CompositionPrincipes actifs
Alemtuzumab.
L'alemtuzumab est un anticorps monoclonal humanisé produit par la technologie de l'ADN recombinant qui cible la glycoprotéine CD52 de 21-28 kD située à la surface des cellules. L'alemtuzumab est un anticorps IgG1 kappa présentant une structure variable et des régions constantes humaines, ainsi que des sites de liaison à l'anticorps (régions CDR, en anglais «Complementary Determining Regions») obtenus à partir d'un anticorps monoclonal murin (rat). L'anticorps possède un poids moléculaire approximatif de 150 kD. L'alemtuzumab est produit à partir d'une suspension de cellules de mammifères (cellules ovariennes de hamster chinois) en culture dans un milieu nutritif.
Excipients
Phosphate disodique dihydraté (E339), édétate disodique, chlorure de potassium (E508), dihydrogénophosphate de potassium (E340), polysorbate 80 (E433), chlorure de sodium, eau pour préparations injectables ad solutionem pro 1,2 ml.
1 flacon contient 4,26 mg de sodium et 0,19 mg de potassium.
Indications/Possibilités d’emploiLemtrada est indiqué en monothérapie comme traitement de fond chez les patients adultes présentant des formes très actives de sclérose en plaques récurrente-rémittente (SEP-RR), malgré un traitement précédent complet et bien conduit par au moins un traitement de fond.
Posologie/Mode d’emploiLe traitement par Lemtrada doit uniquement être instauré et surveillé par un neurologue expérimenté dans la prise en charge des patients atteints de sclérose en plaques (SEP) dans un hôpital disposant d'un accès direct aux soins intensifs. Des spécialistes ainsi que des équipements nécessaires au diagnostic et à la prise en charge rapide des effets indésirables, notamment ischémie myocardique et infarctus du myocarde, réactions indésirables cérébrovasculaires, troubles auto-immuns et infections, doivent être disponibles. Les médicaments et le matériel nécessaires à la prise en charge du syndrome de libération des cytokines, d'éventuelles réactions d'hypersensibilité et/ou anaphylactiques devront être disponibles lors de la perfusion.
Les patients traités par Lemtrada doivent recevoir la Carte et le Guide Patient et être informés des risques de Lemtrada (voir aussi la notice).
Lors du passage d'autres immunosuppresseurs à Lemtrada, la durée et la nature de l'effet de telles substances doivent être soigneusement considérées afin d'éviter tout effet cumulatif sur le système immunitaire. Étant donné qu'une grande partie des immunosuppresseurs utilisés pour le traitement de la sclérose en plaques ont une longue demi-vie, le risque d'une exposition concomitante est grand. C'est pourquoi la prudence est de rigueur lors du passage d'autres immunosuppresseurs à Lemtrada. Dans ces cas-là, il convient de documenter l'immunocompétence par une numération plaquettaire et un statut immunitaire (phénotypage lymphocytaire) avant instauration du traitement par Lemtrada.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Posologie
La posologie de Lemtrada recommandée est de 12 mg/jour administrée en perfusion intraveineuse au cours de 2 cycles de traitement ou plus (sans dépasser 4 cycles).
Traitement initial sur 2 cycles:
·Premier cycle de traitement: 12 mg/jour pendant 5 jours consécutifs (dose totale de 60 mg)
·Deuxième cycle de traitement: 12 mg/jour pendant 3 jours consécutifs (dose totale de 36 mg) administrés 12 mois après le premier cycle de traitement.
Si nécessaire, cycles de traitement supplémentaires:
Troisième ou quatrième cycle: 12 mg/jour pendant 3 jours consécutifs (dose totale de 36 mg) administrés au moins 12 mois après le cycle de traitement précédent.
Lemtrada doit être dilué avant la perfusion tel que décrit dans la rubrique «Remarques particulières». Le Lemtrada dilué doit être administré par voie intraveineuse sur une période d'environ 4 heures. Une surveillance concernant la survenue de réactions liées à la perfusion est recommandée pendant et jusqu'à deux heures après la perfusion de Lemtrada (cf. «Mises en garde et précautions»).
Les doses oubliées ne doivent pas être administrées le même jour qu'une dose planifiée.
Suivi des patients
Administrer Lemtrada dans un hôpital où l'équipement et le personnel sont disponibles pour gérer correctement l'anaphylaxie, les réactions graves à la perfusion, l'ischémie myocardique, l'infarctus du myocarde et les effets indésirables cérébrovasculaires.
Le schéma d'administration recommandé est de 2 cycles de traitement (voir posologie), avec une surveillance particulière des patients depuis l'instauration du traitement et pendant une période allant jusqu'à 48 mois après la dernière perfusion. Pour plus d'informations, veuillez tenir compte de la rubrique «Mises en garde et précautions».
Médication concomitante recommandée
Une prémédication par corticoïdes doit être administrée aux patients juste avant la perfusion de Lemtrada pendant chacun des 3 premiers jours de chaque cycle de traitement. Au cours des études cliniques, les patients ont reçu 1'000 mg de méthylprednisolone pendant les 3 premiers jours de chaque cycle de traitement par Lemtrada. De plus, une prémédication par antihistaminiques et/ou antipyrétiques peut également être envisagée avant la perfusion de Lemtrada.
Une prophylaxie par voie orale contre une infection par le virus de l'herpès doit être administrée à tous les patients dès le premier jour de chaque cycle de traitement et se poursuivre pendant au moins un mois après la fin du traitement par Lemtrada (voir également la rubrique «Mises en garde et précautions» sous «Infections»). Au cours des études cliniques, les patients ont reçu 200 mg d'aciclovir ou une prophylaxie équivalente deux fois par jour.
Tests de laboratoire utiles pour surveiller les patients
Les tests de laboratoire doivent être effectués à intervalles périodiques jusqu'à 48 mois après le dernier traitement de Lemtrada afin de surveiller les signes précoces de maladie auto-immune:
·Numération formule sanguine et plaquettaire avec transaminases sériques (avant le début du traitement et tous les mois par la suite);
·Taux de créatinine sérique (avant le début du traitement et tous les mois par la suite);
·Analyse d'urine avec numération cellulaire dans les urines (avant le début du traitement et tous les mois par la suite);
·Un test de la fonction thyroïdienne, comme le taux de TSH (avant le début du traitement et tous les 3 mois par la suite).
Patients présentant une insuffisance rénale ou hépatique
Lemtrada n'a pas été étudié chez les patients présentant une insuffisance hépatique ou rénale.
Patients âgés
Les études cliniques n'avaient pas inclus suffisamment de patients de plus de 55 ans. Il n'a donc pas été possible de déterminer si ces patients avaient une réponse au traitement différente de celle des patients plus jeunes.
Enfants et adolescents
La sécurité d'emploi et l'efficacité de Lemtrada chez les enfants atteints de SEP et âgés de 0 à 18 ans n'ont pas encore été établies. Lemtrada n'est pas indiqué pour une utilisation chez les enfants et les adolescents (cf. «Contre-indications»).
Mode d'administration
Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique «Instructions concernant la dilution». Chaque perfusion doit être administrée par voie intraveineuse pendant environ 4 heures.
Contre-indicationsLemtrada est contre-indiqué dans les situations suivantes:
·chez les patients avec réactions d'hypersensibilité de type I connues ou réactions anaphylactiques à l'alemtuzumab ou à l'un des excipients conformément à la composition du produit.
·chez les patients avec infection par le virus de l'immunodéficience humaine (VIH).
·chez les patients avec infections sévères actives, infections chroniques graves (p.ex. tuberculose, hépatite B et C).
·chez les patients avec un risque accru d'infections opportunistes, tels que des patients durablement immunodéprimés (y compris ceux actuellement sous immunosuppresseurs ou dont le système immunitaire est affaibli par des traitements antérieurs).
·chez les patients souffrant d'hypertension non contrôlée.
·chez les patients dont l'anamnèse mentionne des dissections des artères cervico-céphaliques ou qui présentent un risque accru de dissections cervicales (syndrome d'Ehlers-Danlos, syndrome de Marfan etc.)
·chez les patients ayant des antécédents d'accident vasculaire cérébral (AVC).
·chez les patients ayant des antécédents d'angine de poitrine ou d'infarctus du myocarde.
·chez les patients qui présentent un risque accru de diathèse hémorragique (due à des anticoagulants par exemple)
·chez les patients avec affections malignes actives existantes, à l'exception des patients atteints d'un carcinome basocellulaire cutané.
·chez les enfants et adolescents de moins de 18 ans.
·Instauration du traitement pendant la grossesse.
Mises en garde et précautionsGénéralités
L'utilisation de Lemtrada n'est pas recommandée chez les patients atteints de SEP sans activité de la maladie ou ceux dont la maladie est stabilisée par un traitement établi.
Avant le début du traitement, les patients doivent être informés des risques d'un traitement, ainsi que de la nécessité d'effectuer un suivi mensuel étroit pendant le traitement et pendant les 48 mois suivants la dernière perfusion du second cycle de traitement par Lemtrada. Aussi, les patients doivent donner leur accord. Si un traitement supplémentaire est administré, il est important de poursuivre le suivi mensuel jusqu'à 48 mois après la dernière perfusion.
À ce jour, on ne dispose pas de données suffisantes concernant plus de 4 cycles de traitement par Lemtrada.
Le profil de sécurité ne semble pas être modifié en cas de phases de traitement supplémentaires. L'intervalle temporel entre des phases de traitement supplémentaires doit être d'au moins 12 mois.
Les patients traités par Lemtrada doivent recevoir la notice, la carte patient et le guide patients.
Auto-immunité
Le traitement par Lemtrada peut entraîner la formation d'auto-anticorps et augmenter le risque de pathologies auto-immunes, qui peut être grave et mettre la vie en danger. Les pathologies auto-immunes signalées, comprennent notamment des troubles thyroïdiens, purpura thrombopénique immunologique (PTI), ou, dans de rares cas, néphropathies (p.ex. maladie des anticorps anti-membrane basale glomérulaire (syndrome de Goodpasture)), hépatite auto-immune (HAI), hémophilie A acquise, purpura thrombotique thrombocytopénique (PTT), sarcoïdose (y compris la neurosarcoïdose) et encéphalite auto-immune. Dans le contexte post-marketing, des patients développant de multiples troubles auto-immuns après traitement par Lemtrada ont été observés. Les patients qui développent une auto-immunité doivent être évalués pour d'autres affections auto-immunes. Les patients et les médecins doivent être informés du potentiel d'apparition ultérieure de troubles auto-immuns après la période de surveillance de 48 mois.
Il convient de faire preuve de prudence chez les patients avec des antécédents de pathologies auto-immunes autres que la SEP, bien que les données disponibles n'indiquent aucune exacerbation des pathologies auto-immunes préexistantes après un traitement par alemtuzumab.
Hémophilie A acquise
Des cas d'hémophilie A acquise (anticorps anti-facteur VIII) ont été signalés à la fois dans les essais cliniques et après la commercialisation. Les patients présentent généralement des hématomes sous-cutanés spontanés et des ecchymoses étendues, bien qu'une hématurie, une épistaxis, des saignements gastro-intestinaux ou d'autres types de manifestation puissent survenir. Un bilan de coagulation incluant le temps de céphaline activée (TCA) doit être pratiqué chez tous les patients présentant de tels symptômes. Les patients doivent être informés des signes et symptômes de l'hémophilie A acquise. Les patients doivent consulter immédiatement un médecin si l'un de ces symptômes apparaît.
Purpura thrombopénique immunologique (PTI)
Des cas graves de PTI ont été observés chez 12 (1%) patients traités par Lemtrada au cours des études cliniques contrôlées dans la SEP (ce qui correspond à un taux annuel de 0,0047 événements/patient/année).
