
-0,36‡
(-0,56, -0,17)
| |
Différence par rapport à la metformine XR (moyenne ajustée) (IC à 95%)†
|
|
-0,06‡
(-0,26, 0,13)
|
-0,11‡
(-0,31, 0,08)
|
-0,46‡
(-0,66, -0,27)
|
-0,48‡
(-0,67, -0,28)
| |
Patients (%) ayant atteint un taux d'HbA1c <7%
|
43
|
39
|
43
|
50§§
|
57§§
| |
Poids corporel
| |
Valeur initiale (moyenne) en kg
|
92,1
|
90,3
|
93,0
|
88,3
|
91,5
| |
% de variation par rapport à la valeur initiale (moyenne ajustée)
|
-2,1
|
-3,0
|
-3,9
|
-3,5
|
-4,2
| |
Différence par rapport à la metformine XR (moyenne ajustée) (IC à 95%)†
|
|
-0,9§§
(-1,6, -0,2)
|
-1,8§
(-2,6, -1,1)
|
-1,4‡
(-2,1, -0,6)
|
-2,1‡
(-2,9, -1,4)
|
* Population en intention de traiter
† Moyenne ajustée des moindres carrés (LS) pour les covariables incluant la valeur initiale et le facteur de stratification
‡ p=0,001 ajusté
§ p<0,01 ajusté
§§ p<0,05 ajusté
Populations spécifiques
Dans quatre études sur des populations spécifiques (enfants à partir de 10 ans et adolescents, patients âgés, patients présentant un DFGe compris entre 30 et <50 ml/min/1,73 m2 et patients présentant une affection cardiovasculaire ou un risque accru d'une telle affection), Invokana a été ajouté au traitement antidiabétique actuel des patients (régime, monothérapie, association thérapeutique).
Enfants et adolescents
L'étude DIA3018 était une étude en groupes parallèles, randomisée, en double aveugle (DA), contrôlée contre placebo, à deux bras, multicentrique, d'une durée totale de 52 semaines, comportant une phase de traitement principal en DA de 26 semaines, suivie d'une phase d'extension en DA de 26 semaines. L'étude a inclus au total 171 adolescents et enfants à partir de 10 ans atteints de diabète sucré de type 2 et ayant un contrôle glycémique insuffisant (HbA1c ≥6,5% à ≤11,0%) qui avaient auparavant exclusivement suivi un régime alimentaire spécifique et fait de l'exercice physique, ou qui avaient reçu en complément d'un régime alimentaire et de l'exercice physique une monothérapie stable par de la metformine (avec ou sans insuline) ou une monothérapie stable par de l'insuline. Les participants à l'étude ont d'abord été randomisés selon un rapport 1:1 afin de recevoir un traitement par la canagliflozine 100 mg ou par le placebo. L'HbA1c moyenne au début de l'étude était de 8,0% et la durée moyenne du diabète sucré de type 2 était de 2 ans. L'âge moyen des patients ayant reçu de la canagliflozine était de 14,3 ans, 47,4% des patients ayant moins de 15 ans. Sur les 84 patients qui ont reçu de la canagliflozine, 33 patients ayant présenté une valeur de HbA1c ≥7,0% et un DFGe ≥60 ml/min/1,73 m2 à la semaine 12 ont été à nouveau randomisés à la semaine 13, 16 patients ayant continué de recevoir 100 mg et 17 ayant reçu une dose plus élevée, à savoir 300 mg. Avant l'étude, 14% des patients ont, comme traitement de fond, uniquement suivi un régime alimentaire et fait de l'exercice physique, 11% ont reçu de l'insuline en monothérapie, 29% de la metformine et de l'insuline, et 46% de la metformine en monothérapie.
