Propriétés/EffetsCode ATC
L04AC10
Mécanisme d'action
Le sécukinumab est un anticorps IgG1 entièrement humain qui se lie de manière sélective à la cytokine pro-inflammatoire interleukine 17A (IL-17A) et la neutralise. Le sécukinumab agit de manière ciblée sur l'IL-17A et inhibe son interaction avec le récepteur de l'IL-17, qui est exprimé sur différents types de cellules, notamment les kératinocytes. En conséquence, le sécukinumab inhibe la libération de cytokines pro-inflammatoires, de chimiokines et de médiateurs de lésions tissulaires et réduit les effets induits par l'IL-17A dans les maladies auto-immunes et inflammatoires. Des taux de sécukinumab cliniquement pertinents atteignent la peau et réduisent les marqueurs inflammatoires locaux. Comme conséquence directe, le traitement par le sécukinumab réduit l'érythème, le durcissement (induration) et la desquamation dans les lésions du psoriasis en plaques. Chez les patients atteints de psoriasis en plaques, la production d'IL-17A est fortement régulée à la hausse dans les zones cutanées touchées par les lésions par rapport aux zones cutanées sans lésion. En outre, des cellules produisant de l'IL-17 ont été observées plus fréquemment dans le liquide articulaire des patients atteints de rhumatisme psoriasique. De plus, chez les patients atteints de spondylarthrite axiale, les cellules produisant de l'IL-17 étaient également nettement plus nombreuses dans la moelle osseuse sous-chondrale des facettes articulaires. L'IL-17A est nettement régulée à la hausse dans les lésions associées à l'hidrosadénite suppurée en comparaison avec les patients atteints de psoriasis et les personnes de contrôle saines, et des taux sériques d'IL-17A significativement plus élevés ont été observés chez les patients concernés. Une augmentation du nombre de lymphocytes produisant de l'IL-17A a également été constatée chez des patients atteints de spondylarthrite axiale non radiographique.
L'IL-17A favorise également la réaction inflammatoire dans les tissus, l'infiltration par les neutrophiles, la destruction des os et des tissus ainsi que le remodelage des tissus, notamment l'angiogenèse et la fibrose.
Pharmacodynamique
Dans une étude sur le sécukinumab, les taux de neutrophiles infiltrants et de divers marqueurs associés aux neutrophiles, qui sont élevés dans les zones de peau affectées par les lésions chez les patients atteints de psoriasis en plaques, ont été significativement réduits après une à deux semaines de traitement.
Il a été démontré que le sécukinumab réduit (en une à deux semaines de traitement) le taux de protéine C-réactive; la protéine C-réactive est un marqueur de l'inflammation dans le rhumatisme psoriasique et la spondylarthrite axiale.
Efficacité clinique
Psoriasis
Patients adultes
La sécurité et l'efficacité de Cosentyx ont été évaluées dans quatre études de phase III randomisées, en double aveugle, contrôlées contre placebo et d'une durée d'un an menées chez des patients atteints de psoriasis en plaques modéré à sévère qui n'ont pas répondu à la photothérapie ou au traitement systémique ou qui n'ont pas toléré un tel traitement [ERASURE, FIXTURE, FEATURE, JUNCTURE]. L'efficacité et la sécurité de 150 mg et 300 mg de Cosentyx ont été évaluées par rapport à un placebo ou à l'étanercept. De plus, une étude a évalué un schéma de traitement continu par rapport à un schéma avec interruption du traitement à la semaine 12 et reprise du traitement «à la demande» en cas d'aggravation clinique [SCULPTURE]. Dans ces études, chaque dose de 300 mg a été administrée sous la forme de deux injections sous-cutanées de 150 mg.
Afin d'obtenir une évaluation non biaisée de l'efficacité du sécukinumab dans le traitement du psoriasis, l'utilisation concomitante d'un traitement systémique ou topique du psoriasis ou d'une photothérapie n'était pas autorisée pendant les études.