Au cours d'une étude clinique contrôlée auprès de patients atteints de SEP, un patient a présenté un PTI n'ayant pas été diagnostiqué avant la mise en place de la surveillance mensuelle systématique de la formule sanguine; ce patient est décédé des suites d'une hémorragie intracérébrale. Douze événements graves de PTI supplémentaires ont été observés au cours d'un suivi médian de 6,1 ans (12 ans maximum) (taux annualisé cumulatif de 0,0028 événements/patient/année). La survenue de PTI intervient généralement entre 14 et 36 mois après la première exposition à Lemtrada. Les signes cliniques d'un PTI peuvent inclure (sans s'y limiter) une tendance aux ecchymoses, des pétéchies, des saignements cutanéo-muqueux spontanés (p.ex. épistaxis, hémoptysie), des méno-métrorragies. Une hémoptysie peut également constituer un symptôme de maladie anti-MBG (voir ci-dessous) et un diagnostic différentiel approprié doit être réalisé.
Rappeler au patient de rester attentif à l'apparition de signes cliniques éventuels et de consulter un médecin en cas de symptômes.
Une numération formule sanguine avec numération plaquettaire doit être réalisée avant l'instauration du traitement puis tous les mois jusqu'à 48 mois après la dernière perfusion. Au bout de ce délai, des analyses doivent être réalisées si des données cliniques suggèrent l'existence d'un PTI. En cas de suspicion de PTI, il convient de réaliser immédiatement une numération formule sanguine avec numération plaquettaire.
Si la survenue d'un PTI est confirmée, une prise en charge médicale appropriée doit immédiatement être mise en œuvre, incluant la consultation immédiate d'un spécialiste. Les données des études cliniques dans la SEP ont montré que l'observation des bilans sanguins mensuels et l'éducation à la reconnaissance des signes et symptômes de PTI ont permis leur détection et leur traitement précoces. L'évolution a été favorable sous traitement de première intention pour la plupart des patients.
Le risque potentiel associé à une reprise du traitement par Lemtrada après la survenue d'un PTI n'est pas connu.
Néphropathies
Des néphropathies, notamment la maladie des anticorps anti-membrane basale glomérulaire (anti-MBG) (syndrome de Goodpasture), ont été observées chez 6 (0,4%) patients au cours d'études cliniques dans la SEP avec un suivi médian de 6,1 ans (maximum 12 ans). Ces néphropathies sont généralement survenues dans les 39 mois suivant la dernière administration de Lemtrada. Dans les études cliniques, deux cas graves de maladie des anticorps anti-MBG (syndrome de Goodpasture) ont été observés, précocement identifiés grâce aux examens cliniques et biologiques, ils ont évolué favorablement sous traitement.
Les manifestations cliniques d'une néphropathie peuvent inclure une élévation de la créatininémie, une hématurie et/ou une protéinurie. Bien qu'aucun cas n'ait été observé au cours des études cliniques, une hémorragie alvéolaire se manifestant sous la forme d'une hémoptysie peut survenir associée à la maladie des anticorps anti-MBG (syndrome de Goodpasture). La maladie anti-MBG (syndrome de Goodpasture) peut entraîner une insuffisance rénale nécessitant une dialyse et/ou une transplantation si elle n'est pas traitée rapidement et peut être mortelle si elle n'est pas traitée. Il faut rappeler au patient de rester vigilant quant aux symptômes qu'il peut ressentir et de consulter immédiatement un médecin s'il a des inquiétudes.
Étant donné qu'une hémoptysie peut également constituer un signe de PTI ou d'hémophilie A acquise (voir ci-dessus), un diagnostic différentiel approprié doit être réalisé.
Une créatininémie doit être réalisée avant l'instauration du traitement puis tous les mois jusqu'à 48 mois après la dernière perfusion. Un examen microscopique des urines doit être réalisé avant l'instauration du traitement puis mensuellement jusqu'à 48 mois après la dernière perfusion.
Des modifications cliniquement significatives de la créatininémie par rapport aux valeurs initiales, une hématurie inexpliquée et/ou une protéinurie doivent inciter à réaliser des examens approfondis pour détecter une néphropathie, et à consulter immédiatement un spécialiste. La détection et la mise en œuvre d'un traitement précoces peuvent améliorer le pronostic d'une néphropathie. Au-delà de cette période, des analyses devront être réalisées si des données cliniques suggèrent une néphropathie.
Le risque potentiel associé à une reprise du traitement par Lemtrada après la survenue d'une néphropathie n'est pas connu.
Troubles thyroïdiens
Des troubles endocriniens incluant des troubles thyroïdiens auto-immuns ont été observés chez 36,8% des patients traités par Lemtrada 12 mg au cours des études cliniques portant sur la SEP jusqu'à 48 mois depuis la première exposition au Lemtrada, dans le cadre d'un suivi médian de 6,1 ans (maximum 12 ans). L'incidence de troubles thyroïdiens était plus élevée chez les patients présentant des antécédents de troubles thyroïdiens, tant dans le groupe Lemtrada que dans le groupe interféron bêta-1a (IFNB-1a). Lemtrada ne doit être administré à des patients atteints de troubles thyroïdiens que si le profil bénéfices-risques est favorable.
Les troubles thyroïdiens auto-immuns observés comprenaient des cas d'hyperthyroïdies et d'hypothyroïdies. La plupart des événements ont été d'intensité légère à modérée. Des événements endocriniens graves sont survenus chez 4,4% des patients, la maladie de Basedow, une hyperthyroïdie, une hypothyroïdie, une thyroïdite auto-immune et un goitre ont affecté plus d'un patient. La plupart des événements thyroïdiens ont été pris en charge par un traitement médicamenteux classique, néanmoins certains patients ont nécessité une intervention chirurgicale.
Environ 5% des patients de la population totale de l'étude ont développé des troubles thyroïdiens au cours de l'année qui a suivi le premier traitement par alemtuzumab et ont été re-traités. Les patients re-traités n'ont généralement pas présenté d'exacerbation des troubles thyroïdiens. La possibilité d'un traitement supplémentaire par Lemtrada doit être envisagée au cas par cas en fonction de l'état clinique du patient.
Un bilan thyroïdien avec mesure du taux de thyrotropine (TSH), doit être réalisé avant l'instauration du traitement puis tous les 3 mois jusqu'à 48 mois après la dernière perfusion. Au bout de ce délai, des examens devront être réalisés en fonction de résultats cliniques correspondants ou en cas de grossesse.
Dans les études cliniques, le statut des anticorps anti-thyroperoxydase (anti-TPO) du patient avant traitement n'était pas prédictif du développement d'un effet indésirable thyroïdien. La moitié des patients positifs et le quart des patients négatifs à l'inclusion pour les anticorps anti-TPO ont présenté un événement thyroïdien. La grande majorité (environ 80%) des patients ayant présenté un événement thyroïdien après le traitement avaient des anticorps anti-TPO négatifs à l'inclusion. Quels que soient les résultats de leur test des anticorps anti-TPO avant le traitement, les patients sont donc susceptibles de développer un effet indésirable thyroïdien et doivent réaliser tous les examens périodiques comme décrits ci-dessus.
Les troubles thyroïdiens représentent un risque particulier chez la femme enceinte (cf. «Grossesse, Allaitement»).
Cytopénies
Des suspicions de cytopénies auto-immunes telles que neutropénie, anémie hémolytique ou pancytopénie, ont été peu fréquemment rapportées au cours des études cliniques dans la SEP. Les résultats de la numération formule sanguine avec numération plaquettaire (voir ci-dessus information sur le PTI) permettent de détecter une cytopénie. Si une cytopénie est confirmée, une prise en charge médicale appropriée doit être rapidement mise en œuvre, incluant l'avis d'un spécialiste.
Hépatite auto-immune (HAI)
Des cas d'hépatite auto-immune (y compris des cas mortels) avec insuffisance hépatique aiguë nécessitant une greffe ont été signalés chez des patients traités par Lemtrada (données post-marketing). Si un patient présente des signes cliniques, tels qu'une élévation inexpliquée des enzymes hépatiques ou des symptômes évoquant une atteinte hépatique (p.ex. nausées, vomissements, douleurs abdominales, fatigue, anorexie, ictère et/ou urine foncée), il faut mesurer rapidement les transaminases sériques et la bilirubine totale et interrompre le traitement par Lemtrada si nécessaire. Des tests de la fonction hépatique doivent être effectués avant le traitement initial et à des intervalles mensuels jusqu'à 48 mois après la dernière perfusion. Les patients doivent être informés du risque d'hépatite auto-immune et des symptômes associés.
Purpura thrombotique thrombocytopénique (PTT)
Lors de l'utilisation post-commercialisation, le PTT, qui peut être mortel, a été signalé chez des patients traités par Lemtrada. Le PTT est une affection grave qui nécessite une évaluation et un traitement urgents. Le PTT peut être caractérisé par une thrombocytopénie (provoquant un purpura et des ecchymoses), une anémie hémolytique microangiopathique, des symptômes neurologiques (p.ex. confusion, coma, convulsions, céphalées, nausées, troubles du langage, hémiparésie, troubles visuels, paresthésies), de la fièvre, des ischémies cardiaques et une insuffisance rénale. Le PTT est associé à des taux élevés de morbidité et de mortalité s'il n'est pas reconnu et traité précocement.
Encéphalite auto-immune
Au cours de l’utilisation post-marketing, des cas d’encéphalite auto-immune ont été rapportés chez des patients traités par Lemtrada. L’encéphalite auto-immune est caractérisée par la présence d’anticorps antineuronaux ainsi que par diverses manifestations cliniques telles que l’apparition subaiguë (avec une progression rapide sur plusieurs mois) de troubles de la mémoire, d’une altération de l’état mental, de symptômes psychiatriques, de signes neurologiques focaux et de crises d’épilepsie.
Les patients chez qui l'on soupçonne une encéphalite auto-immune doivent réaliser une imagerie cérébrale (IRM), un EEG, une ponction lombaire et des tests sérologiques pour les biomarqueurs appropriés (par exemple, les auto-anticorps neuronaux) afin de confirmer le diagnostic et d'exclure d'autres étiologies.
Réactions liées à la perfusion (RAP)
Dans les études cliniques contrôlées, les réactions liées à la perfusion (RAP) ont été définies comme tout évènement indésirable survenant pendant ou dans les 24 heures suivant la perfusion de Lemtrada. La plupart de ces réactions peuvent être dues à la libération de cytokines pendant la perfusion. La plupart des patients traités par Lemtrada au cours des études cliniques sur la SEP ont rapporté des RAP d'intensité légère à modérée pendant l'administration et/ou dans les 24 heures suivant la perfusion de Lemtrada 12 mg. L'incidence de RAP était plus élevée au cours du cycle 1 que lors des cycles ultérieurs. Tout au long des suivis, en incluant les patients ayant reçu des cycles de traitement ultérieurs, les RAP les plus fréquemment observées correspondaient à des céphalées, des éruptions cutanées, de la fièvre, des nausées, une urticaire, un prurit, une insomnie, des frissons, des bouffées vasomotrices, une fatigue, une dyspnée, une dysgueusie, une gêne thoracique, une éruption généralisée, une tachycardie, une bradycardie, une dyspepsie, des étourdissements/vertiges et des douleurs. Des réactions graves sont survenues chez 3% des patients, notamment des cas de céphalée, de fièvre, d'urticaire, de tachycardie, de fibrillation auriculaire, de nausées, de gêne thoracique et d'hypotension. Par ailleurs, dans de rares cas, des réactions anaphylactiques ont été signalées. Les signes cliniques d'anaphylaxie peuvent être similaires aux caractéristiques cliniques des réactions liées à la perfusion, bien qu'ils soient généralement plus sévères ou susceptibles de mettre en jeu le pronostic vital.