Dans l'étude DIA3018, la distribution ethnique était de 42% d'Asiatiques, 42% de Blancs, 11% de Noirs ou Afro-américains, 5% d'autochtones d'Amérique ou d'Alaska et 36% se sont désignés comme étant des Hispaniques ou des Latinos. La valeur initiale moyenne du DFGe était de 157,3 ml/min/1,73 m2, et environ 16% de la population à l'étude présentaient une microalbuminurie.
Tableau 9: Analyse primaire du critère d'évaluation principal (estimation primaire, Treatment-Policy-Strategy): modification de l'HbA1c par rapport à la valeur initiale (%) à la semaine 26 en utilisant le modèle de mélange de motifs (Pattern Mixture Model); tous les participants à l'étude; ensemble complet d'analyses (étude JNJ28431754-DIA3018)
|
|
Placebo
(N=87)
|
Canagliflozine
(N=84)
| |
Valeur observée au début de l'étude (valeur initiale)
| |
N
|
87
|
84
| |
Moyenne (écart type, ET)
|
8,3 (1,35)
|
7,8 (1,31)
| |
Valeur observée à la semaine 26
| |
N
|
80
|
77
| |
Moyenne (ET)
|
8,6 (2,01)
|
7,3 (1,78)
| |
Modification par rapport à la valeur initiale à la semaine 26
| |
N
|
80
|
77
| |
Moyenne (ET)
|
0,3 (1,56)
|
-0,4 (1,44)
| |
Participants présentant des événements intercurrents
|
30
|
10
| |
Participants ayant arrêté le traitement avant la semaine 26
|
9
|
5
| |
Participants ayant reçu un traitement d'urgence avant la semaine 26
|
24
|
5
| |
Résumé des résultats à la semaine 26 en utilisant la règle de Rubin de combinaison de l'ANCOVA sur les ensembles de données calculées sur la base du modèle de mélange de motifs (Pattern Mixture Model)*
| |
Moyenne des moindres carrés (LS)
(erreur standard, ES)
|
0,39 (0,191)
|
-0,37 (0,194)
| |
Différence [cana-placebo] dans les moyennes des moindres carrés (ES)
|
|
-0,76 (0,249)
| |
IC à 95%
|
|
(-1,25, -0,27)
| |
Valeur de p
|
|
0,002
| |
* Les données comptables sont analysées à l'aide d'une analyse de covariance (ANCOVA) avec les termes pour le traitement, les facteurs de stratification (utilisation de l'AHA et groupe d'âge) ainsi que la valeur de l'HbA1c au début de l'étude.
Un participant est considéré comme ayant reçu un traitement d'urgence avant la semaine 26 lorsque le jour du début du traitement précède le début de la fenêtre d'analyse pour le rendez-vous à la semaine 26 (jours 163 à 211). La même règle s'applique pour les participants qui ont arrêté le traitement avant la semaine 26.
|
Patients âgés
Une étude en double aveugle, contrôlée contre placebo, d'une durée de 26 semaines, a été menée chez au total 714 patients âgés de ≥55 à ≤80 ans (dont 227 patients âgés de 65 à <75 ans et 46 patients âgés de 75 à <85 ans) et présentant un contrôle glycémique insuffisant avec le traitement antidiabétique actuel (régime et activité physique seuls ou associés à des principes actifs antihyperglycémiants oraux ou parentéraux). Des modifications statistiquement significatives (p<0,001) du taux d'HbA1c par rapport à la valeur initiale, de -0,57% sous Invokana 100 mg et de -0,70% sous Invokana 300 mg, ont été observées comparativement au placebo. Par ailleurs, une diminution statistiquement significative de la GAJ a été observée et un pourcentage plus élevé de patients a atteint un taux d'HbA1c <7,0%, comparativement au placebo (voir «Posologie/Mode d'emploi» et «Effets indésirables»).