Parmi les 2403 patients inclus dans les études contrôlées contre placebo, 79% n'avaient pas reçu de traitement biologique préalable; 45% présentaient un échec thérapeutique sous un traitement non biologique, 8% un échec thérapeutique sous un traitement biologique, 6% un échec thérapeutique sous un traitement anti-TNF et 2% un échec thérapeutique sous un traitement antip40 (anti-IL-12/IL-23). Les caractéristiques de la maladie à l'inclusion étaient généralement comparables dans tous les groupes de traitement: la valeur médiane du score de l'indice de surface et de sévérité du psoriasis (PASI, Psoriasis-Area-Severity-Index-Score) à l'inclusion était de 19 à 20, le score IGA mod 2011 au moment de l'inclusion était compris entre «modéré» (62%) et «sévère» (38%), la valeur médiane de la surface corporelle (BSA, Body Surface Area) à l'inclusion était ≥27 et la valeur médiane du score de l'indice de qualité de vie en dermatologie (DLQI, Dermatology Life Quality Index) se situait dans la plage allant de 10 à 12. Environ 15 à 25% des patients des études de phase III présentaient un rhumatisme psoriasique (RP) au début de l'étude.
L'étude 1 portant sur le psoriasis (ERASURE) a évalué 738 patients. Les patients randomisés pour recevoir un traitement par Cosentyx ont reçu des doses de 150 mg ou 300 mg aux semaines 0, 1, 2, 3 et 4, suivies de la même dose chaque mois. Les patients randomisés pour recevoir un traitement par placebo et qui étaient non répondeurs à la semaine 12 sont ensuite passés à un traitement par Cosentyx avec des doses de 150 ou 300 mg aux semaines 12, 13, 14 et 15, suivies de la même dose chaque mois à partir de la semaine 16.
L'étude 2 portant sur le psoriasis (FIXTURE) a évalué 1306 patients et a comporté, en plus du groupe placebo, un bras utilisant l'étanercept comme comparateur actif. Le traitement par Cosentyx et placebo était le même que dans l'étude 1. Les patients randomisés pour recevoir un traitement par l'étanercept ont reçu des doses de 50 mg deux fois par semaine pendant 12 semaines, suivies de 50 mg chaque semaine.
Dans l'étude 3 (FEATURE) et l'étude 4 (JUNCTURE) portant sur le psoriasis, 177 patients traités par une seringue préremplie et 182 patients traités par un stylo prérempli ont été comparés à un placebo après 12 semaines de traitement pour évaluer la sécurité, la tolérance et la praticabilité de l'auto-administration de Cosentyx à l'aide de la seringue préremplie. Le traitement par Cosentyx et placebo était le même que dans l'étude 1. Dans l'étude 5 portant sur le psoriasis (SCULPTURE), 966 patients ont été évalués. Tous les patients ont reçu Cosentyx 150 mg ou 300 mg aux semaines 0, 1, 2, 3, 4, 8 et 12, puis ont été randomisés pour suivre soit un traitement d'entretien avec administration continue de la même dose chaque mois, soit, après interruption du traitement, un schéma avec reprise du traitement «à la demande» en cas d'aggravation clinique. Le maintien de la réponse a été moins bon chez les patients ayant interrompu leur traitement et l'ayant repris «à la demande» comparativement aux patients ayant suivi un traitement d'entretien mensuel fixe.
Les critères d'évaluation principaux dans les études contrôlées contre placebo et par agent actif étaient la proportion de patients ayant obtenu une réponse PASI 75 et une réponse «Investigator's Global Assessment (IGA)» mod 2011 dans les catégories «exempt de» ou «presque exempt de» par rapport au placebo à la semaine 12 (voir tableau 2 et tableau 3). Le taux de réponse maximal a été atteint à la semaine 16 et la dose de 300 mg s'est avérée supérieure dans toutes les études.
