CompositionPrincipes actifs
Empagliflozine.
Excipients
Jardiance 10 mg:
·Noyau du comprimé: lactose monohydraté 162,5 mg, cellulose microcristalline, hydroxypropylcellulose, croscarmellose sodique (correspondant à 0,01 mg de sodium), silice colloïdale anhydre, stéarate de magnésium
·Pelliculage: hypromellose 2910, dioxyde de titane (E171), talc, macrogol 400, oxyde de fer jaune (E172)
Jardiance 25 mg:
·Noyau du comprimé: lactose monohydraté 113 mg, cellulose microcristalline, hydroxypropylcellulose, croscarmellose sodique (correspondant à 0,008 mg de sodium), silice colloïdale anhydre, stéarate de magnésium
·Pelliculage: hypromellose 2910, dioxyde de titane (E171), talc, macrogol 400, oxyde de fer jaune (E172)
Indications/Possibilités d’emploiJardiance est indiqué dans le traitement du diabète de type 2 chez les adultes, ainsi que chez les enfants et les adolescents à partir de 10 ans, en plus de mesures diététiques et de l'exercice physique, lorsque ces mesures seules ne permettent pas d'obtenir un contrôle glycémique adéquat:
·en monothérapie chez les patients, pour lesquels la metformine ne peut être utilisée en raison d'une contre-indication ou d'une intolérance.
·en association à un traitement d'appoint par d'autres hypoglycémiants.
Pour les résultats des études concernant les traitements en association, voir «Efficacité clinique».
Jardiance est indiqué pour la prévention d'événements cardiovasculaires chez des patients adultes atteints d'un diabète de type 2 et d'une maladie cardiovasculaire déjà manifeste (voir «Efficacité clinique»).
Jardiance est indiqué chez les patients adultes pour le traitement de l'insuffisance cardiaque ventriculaire gauche symptomatique chronique en association avec d'autres traitements médicamenteux de l'insuffisance cardiaque (voir «Efficacité clinique»).
Jardiance est indiqué pour la réduction du risque de progression d'une maladie rénale chronique chez des patients adultes atteints d'une maladie rénale chronique (voir rubriques «Posologie/Mode d'emploi» et «Efficacité clinique»).
Posologie/Mode d’emploiPosologies recommandées de Jardiance chez les adultes ainsi que chez les enfants et les adolescents âgés de 10 ans et plus:
|
Population
|
Indication
|
Posologie recommandée
| |
Adultes
|
Insuffisance cardiaque
|
·La dose recommandée est de 10 mg une fois par jour
| |
Maladie rénale chronique
| |
Diabète de type 2
|
·La dose recommandée est de 10 mg une fois par jour.
·La dose peut être augmentée à 25 mg une fois par jour chez les patients supportant une dose de 10 mg d'empagliflozine une fois par jour et nécessitant un meilleur contrôle glycémique*
| |
Enfants et adolescents âgés de 10 ans et plus
|
Diabète de type 2
|
·La dose recommandée est de 10 mg une fois par jour.
·La dose peut être augmentée à 25 mg une fois par jour chez les patients supportant une dose de 10 mg d'empagliflozine une fois par jour et nécessitant un meilleur contrôle glycémique
|
* Pour l'utilisation chez les patients présentant une insuffisance rénale, voir rubrique «Patients présentant des troubles de la fonction rénale».
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Voir «Mises en garde et précautions»
Patients présentant des troubles de la fonction rénale
Le traitement d'une insuffisance cardiaque ou d'une maladie rénale chronique est réalisé indépendamment de la fonction rénale en administrant une dose d'empagliflozine de 10 mg.
Étant donné que des données concernant les patients présentant un DFGe < 20 ml/min/1,73 m2 ne sont disponibles dans aucune des indications, l'instauration d'un traitement par Jardiance chez ces patients n'est généralement pas recommandée. Chez les patients atteints de diabète de type 2, l'effet hypoglycémiant de l'empagliflozine est diminué chez les patients présentant un DFGe < 45 ml/min/1,73 m2 et reste probablement sans effet chez les patients présentant un DFGe < 30 ml/min/1,73 m2. Par conséquent, à partir d'un DFGe < 45 ml/min/1,73 m2, pour autant que ceci s'avère nécessaire, un traitement hypoglycémiant complémentaire doit être envisagé.
Patients âgés
Aucune adaptation posologique n'est nécessaire chez les patients âgés.
Enfants et adolescents
La sécurité et l'efficacité de l'empagliflozine chez les enfants et adolescents a été prouvée exclusivement pour le traitement du diabète de type 2 et uniquement pour le groupe d'âge de 10 à 17 ans. Jardiance n'est pas autorisé pour le traitement des enfants de moins de 10 ans. Pour ce groupe d'âge, ainsi que pour les enfants et adolescents présentant un DFGe < 60 ml/min/1,73 m², aucune donnée n'est disponible.
Traitement combiné
Traitement combiné avec une sulfonylurée et/ou de l'insuline
Voir «Mises en garde et précautions», «Interactions» et «Effets indésirables».
Prise retardée
Lorsqu'une dose a été oubliée, le patient devrait la prendre dès qu'il s'en souvient. Il ne doit toutefois pas prendre une double dose le même jour.
Mode d'administration
Jardiance peut être pris au cours ou en dehors des repas.
Interruption temporaire en cas d'opérations
Si possible, Jardiance doit être arrêté au moins 3 jours avant une opération chirurgicale majeure ou des interventions associées à un jeûne prolongé. Jardiance peut être repris si le patient est cliniquement stable et consomme des aliments par voie orale (voir «Mises en garde et précautions»).
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients.
Les comprimés Jardiance 10 mg resp. Jardiance 25 mg contiennent 162,5 mg resp. 113 mg de lactose par comprimé.
L'utilisation du médicament est contre-indiquée chez les patients souffrant de troubles génétiques rares entrainant une incompatibilité avec un des excipients (lactose).
Mises en garde et précautionsJardiance ne doit pas être utilisé chez les patients souffrant de diabète de type 1.
Acidocétose
Des cas d'acidocétose, trouble grave du métabolisme mettant le pronostic vital en jeu et exigeant une hospitalisation immédiate, ont été rapportés pour des patients diabétiques traités par empagliflozine, dont des cas à issue fatale. Dans certains des cas rapportés, la maladie a présenté une forme atypique avec des valeurs de glycémie seulement modérément élevées inférieures à 14 mmol/l (250 mg/dl). Bien que les patients non diabétiques soient moins susceptibles de présenter une acidocétose, des cas ont également été signalés dans cette population.
Le risque d'acidocétose doit être envisagé en cas de symptômes aspécifiques comme nausées, vomissements, manque d'appétit, douleurs abdominales, soif excessive, difficultés à respirer, confusion, épuisement inhabituel ou fatigue chez des patients diabétiques traités par empagliflozine.
Si ces symptômes surviennent, il convient d'effectuer chez ces patients, indépendamment du taux de glycémie, un test de corps cétoniques. En cas de suspicion d'acidocétose, Jardiance doit être arrêté, l'état du patient doit être examiné et un traitement immédiat doit être démarré. L'acidocétose diabétique peut durer plus longtemps chez certains patients après l'arrêt de Jardiance, c'est-à-dire qu'elle peut durer plus longtemps que prévu sur la base de la demi-vie plasmatique de l'empagliflozine. Une glucosurie prolongée a été observée, ainsi qu'une acidocétose diabétique persistante. L'excrétion urinaire de glucose persiste jusqu'à 3 jours après l'arrêt de Jardiance; cependant, il existe des rapports post-commercialisation d'acidocétose diabétique et de glucosurie qui durent plus de 6 jours et parfois jusqu'à 2 semaines après l'arrêt des inhibiteurs du SGLT2.
Un risque accru d'acidocétose peut exister lors de la prise de Jardiance chez les patients ayant une alimentation très pauvre en glucides (étant donné que l'association pourrait accroître davantage la production de corps cétoniques), chez les patients souffrant d'une pathologie aiguë, en cas de maladies du pancréas indiquant un manque d'insuline (p.ex. diabète de type 1, pancréatite ou opération du pancréas dans l'anamnèse), en cas de réduction de la dose d'insuline (y compris défaillance de la pompe d'insuline), en cas d'abus d'alcool et de forte déshydratation ainsi que chez les patients fortement déshydratés et chez les patients qui ont déjà eu, par le passé, une acidocétose. Jardiance doit être utilisé avec précaution chez ces patients. La prudence est de mise lors de la réduction de la dose d'insuline [voir «Posologie/Mode d'emploi»]. Dans les situations cliniques dont on sait qu'elles prédisposent à une acidocétose (p.ex. jeûne prolongé en raison d'une affection aiguë, d'une intervention ou d'une opération chirurgicale), une surveillance de l'acidocétose est indiquée et le traitement par Jardiance doit être temporairement interrompu. Dans ces situations, une surveillance des corps cétoniques doit également être envisagée, même si le traitement par Jardiance est interrompu. Le traitement par Jardiance peut être poursuivi si le patient est cliniquement stable et consomme des aliments par voie orale (voir «Posologie/Mode d'emploi»).
Fasciite nécrosante du périnée (gangrène de Fournier)
Des cas de fasciite nécrosante du périnée (aussi appelée «gangrène de Fournier») ont été rapportés chez des patients de sexe masculin et féminin traités par des inhibiteurs du SGLT2, dont l'empagliflozine également. Il s'agit d'une infection nécrosante rare, mais grave et menaçant le pronostic vital. Parmi les conséquences graves comptaient hospitalisations, multiples opérations et décès.
Les patients traités par Jardiance et rapportant des douleurs ou une sensibilité à la pression, des érythèmes ou une tuméfaction dans la région génitale ou périnéale, de la fièvre ou un malaise doivent être examinés à la recherche d'une fasciite nécrosante. En cas de suspicion de fasciite nécrosante, le traitement par Jardiance doit être arrêté et un traitement (entre autres par des antibiotiques à large spectre et, le cas échéant, par un débridement chirurgical) doit être instauré immédiatement.
Utilisation chez les patients insuffisants rénaux
Des données suffisantes n'étant pas disponibles, l'instauration d'un traitement par l'empagliflozine n'est pas recommandée chez les patients présentant un DFGe < 20 ml/min/1,73 m2.
L'efficacité hypoglycémiante de l'empagliflozine dépend de la fonction rénale et est probablement absente chez les patients présentant un DFGe < 30 ml/min/1,73 m2 (voir «Posologie/Mode d'emploi»).
La fonction rénale doit être régulièrement contrôlée pendant l'utilisation de Jardiance.
Hypovolémie
En raison de la diurèse osmotique, l'empagliflozine entraine une légère baisse de la pression artérielle (la baisse systolique étant plus forte que la diastolique) et peut potentiellement provoquer une hypotension orthostatique, ce qui peut entrainer des effets indésirables tels que vertiges, syncopes ou chutes. La prudence est donc de mise chez les patients avec hypotension orthostatique, les patients sous antihypertenseurs, les patients âgés ainsi que les patients avec affection cardiovasculaire et/ou cérébrovasculaire connue.
L'expérience montre que l'hématocrite augmente de 2% environ.
En cas d'affections pouvant entrainer une perte hydrique (p.ex. les maladies gastro-intestinales), une surveillance attentive de l'état volémique et des électrolytes est recommandée chez les patients recevant de l'empagliflozine. L'interruption temporaire du traitement par empagliflozine doit être envisagée jusqu'à ce que la perte hydrique soit corrigée.
Infections urinaires avec des complications
Dans un ensemble d'études en double insu, contrôlées contre placebo, d'une durée de 18 à 24 semaines, menées chez des patients atteints de diabète de type 2, la fréquence globale des infections urinaires rapportées comme effets indésirables était supérieure sous empagliflozine 25 mg et comparable au placebo (7,6%) et sous empagliflozine 10 mg (9,3%). Comme pour les patients sous placebo, la fréquence des infections urinaires rapportée avec l'empagliflozine était plus élevée chez les patients avec une anamnèse connue d'infections urinaires chroniques ou récidivantes. Les rapports sur la sévérité des infections urinaires correspondaient aux rapports pour le placebo (légère, modérée et sévère). Chez les femmes, les infections urinaires étaient plus fréquentes sous empagliflozine que sous placebo. Ceci n'a pas été observé chez les hommes.
Des cas d'infection des voies urinaires avec des complications incluant des pyélonéphrites et des urosepsis, ont été signalés chez les patients traités par l'empagliflozine (voir «Effets indésirables»). Une interruption temporaire de l'empagliflozine doit être envisagée chez les patients ayant une infection des voies urinaires avec des complications.
Amputations des membres inférieurs
Une augmentation du nombre de cas d'amputation des membres inférieurs (principalement d'un orteil) a été observée au cours d'études cliniques à long terme menées avec un autre inhibiteur du SGLT2. On ignore s'il s'agit d'un effet de classe. Comme pour tous les patients diabétiques, il est important de sensibiliser les patients sur l'importance des soins préventifs de routine pour les pieds.
Patients présentant une insuffisance hépatique
On ne dispose que d'expériences très limitées de l'administration d'empagliflozine chez les patients souffrant d'une insuffisance hépatique sévère ou d'une élévation sensible des transaminases (ayant plus que triplé). L'administration d'empagliflozine chez ces patients n'est pas recommandée.
Traitement associé avec une sulfonylurée et/ou l'insuline
Si Jardiance est utilisé en association avec une sulfonylurée ou de l'insuline, envisager de réduire la dose de sulfonylurée ou d'insuline pour réduire le risque d'hypoglycémie.