Patients présentant un DFGe compris entre 45 et <60 ml/min/1,73 m2
Dans une analyse regroupée des données de patients adultes (N=722) présentant un DFGe initial compris entre 45 ml/min/1,73 m2 et <60 ml/min/1,73 m2, la canagliflozine à la dose de 100 mg a entraîné une réduction du taux d'HbA1c de -0,47% par rapport au placebo. Les patients présentant un DFGe initial compris entre 45 ml/min/1,73 m2 et <60 ml/min/1,73 m2 ont présenté une diminution moyenne du poids corporel de -1,8% par rapport au placebo sous 100 mg de canagliflozine.
La majorité des patients présentant un DFGe initial compris entre 45 et <60 ml/min/1,73 m2 a été traité par l'insuline et/ou des sulfonylurées (85% [614/722]). Dans cette analyse des données regroupées, 27,3% des patients sous placebo, 39,8% des patients sous Invokana 100 mg et 41,8% des patients sous Invokana 300 mg ont eu ≥1 hypoglycémie. Le traitement concomitant par l'insuline et/ou des sulfonylurées explique cette incidence accrue d'hypoglycémies (voir «Effets indésirables»).
Pression artérielle
Dans les études contrôlées contre placebo menées chez des patients adultes, les diminutions moyennes de la pression artérielle systolique par rapport au placebo ont été les suivantes: -3,9 mmHg sous Invokana 100 mg, -5,3 mmHg sous Invokana 300 mg et -0,1 mmHg sous placebo. L'effet sur la pression diastolique a été moins important dans la même population, avec des modifications moyennes de -2,1 mmHg sous Invokana 100 mg, de -2,5 mmHg sous Invokana 300 mg et de -0,3 mmHg sous placebo. Aucune modification notable de la fréquence cardiaque n'a été observée.
Résultats cardiovasculaires dans le programme CANVAS
L'effet de la canagliflozine sur le risque cardiovasculaire chez les adultes atteints de diabète de type 2 et présentant une maladie cardiovasculaire (CV) manifeste ou un risque CV (deux risques CV ou plus) a été évalué au cours du programme CANVAS (étude CANVAS et étude CANVAS-R). Il s'agissait d'études multicentriques, multinationales, randomisées en double aveugle avec des groupes parallèles et avec des critères d'inclusion et d'exclusion ainsi que des collectifs de patients similaires. Ces études ont comparé le risque de survenue d'un événement indésirable cardiovasculaire majeur (ECM, défini comme un décès consécutif à un événement cardiovasculaire, un infarctus du myocarde non fatal ou un accident vasculaire cérébral non fatal) entre la canagliflozine et un placebo dans le contexte d'un traitement standard du diabète et d'une maladie cardiovasculaire athérosclérotique.
Dans l'étude CANVAS les patients ont été randomisés par un procédé aléatoire selon un ratio 1:1:1 et assignés à un traitement par canagliflozine 100 mg, canagliflozine 300 mg ou placebo pertinent. Dans l'étude CANVAS-R les patients ont été randomisés par un procédé aléatoire selon un ratio 1:1 et assignés à un traitement par canagliflozine 100 mg ou placebo pertinent. À la discrétion du médecin investigateur, une augmentation de la dose à 300 mg a été autorisée après la semaine 13, en fonction de la tolérance et des besoins glycémiques. Pour garantir aux sujets de recevoir un traitement contre le diabète et l'athérosclérose conforme au standard de prise en charge, un ajustement des traitements concomitants correspondants a été autorisé selon l'appréciation de l'investigateur.
Au total 10 134 patients adultes (4327 dans l'étude CANVAS et 5807 dans l'étude CANVAS-R; au total 4344 sujets ont été attribués aléatoirement au groupe placebo et 5790 au traitement par canagliflozine) ont été traités sur une période de 149 semaines (valeur moyenne; la durée du traitement a été de 223 semaines dans l'étude CANVAS et de 94 semaines dans l'étude CANVAS-R). Le statut vital a été saisi chez 99,6% des participants dans les deux études. Environ 78% des sujets étaient caucasiens, 13% étaient asiatiques et 3% étaient noirs. L'âge moyen était de 63 ans et environ 64% étaient de sexe masculin.