Tableau 2: Résumé des réponses cliniques PASI 50/75/90/100 & IGA⃰-mod-2011 dans les catégories «guéri» ou «presque guéri» dans les études sur le psoriasis 1, 3 et 4 (ERASURE, FEATURE et JUNCTURE)
|
|
Semaine 12
|
Semaine 16
|
Semaine 52
| |
|
Placebo
n (%)
(IC à 95%)
|
150 mg
n (%)
(IC à 95%)
|
300 mg
n (%)
(IC à 95%)
|
150 mg
n (%)
(IC à 95%)
|
300 mg
n (%)
(IC à 95%)
|
150 mg
n (%)
(IC à 95%)
|
300 mg
n (%)
(IC à 95%)
| |
Étude 1
| |
Nombre de patients
|
246
|
244
|
245
|
244
|
245
|
244
|
245
| |
Réponses PASI 50
|
22 (8,9%)
(5,8, 13,4)
|
203 (83,5%)
(78,1, 87,9)
|
222 (90,6%)
(86,1, 93,8)
|
212 (87,2%)
(82,2, 91,0)
|
224 (91,4%)
(87,0, 94,5)
|
187 (77%)
(71,0, 82,0)
|
207 (84,5%)
(79,2, 88,7)
| |
Réponse PASI 75
|
11 (4,5%)
(2,4, 8,1)
|
174 (71,6%)**
(65,4, 77,1)
|
200 (81,6%)**
(76,1, 86,2)
|
188 (77,4%)
(71,5, 82,4)
|
211 (86,1%)
(81,0, 90,1)
|
146 (60,1%)
(53,6, 66,2)
|
182 (74,3%)
(68,3, 79,5)
| |
Réponse PASI 90
|
3 (1,2%)
(0,3, 3,8)
|
95 (39,1%)**
(33,0, 45,6)
|
145 (59,2%)**
(52,7, 65,3)
|
130 (53,5%)
(47,0, 59,9)
|
171 (69,8%)
(63,6, 75,4)
|
88 (36,2%)
(30,2, 42,6)
|
147 (60,0%)
(53,6, 66,1)
| |
Réponse PASI 100
|
2 (0,8%)
(0,1, 3,2)
|
31 (12,8%)
(9,0, 17,8)
|
70 (28,6%)
(23,1, 34,7)
|
51 (21,0%)
(16,2, 26,8)
|
102 (41,6%)
(35,4, 48,1)
|
49 (20,2%)
(15,4, 25,9)
|
96 (39,2%)
(33,1, 45,6)
| |
Réponse sur l'échelle IGA mod 2011 dans les catégories «exempt de» ou «presque exempt de»
|
6 (2,40%)
(1,0, 5,5)
|
125 (51,2%)**
(44,8, 57,6)
|
160 (65,3%)**
(58,9, 71,2)
|
142 (58,2%)
(51,7, 64,4)
|
180 (73,5%)
(67,4, 78,8)
|
101 (41,4%)
(35,2, 47,9)
|
148 (60,4%)
(54,0, 66,5)
| |
Étude 3
| |
Nombre de patients
|
59
|
59
|
58
|
-
|
-
|
-
|
-
| |
Réponse PASI 50
|
3 (5,1%)
(1,3, 15,1)
|
51 (86,4%)
(74,5, 93,6)
|
51 (87,9%)
(76,1, 94,6)
|
-
|
-
|
-
|
-
| |
Réponse PASI 75
|
0 (0,0%)
(0,0, 7,6)
|
41 (69,5%)**
(56,0, 80,5)
|
44 (75,9%)**
(62,5, 85,7)
|
-
|
-
|
-
|
-
| |
Réponse PASI 90
|
0 (0,0%)
(0,0, 7,6)
|
27 (45,8%)
(32,9, 59,2)
|
35 (60,3%)
(46,6, 72,7)
|
-
|
-
|
-
|
-
| |
Réponse PASI 100
|
0 (0,0%)
(0,0, 7,6)
|
5 (8,5%)
(3,2, 19,4)
|
25 (43,1%)
(30,4, 56,7)
|
-
|
-
|
-
|
-
| |