Au cours de l'utilisation post-marketing, des événements indésirables graves, parfois mortels et imprévisibles de divers systèmes d'organes ont été signalés. Des cas d'hémorragie alvéolaire pulmonaire, d'ischémie myocardique, d'infarctus du myocarde, d'accident vasculaire cérébral (y compris d'accident ischémique et hémorragique), de dissection artérielle cervico-céphalique (par exemple: vertébrale, carotide) et de thrombocytopénie ont été rapportés. Des réactions peuvent survenir après n'importe laquelle des doses au cours du traitement. Dans la majorité des cas, le délai d'apparition était de 1 à 3 jours suivant la perfusion de Lemtrada. Les patients doivent être informés des signes et symptômes et ils doivent consulter immédiatement un médecin si l'un de ces symptômes apparaît.
Ischémie myocardique et infarctus du myocarde
Il a été noté que chez certains patients, la pression artérielle et/ou la fréquence cardiaque étaient temporairement anormales pendant la perfusion. Il n'y avait aucun facteur de risque évident chez la majorité des patients.
Accident vasculaire cérébral (AVC)
Au cours de l'utilisation post-marketing, des cas graves et potentiellement mortels d'accident vasculaire cérébral (y compris des accidents ischémiques et hémorragiques) ont été rapportés, dont certains sont survenus dès les 3 premiers jours suivant l'administration de Lemtrada.
Chez les patients dont la documentation était disponible, il a été noté une augmentation de la pression artérielle par rapport à la valeur initiale avant l'hémorragie. Il n'y avait aucun facteur de risque évident chez la majorité des patients.
Dissection des artères cervico-céphaliques
Au cours de l'utilisation post-marketing, des cas de dissection artérielle cervico-céphalique (p.ex. artère vertébrale, artère carotide), y compris des dissections multiples, ont été signalés dans les premiers jours suivant l'administration de Lemtrada ou plus tard dans le premier mois suivant la perfusion. L'ensemble des données disponibles ne permet pas de conclure sur l'existence d'un lien de causalité entre l'utilisation de Lemtrada et la survenue de cas d'accident vasculaire cérébral ou de dissection artérielle cervico-céphalique.
Éduquer les patients sur les symptômes de l'AVC et de la dissection artérielle cervico-céphalique (p. ex., carotide, vertèbre). Alerter les patients sur l'importance de consulter immédiatement un médecin si des symptômes d'AVC ou de dissection artérielle cervico-céphalique apparaissent.
Hémorragie alvéolaire pulmonaire
Les cas rapportés d'événements temporellement associés n'étaient pas liés à la maladie anti-MBG (syndrome de Goodpasteurs).
Thrombocytopénie
Une thrombocytopénie s'est produite dans les premiers jours suivant la perfusion (contrairement au PTI). Elle était souvent autolimitante et relativement légère, bien que la gravité et l'issue aient été inconnues dans de nombreux cas.
Afin de limiter les réactions liées à la perfusion, il est recommandé d'administrer une prémédication par corticoïdes aux patients juste avant la perfusion de Lemtrada pendant chacun des 3 premiers jours de chaque cycle de traitement. Au cours des études cliniques, les patients ont reçu 1'000 mg de méthylprednisolone pendant les 3 premiers jours de chaque cycle de traitement par Lemtrada. De plus, une prémédication par antihistaminiques et/ou antipyrétiques peut également être envisagée avant la perfusion de Lemtrada.
La plupart des patients inclus dans les études cliniques contrôlées ont reçu au moins une fois une prémédication par antihistaminiques et/ou antipyrétiques avant la perfusion de Lemtrada. Les patients peuvent toutefois présenter des RAP malgré une prémédication. Une surveillance des éventuelles RAP est recommandée pendant et au moins deux heures après la perfusion de Lemtrada. Les médecins devraient informer les patients qu'une RAP peut survenir dans les 48 heures suivant la perfusion. Il est recommandé de surveiller les signes vitaux avant la perfusion et périodiquement pendant la perfusion. Un temps d'observation prolongé doit être envisagé, le cas échéant. En cas de réactions sévères, l'arrêt immédiat de la perfusion IV doit être envisagé. Au cours des études cliniques, les réactions anaphylactiques ou graves ayant nécessité l'arrêt du traitement ont été très rares.
Lemtrada doit être administré dans un hôpital disposant d'équipements et de personnel pour prendre en charge de façon appropriée les chocs anaphylactiques, les réactions sévères à la perfusion, les ischémies myocardiques, les infarctus du myocarde et les effets indésirables cérébrovasculaires.
Instructions de perfusion pour réduire les réactions graves temporairement associées à la perfusion de Lemtrada:
·Evaluations préalables à la perfusion:
·Réaliser un ECG et mesurer les signes vitaux (notamment la fréquence cardiaque et la pression artérielle) lors de la visite initiale. Réaliser des analyses biologiques (hémogramme avec formule leucocytaire, transaminases sériques, créatinine sérique, test de la fonction thyroïdienne et analyse d'urine avec microscopie).
·Pendant la perfusion:
·Réaliser une surveillance continue/fréquente (au moins toutes les heures) de la fréquence cardiaque, de la pression artérielle et de l'état clinique général des patients.
·En cas d'effet indésirable sévère
·Interrompre la perfusion.
·Evaluer médicalement le patient en fonction du profil d'effets indésirables de Lemtrada avant d'envisager la reprise du traitement.
·Fournir un traitement approprié s'il y a lieu.
·Envisager d'interrompre définitivement la perfusion de Lemtrada si le patient présente des symptômes cliniques suggérant le développement d'un effet indésirable sévère associé à la perfusion (ischémie myocardique, accident vasculaire cérébral hémorragique, dissection artérielle cervico-céphalique ou hémorragie alvéolaire pulmonaire).
·Après la perfusion:
·L'observation en vue de détecter d'éventuelles réactions à la perfusion est recommandée pendant au moins 2 heures après la perfusion de Lemtrada. Les patients présentant des symptômes cliniques suggérant le développement d'un effet indésirable sévère temporellement associé à la perfusion (ischémie myocardique, accident vasculaire cérébral hémorragique, dissection artérielle cervico-céphalique ou hémorragie alvéolaire pulmonaire) doivent être étroitement surveillés jusqu'à la disparition complète des symptômes.
La période d'observation doit être prolongée autant que nécessaire. Les patients doivent être informés du potentiel d'apparition tardive de réactions associées à la perfusion, et invités à signaler les symptômes et à consulter un service de soins approprié.
Lymphohistiocytose hémophagocytaire (LHH)
Au cours de l'utilisation post-marketing, des cas de LHH (y compris les cas mortels) ont été rapportés chez des patients traités par Lemtrada. La LHH est un syndrome potentiellement mortel d'activation immunitaire pathologique caractérisé par des signes cliniques et des symptômes d'inflammation systémique extrême. La LHH se caractérise par de la fièvre, une hépatomégalie et des cytopénies. Elle est associée à des taux de mortalité élevés si elle n'est pas reconnue et traitée tôt. Des symptômes ont été signalés dans les quelques mois à quatre ans suivant le début du traitement. Les patients doivent être informés des symptômes de la LHH et du délai d'apparition. Les patients qui développent des manifestations précoces d'activation immunitaire pathologique doivent être évalués immédiatement et un diagnostic de LHH doit être envisagé.
Maladie de Still de l'adulte (MSA)
Au cours de l'utilisation post-marketing, des cas de MSA ont été signalés chez des patients traités par Lemtrada. La maladie de Still de l'adulte est un syndrome inflammatoire rare qui nécessite une évaluation et un traitement urgents. Les patients atteints de MSA peuvent présenter une combinaison des signes et symptômes suivants: fièvre, arthrite, éruption cutanée et leucocytose en l'absence d'infections, de tumeurs malignes et d'autres affections rhumatismales. Envisager l'interruption ou l'arrêt du traitement par Lemtrada si une autre étiologie des signes ou symptômes ne peut être établie.
Infections
Des infections sont survenues chez 71% des patients traités par Lemtrada 12 mg, contre 53% des patients traités par Rebif® (interféron bêta-1a [IFNB-1a]) au cours des études cliniques contrôlées d'une durée de 2 ans dans la SEP. Ces infections étaient généralement d'intensité légère à modérée.
Les infections plus fréquemment observées chez les patients traités par Lemtrada versus celles observées chez les patients traités par IFNB-1a ont été: nasopharyngite, infection des voies urinaires, infection des voies respiratoires supérieures, sinusite, herpès simplex, grippe et bronchite. Des infections graves sont survenues chez 2,7% des patients traités par Lemtrada, contre 1,0% des patients traités par IFNB-1a au cours des études cliniques contrôlées dans la SEP. Les infections graves observées chez les patients traités par Lemtrada ont été: appendicite, gastro-entérite, pneumonie, zona et infection dentaire. Les infections ont été généralement d'une durée habituelle et guéries sous traitement médical habituel.
Le taux annualisé cumulatif des infections était de 0,99 pendant le suivi médian de 6,1 ans (maximum 12 ans) depuis la première exposition à Lemtrada, par rapport à 1,27 dans les essais cliniques contrôlés.
Des infections graves par le virus varicelle-zona (VZV), incluant primo-infection (varicelle) et réactivation du VZV, ont été plus fréquentes chez les patients traités par Lemtrada 12 mg que chez ceux traités par IFNB-1a (0,4% vs 0%) dans les études cliniques. Des infections gynécologiques par le virus papilloma humain (VPH), avec dysplasie du col utérin, ont également été rapportées chez les patientes du groupe Lemtrada 12 mg (2%). Il est recommandé de réaliser un test de dépistage annuel du VPH chez les patientes.
Des cas de tuberculose ont été rapportés chez des patients traités par Lemtrada et IFNB-1a au cours des études cliniques contrôlées. Des cas de tuberculose active ou latente ont été rapportés chez 0,3% des patients traités par Lemtrada, généralement en zone d'endémie. Avant le début du traitement, une éventuelle tuberculose active ou non (latente) doit être recherchée chez tous les patients conformément aux recommandations locales.
Des cas de listériose/méningite à Listeria ont été rapportés chez des patients traités par Lemtrada, généralement durant le premier mois de traitement. Si elle n'est pas traitée, l'infection par listeria peut conduire à une morbidité ou une mortalité importante. Afin de réduire le risque, les patients qui reçoivent Lemtrada doivent éviter d'ingérer de la viande crue ou pas assez cuite, des fromages à pâtes molles et des produits laitiers non pasteurisés pendant au moins un mois suite à la dernière perfusion de Lemtrada.
Des infections fongiques superficielles, en particulier des candidoses orale ou vaginale, ont été plus fréquemment rapportées chez les patients traités par Lemtrada que chez ceux traités par IFNB-1a (12% vs 3%) au cours des études cliniques contrôlées dans la SEP.
Chez les patients présentant une infection active, il est recommandé de retarder le traitement par Lemtrada jusqu'à ce que l'infection soit résolue.
Une prophylaxie par voie orale contre le virus de l'herpès doit être initiée dès le premier jour du traitement par Lemtrada et poursuivie pendant au moins un mois après chaque cycle de traitement. Au cours des études cliniques, les patients ont reçu 200 mg d'aciclovir ou un équivalent deux fois par jour.
Dans le traitement de la SEP, Lemtrada n'a pas été administré en association ou à la suite de traitements antinéoplasiques ou immunosuppresseurs. L'utilisation concomitante de Lemtrada avec l'un de ces traitements pourrait augmenter le risque d'immunosuppression.