Patients âgés
Dans le cadre d'études cliniques, Jardiance a été administré à 992 patients âgés de 75 ans et plus. Un risque accru d'hypovolémie existe éventuellement pour les patients de 75 ans et plus. C'est pourquoi la prudence est de mise lors de la prescription de Jardiance dans ce groupe de patients (voir «Effets indésirables»).
Evénements cérébrovasculaires
Dans le cadre de l'étude EMPA-REG OUTCOME, Jardiance (groupes combinés traités par empagliflozine 10 mg et 25 mg) comparé au groupe placebo était associé à un risque supérieur d'AVC mortels/non mortels à une tendance non significative: HR 1,18 (IC à 95% 0,89; 1,56) (voir «Efficacité clinique»). Un lien causal entre Jardiance et l'AVC n'est pas prouvé. Il est néanmoins recommandé de faire preuve de prudence avec les patients affichant un risque élevé de survenue d'événements cérébrovasculaires.
Lactose
Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé pelliculé, c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsInteractions pharmacocinétiques
Effets des autres médicaments sur l'empagliflozine
Les données in vitro suggèrent que la voie primaire du métabolisme de l'empagliflozine chez l'homme est la glucuronidation par les uridine-5'-diphosphoglucuronosyltransférases UGT1A3, UGT1A8, UGT1A9 et UGT2B7. L'empagliflozine est un substrat des transporteurs humains OAT3, OATP1B1 et OATP1B3, mais pas d'OAT1 et OCT2. L'empagliflozine est un substrat de la glycoprotéine P (P-gp) et de la protéine de résistance du cancer du sein (BCRP).
L'administration concomitante d'empagliflozine et de probénécide, un inhibiteur des enzymes UGT et de l'OAT3, a entraîné une augmentation de 26% du pic de concentration plasmatique d'empagliflozine (Cmax) et une augmentation de 54% de l'aire sous la courbe des concentrations en fonction du temps (ASC).
Ces variations n'ont pas été considérées comme cliniquement significatives.
L'effet de l'induction des UGT sur l'empagliflozine n'a pas été étudié. Un traitement concomitant par des inducteurs connus des enzymes UGT doit être évité en raison du risque potentiel d'une diminution de l'efficacité.
Une étude d'interaction avec le gemfibrozil, un inhibiteur in vitro des transporteurs OAT3 et OATP1B1/1B3, a montré une augmentation de 15% de la Cmax de l'empagliflozine et de 59% de l'ASC en cas d'administration concomitante. Ces variations n'ont pas été considérées comme cliniquement significatives.
L'inhibition des transporteurs OATP1B1/1B3 par l'utilisation concomitante de rifampicine a entrainé une augmentation de 75% de la Cmax de l'empagliflozine ainsi qu'une augmentation de 35% de l'ASC.
Ces variations n'ont pas été considérées comme cliniquement significatives.
L'exposition à l'empagliflozine était similaire avec ou sans administration concomitante de vérapamil, un inhibiteur de la P-gp; ceci indique que l'inhibition de la P-gp n'a pas d'effet cliniquement significatif sur l'empagliflozine.
Les études d'interaction menées chez des volontaires sains suggèrent que la pharmacocinétique de l'empagliflozine n'est pas influencée par l'administration concomitante de metformine, de glimépiride (dose unique), de vérapamil, de ramipril, de simvastatine, de torasémide ou d'hydrochlorothiazide.
Effets de l'empagliflozine sur les autres médicaments
L'empagliflozine n'entraîne pas d'inhibition, d'inactivation ni d'induction des isoenzymes du CYP450. Les UGT1A1, UGT1A3, UGT1A8, UGT1A9 ou UGT2B7 ne sont pas inhibées par l'empagliflozine.
À des doses thérapeutiques, la capacité de l'empagliflozine d'inhiber ou d'inactiver de manière irréversible les principales isoenzymes du CYP450 et de l'UGT est faible. Par conséquent, des interactions médicamenteuses entre les principales isoenzymes du CYP450 et de l'UGT et l'empagliflozine ainsi que des substrats co-administrés de ces enzymes sont considérées comme peu probables.
A des doses thérapeutiques, l'empagliflozine n'inhibe pas la P-gp. Sur la base des études in vitro, il est considéré comme peu probable que l'empagliflozine entraîne des interactions avec des médicaments substrats de la P-gp. L'administration concomitante d'empagliflozine et de digoxine, un substrat de la P-gp, a entraîné une augmentation de 6% de l'ASC et une augmentation de 15% de la Cmax de la digoxine. Il convient donc de surveiller les patients sous digoxine.
A des concentrations cliniquement significatives, l'empagliflozine n'inhibe pas les transporteurs humains OAT3, OATP1B1 etOATP1B3 in vitro. C'est pourquoi les interactions médicamenteuses avec des substrats de ces transporteurs sont considérées comme peu probables.
Les études d'interaction menées chez des volontaires sains suggèrent que l'empagliflozine n'a pas d'influence cliniquement significative sur la pharmacocinétique de la metformine, du glimépiride, de la simvastatine, de la warfarine, du ramipril, de l'hydrochlorothiazide, de la torasémide et des contraceptifs oraux.
Aucune donnée n'est disponible pour l'acécoumarol et la phenprocoumone.
Les inhibiteurs du SGLT2, y compris l'empagliflozine, peuvent augmenter l'excrétion rénale du lithium et entraîner une diminution des concentrations sanguines de lithium. La concentration sérique de lithium doit être contrôlée plus souvent après l'instauration du traitement par l'empagliflozine et en cas de modification de la posologie. Pour surveiller la concentration sérique du lithium, le patient doit être adressé au médecin ayant prescrit le lithium.
Interactions pharmacodynamiques
Diurétiques
L'empagliflozine peut majorer l'effet diurétique des thiazidiques et des diurétiques de l'anse et augmenter le risque de déshydratation et d'hypotension.
Insuline et sécrétagogues de l'insuline
L'insuline et les sécrétagogues de l'insuline, comme les sulfonylurées, peuvent augmenter le risque d'hypoglycémie. Par conséquent, une réduction de la dose d'insuline ou du sécrétagogue de l'insuline peut être nécessaire pour diminuer le risque d'hypoglycémie lorsqu'ils sont utilisés en association avec l'empagliflozine.
Interférence avec le dosage du 1,5-anhydroglucitol (1,5-AG)
Une surveillance de la glycémie au moyen du dosage du 1,5-AG n'est pas recommandée, car les taux de 1,5-AG ne permettent pas une mesure fiable du contrôle glycémique chez les patients traités par des inhibiteurs du SGLT2. D'autres méthodes devraient être utilisées pour surveiller la glycémie.
Enfants et adolescents
Des études d'interaction ont été réalisées uniquement chez l'adulte.
Grossesse, allaitementGrossesse
Il n'existe pas de données suffisantes concernant l'emploi de Jardiance chez la femme enceinte. Les expérimentations animales n'ont pas révélé de toxicité de reproduction (voir «Données précliniques»). En raison d'effets potentiels sur le développement fœtal, Jardiance ne doit pas être utilisé pendant la grossesse, sauf en cas de nécessité absolue.
Allaitement
Aucune information n'est disponible concernant un éventuel passage de l'empagliflozine dans le lait maternel. Les données disponibles provenant des expérimentations animales ont montré que l'empagliflozine passe dans le lait. Les expérimentations animales ont montré des effets indésirables sur le développement post-natal (voir «Données précliniques»). Un risque pour le nouveau-né / le nourrisson ne peut être exclu. C'est pourquoi l'allaitement doit être interrompu pendant le traitement avec Jardiance.
Fertilité
Aucune étude n'a été menée sur les effets de Jardiance sur la fertilité humaine. Les études expérimentales animales n'ont pas mis en évidence d'effets nocifs directs ou indirects sur la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée. Les patients doivent toutefois être prévenus du risque d'hypoglycémie lorsque Jardiance est utilisé comme traitement supplémentaire à l'insuline et/ou une sulfonylurée. Ils doivent aussi être prévenus du risque majoré d'effets indésirables liés à la réduction du volume intravasculaire, p.ex. des vertiges (voir «Mises en garde et précautions» et «Effets indésirables»).
Effets indésirablesDiabète de type 2
Dans des études de sécurité, un total de 15'582 patients avec un diabète de type 2 ont été traités par empagliflozine, dont 10'004 patients recevant de l'empagliflozine seule ou associée à la metformine, une sulfonylurée, la thiazolidinédione, aux inhibiteurs du DPP4 ou à l'insuline.
Dans ces études, le taux d'abandon à cause des effets indésirables était similaire pour tous les groupes de traitement: placebo (5,6%), Jardiance 10 mg (5,0%) et Jardiance 25 mg (4,9%).
Plusieurs études en double aveugle contrôlées contre placebo d'une durée de 18 à 24 semaines ont inclus 3'534 patients, dont 1'183 ont reçu un placebo et 1'185 ont été traités par Jardiance 10 mg et 1166 ont reçu Jardiance 25 mg.
L'effet indésirable le plus fréquent était une hypoglycémie, en fonction du traitement de fond utilisé dans les différentes études (voir la description des effets indésirables spéciaux).
Insuffisance cardiaque
Les études EMPEROR incluaient des patients atteints d'insuffisance cardiaque à fraction d'éjection réduite (n = 3'726) ou à fraction d'éjection préservée (n = 5'985), traités par l'empagliflozine 10 mg ou par un placebo. Environ la moitié des participants à l'étude présentaient un diabète de type 2.
L'effet indésirable le plus fréquent était la déplétion volémique (empagliflozine 10 mg: 11,4%; placebo: 9,7%).
Maladie rénale chronique
L'étude EMPA-KIDNEY incluait des patients atteints de maladie rénale chronique (n = 6609) traités par l'empagliflozine 10 mg ou par un placebo. Environ 44 % des patients présentaient un diabète de type 2.
Aucun nouvel effet indésirable n'a été constaté dans l'étude EMPA-KIDNEY.
Le profil de sécurité général de Jardiance était globalement homogène pour toutes les indications étudiées.
Les effets indésirables suivants ont été rapportés chez les patients traités par empagliflozine dans des études en double aveugle contrôlées contre placebo:
Définition des fréquences utilisées:
«très fréquents» (≥1/10), «fréquents» (≥1/100 à <1/10), «occasionnels» (≥1/1000 à <1/100), «rares» (≥1/10 000 à <1/1000), «très rares» (<1/10 000), «fréquence inconnue» (basée principalement sur les déclarations spontanées issues de la surveillance du marché, la fréquence exacte ne peut être estimée sur la base des données disponibles).
Infections et infestations
Fréquents: Candidose vaginale, vulvovaginite, balanite et autres infections des voies génitalesb, infections des voies urinairesb (incluant des pyélonéphrites et des urosepsis)d.
Rares: Fasciite nécrosante du périnée (gangrène de Fournier)d.
Troubles du métabolisme et de la nutrition
Très fréquentsa: Hypoglycémie (lors de l'association à une sulfonylurée ou à l'insuline)a
Occasionnels: Acidocétosed.
Affections gastro-intestinales
Fréquents: Constipation.
Affections de la peau et du tissu sous-cutané
Fréquents: Prurit, réactions cutanées allergiques (p.ex. éruption cutanée, urticaire)d.
Occasionnels: Angioœdèmed.
Troubles vasculaires
Très fréquents: Hypovolémiea.
Troubles rénaux et urinaires
Fréquents: Augmentation des mictionsa.
Occasionnels: Dysurie.
Très rare: Néphrite tubulo-interstitielle.
Troubles généraux et accidents liés au site d'administration
Fréquents: Soif.
Investigations
Fréquents: Augmentation des lipides sériquesc, augmentation de l'hématocritec.
Occasionnels: Baisse du débit de filtration glomérulairea, augmentation de la créatinine sanguinea.
a Voir les sous-sections ci-dessous pour plus d'informations
b Voir la rubrique «Mises en garde et précautions»
c Voir «Efficacité clinique» pour plus d'informations
d Issu des expériences depuis la mise sur le marché
Description de certains effets indésirables
Les fréquences mentionnées des effets indésirables ci-dessous sont indiquées indépendamment d'une relation causale.
Hypoglycémie
La fréquence des hypoglycémies dépendait du traitement de fond dans les différentes études.
La fréquence des patients avec une hypoglycémie mineure a été similaire dans les groupes empagliflozine ou placebo en monothérapie, comme traitement d'appoint de la metformine et comme traitement d'appoint de la pioglitazone avec ou sans metformine et comme traitement d'appoint de la linagliptine avec ou sans metformine. Une augmentation de la fréquence a été observée lorsque l'empagliflozine était associée à un traitement d'appoint par metformine et sulfonylurée (empagliflozine 10 mg: 16,1%, empagliflozine 25 mg: 11,5%, placebo: 8,4%). Comme traitement d'appoint à l'insuline avec ou sans metformine et avec ou sans sulfonylurée, l'empagliflozine n'a pas montré d'augmentation du risque d'hypoglycémie par rapport au placebo (empagliflozine 10 mg: 19,5%, empagliflozine 25 mg: 28,4%, placebo: 20,6% pendant les premières 18 semaines de traitement, au cours desquelles la dose d'insuline ne pouvait pas être ajustée; empagliflozine 10 mg et 25 mg: 36,1%, placebo 35,3% pendant les 78 semaines de l'étude).