Réponse sur l'échelle IGA mod 2011 dans les catégories «exempt de» ou «presque exempt de»
|
0 (0,0%)
(0,0, 7,6)
|
31 (52,5%)**
(39,2, 65,5)
|
40 (69,0%)**
(55,3, 80,1)
|
-
|
-
|
-
|
-
| |
Étude 4
| |
Nombre de patients
|
61
|
60
|
60
|
-
|
-
|
-
|
-
| |
Réponse PASI 50
|
5 (8,2%)
(3,1, 18,8)
|
48 (80,0%)
(67,3, 88,8)
|
58 (96,7%)
(87,5, 99,4)
|
-
|
-
|
-
|
-
| |
Réponse PASI 75
|
2 (3,3%)
(0,6, 12,4)
|
43 (71,7%)**
(58,4, 82,2)
|
52 (86,7%)**
(74,9, 93,7)
|
-
|
-
|
-
|
-
| |
Réponse PASI 90
|
0 (0,0%)
(0,0, 7,4)
|
24 (40,0%)
(27,8, 53,5)
|
33 (55,0%)
(41,7, 67,7)
|
-
|
-
|
-
|
-
| |
Réponse PASI 100
|
0 (0,0%)
(0,0, 7,4)
|
10 (16,7%)
(8,7, 29,0)
|
16 (26,7%)
(16,5, 39,9)
|
-
|
-
|
-
|
-
| |
Réponse sur l'échelle
IGA mod 2011 dans les catégories «exempt de» ou «presque exempt de»
|
0 (0,0%)
(0,0, 7,4)
|
32 (53,3%)**
(40,1, 66,1)
|
44 (73,3%)**
(60,1, 83,5)
|
-
|
-
|
-
|
-
| |
* L'échelle IGA mod 2011 est une échelle de 5 catégories avec «0 = exempt de», «1 = presque exempt de», «2 = léger», «3 = modéré» et «4 = sévère» et reflète l'évaluation globale par le médecin de la sévérité du psoriasis en termes de durcissement, d'érythème et de desquamation. Les réponses «exempt de» ou «presque exempt de», considérées comme un succès thérapeutique, sont définies comme l'absence de signes de psoriasis ou une coloration normale à rose des lésions, l'absence d'épaississement des plaques et l'absence de desquamation ou en une desquamation focale minimale.
** Valeurs p versus placebo et ajustée pour la multiplicité: p < 0,0001
|
Tableau 3: Résumé de la réponse clinique dans l'étude 2 portant sur le psoriasis (FIXTURE)
|
|
Semaine 12
|
Semaine 16
|
Semaine 52
| |
|
Placebo
n (%)
(IC à 95%)
|
150 mg
n (%)
(IC à 95%)
|
300 mg
n (%)
(IC à 95%)
|
Étanercept
n (%)
(IC à 95%)
|
150 mg
n (%)
(IC à 95%)
|
300 mg
n (%)
(IC à 95%)
|
Étanercept
n (%)
(IC à 95%)
|
150 mg
n (%)
(IC à 95%)
|
300 mg
n (%)
(IC à 95%)
|
Étanercept
n (%)
(IC à 95%)
| |
Nombre de patients
|
324
|
327
|
323
|
323
|
327
|
323
|
323
|
327
|
323
|
323
| |
Réponse
PASI 50
|
49
(15,1%)
(11,5; 19,6)
|
266
(81,3%)
(76,6; 85,3)
|
296
(91,6%)
(87,9; 94,3)
|
226
(70,0%)
(64,6; 74,9)
|
290