Aucune donnée n'est disponible concernant l'administration de Lemtrada et une réactivation du virus de l'hépatite B (VHB) ou de l'hépatite C (VHC), les patients présentant des signes d'infection active ou chronique ayant été exclus des études cliniques. Un dépistage des patients à haut risque d'infection par le VHB et/ou le VHC avant instauration d'un traitement par Lemtrada doit être envisagé. Une attention particulière doit être exercée en cas de prescription de Lemtrada à des patients porteurs du VHB et/ou du VHC, car ils présentent, du fait de leur statut de porteur, un risque d'atteinte irréversible du foie en cas de réactivation du virus.
Des infections dues au cytomégalovirus (CMV) ont été rapportées chez les patients recevant Lemtrada en association à l'utilisation de corticostéroïdes. La plupart des cas sont apparus au cours des 2 mois après administration d'alemtuzumab. Chez les patients présentant des symptômes, une évaluation clinique devrait au minimum être réalisée au cours des deux mois qui suivent chaque cycle de traitement par Lemtrada à la recherche d'une infection par le CMV.
Des infections dues au virus d'Epstein-Barr (VEB) pouvant entrainer notamment des cas d'hépatites graves et parfois mortelles ont été signalés chez les patients traités par Lemtrada.
Leucoencéphalopathie multifocale progressive (LEMP)
La leucoencéphalopathie multifocale progressive (LEMP) est une infection virale opportuniste du cerveau causée par le virus JC (JCV) qui ne survient généralement que chez les patients immunodéprimés et qui entraîne généralement la mort ou une invalidité grave. Les symptômes typiques associés à la LEMP sont divers, évoluent au cours des jours ou des semaines et comprennent une faiblesse progressive d'un côté du corps ou une maladresse des membres, une perturbation de la vision et des changements dans la pensée, la mémoire et l'orientation menant à la confusion et aux changements de personnalité.
Aucun cas de LEMP n'a été signalé dans les études cliniques sur l'alemtuzumab chez les patients atteints de sclérose en plaques. La LEMP a été rapportée dans le contexte post-marketing chez des patients présentant d'autres facteurs de risque, en particulier un traitement antérieur avec des produits de SEP associés à la LEMP.
Les résultats de l'IRM peuvent être apparents avant les signes ou symptômes cliniques. Des cas de LEMP, diagnostiqués sur la base des résultats de l'IRM et de la détection d'ADN JCV dans le liquide céphalorachidien en l'absence de signes ou symptômes cliniques spécifiques à la LEMP, ont été signalés chez des patients traités avec d'autres médicaments contre la SEP associés à la LEMP. Beaucoup de ces patients sont devenus par la suite symptomatiques de la LEMP. Par conséquent, la surveillance par IRM, y compris avant l'initiation de Lemtrada, pour les signes qui peuvent être compatibles avec la LEMP peut être utile, et tout résultat suspect devrait conduire à une enquête plus approfondie pour permettre un diagnostic précoce de la LEMP, le cas échéant. Après l'arrêt d'un autre médicament contre la SEP associé à la LEMP, une mortalité et une morbidité liées à la LEMP plus faibles ont été rapportées chez des patients initialement asymptomatiques au moment du diagnostic par rapport aux patients qui présentaient des signes et symptômes cliniques caractéristiques au moment du diagnostic. On ne sait pas si ces différences sont dues à une détection précoce et à l'arrêt du traitement de la SEP ou à des différences de maladie chez ces patients.
Malignité
Comme c'est le cas pour les autres immunomodulateurs, il convient de faire preuve de prudence lors de l'instauration d'un traitement par Lemtrada chez les patients présentant une pathologie maligne préexistante. On ignore actuellement si l'alemtuzumab augmente le risque d'apparition de cancers de la thyroïde, les manifestations auto-immunes thyroïdiennes pouvant constituer un facteur de risque de malignité thyroïdienne. Les patients souffrant d'affections malignes actives connues, à l'exception des patients atteints d'un carcinome basocellulaire cutané, ne doivent pas être traités par Lemtrada (cf. «Contre-indications»).
Pneumonite
Des cas de pneumonite ont été rapportés chez des patients traités par Lemtrada. La plupart des cas sont survenus au cours du premier mois après traitement par Lemtrada. Les patients devraient être avisés de signaler les symptômes d'une pneumonite, lesquels peuvent inclure essoufflement, toux, respiration sifflante, douleur thoracique ou oppression thoracique et hémoptysie.
Cholécystite aiguë
Lemtrada peut augmenter le risque de cholécystite aiguë. Lors des études contrôlées, 0,2% des patients SEP recevant Lemtrada ont développé une cholécystite aiguë versus 0% des patients traités par interféron bêta-1a.
Au cours de l'utilisation post-marketing, des cas supplémentaires de cholécystites aiguës ont été rapportés chez les patients recevant Lemtrada. La durée d'apparition des symptômes variait entre 24 heures et 2 mois après la perfusion de Lemtrada. La plupart des patients étaient traités de manière conservatrice avec des antibiotiques et ont récupéré sans intervention chirurgicale alors que les autres ont subi une cholécystectomie.
Les symptômes de cholécystite aiguë comprennent douleur abdominale, sensibilité abdominale, fièvre, nausée, vomissements. Si elle n'est pas diagnostiquée à temps ni traitée, la cholécystite aiguë est une condition pouvant être associée à des taux élevés de morbi-mortalité. En cas de suspicion d'une cholécystite aiguë, évaluez et traitez sans attendre.
Contraception
Un passage transplacentaire et une activité pharmacologique potentielle de Lemtrada ont été observés chez la souris pendant la gestation et après la naissance. Les femmes en âge de procréer doivent avoir une contraception efficace pendant le traitement et pendant les 4 mois suivant chaque cycle de traitement par Lemtrada (cf. «Grossesse/Allaitement» et «Données précliniques»).
Vaccins
Il est recommandé aux patients de mettre leurs vaccins à jour selon les recommandations locales au moins 6 semaines avant l'instauration du traitement par Lemtrada. La réponse immunitaire à un vaccin après le traitement par Lemtrada n'a pas été étudiée.
La sécurité d'une vaccination par un virus vivant après un cycle de traitement par Lemtrada n'a pas été formellement étudiée au cours des études cliniques contrôlées dans la SEP. Ce type de vaccin ne doit donc pas être administré à des patients atteints de SEP ayant récemment reçu un cycle de traitement par Lemtrada.
Test de dépistage des anticorps/vaccinations contre le virus du varicelle-zona
Comme pour tout médicament modifiant la réponse immunitaire, une sérologie VZV doit être réalisée avant tout cycle de traitement par Lemtrada chez les patients n'ayant pas d'antécédent de varicelle et n'ayant pas été vaccinés contre le VZV. La vaccination contre le VZV des patients ayant une sérologie négative doit être envisagée avant l'instauration du traitement par Lemtrada. Pour garantir une efficacité optimale du vaccin, le traitement par Lemtrada devra être initié au plus tôt 6 semaines après la vaccination.
Analyses biologiques recommandées pour la surveillance des patients
Des analyses biologiques doivent être réalisées régulièrement pendant les 48 mois après le dernier cycle de traitement par Lemtrada afin de détecter les premiers signes de maladie auto-immune.
·Numération formule sanguine et plaquettaire avec transaminases sériques (avant l'instauration du traitement puis tous les mois)
·Créatininémie (avant l'instauration du traitement puis tous les mois)
·Analyse urinaire et numération cellulaire dans les urines (avant l'instauration du traitement puis tous les mois)
·Bilan thyroïdien avec dosage de TSH (avant l'instauration du traitement puis tous les 3 mois)
Au-delà de cette période, des analyses supplémentaires seront nécessaires en présence de tout résultat clinique suggérant l'existence d'une néphropathie ou de troubles thyroïdiens.
Traitements immunosuppresseurs concomitants ou antérieurs
La sécurité et l'efficacité de Lemtrada associé à d'autres immunosuppresseurs et antinéoplasiques n'ont pas été prouvées. Lors de l'instauration du traitement par Lemtrada, il faut tenir compte du risque de double exposition du système immunitaire et des conséquences (cf. «Posologie/Mode d'emploi»). L'utilisation concomitante de ces substances avec Lemtrada peut augmenter le risque d'infections, également celui d'infections opportunistes, et représente par conséquent une contre-indication (cf. «Contre-indications»).
Outre la prémédication recommandée par corticoïdes au cours des 3 premiers jours de chaque cycle de traitement, on peut également administrer des stéroïdes à court terme pendant le traitement par Lemtrada pour traiter les poussées.
Arrêt du traitement
La sécurité et l'efficacité d'autres traitements de la SEP influençant le système immunitaire n'ont pas été prouvées après un traitement par Lemtrada. Lors du passage de Lemtrada à d'autres médicaments pour le traitement de la SEP, il faut tenir compte de la possibilité d'une exposition concomitante et de ses conséquences sur le système immunitaire. On doit disposer d'un statut immunitaire complet récent, ainsi que d'une numération plaquettaire.
Informations relatives à l'utilisation de l'alemtuzumab avant l'autorisation de mise sur le marché de Lemtrada en dehors des études financées par le titulaire
Les effets indésirables suivants ont été identifiés avant l'autorisation de mise sur le marché dans la SEP de Lemtrada, lors de l'administration de l'alemtuzumab dans le traitement de la leucémie lymphoïde chronique à cellules B (LLC-B) et dans d'autres pathologies, à des doses généralement plus élevées (p.ex. 30 mg) et plus fréquentes que celles recommandées dans le traitement de la SEP. Ces effets indésirables ayant été rapportés spontanément et dans une population difficile à évaluer, il n'a pas toujours été possible d'estimer leur fréquence avec précision, ni d'établir un lien de causalité avec l'exposition à l'alemtuzumab.
Maladies auto-immunes
Des événements auto-immuns comprenant neutropénie, anémie hémolytique (dont un cas fatal), hémophilie acquise, maladie des anticorps anti-MBG (syndrome de Goodpasture) et troubles thyroïdiens ont été rapportés chez des patients traités par alemtuzumab. Des phénomènes auto-immuns graves et parfois fatals, tels que: anémie hémolytique auto-immune, thrombopénie auto-immune, anémie aplasique, syndrome de Guillain-Barré et polyradiculonévrite chronique inflammatoire démyélinisante, ont été rapportés chez les patients traités par alemtuzumab, mais non atteints de SEP. Un test de Coombs positif a été observé chez un patient traité par alemtuzumab en oncologie. Un décès à la suite d'une transfusion dans le cadre d'une réaction du greffon contre l'hôte a été rapporté avec l'alemtuzumab.
Réactions liées à la perfusion
Des RAP graves et parfois fatales, comprenant bronchospasme, hypoxie, syncope, infiltrats pulmonaires, syndrome de détresse respiratoire aiguë, arrêt respiratoire, infarctus du myocarde, arythmies, insuffisance cardiaque aiguë et arrêt cardiaque, ont été rapportés chez des patients non atteints de SEP et traités par alemtuzumab à des doses plus élevées et plus fréquentes que celles recommandées dans la SEP. Une anaphylaxie grave et d'autres réactions d'hypersensibilité, y compris un choc anaphylactique et un angioœdème, ont également été rapportés.
Infections et infestations
Des infections virales, bactériennes, fongiques et à protozoaires, graves et parfois fatales, notamment liées à la réactivation d'infections latentes, ont été rapportées chez les patients non atteints de SEP et traités par alemtuzumab à des doses plus élevées et plus fréquentes que celles recommandées dans la SEP. Des cas de leuco-encéphalopathie multifocale progressive (LEMP) ont été signalés chez des patients atteints de LLC-B avec ou sans traitement par alemtuzumab. La fréquence de survenue de la LEMP chez les patients atteints de LLC-B traités par alemtuzumab n'est pas supérieure à celle observée dans la population de patients atteints de LLC-B.