Dans les études EMPEROR sur l'insuffisance cardiaque, l'hypoglycémie a été observée à une fréquence similaire lors de l'administration de sulfonylurée ou d'insuline en traitement d'appoint (empagliflozine 10 mg: 6,5%, placebo: 6,7%).
Hypoglycémie majeure (hypoglycémie exigeant un traitement)
Le nombre d'épisodes hypoglycémiques sévères n'a pas augmenté sous empagliflozine par rapport au placebo.
Dans les études EMPEROR sur l'insuffisance cardiaque, les cas d'hypoglycémie majeure ont été observés à la même fréquence chez les patients diabétiques lorsque l'empagliflozine était associée à un traitement d'appoint par une sulfonylurée ou par l'insuline (empagliflozine 10 mg: 2,2%, placebo: 1,9%).
Candidose vaginale, vulvovaginite, balanite et autres infections des voies génitales
Dans des études menées chez des patients atteints de diabète de type 2, des candidoses vaginales, vulvovaginites, balanites et autres infections génitales étaient plus fréquentes dans le groupe sous empagliflozine 10 mg (4,1%) et empagliflozine 25 mg (3,7%) par rapport au placebo (0,9%); comparé au groupe placebo, les femmes sous empagliflozine étaient plus souvent atteintes et la différence de fréquence était moins prononcée chez les hommes. Les infections des voies génitales étaient d'intensité légère à modérée.
Des cas de phimosis/phimosis acquis ont été rapportés concomitamment à des infections génitales.
Augmentation des mictions
Ainsi qu'attendu au vu de mécanisme d'action, une augmentation des mictions (comprenant les termes génériques (PT): pollakiurie, polyurie et nycturie) a été observée plus fréquemment chez les patients traités par Jardiance 10 mg (3,45%) et par Jardiance 25 mg (3,3%) que sous placebo (1,4%). L'intensité de cet effet était principalement légère à modérée. La fréquence de la nycturie rapportée était similaire pour le placebo et Jardiance (<1%).
Dans les études EMPEROR sur l'insuffisance cardiaque, une augmentation des mictions a été observée à une fréquence similaire chez les patients traités par l'empagliflozine et chez les patients ayant reçu le placebo (empagliflozine 10 mg: 0,9%, placebo: 0,5%).
Hypovolémie
La fréquence globale des hypovolémies (comprenant les termes génériques de diminution de la pression artérielle (mesure ambulatoire), diminution de la pression artérielle systolique, déshydratation, hypotension [chute de tension], hypovolémie, hypotension orthostatique et syncope) a été similaire à celle sous placebo (Jardiance 10 mg 0,6%, Jardiance 25 mg 0,4% et placebo 0,3%). L'action de l'empagliflozine sur l'élimination du glucose avec l'urine est associée à une diurèse osmotique, ce qui pourrait influencer le statut d'hydratation des patients âgés de 75 ans et plus. La fréquence des événements d'hypovolémie rapportés chez les patients âgés de 75 ans et plus était similaire sous Jardiance 10 mg (2,3%) ou Jardiance 25 mg (4,3%) et sous placebo (2,1%).
Augmentation de la créatinine sanguine et baisse du débit de filtration glomérulaire
La fréquence globale des patients présentant une augmentation de la créatinine sanguine et une baisse du débit de filtration glomérulaire a été comparable dans les groupes recevant l'empagliflozine ou le placebo (augmentation de la créatinine sanguine: empagliflozine 10 mg 0,6 %, empagliflozine 25 mg 0,1 %, placebo 0,5%; baisse du débit de filtration glomérulaire: empagliflozine 10 mg 0,1 %, empagliflozine 25 mg 0 %, placebo 0,3 %).
Dans des études en double aveugle contrôlées par placebo menées sur une période allant jusqu'à 76 semaines, des augmentations initiales temporaires de la créatinine sanguine (variation moyenne par rapport à la valeur initiale à la semaine 12: empagliflozine 10 mg 0,02 mg/dl, empagliflozine 25 mg 0,01 mg/dl) ainsi que des baisses initiales temporaires du débit de filtration glomérulaire estimé (variation moyenne par rapport à la valeur initiale à la semaine 12: empagliflozine 10 mg -1,34 ml/min/1,73 m2, empagliflozine 25 mg -1,37 ml/min/1,73 m2) ont été observées. Ces variations étaient généralement réversibles en cas de poursuite du traitement ou après arrêt de la médication.
Modification des paramètres de laboratoire
- Hématocrite
Par rapport aux valeurs initiales, les modifications de l'hématocrite étaient de -0,8 à -0,1% pour le placebo et de 2,1 à 2,5% pour l'empagliflozine 10 mg.
- Lipides
Par rapport aux valeurs initiales, les modifications des paramètres lipidiques pour l'empagliflozine 10 mg par rapport au placebo étaient de 0,01 à 0,14 mmol/l (cholestérol total), de 0,03 à 0,08 mmol/l (HDL-cholestérol), de 0,01 à 0,12 mmol/l (LDL-cholestérol) et de -0,23 à -0,05 (triglycérides).
Enfants et adolescents
Dans le cadre de l'étude DINAMO, 157 enfants à partir de 10 ans atteints de diabète de type 2 ont reçu un traitement, dont 52 patients l'empagliflozine, 52 patients la linagliptine et 53 patients un placebo (voir le paragraphe sur les études cliniques).
Pendant la phase contrôlée contre placebo, l'effet indésirable le plus fréquent était l'hypoglycémie (empagliflozine 10 mg et 25 mg, poolée: 23,1 %, placebo: 9,4 %).
Aucun de ces événements n'était grave ou n'a nécessité d'assistance.
Dans l'ensemble, le profil de sécurité chez l'enfant était comparable au profil de sécurité chez le patient adulte atteint de DT2.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSignes et symptômes
Dans le cadre d'études cliniques contrôlées avec des volontaires sains, des doses uniques jusqu'à 800 mg d'empagliflozine ont été administrées.
Traitement
En cas de surdosage, un traitement approprié doit être initié en fonction de l'état clinique du patient. L'élimination de l'empagliflozine par hémodialyse n'a pas été étudiée.
Propriétés/EffetsCode ATC
A10BK03
Mécanisme d'action / Pharmacodynamique
L'empagliflozine est un inhibiteur compétitif sélectif, réversible et très puissant (CI50 de 1,3 nmol) du cotransporteur du SGLT2. Il montre une sélectivité 5000 fois supérieure pour SGLT2 par rapport au SGLT1 humain responsable de l'absorption intestinale du glucose (CI50: 6278 nmol).
Un autre effet inhibiteur des transporteurs du glucose GLUT, qui sont responsables du transport du glucose dans les différents tissus, n'a pu être montré.
SGLT-2 est principalement exprimé dans les reins. En tant que transporteur prédominant, il est responsable de la réabsorption du glucose du filtrat glomérulaire vers la circulation générale.
L'empagliflozine réduit la réabsorption rénale du glucose d'une manière indépendante de l'insuline. La quantité de glucose éliminée par le rein via ce mécanisme dépend de la glycémie et du taux de filtration glomérulaire.
L'augmentation de l'excrétion rénale du glucose entraine une diurèse osmotique et, par cet effet diurétique, une baisse de la pression artérielle (4-5 mmHg en moyenne pour la pression systolique et 1-2 mmHg pour la diastolique) ainsi qu'une augmentation de l'hématocrite (environ 2-3%). L'empagliflozine a également un effet uricosurique et réduit ainsi le taux plasmatique d'urée (environ 50 µmol/L). L'élimination rénale du glucose augmente le risque d'infections urogénitales, particulièrement chez les femmes et les personnes âgées.
L'élimination rénale du glucose après la prise de 10 mg d'empagliflozine est de 64 g par jour (correspondant à environ 256 kcal). Suite à la prise de 25 mg d'empagliflozine, l'élimination rénale du glucose s'élève à environ 78 g par jour (correspondant à environ 312 kcal).
Chez les patients avec diabète de type 2, l'élimination rénale du glucose dans l'urine augmente après la première dose d'empagliflozine pour se maintenir pratiquement au même niveau pendant tout l'intervalle de dosage de 24 heures.
L'augmentation de la glycosurie a mené à une réduction de la concentration plasmatique de glucose chez les patients avec un diabète de type 2.
L'effet hypoglycémiant de l'empagliflozine est indépendant de la fonction des cellules bêta et des voies d'activation de l'insuline.
L'empagliflozine réduit en outre la réabsorption de sodium et augmente l'apport en sodium au niveau du tubule distal. Ceci peut avoir des répercussions sur différentes fonctions physiologiques et entraîner notamment: augmentation du rétrocontrôle tubuloglomérulaire et réduction de la pression intraglomérulaire, diminution de la pré- et postcharge du cœur, régulation négative de l'activité sympathique et réduction de la contrainte de la paroi ventriculaire gauche, ce qui est démontré par des valeurs de NT-proBNP plus faibles avec des effets positifs éventuels sur le remodelage cardiaque, la pression de remplissage et la fonction diastolique ainsi que la préservation de la structure et de la fonction rénales.
Efficacité clinique
Diabète de type 2
Un total de 11'250 patients avec diabète de type 2 ont été traités dans 10 études en double aveugle avec contrôle placebo et comparateur actif; parmi ceux-ci, 3'021 patients ont reçu de l'empagliflozine 10 mg. Dans quatre études, la durée du traitement était de 24 semaines; avec l'extension de ces études ainsi que d'autres, les patients ont été traités par empagliflozine pendant 102 semaines au total.
Le traitement par empagliflozine en monothérapie et en association avec la metformine, une sulfonylurée, des inhibiteurs de la DPP-4 ou l'insuline, a entrainé des améliorations cliniquement significatives de l'HbA1c, de la glycémie à jeun (GAJ), du poids corporel ainsi que de la pression artérielle systolique et diastolique.
Monothérapie
L'efficacité et la tolérance de l'empagliflozine en monothérapie ont été évaluées chez des patients naïfs de traitement antidiabétique au cours d'une étude en double aveugle, avec contrôle placebo et comparateur actif, d'une durée de 24 semaines. Par rapport au placebo, le traitement par empagliflozine a entrainé une réduction statistiquement significative (p<0,0001) de l'HbA1c (Tableau 1) et une diminution cliniquement significative de la GAJ.
Dans une analyse prédéfinie de patients (N=201) avec une valeur initiale d'HbA1c ≥8,5%, le traitement a entrainé une réduction de l'HbA1c par rapport à la valeur initiale de -1,44% pour l'empagliflozine 10 mg, -1,43% pour l'empagliflozine 25 mg, -1,04% pour la sitagliptine et une augmentation de 0,01% pour le placebo.
Dans la période d'extension menée en double aveugle et avec contrôle placebo de cette étude, les réductions de l'HbA1c, du poids corporel et de la pression artérielle ont été maintenues jusqu'à la semaine 76.
Tableau 1: Résultats d'une étude de 24 semaines (LOCF) contrôlée contre placebo avec Jardiance comme monothérapie
|
|
Placebo
|
Empagliflozine
10 mg
|
Empagliflozine
25 mg
|
Sitagliptine
100 mg
| |
N
|
228
|
224
|
224
|
223
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
7,91
|
7,87
|
7,86
|
7,85
| |
Variation par rapport à la valeur initiale1
|
0,08
|
-0,66
|
-0,78
|
-0,66
| |
Différence par rapport au placebo1 (IC 97,5%)
|
|
-0,74*
(-0,90, -0,57)
|
-0,85*
(-1,01, -0,69)
|
-0,73
(-0,88, -0,59)3
| |
N
|
208
|
204
|
202
|
200
| |
Patients (%) atteignant une valeur d'HbA1c <7% (valeur initiale d'HbA1c ≥7%)4
|
12,0
|
35,3
|
43,6
|
37,5
| |
N
|
228
|
224
|
224
|
223
| |
Poids corporel (kg)
| |
Valeur initiale (moyenne)
|
78,23
|
78,35
|
77,80
|
79,31
| |
Variation par rapport à la valeur initiale1
|
-0,33
|
-2,26
|
-2,48
|
0,18
| |
Différence par rapport au placebo1
(IC 97,5%)
|
|
-1,93*
(-2,48, -1,38)
|
-2,15*
(-2,70,-1,60)
|
0,52
(-0,04, 1,00)3
| |
N
|
228
|
224
|
224
|
223
| |
PAS (mmHg)2
| |
Valeur initiale (moyenne)
|
130,4
|
133,0
|
129,9
|
132,5
| |
Variation par rapport à la valeur initiale1
|
-0,3
|
-2,9
|
-3,7
|
0,5
| |
Différence par rapport au placebo1
(IC 95%)
|
|
-2,6* (-5,2, -0,0)
|
-3,4* (-6,0, -0,9)
|
0,8 (-1,4, 3,1)3
|
1 Moyenne ajustée par rapport à la valeur initiale
2 Last Observation Carried Forward (LOCF), valeurs censurées après traitement de secours antihypertenseur
3 CI 95%
4 Non évalué en termes de signification statistique du fait de la procédure d'analyse de confirmation séquentielle
* p<0,0001
Association thérapeutique
Empagliflozine comme traitement d'appoint à la metformine et à une sulfonylurée
Par rapport au placebo, l'empagliflozine comme traitement d'appoint à la metformine ou la metformine avec une sulfonylurée a entrainé une réduction statistiquement significative (p<0,0001) de l'HbA1c (Tableau 1) et du poids corporel (Tableau 2). De plus, elle a entrainé une réduction cliniquement significative de la glycémie à jeun et de la pression artérielle systolique et diastolique par rapport au placebo.