(88,7%)
(84,6; 91,8)
|
302
(93,5%)
(90,1; 95,8)
|
257
(79,6%)
(74,7; 83,7)
|
249
(76,1%)
(71,1; 80,6)
|
274
(84,8%)
(80,3; 88,5)
|
234
(72,4%)
(67,2; 77,2)
| |
Réponse
PASI 75
|
16 (4,9%)
(2,9; 8,1)
|
219 (67,0%)**
(61,5; 72,0)
|
249 (77,1%)**
(72,0; 81,5)
|
142
(44,0%)
(38,5; 49,6)
|
247
(75,5%)
(70,4; 80,0)
|
280
(86,7%)
(82,4; 90,1)
|
189
(58,5%)
(52,9; 63,9)
|
215 (65,7%)**
(60,3; 70,8)
|
254 (78,6%)**
(73,7; 82,9)
|
179
(55,4%)
(49,8; 60,9)
| |
Réponse
PASI 90
|
5
(1,5%)
(0,6; 3,8)
|
137
(41,9%)
(36,5; 47,5)
|
175
(54,2%)
(48,6; 59,7)
|
67
(20,7%)
(16,5; 25,7)
|
176
(53,8%)
(48,3; 59,3)
|
234
(72,4%)
(67,2; 77,2)
|
101
(31,3%)
(26,3; 36,7)
|
147
(45,0%)
(39,5; 50,5)
|
210
(65,0%)
(59,5; 70,2)
|
108
(33,4%)
(28,4; 38,9)
| |
Réponse
PASI 100
|
0
(0%)
(0,0; 1,5)
|
47
(14,4%)
(10,8; 18,8)
|
78
(24,1%)
(19,7; 29,3)
|
14
(4,3%)
(2,5; 7,3)
|
84
(25,7%)
(21,1; 30,8)
|
119
(36,8%)
(31,6; 42,4)
|
24
(7,4%)
(4,9; 11,0)
|
65
(19,9%)
(15,8; 24,7)
|
117
(36,2%)
(31,0; 41,8)
|
32
(9,9%)
(7,0; 13,8)
| |
Réponse sur l'échelle IGA mod 2011 dans les catégories «exempt de» ou «presque exempt de»
|
9
(2,8%)
(1,4; 5,4)
|
167 (51,1%)**
(45,5; 56,6)
|
202 (62,5%)**
(57,0; 67,8)
|
88
(27,2%)
(22,5; 32,5)
|
200
(61,2%)
(55,6; 66,4)
|
244
(75,5%)
(70,4; 80,1)
|
127
(39,3%)
(34,0; 44,9)
|
168 (51,4%)**
(45,8; 56,9)
|
219 (67,8%)**
(62,4; 72,8)
|
120
(37,2%)
(31,9; 42,7)
| |
** Valeurs p ajustées versus étanercept: p = 0,0250
|
Une étude supplémentaire portant sur le psoriasis (CLEAR) a évalué 676 patients. Le sécukinumab 300 mg a atteint les critères d'évaluation principaux et secondaires grâce à sa supériorité sur l'ustékinumab en termes d'ampleur de la réponse PASI 90 à la semaine 16 (critère d'évaluation principal) et de réponse PASI 90 à long terme à la semaine 52. Une efficacité supérieure du sécukinumab par rapport à l'ustékinumab pour les critères d'évaluation réponses PASI 75/90/100 et IGA mod 2011 0 ou 1 («exempt de» ou «presque exempt de») a été rapidement visible et s'est poursuivie jusqu'à la semaine 52. Dans cette étude, chaque dose de 300 mg a été administrée sous la forme de deux injections sous-cutanées de 150 mg.