Au moment de la sélection, tous les patients de l'étude présentaient un diabète sucré de type 2 insuffisamment contrôlé (HbA1c ≥7,0% à ≤10,5%). La valeur moyenne de l'HbA1c au début de l'étude était de 8,2% et la durée moyenne du diabète était de 13,5 ans. Environ 31%, 21% et respectivement 18% des sujets présentaient des antécédents de neuropathie, de rétinopathie et de néphropathie. 80% des patients présentaient une fonction rénale normale ou légèrement insuffisante au début de l'étude et 20% présentaient une insuffisance rénale modérée (DFGe moyen: 77 ml/min/1,73 m2). Au début de l'étude les patients suivaient un traitement par un antidiabétique au moins, incluant la metformine (77%), l'insuline (50%) et une sulfonylurée (43%).
66 pour-cent des sujets présentaient des antécédents de maladie cardiovasculaire manifeste, dont 56% une maladie coronarienne, 19% une affection cérébrovasculaire et 21% une maladie vasculaire périphérique; 14% présentaient des antécédents d'insuffisance cardiaque. Au début de l'étude la pression artérielle systolique moyenne était de 137 mmHg et la pression artérielle diastolique moyenne de 78 mmHg, le taux moyen de LDL s'élevait à 89 mg/dl, le taux moyen de HDL à 46 mg/dl et le rapport albuminurie/créatininurie (ACR) à 115 mg/g. Au début de l'étude près de 80% des patients étaient sous traitement par des inhibiteurs du système rénine-angiotensine, 54% sous traitement par des bêtabloquants, 13% sous traitement par des diurétiques de l'anse, 36% sous traitement par d'autres diurétiques, 75% sous traitement par des statines et 74% sous traitement par des antiagrégants plaquettaires (principalement de l'acide acétylsalicylique).
Le critère d'évaluation principal du programme CANVAS a été la durée de la période écoulée jusqu'à la première apparition d'un ECM. Les HR pour l'EMC et l'IC à 95% chez les patients traités par la canagliflozine, comparés aux patients du groupe placebo, ont été évalués à l'aide d'un modèle de régression de Cox constant, avec une stratification selon l'étude et une maladie cardiovasculaire manifeste.
La canagliflozine a entraîné une réduction significative du risque de première survenue du critère d'évaluation principal combiné ECM (HR: 0,86; IC à 95% 0,75, 0,97) auquel chaque composante ECM a contribué. Les résultats pour les deux dosages recommandés de canagliflozine (100 mg et 300 mg) étaient cohérents avec les résultats obtenus pour les données regroupées. L'efficacité de la canagliflozine concernant les ECM était différente selon que les patients présentaient déjà au début de l'étude une maladie cardiovasculaire manifeste (HR: 0,82; IC à 95% 0,72, 0,95) ou seulement des facteurs de risque cardiovasculaire (HR: 0,98; IC à 95% 0,74, 1,30).
2011 patients adultes présentaient un DFGe situé entre 30 et <60 ml/min/1,73 m2. Les résultats portant sur l'ECM dans ce sous-groupe ont été en corrélation avec les résultats globaux.