Affections hématologiques et du système lymphatique
Des hémorragies sévères ont été rapportées chez des patients non atteints de SEP.
Affections cardiaques
Des cas d'insuffisance cardiaque congestive, de cardiomyopathie et de diminution de la fraction d'éjection ont été signalés chez les patients traités par alemtuzumab et non atteints de SEP ayant été traités préalablement par des molécules potentiellement cardiotoxiques.
Affections lymphoprolifératives liées au virus d'Epstein-Barr
Des affections lymphoprolifératives liées au virus d'Epstein-Barr ont été rapportées en dehors des études financées par le titulaire.
Informations importantes relatives à certains autres composants de Lemtrada
Ce médicament contient du sodium et du potassium, mais moins de 1 mmol (23 mg) de sodium et moins de 1 mmol (39 mg) de potassium par flacon.
InteractionsAucune étude d'interaction médicamenteuse formelle n'a été menée avec Lemtrada administré à la dose recommandée dans le traitement de la SEP. Au cours d'une étude clinique contrôlée dans la SEP, les patients récemment traités par interféron bêta et par acétate de glatiramère devaient interrompre leur traitement 28 jours avant l'instauration du traitement par Lemtrada.
Grossesse, allaitementFemmes en âge de procréer
Les concentrations sériques en médicament étaient légèrement supérieures ou inférieures au seuil de détection environ 30 jours après chaque cycle de traitement. Les femmes en âge de procréer doivent par conséquent utiliser une contraception efficace pendant un cycle de traitement par Lemtrada ainsi que pendant les 4 mois suivants.
Grossesse
Les données d'utilisation de Lemtrada chez la femme enceinte sont limitées. Aucune étude formelle n'a été conduite. Lemtrada ne doit être administré pendant la grossesse, à moins que cela s'avère clairement nécessaire. En revanche, il ne faut pas instaurer de traitement pendant la grossesse (cf. «Contre-indications»).
L'IgG humaine traversant la barrière placentaire, l'alemtuzumab est également susceptible de traverser la barrière placentaire et peut donc entraîner un risque pour le fœtus. Les études de toxicité effectuées chez l'animal ont mis en évidence une toxicité sur la fonction de reproduction (cf. rubrique «Données précliniques»). L'effet de l'alemtuzumab sur le fœtus lors de l'administration à une femme enceinte ou son effet sur la fonction de reproduction ne sont pas connus.
Les troubles thyroïdiens (cf. rubrique «Mises en garde et précautions, Troubles thyroïdiens») représentent un risque particulier pour la femme enceinte. Une hypothyroïdie non traitée pendant la grossesse peut accroître le risque de fausse couche et le risque pour le fœtus tel que retard mental et nanisme. Chez les femmes enceintes atteintes de la maladie de Basedow, les anticorps maternels anti-récepteur de la thyrotropine peuvent être transmis au fœtus et entraîner une maladie de Basedow néonatale transitoire.
L'instauration du traitement par Lemtrada pendant la grossesse est contre-indiquée (cf. «Contre-indications»).
Allaitement
Lemtrada a été détecté dans le lait maternel et chez la descendance de souris femelles allaitantes ayant reçu 10 mg/kg par jour pendant 5 jours consécutifs après la mise bas.
Le passage de Lemtrada dans le lait maternel humain n'est pas connu. Un risque pour le nouveau-né allaité ne peut pas être exclu. L'allaitement doit donc être interrompu pendant chaque cycle de traitement par Lemtrada et pendant les 4 mois suivant la dernière perfusion. Il n'existe aucune donnée portant sur la détection de Lemtrada dans le lait maternel après un cycle de traitement par Lemtrada.
Effet sur l’aptitude à la conduite et l’utilisation de machinesLes effets de Lemtrada sur l'aptitude à la conduite et à l'utilisation de machines n'ont pas été étudiés.
La plupart des patients présentent des réactions liées à la perfusion pendant ou dans les 24 heures suivant le traitement par Lemtrada. Certaines réactions liées à la perfusion (p.ex. étourdissements/vertiges) pourraient temporairement affecter la capacité du patient à conduire des véhicules ou à utiliser des machines et il convient de faire preuve de prudence jusqu'à leur résolution.
Effets indésirablesRésumé du profil de sécurité d'emploi
La sécurité d'emploi a été évaluée chez 1 486 patients traités par Lemtrada (12 mg ou 24 mg) selon une analyse poolée des études cliniques contrôlées dans la SEP avec un suivi médian de 6,1 ans (maximum 12 ans), cette population correspond à 8 635 années-patients de suivi de sécurité d'emploi.
Les principaux effets indésirables ont été de nature auto-immune (PTI, troubles thyroïdiens, néphropathies, cytopénies), des réactions liées à la perfusion et des infections. Ils sont décrits à la rubrique «Mises en garde et précautions».
Les effets indésirables les plus fréquents avec Lemtrada (survenant chez ≥20% des patients) sont: éruptions cutanées, céphalées, fièvre et infections des voies respiratoires.
Tableau des effets indésirables
Le tableau suivant repose sur les données poolées de sécurité d'emploi de quatre études: les études 1 et 2 étaient jusqu'à 24 mois chez des patients atteints de SEP récurrente-rémittente traités par Lemtrada 12 mg/jour pendant 5 jours consécutifs à l'inclusion et pendant 3 jours consécutifs au 12e mois de l'étude. L'étude 3 (CAMMS223) a évalué la sécurité et l'efficacité de Lemtrada chez les patients atteints de SEP-RR sur une durée de 3 ans. L'étude 4 (CAMMS03409) était une étude de prolongation non contrôlée visant à évaluer la sécurité et l'efficacité à long terme (4 années supplémentaires) de Lemtrada chez les patients des études 1, 2 ou 3.
Les effets indésirables survenant chez ≥0,5% des patients sont présentés selon MedDRA (principales classes de systèmes d'organes selon Medical Dictionary for Regulatory Activities) et les termes préférentiels (preferred term, PT).
Tableau 1: Effets indésirables observés au cours des études 1, 2, 3 et 4 chez ≥0,5% des patients traités par Lemtrada 12 mg
|
Classe de systèmes d'organes
|
Très fréquents
(≥1/10)
|
Fréquents
(≥1/100, <1/10)
|
Peu fréquents
(≥1/1'000, <1/100)
| |
Infections et infestations
|
Infections des voies respiratoires (36,0%), infections des voies urinaires (14,2%)
|
Infections des voies respiratoires inférieures, zona, gastro-entérite, herpès oral, candidose orale, candidose vulvo-vaginale, grippe, infection de l'oreille, pneumonie, infection vaginale
|
Infection dentaire, abcès dentaire, onychomycose, gastroentérite virale, gingivite, infection fongique de la peau, amygdalite, sinusite aiguë, vaginose bactérienne, infection par le virus de l'herpès, herpès génital, cellulite, pneumonite
| |
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes)
|
|
|
Papillome cutané
| |
Affections hématologiques et du système lymphatique
|
Lymphopénie (32,8%), leucopénie (14,8%)
|
Lymphadénopathie, purpura thrombopénique immunologique (PTI), thrombopénie, hausse de la numération leucocytaire, anémie, diminution de l'hématocrite, neutrophilie, hausse de la numération des éosinophiles, monocytose, hausse de la numération lymphocytaire
|
| |
Affections du système immunitaire
|
|
Syndrome de libération des cytokines
|
Hypersensibilité
| |
Affections endocriniennes
|
Maladie de Basedow (13,1%), hyperthyroïdie (14,4%), hypothyroïdie (14,1%)
|
Thyroïdite auto-immune, goitre, anticorps anti-thyroïde positifs
|
| |
Troubles du métabolisme et de la nutrition
|
|
|
Diminution de l'appétit
| |
Affections psychiatriques
|
|
Insomnie*, anxiété, dépression
|
| |
Affections du système nerveux
|
Céphalées* (46,4%)
|
Poussée de SEP, étourdissement*, hypoesthésie, paresthésies, tremblements, dysgueusie
|
Hyperesthésie, trouble sensoriel
| |
Affections oculaires
|
|
Conjonctivite, ophtalmopathie endocrinienne, vision trouble
|
Diplopie
| |
Affections de l'oreille et du labyrinthe
|
|
Vertige
|
Douleur d'oreille
| |
Affections cardiaques
|
Tachycardie* (10,8%)
|
Bradycardie*, palpitations
|
| |
Affections vasculaires
|
Bouffées vasomotrices* (11,8%)
|
Hypotension*, hypertension
|
| |
Affections respiratoires, thoraciques et médiastinales
|
|
Dyspnée*, toux, épistaxis, hoquet, douleur oropharyngée
|
Sensation de gorge serrée, irritation de la gorge, asthme, toux productive
| |
Affections gastro-intestinales
|
Nausées* (18,0%)
|
Douleur abdominale, vomissements, diarrhées, dyspepsie*, stomatite
|
Constipation, reflux gastro-œsophagien, saignement gingival, bouche sèche, dysphagie, troubles gastro-intestinaux, hématochézie
| |
Affections hépatobiliaires
|
|
Hausse de l'aspartate aminotransférase (ASAT), hausse de l'alanine aminotransférase (ALAT)
|
| |
Affections de la peau et du tissu sous-cutané
|
Urticaire* (16,4%), rash* (50,1%), prurit* (18,6%), rash généralisé* (13,2%)
|
Erythème, ecchymose, alopécie, hyperhidrose, acné
|
Vésicules cutanées, sueurs nocturnes, lésion cutanée, gonflement du visage, eczéma, dermatite
| |
Affections musculo-squelettiques et du tissu conjonctif
|
|
Myalgie, faiblesse musculaire, arthralgie, dorsalgie, extrémités douloureuses, contractures musculaires, cervicalgie
|
Douleur musculo-squelettique, raideur musculo-squelettique, douleur thoracique musculo-squelettique, inconfort dans les membres
| |
Affections du rein et des voies urinaires
|
|
Protéinurie, hématurie, hausse de la créatininémie, estérase leucocytaire positive dans l'urine
|
Néphrolithiase, cétonurie
| |
Affections des organes de reproduction et du sein
|
|
Ménorragies, règles irrégulières
|
Dysplasie du col utérin, aménorrhée
| |
Troubles généraux et anomalies au site d'administration
|
Fièvre* (37,5%), fatigue* (11,3%), frissons* (11,2%)
|
Gêne thoracique*, douleur*, œdèmes périphériques, asthénie, syndrome pseudo-grippal, malaise, douleur au point d'injection
|
| |
Investigations
|
|
|
Perte de poids, prise de poids, diminution de la numération des globules rouges, test bactérien positif, diminution du rapport CD4/CD8, hausse de la glycémie, augmentation du volume globulaire moyen (VGM)
| |
Lésions, intoxications et complications liées aux procédures d'intervention
|
|
Contusions, réaction liée à la perfusion
|
|
Les termes marqués par un astérisque (*) désignent les effets indésirables rapportés comme étant liés à la perfusion (RAP). Les RAP comprennent fibrillation auriculaire et anaphylaxie avec, pour les événements liés au traitement, une fréquence inférieure au seuil de fréquence de 0,5% (cf. rubrique «Mises en garde et précautions»).
Effets indésirables identifiés après la mise sur le marché (fréquence inconnue – la fréquence ne peut pas être estimée sur la base des données disponibles):
Infections et infestations: infections dues au cytomégalovirus (CMV), infections dues au virus d'Epstein-Barr (VEB).
Affections hématologiques et du système lymphatique: cas de neutropénie sévère (y compris mortelle), hémophilie A acquise, purpura thrombotique thrombocytopénique (PTT).