Dans la période d'extension menée en double aveugle et avec contrôle placebo de ces études, les réductions de l'HbA1c, du poids corporel et de la pression artérielle ont été maintenues jusqu'à la semaine 52.
Tableau 2: Résultats d'une étude de 24 semaines (LOCF) contrôlée contre placebo avec Jardiance comme traitement d'appoint à la metformine ou à la metformine et une sulfonylurée (Full Analysis Set)
|
Jardiance comme appoint à la metformine
|
Placebo
|
Empagliflozine
10 mg
|
Empagliflozine
25 mg
| |
N
|
207
|
217
|
213
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
7,90
|
7,94
|
7,86
| |
Variation par rapport à la valeur initiale1
|
-0,13
|
-0,70
|
-0,77
| |
Différence par rapport au placebo1
(IC 97,5%)
|
|
-0,57*
(-0,72, -0,42)
|
-0,64*
(-0,79, -0,48)
| |
N
|
184
|
199
|
191
| |
Patients (%) atteignant une valeur d'HbA1c <7% (valeur initiale d'HbA1c ≥7%)2
|
12,5
|
37,7
|
38,7
| |
N
|
207
|
217
|
213
| |
Poids corporel (kg)
| |
Valeur initiale (moyenne)
|
79,73
|
81,59
|
82,21
| |
Variation par rapport à la valeur initiale1
|
-0,45
|
-2,08
|
-2,46
| |
Différence par rapport au placebo1
(IC 97,5%)
|
|
-1,63*
(-2,17, -1,08)
|
-2,01*
(-2,56, -1,46)
| |
N
|
207
|
217
|
213
| |
PAS (mmHg)2
| |
Valeur initiale (moyenne)
|
128,6
|
129,6
|
130,0
| |
Variation par rapport à la valeur initiale1
|
-0,4
|
-4,5
|
-5,2
| |
Différence par rapport au placebo1
(IC 95%)
|
|
-4,1*
(-6,2, -2,1)
|
-4,8*
(-6,9, -2,7)
|
|
Jardiance comme appoint à la metformine et une sulfonylurée
|
Placebo
|
Empagliflozine
10 mg
|
Empagliflozine
25 mg
| |
N
|
225
|
225
|
216
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,15
|
8,07
|
8,10
| |
Variation par rapport à la valeur initiale1
|
-0,17
|
-0,82
|
-0,77
| |
Différence par rapport au placebo1
(IC 97,5%)
|
|
-0,64*
(-0,79, -0,49)
|
-0,59*
(-0,74, -0,44)
| |
N
|
216
|
209
|
202
| |
Patients (%) atteignant une valeur d'HbA1c <7% (valeur initiale d'HbA1c≥7%)2
|
9,3
|
26,3
|
32,2
| |
N
|
224
|
225
|
215
| |
GAJ (mg/dl) [mmol/l]
| |
Valeur initiale (moyenne)
|
151,7 [8,42]
|
151,0 [8,38]
|
156,5 [8,68]
| |
Variation par rapport à la valeur initiale1
|
5,5 [0,31]
|
-23,3 [-1,29]
|
-23,3 [-1,29]
| |
Différence par rapport au placebo1
(IC 95%)
|
|
-28,8
(-34,3, -23,4)
[-1,60
(-1,90, -1,30)]
|
-28,8
(-34,3, -23,3)
[-1,60
(-1,90, -1,29)]
| |
N
|
225
|
225
|
216
| |
Poids corporel (kg)
| |
Valeur initiale (moyenne)
|
76,23
|
77,08
|
77,50
| |
Variation par rapport à la valeur initiale1
|
-0,39
|
-2,16
|
-2,39
| |
Différence par rapport au placebo1
(IC 97,5%)
|
|
-1,76*
(-2,25, -1,28)
|
-1,99*
(-2,48, -1,50)
| |
N
|
225
|
225
|
216
| |
PAS (mmHg)2
| |
Valeur initiale (moyenne)
|
128,8
|
128,7
|
129,3
| |
Variation par rapport à la valeur initiale1
|
-1,4
|
-4,1
|
-3,5
| |
Différence par rapport au placebo1
(IC 95%)
|
|
-2,7
(-4,6, -0,8)
|
-2,1
(-4,0, -0,2)
|
1 Moyenne ajustée par rapport à la valeur initiale
2 Non évalué en termes de signification statistique du fait de la procédure d'analyse de confirmation séquentielle
* p<0,0001
Empagliflozine par rapport à un placebo chez des patients présentant un contrôle glycémique insuffisant sous metformine et linagliptine
Chez des patients présentant un contrôle glycémique insuffisant sous metformine et linagliptine, un traitement de 24 semaines avec 10 mg d'empagliflozine a entraîné des améliorations statistiquement significatives de la valeur HbA1c, de la GJ ainsi que du poids corporel par rapport à un placebo (traitement de fond linagliptine 5 mg et metformine). (Tableau 3).
Tableau 3: Paramètres d'efficacité pour la comparaison entre empagliflozine et placebo en traitement d'appoint chez des patients présentant un contrôle glycémique insuffisant sous metformine et linagliptine 5 mg
|
|
Metformine + linagliptine 5 mg
| |
|
Empagliflozine
10 mg1
|
Placebo2
| |
HbA1c (%) – 24 semaines3
| |
N
|
109
|
106
| |
Valeur initiale (moyenne)
|
7,97
|
7,96
| |
Variation par rapport à la valeur initiale
(moyenne ajustée)
|
-0,65
|
0,14
| |
Différence par rapport à la moyenne ajustée du placebo
(IC à 95 %)2
|
-0,79
(-1,02, -0,55)
p<0,0001
|
| |
GJ (mg/dL) – 24 semaines3
| |
N
|
109
|
106
| |
Valeur initiale (moyenne)
|
167,9
|
162,9
| |
Variation par rapport à la valeur initiale
(moyenne ajustée)
|
-26,3
|
6,1
| |
Différence par rapport à la moyenne ajustée du placebo (IC à 95 %)
|
-32,4 (-41,7, -23,0)
p<0,0001
|
| |
Poids corporel – 24 semaines3
| |
N
|
109
|
106
| |
Valeur initiale (moyenne) en kg
|
88,4
|
82,3
| |
Variation par rapport à la valeur initiale (moyenne ajustée)
|
-3,1
|
-0,3
| |
Différence par rapport au placebo (moyenne ajustée) (IC à 95 %)1
|
-2,8 (-3,5, -2,1)
p<0,0001
|
|
1 Les patients randomisés dans le groupe avec empagliflozine 10 mg ont reçu de l'empagliflozine 10 mg/linagliptine 5 mg avec metformine en traitement de fond
2 Les patients randomisés dans le groupe placebo ont reçu un placebo plus de la linagliptine 5 mg avec metformine en traitement de fond
3 Le modèle MMRM appliqué pour le Full Analysis Set (OC) englobe la valeur initiale HbA1c, la valeur initiale DFGe (formule MDRD), la région géographique, la date d'examen, le traitement et l'effet du traitement en fonction du temps (treatment-by-visit-interaction). La GJ inclut également la valeur initiale de GJ. Le poids inclut également la valeur initiale de poids.
Traitement d'appoint à l'insuline
Empagliflozine comme traitement d'appoint à l'insuline basale
L'efficacité et la tolérance de l'empagliflozine comme traitement d'appoint à l'insuline basale avec ou sans metformine et/ou une sulfonylurée ont été évaluées au cours d'une étude en double aveugle avec contrôle placebo d'une durée de 78 semaines. Pendant les 18 premières semaines, la dose d'insuline a été maintenue constante, mais elle a été ajustée au cours des 60 semaines suivantes pour obtenir une glycémie à jeun <110 mg/dl.
Jusqu'à la semaine 18, l'empagliflozine a entrainé une amélioration statistiquement significative de l'HbA1c (Tableau 4).
Après la semaine 78, le traitement par empagliflozine a entrainé une réduction statistiquement significative de l'HbA1c ainsi qu'une épargne insulinique par rapport au placebo. De plus l'empagliflozine a également entrainé une réduction de la GAJ, du poids corporel et de la pression artérielle.
Tableau 4: Résultats d'une étude avec contrôle placebo sur Jardiance comme appoint à l'insuline basale avec ou sans metformine ou une sulfonylurée (Full Analysis Set – Completer) après 18 ou 78 semaines (LOCF)
|
|
Placebo
|
Empagliflozine
10 mg
|
Empagliflozine
25 mg
| |
N
|
125
|
132
|
117
| |
HbA1c (%) à la semaine 18
| |
Valeur initiale (moyenne)
|
8,10
|
8,26
|
8,34
| |
Variation par rapport à la valeur initiale1
|
-0,01
|
-0,57
|
-0,71
| |
Différence par rapport au placebo1
(IC 97,5%)
|
|
-0,56* (-0,78, -0,33)
|
-0,70* (-0,93, -0,47)
| |
N
|
112
|
127
|
110
| |
HbA1c (%) à la semaine 78
| |
Valeur initiale (moyenne)
|
8,09
|
8,27
|
8,29
| |
Variation par rapport à la valeur initiale1
|
-0,02
|
-0,48
|
-0,64
| |
Différence par rapport au placebo1
(IC 97,5%)
|
|
-0,46* (-0,73, -0,19)
|
-0,62* (-0,90, -0,34)
| |
N
|
112
|
127
|
110
| |
Dose d'insuline basale (UI/jour) à la semaine 78
| |
Valeur initiale (moyenne)
|
47,84
|
45,13
|
48,43
| |
Variation par rapport à la valeur initiale1
|
5,45
|
-1,21
|
-0,47
| |
Différence par rapport au placebo1
(IC 97,5%)
|
|
-6,66** (-11,56, -1,77)
|
-5,92** (-11,00, -0,85)
|
1 Moyenne ajustée par rapport à la valeur initiale
* p <0,0001
** p <0,025
Empagliflozine comme traitement d'appoint à l'insuline injectée plusieurs fois par jour
L'efficacité et la tolérance de l'empagliflozine comme traitement d'appoint à l'insuline administrée plusieurs fois par jour avec ou sans traitement associé par metformine ont été évaluées au cours d'une étude en double aveugle avec contrôle placebo d'une durée de 52 semaines. Pendant les 18 premières semaines et les 12 dernières semaines, la dose d'insuline a été maintenue constante, mais elle a été ajustée entre les semaines 19 et 40 pour obtenir des glycémies préprandiales <100 mg/dl [5,5 mmol/l], et des glycémies postprandiales <140 mg/dl [7,8 mmol/l].
Jusqu'à la semaine 18, l'empagliflozine a entrainé une amélioration statistiquement significative de l'HbA1c par rapport au placebo (Tableau 5).
Après la semaine 52, le traitement par empagliflozine a entrainé une réduction statistiquement significative de l'HbA1c ainsi qu'une épargne insulinique par rapport au placebo. De plus l'empagliflozine a également entrainé une réduction de la GAJ et du poids corporel.
Tableau 5: Résultats d'efficacité aux semaines 18 et 52 d'une étude sur l'empagliflozine avec contrôle placebo comme traitement d'appoint à plusieurs doses d'insuline par jour avec ou sans metformine
|
|
Placebo
|
Empagliflozine
10 mg
|
Empagliflozine
25 mg
| |
N
|
188
|
186
|
189
| |
HbA1c (%) à la semaine 18
| |
Valeur initiale (moyenne)
|
8,33
|
8,39
|
8,29
| |
Variation par rapport à la valeur initiale1
|
-0,50
|
-0,94
|
-1,02
| |
Différence par rapport au placebo1 (IC 97,5%)
|
|
-0,44* (-0,61, -0,27)
|
-0,52* (-0,69, -0,35)
| |
N
|
115
|
119
|
118
| |
HbA1c (%) à la semaine 522
| |
Valeur initiale (moyenne)
|
8,25
|
8,40
|
8,37
| |
Variation par rapport à la valeur initiale1
|
-0,81
|
-1,18
|
-1,27
| |
Différence par rapport au placebo1 (IC 97,5%)
|
|
-0,38*** (-0,62, -0,13)
|
-0,46* (-0,70, -0,22)
| |
N
|
113
|
118
|
118
| |
Patients (%) atteignant une valeur d'HbA1c <7% avec une valeur initiale d'HbA1c ≥7% à la semaine 52
|
26,5
|
39,8
|
45,8
| |
N
|
115
|
118
|
117
| |
Dose d'insuline (UI/jour) à la semaine 522
| |
Valeur initiale (moyenne)
|
89,94
|
88,57
|
90,38
| |
Variation par rapport à la valeur initiale1
|
10,16
|
1,33
|
-1,06
| |
Différence par rapport au placebo1 (IC 97,5%)
|
|
-8,83# (-15,69, -1,97)
|
-11,22**(-18,09, -4,36)
| |
N
|
115
|
119
|
118
| |
Poids corporel (kg) à la semaine 522
| |
Valeur initiale (moyenne)
|
96,34
|
96,47
|
95,37
| |
Variation par rapport à la valeur initiale1
|
0,44
|
-1,95
|
-2,04
| |
Différence par rapport au placebo1 (IC 97,5%)
|
|
-2,39* (-3,54, -1,24)
|
-2,48* (-3,63, -1,33)
|
1 Moyenne ajustée par rapport à la valeur initiale
2 semaine 19-40: régime Treat-to-Target pour l'ajustement de la dose d'insuline, pour l'atteinte des valeurs cibles prédéfinies de la glycémie (préprandiale <100 mg/dl (5,5 mmol/l), postprandiale <140 mg/dl (7,8 mmol/l)
* valeur de p <0,0001
** valeur de p = 0,0003
*** valeur de p = 0,0005
# valeur de p = 0,0040
Populations spécifiques
Patients avec insuffisance rénale, données d'une étude avec contrôle placebo de 52 semaines
L'efficacité et la tolérance de l'empagliflozine comme traitement d'appoint à un traitement antidiabétique de patients insuffisants rénaux ont été évaluées au cours d'une étude en double aveugle avec contrôle placebo d'une durée de 52 semaines. Par rapport au placebo, le traitement par empagliflozine a entrainé une réduction statistiquement significative (p<0,0001) de l'HbA1c (Tableau 6) à la semaine 24 et une diminution cliniquement significative de la GAJ. L'amélioration de l'HbA1c, du poids corporel et de la pression artérielle a été maintenue jusqu'à 52 semaines.