Tableau 10: Effet du traitement pour le critère d'évaluation principal et ses composantes
|
|
Placebo
N=4347
Taux d'événements pour 1000 années-patients
|
Canagliflozine
N=5795
Taux d'événements pour 1000 années-patients
|
Hazard Ratio
(IC à 95%)
| |
Combinaison de décès consécutif à un événement cardiovasculaire, d'infarctus du myocarde non fatal, ou d'accident vasculaire cérébral non fatal (durée de l'intervalle jusqu'à la première apparition; groupe d'analyse en intention de traiter)1
|
31,48
|
26,93
|
0,86 (0,75-0,97)
| |
Décès consécutif à un événement cardiovasculaire
|
12,84
|
11,60
|
0,87 (0,72-1,06)
| |
Infarctus du myocarde non fatal
|
11,61
|
9,74
|
0,85 (0,69-1,05)
| |
Accident vasculaire cérébral non fatal
|
8,39
|
7,12
|
0,90 (0,71-1,15)
|
1 Valeur de p pour la supériorité (bilatérale) = 0,0158
Résultats selon les sous-groupes présence ou absence d'une maladie cardiovasculaire manifeste:
L'analyse primaire de l'efficacité s'est basée sur la population globale. Une analyse par sous-groupes prédéfinie pour les patients adultes avec ou sans maladie cardiovasculaire manifeste antérieure n'a certes révélé aucun indice net d'hétérogénéité de l'efficacité entre ces deux groupes (valeur de p d'interaction = 0,1803); toutefois, une réduction cliniquement pertinente du risque cardiovasculaire n'a été observée que chez les patients présentant une maladie cardiovasculaire manifeste antérieure (HR: 0,82; IC à 95% 0,72, 0,95) et non chez les patients sans maladie manifeste antérieure (HR: 0,98; IC à 95%: 0,74, 1,30).
Basé sur l'estimateur de Kaplan-Meier de la première apparition d'un ECM, une diminution de l'ECM dans le groupe traité par la canagliflozine a déjà été observée à la semaine 26 et s'est poursuivie pendant toute l'étude restante.
Mortalité globale
Dans le groupe combiné de canagliflozine, le HR pour la mortalité globale a été de 0,87 (0,74; 1,01), comparé au placebo.
Insuffisance cardiaque traitée à l'hôpital
En comparaison avec le placebo, la canagliflozine a diminué le risque d'insuffisance cardiaque nécessitant une hospitalisation (HR: 0,67; IC à 95% [0,52; 0,87]).
Critères d'évaluation rénaux dans le programme CANVAS
Dans le programme CANVAS, le HR pour le temps jusqu'à la survenue de la première néphropathie confirmée (doublement de la créatinine sérique, nécessité d'une thérapie de remplacement rénal, décès d'origine rénale) était de 0,53 (IC à 95%: 0,33; 0,84) pour la canagliflozine (1,5 événement pour 1000 années-patients) par rapport au placebo (2,8 événements pour 1000 années-patients). De plus, la canagliflozine a réduit la progression de l'albuminurie de 25,8% contre 29,2% sous placebo (HR: 0,73; IC à 95%: 0,67, 0,79), chez des patients adultes présentant une normo- ou microalbuminurie avant le début du traitement. Bien que le traitement par des inhibiteurs de l'ECA et/ou un ARA ne représentait pas un critère d'inclusion dans le programme CANVAS, 80% des participants prenaient un inhibiteur de l'ECA et/ou un ARA au moment de la randomisation.
Résultats rénaux de l'étude CREDENCE
L'effet de 100 mg de canagliflozine sur les résultats rénaux observés chez des adultes atteints de diabète sucré de type 2 et d'une néphropathie diabétique (ND) définie par un taux de filtration glomérulaire estimé (DFGe) de 30 à <90 ml/min/1,73 m2 ou une ClCr de 30 à <90 ml/min et une albuminurie (>300 à 5000 mg/g de créatinine) a été estimé au cours de l'étude CREDENCE («Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation Trial»). Il s'agissait d'une étude en groupes parallèles contrôlés par placebo, multicentrique, internationale, randomisée (1:1), en double aveugle, guidée par les événements. Au total, 4401 participants adultes ont été randomisés afin de former l'ensemble des données d'analyse en ITT (Intent-To-Treat), utilisé pour les analyses d'efficacité principales, alors que 4397 participants traités étaient disponibles pour l'évaluation de la sécurité. Au cours de l'étude CREDENCE, le risque de survenue d'une néphropathie diabétique (définie comme critère d'évaluation composite par l'insuffisance rénale terminale, le doublement de la créatinine sérique et le décès d'origine rénale ou cardiovasculaire) a été examiné et comparé lors de la prise de 100 mg de canagliflozine ou d'un placebo sur fond de traitement standard d'une néphropathie diabétique (notamment inhibiteur de l'enzyme de conversion de l'angiotensine [inhibiteur de l'ECA] et antagoniste du récepteur de l'angiotensine [ARA]).