Affections du système immunitaire: lymphohistiocytose hémophagocytaire, sarcoïdose.
Affections du système nerveux: encéphalite auto-immune.
Affections cardiaques: ischémie myocardique*, infarctus du myocarde*.
Affections vasculaires: accident vasculaire cérébral (AVC)*, dissection artérielle cervico-céphalique*, hémorragie alvéolaire pulmonaire*.
Affections hépatobiliaires: cholécystite aigue, hépatite auto-immune, hépatite due au virus d'Epstein-Barr (VEB).
Affections de la peau et du tissu sous-cutané: vitiligo.
Affections musculo-squelettiques et du tissu conjonctif: maladie de Still de l'adulte (MSA).
Le type d'événements indésirables, incluant l'importance et la gravité dans les groupes de traitement sous Lemtrada pendant toute la durée du suivi disponible, incluant les patients ayant reçu les cycles de traitement supplémentaires, étaient similaires à ceux observés au cours des études contrôlées avec traitement actif.
Chez les patients poursuivant le traitement après des études cliniques contrôlées et n'ayant pas reçu de Lemtrada supplémentaire après les deux cycles de traitement initiaux, le taux (événements par année-personne) de la plupart des réactions indésirables était comparable ou inférieur au cours des années 3 à 6 par rapport aux années 1 et 2. Le taux le plus élevé de réactions indésirables touchant la thyroïde était enregistré en année 3 et baissait par la suite.
Chez les patients qui ont reçu des cycles de traitement supplémentaires dans le cadre de l'étude d'extension de 4 ans, on a observé une différence numérique du faible risque total d'infections graves par rapport aux patients sans traitement supplémentaire (taux 0,027 vs. 0,012). Ces infections impliquaient différents organes et/ou pathogènes; aucune tendance n'a été observée.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDans les études cliniques contrôlées, deux patients atteints de SEP ont accidentellement reçu jusqu'à 60 mg de Lemtrada (c'est-à-dire la dose totale du cycle de traitement initial) en une seule perfusion et ont présenté des réactions graves (céphalées, éruption cutanée et soit une hypotension soit une tachycardie sinusale). Des doses de Lemtrada supérieures à celles évaluées dans les études cliniques peuvent augmenter l'intensité et/ou la durée des réactions liées à la perfusion ou ses effets sur l'immunité.
Il n'existe pas d'antidote connu en cas de surdosage d'alemtuzumab. Le traitement consiste à cesser immédiatement l'administration du médicament et à mettre en œuvre un traitement symptomatique.
Propriétés/EffetsCode ATC
L04AA34
Classe pharmacothérapeutique: immunosuppresseurs sélectifs.
Mécanisme d'action
Lemtrada est un anticorps monoclonal humanisé produit par ADN recombinant qui cible la glycoprotéine CD52 de 21-28 kD située à la surface des cellules. Lemtrada est un anticorps IgG1 kappa présentant une structure variable et des régions constantes humaines, ainsi que des régions déterminant la complémentarité obtenues à partir d'un anticorps monoclonal murin (rat). L'anticorps possède un poids moléculaire approximatif de 150 kD.
Lemtrada se lie à l'antigène CD52, présent en grandes quantités à la surface des lymphocytes T (CD3+) et B (CD19+) et en moindre quantité sur les cellules tueuses naturelles, les monocytes et les macrophages. L'antigène CD52 est peu ou pas présent sur les granulocytes neutrophiles, les plasmocytes et les cellules souches de la moelle osseuse. Après sa liaison à la surface des lymphocytes T et B, Lemtrada provoque la lyse des lymphocytes anticorps-dépendante et la lyse médiée par le complément.
Le mécanisme d'action de Lemtrada dans la SEP n'est pas totalement élucidé. Cependant, des travaux de recherche suggèrent des effets immunomodulateurs avec une déplétion initiale suivie d'une repopulation lymphocytaire, notamment:
·Modifications du nombre, des proportions et des propriétés de certaines sous-populations lymphocytaires après traitement
·Augmentation des lymphocytes T régulateurs
·Augmentation des lymphocytes T et B mémoire
·Effets transitoires sur certaines cellules de l'immunité innée (à savoir, granulocytes neutrophiles, macrophages, cellules NK)
La déplétion en lymphocytes B et T périphériques induite par Lemtrada et suivie d'une repopulation peuvent réduire le risque de poussée et donc ralentir la progression de la maladie.
Pharmacodynamique
Lemtrada réduit le nombre de lymphocytes T et B périphériques après chaque cycle de traitement, les valeurs les plus basses étant observées environ un mois après (soit à la première évaluation post-traitement dans les études cliniques de phase 3). La population lymphocytaire se reconstitue au fil du temps, les lymphocytes B étant généralement en nombre similaire aux valeurs initiales après 6 mois. Le nombre de lymphocytes T revient plus lentement à la normale, et généralement sans retour aux valeurs initiales après 12 mois de traitement. Environ 40% des patients ont présenté un nombre total de lymphocytes à la limite inférieure de la normale (LIN) 6 mois après chaque cycle de traitement, et environ 80% au bout de 12 mois.
Les taux de granulocytes neutrophiles, monocytes, granulocytes éosinophiles, granulocytes basophiles et cellules tueuses naturelles ne sont que transitoirement modifiés par Lemtrada.
Efficacité clinique
La sécurité d'emploi et l'efficacité de Lemtrada chez des patients atteints de SEP-RR ont été évaluées au cours de 3 études cliniques randomisées, avec évaluateur en aveugle, versus comparateur actif et lors d'une étude de prolongation non contrôlée avec évaluateur en aveugle menée auprès de patients atteints de SEP.
Les études 1 et 2 (CAMMS323/CARE-MS I menée chez des patients non pré-traités et CAMMS32400507/CARE-MS II menée chez des patients avec une réponse insuffisante au traitement précédent) ont inclus des patients avec une SEP rémittente-récurrente active ayant eu moins deux poussées cliniques au cours des 2 années précédentes. Des examens neurologiques étaient effectués toutes les 12 semaines et en cas de suspicion de poussée. Une évaluation IRM était réalisée une fois par an. La durée d'observation était de 2 ans. Dans les deux études, les patients ont été randomisés pour recevoir soit Lemtrada 12 mg/jour sous forme de perfusion IV une fois par jour pendant 5 jours au mois 0 et pendant 3 jours au mois 12 (le groupe 12 mg) soit un IFNB-1a 44 µg sous forme d'injections SC effectuées trois fois par semaine. L'étude 2 comprenait en plus un bras de traitement exploratoire avec Lemtrada 24 mg/jour sous forme de perfusion IV une fois par jour pendant 5 jours au mois 0 et pendant 3 jours au mois 12 (le groupe 24 mg). Le critère d'évaluation principal des études 1 et 2 était le taux annualisé de poussées (TAP) sur 2 ans et le délai d'aggravation confirmée du handicap (CDW, en anglais «confirmed disability worsening»), définie comme une augmentation confirmée à 6 mois d'au moins 1 point du score EDSS (Expanded Disability Status Scale) pour un score EDSS initial ≥1,0 (augmentation de 1,5 point chez les patients présentant un score EDSS initial de 0).
L'étude 1 (CAMMS323) a inclus des patients atteints d'une SEP-RR active (au moins 2 poussées au cours des 2 années précédentes) et un score EDSS de 0-3,0 (n=376 pour Lemtrada 12 mg et n=187 pour l'IFNB-1a). À l'inclusion, l'âge moyen était de 33 ans, la durée moyenne de la maladie était de 2 ans et le score EDSS moyen était de 2,0. Avant leur inclusion dans l'étude, les patients n'avaient pas encore reçu de traitement contre la SEP.
Après 2 ans, par rapport aux patients traités par IFNB-1a, le TAP était significativement réduit de 55% chez les patients sous Lemtrada. L'aggravation du handicap confirmée (CDW) à 6 mois n'était pas statistiquement différente entre les deux groupes de traitement: 8% des patients traités par Lemtrada et 11% des patients traités par IFNB-1a ont présenté une augmentation constante du score EDSS. L'influence du traitement sur les critères d'évaluation cliniques a été étayée par un effet significatif sur les paramètres IRM de l'inflammation et de la progression de la maladie, tels que notamment le volume cérébral.
Les résultats sont présentés dans le tableau 2.
Tableau 2: principaux résultats cliniques et IRM de l'étude 1
|
Critère d'évaluation
|
Lemtrada
(n=376)
|
IFNB-1a SC
(n=187)
| |
Critères d'évaluation cliniques
| |
Taux de poussées (co-critère d'évaluation principal)1
| |
Patients avec événements (nombre d'événements)
|
82 (119)
|
75 (122)
| |
TAP (IC à 95%)
|
0,18
(0,13; 0,23)
|
0,39
(0,29; 0,53)
| |
Risque relatif (IC à 95%)
|
0,45
(0,32; 0,63)
|
| |
Réduction du risque
|
54,88
|
| |
p
|
<0,0001
|
| |
Handicap (aggravation du handicap confirmée ≥6 mois; co-critère d'évaluation principal)2
| |
Estimation des patients avec une aggravation du handicap confirmée à 6 mois (IC à 95%)
|
8,00
(5,66; 11,24)
|
11,12
(7,32; 16,71)
| |
Risque relatif (IC à 95%)
|
0,70
(0,40; 1,23)
|
| |
p
|
0,2173
|
| |
Pourcentage de patients sans poussée à 2 ans (%)
| |
Estimation (IC à 95%)
|
77,59
(72,87; 81,60)
|
58,69
(51,12; 65,50)
| |
p
|
<0,0001
|
| |
Évolution du score EDSS à 2 ans par rapport à l'inclusion
| |
Estimation (IC à 95%)
|
-0,14
(-0,25; -0,02)
|
-0,14
(-0,29; 0,01)
| |
p
|
0,4188
|
| |
Critères d'évaluation IRM
| |
Variation du volume des lésions en T2 à 2 ans par rapport à l'inclusion (%)
|
-9,3
(-19,6; -0,2)
|
-6,5
(-20,7; 2,5)
| |
p
|
0,3080
|
| |
Patients présentant de nouvelles lésions/lésions élargies en T2 à 2 ans (%)
|
48,5 (23,1)
|
57,6 (40,6)
| |
p
|
0,0352
|
| |
Patients avec des lésions rehaussées par le gadolinium à 2 ans (%)
|
15,4
|
27,0
| |
p
|
0,0008
|
| |
Patients avec de nouvelles lésions hypointenses en T1 à 2 ans (%)
|
24,0
|
31,4
| |
p
|
0,0545
|
| |
Modification de l'atrophie cérébrale à 2 ans par rapport à l'inclusion (%)
|
-0,867
|
-1,488
| |
p
|
<0,0001
|
|
Pour le score EDSS, c'est la modification moyenne qui est indiquée, en utilisant un modèle mixte pour les mesures répétées. Pour le volume des lésions en T2 à l'IRM et pour la Fraction de parenchyme cérébral, ce sont les modifications médianes qui sont indiquées.
1 Co-critère d'évaluation principal: taux annualisé de poussées et progression du handicap. L'étude était jugée positive si au moins un des deux co-critères principaux était atteint.
2 Délai d'apparition d'une progression du handicap définie comme une augmentation confirmée à 6 mois d'au moins 1 point du score sur l'échelle étendue de Kurtzke (Expanded Disability Status Scale, EDSS) pour un score EDSS initial ≥1,0 (augmentation de 1,5 point chez les patients présentant un score EDSS initial de 0).