Tableau 6: Résultats d'une étude avec contrôle placebo avec Jardiance sur des patients avec diabète de type2 et insuffisance rénale (Full Analysis Set) après 24 semaines (LOCF)
|
|
Placebo
|
Empagliflozine
10 mg
|
Empagliflozine
25 mg
|
Placebo
|
Empagliflozine
25 mg
| |
|
DFGe ≥60 à <90 ml/min/1,73 m²
|
DFGe ≥30 à <60 ml/min/1,73 m²
| |
N
|
95
|
98
|
97
|
187
|
187
| |
HbA1c (%)
| |
Valeur initiale (moyenne)
|
8,09
|
8,02
|
7,96
|
8,04
|
8,03
| |
Variation par rapport à la valeur initiale1
|
0,06
|
-0,46
|
-0,63
|
0.05
|
-0,37
| |
Différence par rapport au placebo1
(IC 95%)
|
|
-0,52*
(-0,72, -0,32)
|
-0,68*
(-0,88; -0,49)
|
|
-0,42*
(-0,56, -0,28)
| |
N
|
89
|
94
|
91
|
178
|
175
| |
Patients (%) atteignant une valeur d'HbA1c <7% (valeur initiale d'HbA1c ≥7%)2
|
6,7
|
17,0
|
24,2
|
7,9
|
12,0
| |
N
|
95
|
98
|
97
|
187
|
187
| |
Poids corporel (kg)2
| |
Valeur initiale (moyenne)
|
86,00
|
92,05
|
88,06
|
82,49
|
83,22
| |
Variation par rapport à la valeur initiale1
|
-0,33
|
-1,76
|
-2,33
|
-0,08
|
-0,98
| |
Différence par rapport au placebo1
(IC 95%)
|
|
-1,43
(-2,09, -0,77)
|
-2,00
(-2,66; -1,34)
|
|
-0,91 (-1,41
-0,41)
| |
N
|
95
|
98
|
97
|
187
|
187
| |
PAS (mmHg)2
| |
Valeur initiale (moyenne)
|
134,69
|
137,37
|
133,68
|
133,38
|
136,64
| |
Variation par rapport à la valeur initiale1
|
0,65
|
-2,92
|
-4,47
|
0,40
|
-3,88
| |
Différence par rapport au placebo1
(IC 95%)
|
|
-3,57
(-6,86, -0,29)
|
-5,12
(-8,41; -1,82)
|
|
-4,28 (-6,88; -1,68)
|
1 Moyenne ajustée par rapport à la valeur initiale
2 Non évalué en termes de signification statistique du fait de la procédure d'analyse de confirmation séquentielle
* p<0,0001
Evénements cardiovasculaires chez des patients atteints d'une maladie cardiovasculaire manifeste
L'étude EMPA-REG OUTCOME a comparé le risque de survenue d'événements cardiovasculaires chez des patients atteints d'un diabète de type 2 et d'une maladie cardiovasculaire préexistante traités par empagliflozine par rapport à un placebo. Cette étude a inclus au total 7020 patients atteints d'une maladie coronarienne (affectant un ou plusieurs vaisseaux), présentant un état consécutif à un infarctus du myocarde (IM), un état consécutif à un accident vasculaire cérébral et/ou une artériopathie oblitérante périphérique qui ont été traités pendant jusqu'à 4,5 ans (durée de traitement médiane: 2,6 ans et durée d'observation médiane: 3,1 ans) en plus d'un traitement antérieur par empagliflozine 10 mg (n=2'345), empagliflozine 25 mg (n=2'342) ou placebo (n=2'333).
La population se composait à 72,4% de personnes de type caucasien, 21,6% de type asiatique et 5,1% de type noir. L'âge moyen s'élevait à 63 ans (9,3% des patients étaient âgés de ≥75 ans), 71,5% des patients étaient masculins. Au début de l'étude, près de 81% des patients ont reçu un inhibiteur du système rénine angiotensine, 65% des bêtabloquants, 43% des diurétiques, 89% des anticoagulants et 81% un médicament hypolipémiant. Environ 74% des patients prenaient de la metformine au début de l'étude, 48% s'administraient de l'insuline et 43% une sulfonylurée. L'étude EMPA-REG n'a pas été conçue pour la mesure de l'efficacité glycémique, à l'exception des 12 premières semaines, mais a servi à titre d'étude sur l'équilibre glycémique destinée à évaluer le résultat cardiovasculaire final. Au cours des 12 premières semaines, le traitement de base servant à diminuer la glycémie devait être maintenu sans changement. Suite à cette période initiale, le traitement de diminution de la glycémie pouvait être ajusté conformément aux directives de traitement actuelles. D'autres médicaments concomitants (servant notamment à traiter l'hypertension et la dyslipidémie) pouvaient être ajoutés ou modifiés au cours de l'ensemble de l'étude afin d'atteindre le meilleur traitement standard.
Le critère d'évaluation primaire (défini pour les deux bras de traitement par empagliflozine) était la durée jusqu'au premier événement du critère d'évaluation combiné composé de décès cardiovasculaires, d'IM non mortels et d'AVC non mortels (3-point MACE [Major Adverse Cardiovascular Events]). L'ensemble des événements du critère d'évaluation et des décès ont été évalués par un comité d'experts (CEC) indépendant, externe et en aveugle. Conformément au règlement d'évaluation, les décès cardiovasculaires ont été répartis dans les catégories suivantes: IM mortel, AVC mortel, décès dû à une insuffisance cardiaque (par la progression ou un choc cardiogénique), mort subite ou autres événements cardiovasculaires mortels.
Réduction du risque de décès cardiovasculaires et mortalité totale
L'empagliflozine était supérieure au placebo en termes de critère d'évaluation primaire (c'est-à-dire qu'elle a permis une diminution du risque de survenue d'événements cardiovasculaires). La réduction significative du critère d'évaluation primaire traduisait en grande partie la diminution nette de l'incidence de décès cardiovasculaires (Tableau 7 et Figure 1) et ne pouvait pas s'expliquer entièrement par l'existence de différences dans le contrôle glycémique des groupes de traitement. Un effet préventif a été observé pour toutes les catégories de décès cardiovasculaires. Dans un même temps, le risque de survenue d'infarctus du myocarde non mortels n'a pas diminué de manière significative d'un point de vue statistique. L'empagliflozine n'exerçait pas d'effet préventif sur les AVC, une réduction numérique toutefois non significative a été observée en ce qui concerne les AVC mortels (HR 0,72 (IC à 95% 0,33-1,55). Une hausse numérique toutefois non significative des AVC non mortels a été observée (HR 1,24 (IC à 95% 0,92-1,67) (voir «Mises en garde et précautions»). Le traitement par empagliflozine a aussi permis une amélioration de la mortalité totale résultant principalement de la réduction des décès cardiovasculaires (l'empagliflozine n'a pas permis de réduction significative d'un point de vue statistique de la mortalité non cardiovasculaire).
Tableau 7: Effet thérapeutique par rapport au critère d'évaluation primaire composé, ses composants et la mortalité (population traitée*)
|
|
Placebo
|
Empagliflozine
(10 et 25 mg, poolée)
| |
N
|
2333
|
4687
| |
Durée écoulée jusqu'à la première survenue d'un décès cardiovasculaire, d'un IM non mortel ou d'un AVC non mortel
|
282 (12,1)
|
490 (10,5)
| |
Hazard ratio vs placebo (IC à 95,02%)**
|
|
0,86 (0,74; 0,99)
| |
Valeur p pour la supériorité
|
|
0,0382
| |
Décès cardiovasculaires N (%)
|
137 (5,9)
|
172 (3,7)
| |
Hazard ratio vs placebo (IC à 95%)
|
|
0,62 (0,49; 0,77)
| |
Valeur p
|
|
<0,0001
| |
IM non mortel N (%)
|
121 (5,2)
|
213 (4,5)
| |
Hazard ratio vs placebo (IC à 95%)
|
|
0,87 (0,70; 1,09)
| |
Valeur p
|
|
0,2189
| |
AVC non mortels N (%)
|
60 (2,6)
|
150 (3,2)
| |
Hazard ratio vs placebo (IC à 95%)
|
|
1,24 (0,92; 1,67)
| |
Valeur p
|
|
0,1638
| |
Mortalité totale N (%)
|
194 (8,3)
|
269 (5,7)
| |
Hazard ratio vs placebo (IC à 95%)
|
|
0,68 (0,57; 0,82)
| |
Valeur p
|
|
<0,0001
| |
Mortalité non cardiovasculaire N (%)
|
57 (2,4)
|
97 (2,1)
| |
Hazard ratio vs placebo (IC à 95%)
|
|
0,84 (0,60; 1,16)
|
* c.-à-d. les patients ayant reçu au moins une dose du médicament à l'étude
** Etant donné la prise en compte des données de l'étude dans le cadre d'une analyse intermédiaire, un intervalle de confiance bilatéral à 95,02% correspondant à une valeur p inférieure à 0,0498 pour la pertinence
Figure 1: Durée écoulée jusqu'à la première survenue d'un événement du critère d'évaluation primaire composé (décès cardiovasculaire/IM non mortel/AVC non mortel)

Dans une étude croisée randomisée avec contrôle placebo et comparateur actif portant sur 30 volontaires sains, aucune augmentation de l'intervalle QTc n'a été observée sous 25 mg ou 200 mg d'empagliflozine.
Glycémie à jeun (GAJ)
Dans quatre études contrôlées versus placebo, le traitement par empagliflozine en monothérapie ou en appoint à la metformine ou la metformine plus une sulfonylurée a entrainé des variations moyennes de la GAJ par rapport à la valeur initiale de -20,5 mg/dl [-1,14 mmol/l] pour l'empagliflozine 10 mg et de -23,2 mg/dl [1,29 mmol/l] pour l'empagliflozine 25 mg par rapport au placebo (7,4 mg/dl [0,41 mmol/l]). Cet effet a été observé après 24 semaines et s'est maintenu pendant 76 semaines.
Glycémie postprandiale à 2 heures
Le traitement par empagliflozine en appoint à la metformine ou à la metformine et une sulfonylurée a entrainé une réduction cliniquement significative de la glycémie postprandiale à 2 heures (test de tolérance au repas) à 24 semaines (traitement d'appoint à la metformine: placebo +5,9 mg/dl [+0,33 mmol/l], empagliflozine 10 mg: -46,0 mg/dl) [-2,56 mmol/l], empagliflozine 25 mg: -44,6 mg/dl [-2,5 mmol/l]. Traitement d'appoint à la metformine et à une sulfonylurée: placebo -2,3 mg/dl [-0,13 mmol/l], empagliflozine 10 mg: -35,7 mg/dl [1,98 mmol/l], empagliflozine 25 mg: -36,6 mg/dl [2 mmol/l].
Poids corporel
Dans une analyse poolée prédéfinie de 4 études contrôlées versus placebo, le traitement par empagliflozine a entrainé une réduction du poids corporel (-0,24 kg pour le placebo, -2,04 kg pour l'empagliflozine 10 mg, -2,26 kg pour l'empagliflozine 25 mg) à la semaine 24, qui s'est maintenue jusqu'à la semaine 52 (-0,16 kg pour le placebo, -1,96 kg pour l'empagliflozine 10 mg et -2,25 kg pour l'empagliflozine 25 mg).
Pression artérielle
L'efficacité et la tolérance de l'empagliflozine ont été évaluées au cours d'une étude en double aveugle avec contrôle placebo d'une durée de 12 semaines portant sur des patients avec diabète de type 2 et une hypertension artérielle, traités par divers médicaments antidiabétiques et jusqu'à 2 médicaments antihypertenseurs. Le traitement par empagliflozine une fois par jour a entrainé une amélioration statistiquement significative de l'HbA1c et de la pression artérielle systolique et diastolique moyenne sur 24 heures, déterminée par un suivi ambulatoire de la pression artérielle (Tableau 8). Le traitement par empagliflozine a entrainé une réduction de la PAS et de la PAD en position assise.