Au cours de l'étude CREDENCE les sujets ont été traités par 100 mg de canagliflozine ou le placebo, stratifiés d'après le DFGe au moment du dépistage (30 à <45, 45 à <60, 60 à <90 ml/min/1,73 m2) ou la ClCr (30 à <45, 45<60, 60 à <90 ml/min) jusqu'au commencement de la dialyse ou jusqu'à la transplantation rénale.
Au total 4397 sujets ont été traités, la durée moyenne d'exposition était de 115 semaines. Le statut vital a été saisi chez 99,9% des sujets de l'étude. L'âge moyen était de 63 ans, 66% étaient de sexe masculin.
La valeur initiale moyenne de l'HbA1c était de 8,3%, la valeur initiale médiane du quotient albumine/créatinine dans l'urine était de 927 mg/g. Les principes actifs antihyperglycémiants les plus fréquemment utilisés en début d'étude étaient l'insuline (65,5%), les dérivés biguanides (57,8%) et les sulfonylurées (28,8%). Presque tous les sujets (99,9%) recevaient un inhibiteur de l'ECA ou un ARA au moment de la randomisation. Environ 92% des sujets recevaient un traitement cardiovasculaire au début de l'étude (en dehors des inhibiteurs de l'ECA et des ARA), environ 60% étaient traités par un principe actif antithrombotique (y compris l'acide acétylsalicylique) et 69% par des statines.
Au début de l'étude, le DFGe moyen était de 56,2 ml/min/1,73 m2 (environ 60% de la population de l'étude présentait un DFGe <60 ml/min/1,73 m2). En moyenne, les participants étaient diabétiques depuis environ 16 ans. Le pourcentage de patients présentant des antécédents d'affection cardiovasculaire était de 50,4%; un antécédent d'insuffisance cardiaque était présent chez 14,8% des patients. En plus de la néphropathie diabétique, environ 64% de la population souffrait d'au moins une autre complication microvasculaire.
Pour le critère d'évaluation principal et pour les critères d'évaluation secondaires, les HR et l'IC à 95% correspondant ont été évalués à l'aide d'un modèle stratifié de régression aléatoire proportionnelle de Cox (avec le traitement comme variable explicative et stratifiés d'après le DFGe au moment du dépistage [30 à <45, 45 à <60, 60 à <90 ml/min/1,73 m2] ou la ClCr [30 à <45, 45 à <60, 60 à <90 ml/min]).
Le critère d'évaluation principal composite de l'étude CREDENCE était le délai jusqu'à/au a) la première survenue d'une insuffisance rénale terminale (IRT; définie par la chute du DFGe en dessous de 15 ml/min/1,73 m2 ou de la ClCr sous 15 ml/min) ou le début de la dialyse chronique ou une transplantation rénale, b) doublement de la créatinine sérique, c) décès d'origine rénale, ou d) décès d'origine cardiovasculaire.