Des analyses complémentaires ont montré que, conformément à son effet sur le taux de poussées, l'administration de 12 mg/jour de Lemtrada a également entraîné une réduction significative du nombre de patients présentant des poussées sévères (réduction de 61%, p=0,0056), ainsi que du nombre de poussées ayant nécessité un traitement par corticoïdes (réduction de 58%, p<0,0001) par rapport à l'IFNB-1a.
L'étude 2 (CAMMS32400507) a inclus des patients atteints d'une SEP-RR active (au moins 2 poussées au cours des 2 années précédentes) et un score EDSS de 0-5 (n=426 pour Lemtrada 12 mg et n=202 pour IFNB-1a). Avant leur inclusion dans l'étude, les patients avaient présenté au moins 1 poussée sous traitement par bêta-interféron ou acétate de glatiramère, après avoir préalablement reçu le médicament pendant au moins 6 mois (réponse insuffisante au précédent traitement). À l'inclusion, l'âge moyen était de 35 ans, la durée moyenne de la maladie était de 4,5 ans et le score EDSS moyen était de 2,7. Au début de l'étude, la durée d'exposition moyenne par de précédents traitements de la SEP (≥1 médicament) était de 35 mois dans le groupe sous 12-mg de Lemtrada et 29% avaient reçu ≥2 traitements antérieurs de la SEP.
Chez les patients traités par 12 mg de Lemtrada, à 2 ans le TAP présentait une réduction significative de 49% par rapport aux patients traités par IFNB-1a. De plus, par rapport au traitement IFNB-1a, sur une période de 2 ans le traitement par Lemtrada a entraîné une réduction significative de 42% du risque d'aggravation du handicap confirmée à 6 mois. Les critères d'évaluation secondaires importants étaient la modification du score EDSS par rapport à l'inclusion et les paramètres IRM. À 2 ans, les patients traités par Lemtrada ont présenté une réduction significative du score EDSS moyen, tandis que chez les patients sous IFNB-1a le score EDSS moyen avait significativement augmenté par rapport à la valeur initiale. Par rapport aux patients sous IFNB-1a, les patients traités par Lemtrada avaient une probabilité 2,6 fois plus importante de présenter une amélioration du handicap confirmée (CDI, en anglais «Confirmed Disability Improvement»). L'influence du traitement sur les critères d'évaluation cliniques a été étayée par un effet significatif sur les paramètres IRM de l'inflammation et de la progression de la maladie, tels que notamment le volume cérébral.
Les résultats sont présentés dans le tableau 3 et sur la figure 1.
Tableau 3: principaux résultats cliniques et IMR de l'étude 2
|
Critère d'évaluation
|
Lemtrada
(n=426)
|
IFNB-1a SC
(n=202)
| |
Critères d'évaluation cliniques
| |
Taux de poussées (co-critère d'évaluation principal)1
| |
Patients avec événements (nombre d'événements)
|
147 (236)
|
104 (201)
| |
TAP (IC à 95%)
|
0,26
(0,21; 0,33)
|
0,52 (0,41; 0,66)
| |
Risque relatif (IC à 95%)
|
0,51
(0,39; 0,65)
|
| |
Réduction du risque
|
49,40
|
| |
p
|
<0,0001
|
| |
Handicap (aggravation du handicap confirmée ≥6 mois; co-critère d'évaluation principal)2
| |
Estimation des patients avec une aggravation du handicap confirmée à 6 mois (IC à 95%)
|
12,71
(9,89; 16,27)
|
21,13
(15,95; 27,68)
| |
Risque relatif (IC à 95%)
|
0,58
(0,38; 0,87)
|
| |
p
|
0,0084
|
| |
Pourcentage de patients sans poussée à 2 ans (%)
| |
Estimation (IC à 95%)
|
65,38
(60,65; 69,70)
|
46,70
(39,53; 53,54)
| |
p
|
<0,0001
|
| |
Évolution du score EDSS à 2 ans par rapport à l'inclusion
| |
(IC à 95%)
|
-0,17
(-0,29; -0,05)
|
0,24
(0,07; 0,41)
| |
p
|
<0,0001
|
| |
Amélioration du handicap confirmée (CDI)
| |
Estimation des patients avec une amélioration du handicap confirmée à 6 mois (IC à 95%)
|
28,82
(24,18; 34,13)
|
12,93
(8,34; 19,77)
| |
Risque relatif (IC à 95%)
|
2,57
(1,57; 4,20)
|
| |
p
|
0,0002
|
| |
Critères d'évaluation IRM
| |
Variation du volume des lésions en T2 à 2 ans par rapport à l'inclusion (%)
|
-1,27
|
-1,23
| |
p
|
0,1371
|
| |
Patients présentant de nouvelles lésions/lésions élargies en T2 à 2 ans (%)
|
46,2
|
67,9
| |
p
|
<0,0001
|
| |
Patients avec des lésions rehaussées par le gadolinium à 2 ans (%)
|
18,5
|
34,2
| |
p
|
<0,0001
|
| |
Patients avec de nouvelles lésions hypointenses en T1 à 2 ans (%)
|
19,9
|
38,0
| |
P
|
<0,0001
|
| |
Modification de l'atrophie cérébrale à 2 ans par rapport à l'inclusion (%)
|
-0,615
|
-0,810
| |
p
|
0,0121
|
|
Pour le score EDSS, c'est la modification moyenne qui est indiquée en utilisant un modèle mixte pour les mesures répétées. Pour le volume des lésions en T2 à l'IRM et l'atrophie cérébrale, ce sont les modifications médianes qui sont indiquées.
1 Co-critère d'évaluation principal: taux annualisé de poussées et progression du handicap. L'étude était jugée positive si au moins un des deux co-critères principaux était atteint.
2 Aggravation du handicap confirmée (CDW) définie comme une augmentation confirmée à 6 mois d'au moins 1 point du score sur l'échelle étendue de Kurtzke (Expanded Disability Status Scale, EDSS) pour un score EDSS initial ≥1,0 (augmentation de 1,5 point chez les patients présentant un score EDSS initial de 0).
Figure 1: délai d'apparition d'une aggravation du handicap confirmée à 6 mois dans l'étude 2
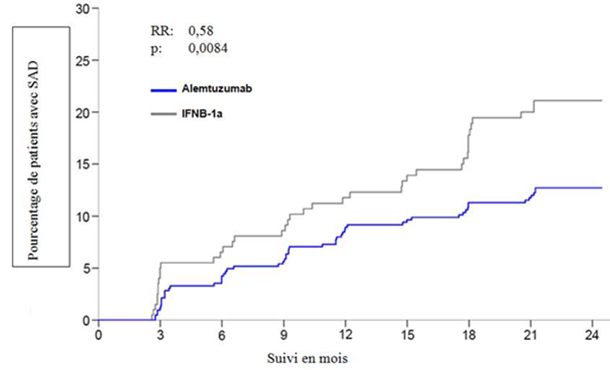
Des analyses complémentaires ont montré que, conformément à son effet sur le taux de poussées, l'administration de 12 mg/jour de Lemtrada réduit significativement le nombre de patients présentant des poussées sévères (réduction de 48%, p=0,0121), ainsi que le nombre de poussées ayant nécessité un traitement par corticoïdes (réduction de 56%, p<0,0001) ou une hospitalisation (réduction de 55%, p=0,0045) par rapport à l'IFNB-1a.
L'amélioration du handicap confirmée (Confirmed disability improvement, CDI) à 6 mois a été définie comme une baisse d'au moins un point sur l'échelle EDSS à partir d'un score EDSS initial ≥2.0. La régression du handicap confirmée mesure une réduction durable du handicap. 29% des patients traités par Lemtrada ont présenté une régression du handicap confirmée dans l'étude 2, contre seulement 13% des patients traités par IFNB-1a sous-cutané. Cette différence était statistiquement significative (p=0,0002).
L'étude 3 (étude CAMMS223 de phase 2) a évalué la sécurité d'emploi et l'efficacité de Lemtrada chez des patients atteints de SEP récurrente-rémittente pendant 3 ans. À l'inclusion, les patients présentaient un score EDSS compris entre 0 et 3,0, avaient eu au moins 2 poussées au cours des 2 années précédentes et au moins 1 lésion rehaussée par le gadolinium. Avant leur inclusion dans l'étude, les patients n'avaient jamais reçu de traitement de la SEP. Les patients ont reçu du Lemtrada 12 mg/jour (n=108) ou 24 mg/jour (n=108) administré par perfusion IV une fois par jour pendant 5 jours au mois 0 et pendant 3 jours au mois 12 ou de l'IFNB-1a sous-cutané 44 μg (n=107) administré 3 fois par semaine pendant 3 ans. Quarante-six patients ont reçu un troisième cycle de traitement par Lemtrada à raison de 12 mg/jour ou 24 mg/jour pendant 3 jours au mois 24.
À 3 ans, Lemtrada a réduit le risque d'aggravation du handicap confirmée à 6 mois de 76% (risque relatif 0,24 [IC à 95%: 0,110; 0,545], p<0,0006) et le taux annualisé de poussées de 67% (risque relatif 0,33 [IC à 95%: 0,196; 0,552], p<0,0001) par rapport à l'IFNB-1a. L'alemtuzumab 12 mg/jour a significativement réduit les scores EDSS (amélioration par rapport à l'inclusion) sur un suivi de 2 ans par rapport à l'IFNB-1a (p<0,0001).
Dans le sous-groupe de patients atteints de SEP-RR avec 2 rechutes ou plus au cours de l'année précédente et au moins 1 lésion T1 rehaussée par Gd au départ, le taux de rechute annualisé était de 0,26 (IC à 95%: 0,20, 0,34) dans le groupe traité par Lemtrada (n = 205) et 0,51 (IC à 95%: 0,40, 0,64) dans le groupe IFNB-1a (n = 102) (p <0,0001). Cette analyse comprend uniquement les données des études de phase 3 (CAMMS324 et CAMMS323) en raison des différences dans les algorithmes d'acquisition d'IRM entre les études de phase 2 et de phase 3. Ces résultats ont été obtenus à partir d'une analyse post hoc et doivent être interprétés avec prudence.
À 5 ans, Lemtrada a réduit le risque de progression du handicap de 69% (risque relatif 0,31 [IC à 95%: 0,161; 0,598], p=0,0005) et le taux annualisé de poussées de 66% (risque relatif 0,34 [IC à 95%: 0,202; 0,569], p<0,0001) par rapport à l'IFNB-1a sous-cutané.
L'étude 4 (CAMMS03409) était une étude d'extension multicentrique ouverte de phase III sur l'efficacité et la sécurité à long terme de Lemtrada, menée en aveugle pour l'évaluateur, chez des patients atteints de SEP-RR ayant participé à l'étude 1, 2 ou 3 (études antérieures de phase III et II). L'efficacité et la sécurité ont été étudiées pendant une durée médiane de 6 ans, depuis l'entrée des patients dans les études 1 et 2.
Dans l'étude 4, les patients étaient éligibles à recevoir un cycle supplémentaire de traitement par Lemtrada s'ils répondaient au moins à l'un des critères suivants:
a.a fait une rechute clinique ou plus,
b.a présenté deux lésions uniques ou plus dans l'imagerie (IRM) du cerveau ou de la moelle épinière sur une durée d'un an, composé d'une combinaison des éléments suivants:
·Lésion(s) réhaussée(s) par le gadolinium (en général ≥3 mm dans chaque dimension)
·Nouvelles lésions ou lésions élargies en T2 (en général ≥3 mm dans chaque dimension ou accroissement ≥3 mm
Le(s) cycle(s) supplémentaire(s) de traitement par Lemtrada était/étaient administré(s) à 12 mg/jour pendant 3 jours consécutifs (dose totale de 36 mg) au moins 12 mois après le cycle de traitement précédent par Lemtrada.