Tableau 8: Résultats d'une étude avec contrôle placebo avec Jardiance sur des patients avec diabète de type2 et pression artérielle non contrôlée (Full Analysis Set) après 12 semaines (LOCF)
|
|
Placebo
|
Empagliflozine
10 mg
|
Empagliflozine
25 mg
| |
N
|
271
|
276
|
276
| |
HbA1c (%) à la semaine 12
| |
Valeur initiale (moyenne)
|
7,90
|
7,87
|
7,92
| |
Variation par rapport à la valeur initiale1
|
0,03
|
-0,59
|
-0,62
| |
Différence par rapport au placebo1
(IC 95%)
|
|
-0,62* (-0,72, -0,52)
|
-0,65* (-0,75, -0,55)
| |
PAS sur 24 heures à la semaine 12
| |
Valeur initiale (moyenne)
|
131,72
|
131,34
|
131,18
| |
Variation par rapport à la valeur initiale1
|
0,48
|
-2,95
|
-3,68
| |
Différence par rapport au placebo1
(IC 95%)
|
|
-3,44* (-4,78, -2,09)
|
-4,16* (-5,50, -2,83)
| |
PAD sur 24 heures à la semaine 12
| |
Valeur initiale (moyenne)
|
75,16
|
75,13
|
74,64
| |
Variation par rapport à la valeur initiale1
|
0,32
|
-1,04
|
-1,40
| |
Différence par rapport au placebo1
(IC 95%)
|
|
-1,36** (-2,15, -0,56)
|
-1,72* (-2,51, -0,93)
|
1 Moyenne ajustée par rapport à la valeur initiale
* p <0,0001
** p < 0,001
Dans une analyse poolée prédéfinie de quatre études contrôlées versus placebo, le traitement par empagliflozine a entrainé une réduction de la pression artérielle systolique (empagliflozine 10 mg: -3,9 mmHg, empagliflozine 25 mg: -4,3 mmHg) par rapport au placebo (-0,5 mmHg) et une réduction de la pression artérielle diastolique (empagliflozine 10 mg: -1,8 mmHg, empagliflozine 25 mg: -2,0 mmHg) par rapport au placebo (-0,5 mmHg) à la semaine 24; ces améliorations étaient maintenues jusqu'à la semaine 52.
Paramètres de laboratoire
Augmentation de l'hématocrite
Dans une analyse de la sécurité (toutes les données combinées des patients avec diabète, n = 13402), les modifications moyennes de l'hématocrite par rapport aux valeurs initiales étaient de 3,4% pour l'empagliflozine 10 mg et de 3,6% pour l'empagliflozine 25 mg par rapport à -0,1% pour le placebo. Dans l'étude EMPA-REG Outcome, les valeurs d'hématocrite étaient redescendues jusqu'aux valeurs initiales à la fin de la phase de suivi de 30 jours après l'arrêt du médicament.
Augmentation des lipides sériques
Dans une analyse de la sécurité (toutes les données combinées des patients avec diabète, n = 13402), le pourcentage d'augmentation moyen par rapport aux valeurs initiales pour l'empagliflozine 10 mg ou 25 mg comparé au placebo était de 4,9% ou 5,7% versus 3,5% pour le cholestérol total; de 3,3% ou 3,6% versus 0,4% pour le cholestérol HDL; de 9,5% ou 10,0% versus 7,5% pour le cholestérol LDL; de 9,2% ou 9,9% versus 10,5% pour les triglycérides.
Insuffisance cardiaque
Empagliflozine chez les patients atteints d'insuffisance cardiaque à fraction d'éjection réduite
Une étude randomisée, en double aveugle, contrôlée contre placebo (EMPEROR-Reduced) a été menée chez 3730 patients atteints d'insuffisance cardiaque chronique (New York Heart Association [NYHA] II-IV) à fraction d'éjection réduite (FEVG ≤40%), afin d'évaluer l'efficacité et la sécurité de l'empagliflozine 10 mg une fois par jour en appoint au traitement standard de l'insuffisance cardiaque.
Les patients avec un DFGe <20 ml/min/1,73 m2, les personnes présentant une obésité extrême (IMC ≥45 kg/m2) et les patients porteurs d'un implant DAVG [dispositif d'assistance ventriculaire gauche] ont été exclus de l'étude.
Le critère d'évaluation primaire était la durée écoulée jusqu'à la première survenue d'un événement du critère d'évaluation combiné (décès cardiovasculaire et hospitalisation pour insuffisance cardiaque). Les critères secondaires confirmatoires étaient la survenue d'hospitalisations pour insuffisance cardiaque [HIC] (première survenue et récidive) ainsi que la vitesse de déclin du DFGe au cours du temps.
Le traitement de base comprenait des inhibiteurs de l'ECA/antagonistes des récepteurs de l'angiotensine/inhibiteur du récepteur de l'angiotensine-néprilysine (88,3%), des bêtabloquants (94,7%), des antagonistes des récepteurs des minéralocorticoïdes (71,3%), des diurétiques (95,0%), un défibrillateur cardioverteur implantable (DCI) (31,4 %) et un traitement de resynchronisation cardiaque (CRT) (11,8%).
1863 patients ont été randomisés pour recevoir l'empagliflozine 10 mg et 1867 patients pour recevoir le placebo; ils ont été suivis pendant une durée médiane de 15,7 mois. L'âge moyen de la population de l'étude (76,1% de sexe masculin) était de 66,8 ans (intervalle: 25–94 ans; 26,8% ≥75 ans). La majorité des patients étaient blancs (70,5%), 18,0% étaient asiatiques et 6,9% étaient noirs/afro-américains. La plupart des patients avaient une IC de classe NYHA II (75,1%; 24,4% de classe III et 0,5% de classe IV). Au début de l'étude, la FEVG moyenne était de 27,5%, le DFGe moyen de 62,0 ml/min/1,73 m2 et le rapport médian albuminurie-créatininurie (urinary albumin to creatinine ratio, UACR) de 22 mg/g. Environ la moitié des patients (51,7%) avait un DFGe ≥60 ml/min/1,73 m2. Le DFGe était compris entre 45 et <60 ml/min/1,73 m2 chez 24,1% des patients, entre 30 et <45 ml/min/1,73 m2 chez 18,6% des patients et entre 20 et < 30 ml/min/1,73 m2 chez 5,3% des patients.
Le traitement par l'empagliflozine était supérieur au traitement par le placebo: l'empagliflozine a réduit significativement le risque du critère d'évaluation primaire combiné et le nombre total d'HIC (première survenue et récidive) (Tableau 9).
Tableau 9: Effet thérapeutique pour le critère d'évaluation primaire combiné, ses composantes et les deux principaux critères d'évaluation secondaires inclus dans les tests de confirmation prédéfinis
|
|
Placebo
|
Empagliflozine
10 mg
| |
N
|
1867
|
1863
| |
Durée écoulée jusqu'à la première survenue d'un décès cardiovasculaire (CV) ou d'une HIC, N (%)
|
462 (24,7)
|
361 (19,4)
| |
Hazard ratio vs placebo (IC à 95,04%)**
|
|
0,75 (0,65, 0,86)
| |
Valeur de p pour la supériorité
|
|
< 0,0001
| |
Décès cardiovasculaire (CV), N (%)*
|
202 (10,8)
|
187 (10,0)
| |
Hazard ratio vs placebo (IC à 95%)
|
|
0,92 (0,75, 1,12)
| |
Valeur de p
|
|
0,4113
| |
HIC (première survenue), N (%)*
|
342 (18,3)
|
246 (13,2)
| |
Hazard ratio vs placebo (IC à 95%)
|
|
0,69 (0,59, 0,81)
| |
Valeur de p
|
|
< 0,0001
| |
HIC (première survenue et récidive), N d'événements
|
553
|
388
| |
Hazard ratio vs placebo (IC à 95,04%)**
|
|
0,70 (0,58, 0,85)
| |
Valeur de p
|
|
0,0003
| |
Pente du DFGe (créatininémie CKD EPI), vitesse du déclin (ml/min/1,73 m2/an)
|
-2,28
|
-0,55
| |
Différence thérapeutique vs placebo (IC à 99,9%)***
|
|
1,73 (0,67, 2,80)
| |
Valeur de p
|
|
p < 0,0001
|
CV = cardiovasculaire, HIC = hospitalisation pour insuffisance cardiaque, DFGe = débit de filtration glomérulaire estimé, CKD EPI = formule élaborée par la collaboration Chronic Kidney Disease Epidemiology
* Non évalué quant aux erreurs de type I
** Sur la base d'une analyse intermédiaire, un intervalle de confiance bilatéral à 95,04% a été utilisé, ce qui correspond à une valeur de p inférieure à 0,0496 pour la significativité. Les événements de décès CV et d'HIC étaient confirmés par un comité indépendant évaluant les événements cliniques et analysés sur la base du groupe randomisé.
*** Comme prédéfini dans la procédure de test statistique, un intervalle de confiance bilatéral à 99,9% a été utilisé, ce qui correspond à une valeur de p inférieure à 0,001 pour la significativité; la vitesse de déclin du DFGe a été analysée sur la base du groupe traité.
L'analyse en sous-groupes n'a montré aucune différence significative de l'effet thérapeutique pour le critère d'évaluation primaire (HR constamment <1) en fonction des paramètres prédéfinis, tels que le statut glycémique (présence ou non d'un diabète de type 2) et le degré d'insuffisance rénale (y compris insuffisance sévère, DFGe <30 ml/min/1,73 m2 [voir critères d'exclusion]).
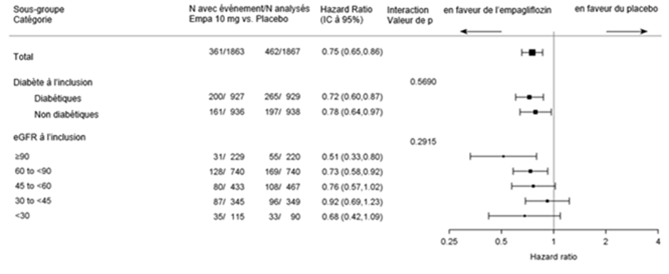
Résultat sur le plan rénal
Le traitement par l'empagliflozine a ralenti la progression de l'insuffisance rénale (baisse du DFGe pendant la durée du traitement, voir Tableau 9).
Empagliflozine chez les patients atteints d'insuffisance cardiaque à fraction d'éjection préservée
Une étude multicentrique, randomisée, en double aveugle et contrôlée par placebo (EMPEROR-Preserved) a évalué l'efficacité et la sécurité de l'empagliflozine 10 mg une fois par jour comme traitement d'appoint du traitement standard chez des patients atteints d'insuffisance cardiaque chronique symptomatique (classe fonctionnelle NYHA II-IV) et présentant une fraction d'éjection (FEVG) > 40 %. Les personnes extrêmement obèses (IMC ≥45 kg/m2) et les patients porteurs de LVAD [systèmes d'assistance ventriculaire gauche] étaient exclus de l'étude.
Le critère d'évaluation principal (composite) était le délai avant la première apparition de l'un des événements suivants: décès d'origine cardiovasculaire (MCV) ou hospitalisation pour insuffisance cardiaque (HIC). Les principaux critères secondaires étaient l'HIC en général (première survenue et récidive) et la pente de la baisse du DFGe (ECG-EPI)cr pendant l'étude (changement par rapport à la référence). La majorité des participants de l'étude recevaient déjà un inhibiteur de l'ECA/antagoniste des récepteurs de l'angiotensine/antagoniste des récepteurs de l'angiotensine et de la néprilysine (80,7%), un bêtabloquant (86,3%), un antagoniste des récepteurs des minéralocorticoïdes (37,5%) et des diurétiques (86,2%).
Au total, 5'988 patients (55,3% d'hommes; 75,9% de personnes de race blanche, 13,8% de personnes d'origine asiatique et 4,3% de personnes de race noire) d'un âge moyen de 71,9 ans (43,0% ≥75 ans) ont été randomisés pour recevoir l'empagliflozine 10 mg (N = 2'997) ou un placebo (N = 2'991) et ont été suivis pendant une période médiane de 26,2 mois.
Au début de l'étude, environ un tiers des patients présentaient une FEVG <50% (33,1%), 50 à <60% (34,4%) ou ≥60% (32,5%). La grande majorité des patients (81,5%) présentaient une IC de classe II de la NHYHA (18,1% et 0,3% respectivement, une IC de classe III et IV de la NYHA). Le DFGe moyen et le ratio albumine/créatinine urinaire (ACR) étaient respectivement de 60,6 ml/min/1,73 m2 et de 21 mg/g. Environ la moitié des patients (50,1%) présentaient un DFGe ≥60 ml/min/1,73 m2, 26,1% un DFGe de 45 à <60 ml/min/1,73 m2, 18,6% un DFGe de 30 à <45 ml/min/1,73 m2 et 4,9% un DFGe de 20 à <30 ml/min/1,73 m2.
L'empagliflozine a été supérieure au placebo en termes de réduction du risque pour le principal critère d'évaluation composite (décès cardiovasculaire ou hospitalisation pour insuffisance cardiaque). En outre, l'empagliflozine a réduit de manière significative le risque de survenue d'une HIC (première survenue et récidive) et a également réduit de manière significative le taux de diminution du DFGe (voir tableau 10).