Tableau 4: Résumé de la réponse clinique dans l'étude CLEAR
|
|
Semaine 16
|
Semaine 52
| |
|
Sécukinumab 300 mg
|
Ustékinumab*
|
Sécukinumab 300 mg
|
Ustékinumab*
| |
Nombre de patients
|
334
|
335
|
334
|
335
| |
Réponse PASI 75 n (%)
|
311 (93,1%)
|
276 (82,4%)
|
306 (91,6%)
|
262 (78,2%)
| |
Réponse PASI 90 n (%)
|
264 (79,0%)**
|
192 (57,3%)
|
250 (74,9%)***
|
203 (60,6%)
| |
Réponse PASI 100 n (%)
|
148 (44,3%)
|
95 (28,4%)
|
150 (44,9%)
|
123 (36,7%)
| |
Réponse sur l'échelle IGA mod 2011 dans les catégories «exempt de» ou «presque exempt de»
|
278 (83,2%)
|
226 (67,5%)
|
261 (78,1%)
|
213 (63,6%)
| |
*Les patients traités par le sécukinumab ont reçu une dose de 300 mg aux semaines 0, 1, 2, 3 et 4, suivie de la même dose chaque mois jusqu'à la semaine 52. Les patients traités par l'ustékinumab ont reçu 45 mg ou 90 mg aux semaines 0 et 4, puis toutes les 12 semaines jusqu'à la semaine 52 (dose déterminée en fonction du poids et selon la posologie approuvée).
** Valeurs p versus ustékinumab: p < 0,0001 pour le critère d'évaluation principal PASI 90 à la semaine 16
*** Valeurs p versus ustékinumab: p = 0,0001 pour le critère d'évaluation secondaire PASI 90 à la semaine 52
|
Cosentyx a été efficace chez les patients non prétraités par un médicament biologique, chez les patients prétraités par un médicament biologique et chez les patients en échec thérapeutique sous un médicament biologique. Les taux de réponse relatifs aux critères d'évaluation principaux, PASI 75 et IGA 0 ou 1, avec Cosentyx 300 mg étaient de 67,7% et 54,1% chez les patients après échec d'un traitement antérieur par inhibiteur du TNF, contre 78,5% et 56,9% chez les patients sans traitement antérieur par inhibiteur du TNF.
Cosentyx, à la dose de 300 mg, a été associé à un début d'action rapide avec une réduction de 50% du PASI moyen à la semaine 3.
Dans toutes les études de phase III portant sur le psoriasis en plaques, environ 15 à 25% des patients inclus présentaient un rhumatisme psoriasique concomitant au début de l'étude. Les améliorations du PASI 75 dans ce groupe de patients ont été comparables à celles observées dans le groupe global de patients atteints de psoriasis en plaques.
Localisation spécifique/formes de psoriasis en plaques
Dans une étude supplémentaire contrôlée contre placebo, une amélioration a été constatée dans le psoriasis unguéal (TRANSFIGURE, 198 patients). Dans l'étude TRANSFIGURE, le sécukinumab a montré un effet supérieur statistiquement significatif par rapport au placebo à la semaine 16 (46,1% pour 300 mg, 38,4% pour 150 mg vs 11,7% pour le placebo) en termes d'amélioration de l'indice de sévérité du psoriasis unguéal (réponse NAPSI, Nail Psoriasis Severity Index) chez des patients atteints de psoriasis en plaques modéré à sévère avec atteinte des ongles.
Dans d'autres études cliniques, des améliorations ont également été observées dans le psoriasis unguéal, l'atteinte du cuir chevelu et l'atteinte palmo-plantaire.
Dans ces études, chaque dose de 300 mg a été administrée sous la forme de deux injections sous-cutanées de 150 mg.
Qualité de vie / résultats rapportés par les patients
Pour le DLQI (Dermatology Life Quality Index), des améliorations statistiquement significatives ont été montrées à la semaine 12 (études 1 à 4) par rapport à la valeur initiale; les améliorations se sont maintenues pendant 52 semaines (études 1 et 2).
Des améliorations statistiquement significatives des signes et symptômes rapportés par les patients - démangeaisons, douleurs et desquamation - à la semaine 12 par rapport à la valeur initiale, en comparaison avec le placebo (études 1 et 2), ont été montrées à l'aide du Psoriasis Symptom Diary© validé.
Dans le DLQI, des améliorations statistiquement significatives par rapport à la valeur initiale ont été observées à la semaine 4 chez les patients traités par le sécukinumab par rapport à ceux traités par l'ustékinumab (CLEAR) et ces améliorations se sont maintenues jusqu'à 52 semaines.