Parmi les patients traités par Lemtrada à 12 mg dans les études 1 et 2, 91,8% sont entrés dans l'étude 4. Parmi ces patients, 82,7% ont participé jusqu'au bout. Parmi les patients initialement traités par Lemtrada à 12 mg/jour dans l'étude 1 ou 2 et recrutés dans l'étude 4, environ la moitié (51,2%) n'a reçu que les 2 premiers cycles de traitement par Lemtrada et aucun autre traitement modificateur de la maladie au cours des 6 années de suivi.
Pendant les 6 années suivant l'administration du premier traitement par Lemtrada, on a observé, chez les patients participant au suivi, des taux de poussées de la SEP, de formation de lésions cérébrales à l'IRM et de perte du volume cérébral correspondant aux effets du traitement par Lemtrada pendant les études 1 et 2, ainsi que des scores de handicap majoritairement stables ou meilleurs. Les patients initialement traités par Lemtrada dans les études 1 et 2, y compris ceux participant au suivi de l'étude 4, présentaient, respectivement, un taux annualisé de poussées (TAP) de 0,17 et 0,23; on a observé une aggravation confirmée du handicap (CDW) chez 22,3% et 29,7% des patients tandis que 32,7% et 42,5% d'entre eux ont obtenu une amélioration du handicap confirmée (CDI). Parmi les patients ayant reçu un ou plusieurs cycles de traitement supplémentaires par Lemtrada, on a observé des améliorations du taux de poussées, de l'activité à l'IRM et des scores moyens de handicap après un premier ou un deuxième re-traitement (cycles 3 et 4) par rapport aux résultats de l'année précédente. Étant donné que l'étude 4 reposait sur un schéma ouvert et non contrôlé, les données de l'étude 4 ne sont cependant pas adaptées pour fournir une preuve définitive de l'efficacité.
Les bénéfices et les risques de 5 cycles ou plus de traitement n'ont pas été établis.
Immunogénicité
Comme avec toutes les protéines thérapeutiques, il existe un risque d'immunogénicité. Les données correspondent au pourcentage de patients dont les tests par immunoadsorption lié aux enzymes (ELISA) ont été positifs aux anticorps anti-alemtuzumab et confirmés par un test de liaison compétitive. Les échantillons positifs ont été évalués afin de détecter des signes d'inhibition in vitro par cytométrie de flux. Les patients inclus dans les études cliniques contrôlées dans la SEP avaient des échantillons sanguins prélevés aux mois 1, 3 et 12 après chaque cycle de traitement afin de doser les anticorps anti-alemtuzumab. Environ 85% des patients sous Lemtrada ont obtenu des résultats positifs aux anticorps anti-alemtuzumab pendant l'étude, >90% d'entre eux ont présenté également un résultat positif aux anticorps inhibiteurs de la liaison avec Lemtrada in vitro. Lorsque des anticorps anti-alemtuzumab étaient détectés, ils se formaient dans les 15 mois suivant l'exposition initiale. Au cours de deux cycles de traitement, aucune corrélation n'a été établie entre la présence d'anticorps anti-alemtuzumab ou d'anticorps inhibiteurs anti-alemtuzumab et une diminution de l'efficacité ou l'apparition d'effets indésirables, notamment de réactions liées à la perfusion. La présence d'anticorps anti-alemtuzumab de titre élevé observée chez certains patients a été associée à une déplétion lymphocytaire incomplète suite à un troisième ou quatrième cycle de traitement. Toutefois, il n'y avait pas d'impact évident des anticorps antialemtuzumab sur l'efficacité clinique ou le profil de sécurité de Lemtrada.
Population pédiatrique
La sécurité d'emploi et l'efficacité de Lemtrada chez les enfants atteints de SEP et âgés de 0 à 18 ans n'ont pas encore été établies. Lemtrada n'est pas indiqué pour une utilisation chez les enfants et les adolescents (cf. «Contre-indications»).
PharmacocinétiqueAbsorption
Les propriétés pharmacocinétiques de Lemtrada ont été établies chez un total de 216 patients atteints de SEP récurrente-rémittente ayant reçu des perfusions intraveineuses de 12 mg/jour ou 24 mg/jour pendant 5 jours consécutifs, puis pendant 3 jours consécutifs 12 mois après le cycle de traitement initial. Les concentrations sériques augmentent après chaque dose consécutive dans un même cycle de traitement, les concentrations maximales sont observées après la dernière perfusion d'un cycle. Les concentrations sériques en médicament sont légèrement supérieures ou inférieures au seuil de détection environ 30 jours après chaque cycle de traitement. L'administration de 12 mg/jour a entraîné une Cmax de 3014 ng/ml le cinquième jour du traitement initial et de 2276 ng/ml le troisième jour du deuxième traitement.
Distribution
Après les perfusions intraveineuses, le volume de distribution était proportionnel au poids du corps et se rapprochait du volume de liquide extracellulaire (14,1 L), ce qui suggère que Lemtrada est largement contenu dans le sang et l'espace interstitiel.
Métabolisme
L'alemtuzumab est une protéine dont le métabolisme attendu correspond à une dégradation en petits peptides et en acides aminés par des enzymes protéolytiques ubiquitaires. Aucune étude de biotransformation classique n'a été conduite.
Élimination
La demi-vie alpha était d'environ 2 jours et était comparable entre les cycles conduisant à des concentrations sériques faibles ou indétectables dans les 30 jours environ suivant chaque cycle de traitement.
La pharmacocinétique de population de Lemtrada a été décrite au mieux par un modèle linéaire à deux compartiments. La clairance systémique diminuait avec le nombre de lymphocytes en raison de la perte de l'antigène CD52 en périphérie; cependant, la diminution du premier au deuxième cycle était inférieure à 20 %.
Cinétique pour certains groupes de patients
Il n'est pas possible de tirer des conclusions à partir des données disponibles concernant l'influence de la race ou du sexe sur les propriétés pharmacocinétiques de Lemtrada. En outre, ces propriétés n'ont pas été étudiées chez les patients âgés de 55 ans et plus.
Données précliniquesCarcinogenèse et mutagenèse
Aucune étude n'a été réalisée pour évaluer le potentiel carcinogène ou mutagène de l'alemtuzumab.
Fécondité et reproduction
Un traitement par Lemtrada par voie intraveineuse à des doses maximales de 10 mg/kg/jour, administrées pendant 5 jours consécutifs (ASC 7,1 fois supérieure à l'exposition humaine à la dose quotidienne recommandée) n'a pas eu d'effet sur la fécondité ni sur les capacités de reproduction chez des souris mâles. Le nombre de spermatozoïdes normaux (<10%) était significativement réduit par rapport au groupe témoin et le pourcentage de spermatozoïdes anormaux (queues ou têtes absentes) avait nettement augmenté (jusqu'à 3%). Ces changements n'ont cependant pas affecté la fécondité.
Chez les souris femelles recevant des doses intraveineuses de Lemtrada allant jusqu'à 10 mg/kg/jour (ASC 4,7 fois supérieure à l'exposition humaine à la dose quotidienne recommandée) pendant 5 jours consécutifs avant la mise en présence avec des souris mâles de type sauvage, le nombre moyen de corps jaunes et de sites d'implantation par souris était significativement réduit par rapport aux animaux qui avaient reçu un placebo. La prise de poids gestationnelle chez les souris gravides ayant reçu 10 mg/kg/jour a été plus faible par rapport au groupe témoin sous placebo. Lemtrada à des doses allant jusqu'à 10 mg/kg/jour n'a pas eu d'influence sur d'autres paramètres de l'accouplement et de la fécondité.
Une étude de toxicité sur la reproduction conduite chez des souris gravides exposées à des doses intraveineuses de Lemtrada allant jusqu'à 10 mg/kg/jour (ASC 2,4 fois supérieure à l'exposition humaine à la dose recommandée de 12 mg/jour) pendant 5 jours consécutifs au cours de la gestation a révélé une augmentation significative du nombre de mères portant des embryons morts ou résorbés, ainsi qu'une réduction du nombre de mères portant des fœtus viables. Aucune malformation/variation fœtale externe, viscérales ou squelettiques n'a été observée à des doses maximales de 10 mg/kg/jour.
Le passage transplacentaire et l'activité pharmacologique potentielle de Lemtrada ont été observés chez la souris pendant et après la gestation. Dans les études sur la souris, des modifications des numérations lymphocytaires ont été observées chez les souriceaux exposés au Lemtrada au cours de la gestation à des doses de 3 mg/kg/jour pendant 5 jours consécutifs (ASC 0,6 fois supérieure à l'exposition humaine à la dose recommandée de 12 mg/jour). Le développement cognitif, physique et sexuel des souriceaux exposés au Lemtrada pendant l'allaitement n'était pas affecté à des doses maximales de 10 mg/kg/jour.
On ne connaît pas l'importance des résultats observés chez des souris transgéniques sur la toxicité pour la reproduction chez l'homme.
Remarques particulièresLemtrada est une solution à diluer pour perfusion stérile, limpide, incolore à légèrement jaune avec un pH de 7,0-7,4.
Incompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments. Ne pas ajouter d'autres médicaments à la solution pour perfusion de Lemtrada et ne pas les perfuser simultanément à l'aide de la même tubulure IV.
Le médicament doit être dilué uniquement avec les solutions mentionnées dans la rubrique «Remarques concernant la manipulation».
Aucune incompatibilité entre l'alemtuzumab et les poches de perfusion en polychlorure de vinyle (PVC), le nécessaire à perfusion en PVC ou en PVC recouvert de polyéthylène et les filtres à faible taux d'adsorption des protéines.
Stabilité
Solution
4 ans.
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Solution diluée
La préparation diluée doit être administrée dans les 8 heures après la dilution.
L'alemtuzumab dilué peut être conservé à température ambiante (15°C - 25°C) ou au réfrigérateur (2°C - 8°C).
La préparation diluée doit être conservée à l'abri de la lumière.
Remarques particulières concernant le stockage
Solution
Conserver au réfrigérateur (2°C - 8°C).
Ne pas congeler ni secouer.
Conserver à l'abri de la lumière.
Conserver hors de la portée des enfants.
Pour les conditions de conservation du médicament après dilution, lire la rubrique «Stabilité».
Remarques concernant la manipulation
Le contenu du flacon doit être inspecté afin de vérifier l'absence de particules et de coloration anormale avant administration. La solution ne doit pas être utilisée si des particules sont présentes ou si la coloration est anormale.
Ne pas congeler ou agiter le flacon avant utilisation. La solution et la préparation diluée doivent être conservées à l'abri de la lumière.
Pour une administration intraveineuse, prélever 1,2 ml de Lemtrada dans le flacon à l'aide d'une seringue, en respectant des conditions d'asepsie habituelles et injecter dans 100 ml de solution pour perfusion de chlorure de sodium à 9 mg/ml (0,9%) ou de glucose (5%). La poche doit être retournée doucement pour bien mélanger la solution.
Lemtrada ne contenant pas de conservateurs antimicrobiens, il convient de garder la solution préparée stérile. Chaque flacon est à usage unique.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Numéro d’autorisation63025 (Swissmedic).
PrésentationLemtrada se présente en flacon en verre transparent de 2 ml pour une utilisation unique, muni d'un bouchon exempt de latex. Chaque flacon de 2 ml de Lemtrada est rempli de manière à ce qu'une quantité de 1,2 ml puisse être prélevée dans une solution de 10 mg/ml (12 mg d'alemtuzumab) (A).
Taille du conditionnement: boîte de 1 flacon.
Titulaire de l’autorisationsanofi-aventis (suisse) sa, 1214 Vernier/GE
Mise à jour de l’informationNovembre 2023
|