Tableau 10: Effet du traitement pour le principal critère d'évaluation composite, ses composantes et les deux principaux critères d'évaluation secondaires inclus dans les tests de confirmation prédéfinis
|
|
Placebo
|
Empagliflozine 10 mg
| |
n
|
2'991
|
2'997
| |
Délai avant le premier événement (MCV ou ICC), n (%)
|
511 (17,1)
|
415 (13,8)
| |
Hazard ratio vs placebo (IC à 95,03%)**
|
|
0,79 (0,69, 0,90)
| |
Valeur p pour la supériorité
|
|
0,0003
| |
MCV, n (%)*
|
244 (8,2)
|
219 (7,3)
| |
Hazard ratio vs placebo (IC à 95%)
|
|
0,91 (0.76, 1,09)
| |
Valeur p
|
|
0,2951
| |
HIC (première survenue), n (%)*
|
352 (11,8)
|
259 (8,6)
| |
Hazard ratio vs placebo (IC à 95%)
|
|
0,71 (0,60, 0,83)
| |
Valeur p
|
|
<0,0001
| |
HIC (première survenue et récidive), nombre d'événements
|
541
|
407
| |
Hazard ratio vs placebo (IC à 95,03%)**
|
|
0,73 (0,61, 0,88)
| |
Valeur p
|
|
0,0009
| |
Pente du DFGe (CKD-EPI)cr, taux de diminution (ml/min/1,73m2/année)
|
−2,62
|
−1,25
| |
Différence du traitement vs placebo (IC à 99,9 %)***
|
|
1,36 (0,86, 1,87)
| |
Valeur p
|
|
<0,0001
|
MCV: mort cardiovasculaire, HIC: hospitalisation en raison d'une insuffisance cardiaque, DFGe: débit de filtration glomérulaire estimé, CKD-EPI = formule Chronic Kidney Disease Epidemiology Collaboration
* non contrôlé pour l'erreur de type 1
** Sur la base d'une analyse intermédiaire, un intervalle de confiance bilatéral de 95,03% a été appliqué, ce qui correspond à une valeur p inférieure à 0,0497 pour la significativité. Les événements MCV et d'HIC ont été évalués par une commission indépendante pour les événements cliniques et analysés sur la base de l'ensemble des patients randomisés.
*** Comme prédéfini dans la procédure de test statistique, un intervalle de confiance bilatéral de 99,9% a été appliqué, ce qui correspond à une valeur p inférieure à 0,001 pour la significativité. La pente du DFGe a été analysée sur la base de l'ensemble des patients traités.
Figure 2: Délai avant le premier événement (MCV adjudiqué ou HIC adjudiquée)
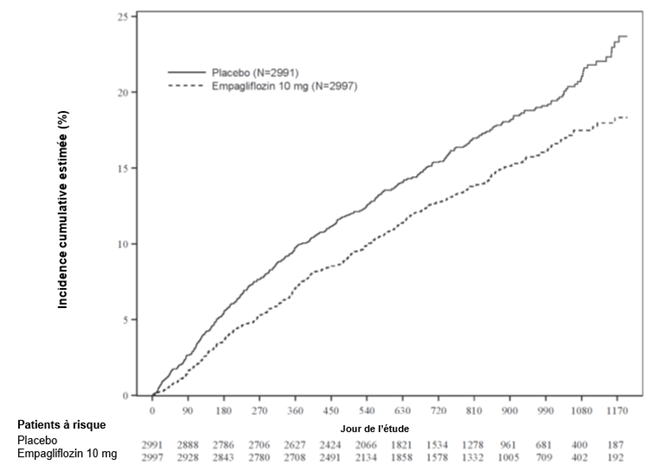
Les résultats pour le principal critère d'évaluation composite concordaient pour tous les sous-groupes prédéfinis, y compris pour les sous-groupes en fonction de la FEVG (voir figure 3).
Figure 3: Analyses de sous-groupes pour le délai avant le premier événement (MCV adjudiqué ou HIC adjudiquée)

^4 patients atteints d'IC de classe fonctionnelle I de la NYHA ont été comptés dans le sous-groupe de classe fonctionnelle NYHA II
*Test de tendance
Critère d'évaluation rénal
Pendant la phase de traitement, le DFGe a diminué significativement plus lentement dans le groupe empagliflozine que dans le groupe placebo (voir tableau 10).
Maladie rénale chronique
Une étude randomisée, en double aveugle, contrôlée contre placebo sur l’empagliflozine 10 mg une fois par jour (EMPA-KIDNEY) a été effectuée chez 6609 patients atteints de maladie rénale chronique (DFGe ≥20–< 45 ml/min/1,73 m2 ou DFGe ≥45–< 90 ml/min/1,73 m2 avec un rapport albumine/créatinine urinaire [urinary albumin to creatinine ratio, UACR] ≥200 mg/g) afin d’évaluer les critères d’évaluation cardio-rénaux en tant que traitement d’appoint au traitement standard. Chez les patients ayant nécessité une dialyse pendant l’étude, le traitement a pu être poursuivi. Le principal critère d’évaluation composite était la durée écoulée jusqu’à la première survenue d’une progression de la maladie rénale chronique (MRC; diminution constante du DFGe ≥40 % après randomisation, DFGe constant < 10 ml/min/1,73 m², insuffisance rénale terminale ou décès rénal) ou jusqu’au décès cardiovasculaire (CV). L’hospitalisation indépendamment de la cause (première survenue et récidive), la première survenue d’une hospitalisation pour insuffisance cardiaque, le décès cardiovasculaire et la mortalité totale ont été inclus dans la procédure d’analyse de confirmation. La majorité des patients inclus ont été traités au début de l’étude par des inhibiteurs du SRAA (85,2 % inhibiteurs de l’ECA ou antagonistes des récepteurs de l’angiotensine).
Au total, 3304 resp. 3305 patients ont été randomisés et traités par l’empagliflozine 10 mg, resp. le placebo et observés pendant une durée médiane de 24,3 mois. La population de l’étude comprenait 66,8 % d’hommes et 33,2 % de femmes d’un âge moyen de 63,3 ans (intervalle: 18–94 ans). Parmi la population de l’étude, 58,4 % des patients étaient blancs, 36,2 % asiatiques et 4,0 % noirs/afro-américains.
Au début de l’étude, le DFGe moyen était de 37,3 ml/min/1,73 m2 (~78 % avec un DFGe < 45 ml/min/1,73 m2). L’UACR médian était de 329 mg/g. Les causes principales de la maladie rénale chronique étaient la néphropathie diabétique/maladie rénale diabétique (31 %), la glomérulopathie (25 %), la maladie hypertensive/rénovasculaire (22 %).
L'empagliflozine était supérieure au placebo en ce qui concerne la baisse du risque du principal critère d'évaluation composite comprenant la progression de l'IRC ou le décès CV. Dans une analyse prédéfinie, le traitement par l'empagliflozine a réduit le risque d'insuffisance rénale terminale ou de décès CV de 28 % (HR 0,72, IC à 95 % 0,59 à 0,89, p nominal = 0,0017) par comparaison avec le placebo. En outre, l'empagliflozine a réduit significativement le risque d'hospitalisation indépendamment de la cause (première survenue et récidive) (voir Tableau 11).
Tableau 11: Effet thérapeutique pour le principal critère d’évaluation composite et les principaux critères d’évaluation secondaires (Empa-KIDNEY) inclus dans les tests de confirmation prédéfinis et leurs composantes
|
|
Placebo
|
Empagliflozine 10 mg
| |
n
|
3305
|
3304
| |
Durée écoulée jusqu'à la première survenue d'une progression de la maladie rénale (diminution constante du DFGe ≥40 % après randomisation, DFGe constant < 10 ml/min/1,73 m², insuffisance rénale terminale* ou décès rénal) ou jusqu'au décès cardiovasculaire, n (%)
|
558 (16,9)
|
432 (13,1)
| |
Hazard ratio vs placebo (IC à 99,83 %)
|
|
0,72 (0,59, 0,89)
| |
Valeur p pour la supériorité
|
|
< 0,0001
| |
Diminution constante du DFGe ≥40 % après randomisation, n (%)
|
474 (14,3)
|
359 (10,9)
| |
Hazard ratio vs placebo (IC à 95 %)
|
|
0,70 (0,61, 0,81)
| |
Valeur p
|
|
< 0,0001
| |
Insuffisance rénale* terminale ou DFGe constant < 10 ml/min/1,73 m², n (%)
|
222 (6,7)
|
157 (4,8)
| |
Hazard ratio vs placebo (IC à 95 %)
|
|
0,68 (0,55, 0,84)
| |
Valeur p
|
|
0,0002
| |
Décès rénal, n (%)**
|
4 (0,1)
|
4 (0,1)
| |
Hazard ratio vs placebo (IC à 95 %)
|
|
| |
Valeur p
|
|
| |
Décès cardiovasculaire, n (%)
|
70 (2,1)
|
59 (1,8)
| |
Hazard ratio vs placebo (IC à 95 %)
|
|
0,83 (0,59, 1,17)
| |
Valeur p
|
|
0,2932
| |
Survenue de l'hospitalisation indépendamment de la cause (première survenue et récidive), n des événements
|
1895
|
1612
| |
Hazard ratio vs placebo (IC à 99,03 %)
|
|
0,86 (0,76, 0,98)
| |
Valeur p
|
|
0,0022
| |
Durée écoulée jusqu'au décès toutes causes confondues, n (%)
|
168 (5,1)
|
149 (4,5)
| |
Hazard ratio vs placebo (IC à 97,1 %)
|
|
0,87 (0,68, 1,11)
| |
Valeur p
|
|
0,2122
|
HIC = Hospitalisation pour insuffisance cardiaque, DFGe = débit de filtration glomérulaire estimé
* L'insuffisance rénale terminale est définie comme le début de la nécessité de la dialyse permanente ou la réalisation d'une transplantation rénale.
** Trop peu de cas de décès rénaux sont survenus pour permettre un calcul exact du hazard ratio.
Figure 4: Durée écoulée jusqu'à la première survenue de la progression de la maladie rénale ou du décès cardiovasculaire adjudiqué*
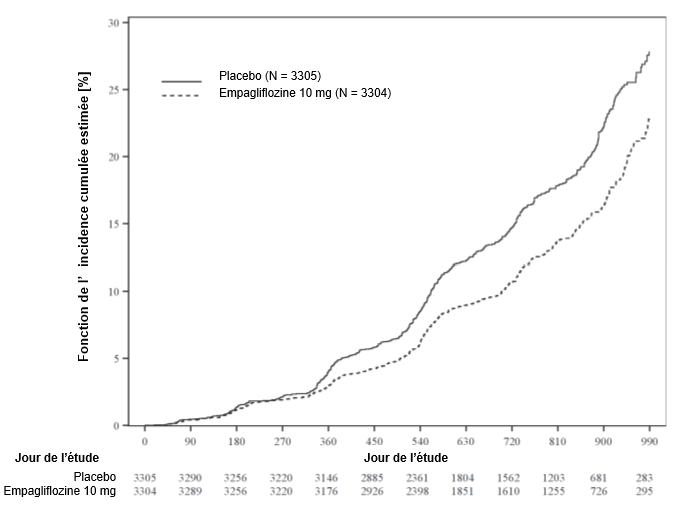
* Fonction de l'incidence cumulée estimée
Les résultats du principal critère d'évaluation composite relatifs aux sous-groupes prédéfinis, y compris les catégories du DFGe, la cause de la maladie rénale, le statut diabétique ou l'utilisation concomitante d'inhibiteurs du SRAA étaient globalement cohérents. Le bénéfice du traitement était plus prononcé chez les patients présentant une albuminurie élevée.
Par comparaison avec le placebo, l'empagliflozine a ralenti le taux annuel de diminution du DFGe de 1,38 ml/min/1,73 m2/an (IC à 95 % 1,16, 1,59) sur la base d'une analyse prédéfinie de toutes les mesures du DFGe effectuées à partir de la visite du mois 2 jusqu'à la dernière visite de suivi.
Enfants et adolescents
L'efficacité et la sécurité cliniques de l'empagliflozine 10 mg avec une augmentation de dose potentielle à 25 mg ou de la linagliptine 5 mg une fois par jour a été étudiée chez des enfants et adolescents âgés de 10 à 17 ans atteints de DT2 dans le cadre d'une étude randomisée, en double aveugle, à groupes parallèles, contrôlée contre placebo (DINAMO) sur une période de 26 semaines, avec une phase de prolongation incluant un traitement à base de principes actifs en double aveugle afin d'évaluer la sécurité jusqu'à 52 semaines.
Dans l'ensemble, 157 patients ont été traités par empagliflozine (10 mg ou 25 mg; N = 52), par linagliptine (N = 52) ou par placebo (N = 53). Parmi les traitements de fond, en tant que complément de mesures diététiques et d'une activité physique, on comptait la metformine (51 %), une association de metformine et d'insuline (40,1 %), l'insuline (3,2 %), ou bien aucun traitement de fond n'a été administré (5,7 %). La valeur initiale moyenne de l'HbA1c était de 8,03 %. La population de l'étude comprenait 38,2 % de patients masculins et 61,8 % de patients féminins d'un âge moyen de 14,5 ans (plage: de 10 à 17 ans); 51,6 % ayant au moins 15 ans. La population de l'étude était en outre composée comme suit: 49,7 % étaient d'origine caucasienne, 5,7 % d'origine asiatique et 31,2 % d'origine africaine/afro-américaine. L'IMC moyen était de 36,04 kg/m², le poids moyen de 99,92 kg. Seuls des patients présentant un DFG ≥60 ml/min/1,73 m² ont été inclus dans l'étude DINAMO.
L'empagliflozine était supérieure au placebo en ce qui concerne le critère d'évaluation principal – la réduction de l'HbA1c après 26 semaines par rapport à la valeur initiale – indépendamment d'un traitement en fonction des besoins ou de l'arrêt d'un traitement. Par ailleurs, le traitement par Jardiance a entraîné une réduction cliniquement pertinente de la FPG (tableau 12).