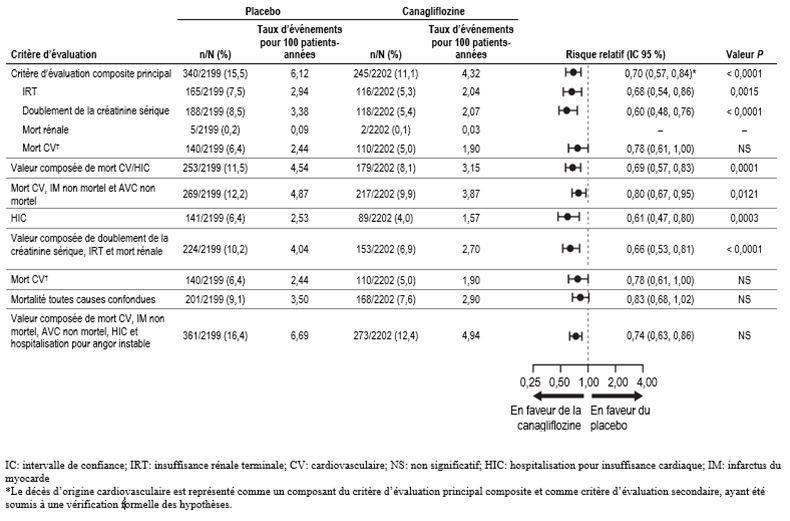
La canagliflozine à 100 mg a diminué le risque de première survenue du critère d'évaluation principal composite (IRT, doublement de la créatinine sérique et décès d'origine rénale ou cardiovasculaire), de manière significative [p <0,0001; HR: 0,70; IC à 95%: 0,59, 0,82] (voir Figure 1). L'effet du traitement était comparable dans tous les sous-groupes, dont les trois sous-groupes définis par le DFGe et chez les sujets présentant ou non des antécédents d'affection cardiovasculaire.
Sur la base de la courbe de Kaplan-Meier du délai jusqu'à la première survenue du critère d'évaluation principal composite, l'effet sur le traitement de 100 mg de canagliflozine était reconnaissable à partir de la semaine 52 et se poursuivait jusqu'à la fin de l'étude.
Les effets de 100 mg de canagliflozine sur les critères d'évaluation secondaires sont résumés dans la Figure 1.
Figure 1: Effet du traitement sur les critères d'évaluation principaux et secondaires et leurs composantes
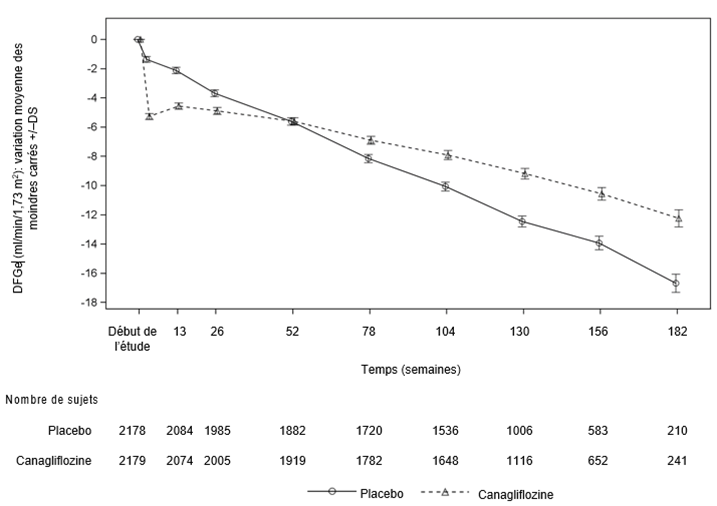
Comme le représente la Figure 2, le DFGe a baissé linéairement et progressivement chez les patients traités par placebo; en revanche, dans le groupe canagliflozine, on a observé une réduction abrupte à la semaine 3, qui s'estompait par la suite; la réduction moyenne calculée par la méthode des moindres carrés était inférieure après la semaine 52 dans le groupe canagliflozine par rapport au groupe placebo, l'effet du traitement se poursuivait jusqu'à la fin du traitement.
Figure 2: Variation moyenne des moindres carrés du DFGe par rapport aux valeurs initiales au fil du temps (analyses des patients sous traitement)
Au cours de l'étude CREDENCE, le taux d'incidence des effets indésirables rénaux du groupe 100 mg de canagliflozine était inférieur à celui du groupe placebo (57 pour 1000 années-patients dans le groupe canagliflozine et 79 pour 1000 années-patients dans le groupe placebo).
|