Tableau 12: Résultats d'une étude contrôlée contre placebo de 26 semaines sur l'empagliflozine chez les enfants et les adolescents atteints de diabète de type 2 (analyse en intention de traiter modifiée)
|
|
Placebo
|
Empagliflozine
(10 et 25 mg, poolée)
| |
N
|
53
|
52
| |
HbA1c (%)¹
| |
Valeur initiale (valeur moyenne)
|
8,05
|
8,00
| |
Variation par rapport à la valeur initiale²
|
0,68
|
-0,17
| |
Différence par rapport au placebo² (IC à 95 %)
|
|
-0,84
(-1,50, -0,19)
| |
Valeur p pour la supériorité
|
|
0,0116
| |
N
|
52
|
48
| |
FPG (mg/dl) [mmol/L] 3,4
| |
Valeur initiale (valeur moyenne)
|
158,6 [8,80]
|
154,4 [8,57]
| |
Variation par rapport à la valeur initiale²
|
15,7 [0,87]
|
-19,5 [-1,08]
| |
Différence par rapport au placebo² (IC à 95 %)
|
|
-35,2 (-58,6, -11,7)
[-1,95 (-3,25, -0,65)]
| |
Valeur p nominale
|
|
0,0035
|
¹ Imputation multiple avec 500 itérations pour les données manquantes
² En ce qui concerne la valeur initiale et la stratification de la valeur moyenne ajustée
³ Last Observation Carried Forward (LOCF), y compris les valeurs initiales
4 Non évalué en termes de signification statistique; ne fait pas partie de la procédure d'analyse séquentielle
PharmacocinétiqueAbsorption
La pharmacocinétique de l'empagliflozine a été largement étudiée chez les volontaires sains et les patients présentant un diabète de type 2. Après administration par voie orale, l'empagliflozine a été rapidement absorbée, avec des concentrations plasmatiques maximales survenant 1,5 heure (tmax médian) après la prise de la dose. Ensuite, les concentrations plasmatiques ont diminué de manière biphasique avec une phase de distribution rapide et une phase terminale relativement lente. Pour une administration une fois par jour (qd) de 10 mg d'empagliflozine, l'ASC plasmatique moyenne à l'état d'équilibre était de 1,870 nmol.h/l et la Cmax 259 nmol/l. Pour une administration une fois par jour (qd) de 25 mg d'empagliflozine, l'ASC plasmatique moyenne à l'état d'équilibre était de 4740 nmol.h/l et la Cmax 687 nmol/l. L'exposition systémique à l'empagliflozine augmenté proportionnellement à la dose (domaine de 10 mg-100 mg). La pharmacocinétique de l'empagliflozine est linéaire par rapport au temps. La pharmacocinétique de l'empagliflozine n'a pas montré de différence cliniquement significative entre les volontaires sains et les patients avec un diabète de type 2.
L'administration d'empagliflozine 25 mg après la prise d'un repas à forte teneur en graisse et en calories a entraîné une faible baisse de la biodisponibilité; l'ASC a diminué d'environ 16% et la Cmax d'environ 37% par rapport à une prise à jeun. L'effet observé des aliments sur la pharmacocinétique de l'empagliflozine n'a pas été jugé cliniquement significatif et l'empagliflozine peut être administrée au cours ou en dehors des repas.
Distribution
D'après l'analyse pharmacocinétique de population, le volume de distribution apparent à l'état d'équilibre a été estimé à 73,8 l. Après l'administration orale d'une solution d'empagliflozine marquée au [14C] à des volontaires sains, la répartition érythrocytaire était de 36,8% environ et la liaison aux protéines plasmatiques de 86,2%.
Métabolisme
Dans une étude avec de l'empagliflozine marqué au [14C], la plus grande partie de la radioactivité totale dans le plasma a pu être attribuée à l'empagliflozine inchangée.
Les métabolites les plus fréquents étaient trois glucuronides conjugués (2 O , 3 O et 6 O glucuronides). L'exposition systémique à chaque métabolite était inférieure à 10% de la radioactivité totale dans le plasma. Les données in vitro suggèrent que la voie primaire du métabolisme de l'empagliflozine chez l'homme est la glucuronidation par les uridine-5'-diphosphoglucuronosyltransférases UGT2B7, UGT1A3, UGT1A8 et UGT1A9.
Élimination
D'après l'analyse pharmacocinétique de population, la demi-vie d'élimination terminale apparente de l'empagliflozine a été estimée à 12,4 heures et la clairance orale apparente de 10,6 l/heure. Les variabilités inter-individuelles et résiduelles pour la clairance orale de l'empagliflozine étaient de 39,1%. Avec une administration une fois par jour, les concentrations plasmatiques d'empagliflozine à l'état d'équilibre étaient atteintes à la cinquième dose. En cohérence avec la demi-vie, une accumulation allant jusqu'à 22% a été observée à l'état d'équilibre pour l'ASC plasmatique. Après l'administration orale d'une solution d'empagliflozine marquée au [14C] à des volontaires sains, environ 41,2%% de la radioactivité était éliminée dans les fèces et 54,4% dans l'urine. La substance mère sous forme inchangée formait la majorité de la radioactivité retrouvée dans les fèces (82,9%) et 43,5% de la radioactivité excrétée dans l'urine.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Chez les sujets présentant une insuffisance hépatique légère, modérée ou sévère selon la classification de Child-Pugh, l'ASC de l'empagliflozine a augmenté d'environ 23%, 47% et 75% et la Cmax d'environ 4%, 23% et 48% respectivement, par rapport aux sujets avec une fonction hépatique normale. Sur la base des données pharmacocinétiques, un ajustement de la dose n'est donc pas nécessaire chez les patients insuffisants hépatiques.
Troubles de la fonction rénale
Chez les patients avec une insuffisance rénale légère (DFGe: 60 - <90 ml/min/1,73 m2), moyenne (DFGe: 30 - <60 ml/min/1,73 m2) et sévère (DFGe: <30 ml/min/1,73 m2) et les patients présentant une insuffisance rénale terminale, l'ASC de l'empagliflozine a augmenté d'environ 18%, 20%, 66% et 48% respectivement, par rapport aux patients avec une fonction rénale normale. Les concentrations plasmatiques maximales d'empagliflozine étaient similaires chez les patients présentant une insuffisance rénale modérée et une insuffisance rénale terminale par rapport aux patients ayant une fonction rénale normale. Les concentrations plasmatiques maximales d'empagliflozine étaient environ 20% plus élevées chez les patients présentant une insuffisance rénale légère et modérée par rapport aux patients ayant une fonction rénale normale. En cohérence avec l'étude de phase I, l'analyse pharmacocinétique de population a montré que la baisse du DFGe allait de pair avec une réduction apparente de la clairance orale de l'empagliflozine et entraînait ainsi une augmentation de l'exposition systémique. Sur la base des données pharmacocinétiques, un ajustement de la dose n'est donc pas nécessaire chez les patients insuffisants rénaux.
Indice de masse corporelle (IMC)
Un ajustement de la dose sur la base de l'IMC n'est pas nécessaire. Selon l'analyse pharmacocinétique de population, l'indice de masse corporelle n'a aucun effet cliniquement significatif sur la pharmacocinétique de l'empagliflozine.
Sexe
Un ajustement de la dose sur la base du sexe n'est pas nécessaire. Selon l'analyse pharmacocinétique de population, le sexe n'a aucun effet cliniquement significatif sur la pharmacocinétique de l'empagliflozine.
Origine ethnique
Selon l'analyse pharmacocinétique de population, l'estimation de l'ASC était 13,5% plus élevée chez les sujets asiatiques avec un IMC de 25 kg/m2 que chez les sujets non asiatiques avec un IMC de 25 kg/m2.
Patients âgés
Selon l'analyse pharmacocinétique de population, l'âge n'a aucun effet cliniquement significatif sur la pharmacocinétique de l'empagliflozine.
Enfants et adolescents
La pharmacocinétique et la pharmacodynamique d'une dose unique d'empagliflozine 5 mg, 10 mg et 25 mg ont été étudiées chez des enfants et adolescents âgés de 10 à 17 ans atteints de DT2. La pharmacocinétique observée et le rapport pharmacocinétique-pharmacodynamique (excrétion du glucose dans l'urine) chez les patients adultes et pédiatriques était comparable après avoir pris en considération des covariables significatives.
La pharmacocinétique et la pharmacodynamique (variation de l'HbA1c par rapport à la valeur initiale) de l'empagliflozine 10 mg avec une augmentation de dose à 25 mg ont été étudiées chez des enfants et adolescents âgés de 10 à 17 ans atteints de DT2. La relation exposition-effet observée était en général comparable chez les adultes, les enfants et les adolescents.
L'administration perorale d'empagliflozine a entraîné une exposition dans la plage observée chez les patients adultes. Les valeurs moyennes géométriques observées pour la concentration minimale et la concentration 1,5 heure après l'administration dans un état stable étaient de 26,6 nmol/L ou 308 nmol/L sous empagliflozine 10 mg une fois par jour et de 67,0 nmol/L ou 525 nmol/L sous empagliflozine 25 mg une fois par jour.
Données précliniquesDans les études de toxicité à long terme sur les rongeurs et les chiens, des signes de toxicité ont été observés à des expositions supérieures ou égales à 10 fois la dose de 25 mg. La toxicité était généralement cohérente avec la pharmacologie secondaire liée à l'excrétion urinaire de glucose, notamment la perte de poids et de graisse corporelle, l'augmentation de la consommation alimentaire, la diarrhée, la déshydratation, la baisse du glucose sérique et les augmentations d'autres paramètres sériques reflétant une augmentation du métabolisme protéique, une gluconéogenèse et un déséquilibre électrolytique, des modifications urinaires (polyurie et glycosurie) et des modifications microscopiques des reins (dont une dilatation tubulaire et une minéralisation tubulaire et pelvienne).
L'empagliflozine n'est pas génotoxique.
L'empagliflozine n'a pas augmenté l'incidence de tumeurs chez des rats femelles jusqu'à la dose maximale de 700 mg/kg/jour (correspondant à environ 72 fois l'exposition clinique sur la base de l'ASC pour une dose de 25 mg). Chez les rats mâles, des lésions prolifératives vasculaires bénignes (hémangiomes) du ganglion lymphatique mésentérique, liées au traitement, ont été observées à la dose de 700 mg/kg/jour (correspondant à environ 42 fois l'exposition clinique pour une dose de 25 mg). Ces tumeurs sont fréquentes chez le rat et ne sont vraisemblablement pas pertinentes pour l'homme.
Des tumeurs à cellules interstitielles (cellules de Leydig) ont été observées dans les testicules à la dose de 300 mg/kg/jour (correspondant à environ 26 fois l'exposition clinique (ASC) pour une dose de 25 mg) et 700 mg/kg/jour. Ces tumeurs sont considérées comme vraisemblablement non pertinentes pour l'homme.
L'empagliflozine n'a pas augmenté l'incidence de tumeurs chez des souris CD-1 femelles jusqu'à la dose maximale de 1000 mg/kg/jour (correspondant à environ 62 fois l'exposition clinique sur la base de l'ASC pour une dose de 25 mg).
Chez des souris mâles, l'empagliflozine a induit des tumeurs rénales à la dose de 1000 mg/kg/jour (correspondant à environ 44 fois l'exposition clinique pour une dose de 25 mg). Le mécanisme d'action entrainant l'apparition de ces tumeurs, est dépendant de la prédisposition naturelle de la souris CD-1 mâle aux pathologies rénales et à une voie métabolique non superposable à l'homme. C'est pourquoi les tumeurs rénales des souris CD-1 mâles sont considérées comme vraisemblablement non pertinentes pour l'homme.
L'empagliflozine administrée à des rats et des lapins pendant l'organogenèse à des doses jusqu'à 300 mg/kg (correspondant à environ 48 fois resp. 128 fois l'exposition clinique sur la base de l'ASC pour une dose de 25 mg) n'était pas tératogène. L'exposition à des doses d'empagliflozine toxiques pour la mère et correspondant à environ 155 fois l'exposition clinique pour une dose de 25 mg, a également causé une incurvation des os des membres chez le rat. L'exposition à des doses d'empagliflozine toxiques pour la mère et correspondant à environ 139 fois l'exposition clinique pour une dose de 25 mg, a également causé une augmentation des pertes embryo-foetales chez le lapin.
Dans les études de toxicité pré- et post-natales chez le rat, une diminution du gain de poids de la progéniture a été observée à des expositions maternelles environ 4 fois supérieures à l'exposition clinique pour une dose de 25 mg.
Dans une étude de toxicité menée chez de jeunes rats, une minéralisation rénale des tubules minimale non nocive, mais dose-dépendante et non réversible, a été observée chez les mâles et les femelles à partir de 10 mg/kg/jour (correspondant à environ 0,6 fois l'exposition clinique à une dose de 25 mg) lors de l'administration d'empagliflozine du jour 21 au jour 90 après la naissance. Une dilatation réno-tubulaire et pelvienne minimale à légère n'a été observée qu'à la dose de 100 mg/kg/jour (correspondant environ à 13 fois la dose clinique de 25 mg) qui n'était cependant plus décelable au bout d'une phase de régénération de 13 semaines exempte d'administration de médicaments.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Conserver à température ambiante (15-25°C). Tenir hors de portée des enfants.
Numéro d’autorisation63227 (Swissmedic)
PrésentationComprimés pelliculés de 10 mg: 30 et 90 (B)
Comprimés pelliculés de 25 mg: 30 et 90 (B)
Titulaire de l’autorisationBoehringer Ingelheim (Suisse) GmbH, Bâle
Mise à jour de l’informationJuillet 2025
|