Dans le Psoriasis Symptom Diary, des améliorations statistiquement significatives des signes et symptômes rapportés par les patients - démangeaisons, douleurs et desquamation - ont été observées à la semaine 16 et à la semaine 52 (CLEAR) chez les patients traités par le sécukinumab par rapport à ceux traités par l'ustékinumab.
Patients pédiatriques
Psoriasis en plaques sévère
Une étude de phase III de 52 semaines, randomisée, en double aveugle, contrôlée contre placebo et étanercept a été menée chez 162 patients pédiatriques âgés de 6 à < 18 ans atteints de psoriasis en plaques sévère (défini par un score PASI ≥20, un score IGA mod 2011 de 4 et une surface corporelle affectée ≥10%), pour lesquels un traitement systémique était envisageable. Environ 43% des patients avaient été traités auparavant par photothérapie, 53% par des thérapies systémiques conventionnelles et 3% par des médicaments biologiques. 9% des patients avaient un rhumatisme psoriasique concomitant.
Les patients ont été randomisés pour recevoir l'un des quatre traitements suivants:
·Faible dose de sécukinumab (75 mg pour un poids corporel < 50 kg ou 150 mg pour un poids corporel ≥50 kg) aux semaines 0, 1, 2, 3 et 4, suivie de la même dose toutes les 4 semaines
·Dose élevée de sécukinumab (75 mg pour un poids corporel < 25 kg, 150 mg pour un poids corporel ≥25 kg et < 50 kg ou 300 mg pour un poids corporel ≥50 kg) aux semaines 0, 1, 2, 3 et 4, suivie de la même dose toutes les 4 semaines
·Placebo aux semaines 0, 1, 2, 3 et 4, suivi de la même dose toutes les 4 semaines
·Etanercept (0,8 mg/kg) chaque semaine (jusqu'à un maximum de 50 mg)
Les patients randomisés pour recevoir un placebo et qui n'avaient pas répondu à la semaine 12 ont été placés dans le groupe sécukinumab à faible dose ou à dose élevée (dose basée sur le groupe de poids corporel) et ont reçu le médicament à l'étude aux semaines 12, 13, 14 et 15, puis la même dose toutes les 4 semaines, en commençant à la semaine 16.
Les co-critères d'évaluation principaux étaient la proportion de patients ayant obtenu une amélioration d'au moins 75% du score PASI (réponse PASI 75) et un score IGA mod 2011 «sans symptômes» ou «presque sans symptômes» (0 ou 1) avec une amélioration d'au moins 2 points entre l'inclusion et la semaine 12.
Au cours de la période de 12 semaines contrôlée contre placebo, l'efficacité du sécukinumab à faible dose et à dose élevée était comparable en ce qui concerne les co-critères d'évaluation principaux. Les estimations du rapport des risques en faveur des deux doses de sécukinumab étaient cliniquement pertinentes et statistiquement significatives, tant pour la réponse PASI 75 que pour la réponse IGA mod 2011 de «sans symptôme» ou «presque sans symptôme» (0 ou 1).
Tous les patients ont été suivis pendant 52 semaines après la première dose en ce qui concerne l'efficacité et la sécurité. La proportion de patients ayant obtenu une réponse PASI 75 et une réponse IGA mod 2011 «sans symptôme» ou «presque sans symptôme» (0 ou 1) montrait déjà une différence entre les groupes de traitement par le sécukinumab et le placebo à la semaine 4, la différence s'accentuant à la semaine 12. La réponse s'est maintenue tout au long de la période de 52 semaines. Les scores PASI 50, PASI 90 et PASI 100 et la proportion de patients ayant un score au Children's Dermatology Life Quality Index (CDLQI) de 0 ou 1 se sont également améliorés et ont été maintenus tout au long de la période de 52 semaines.
Après la semaine 12, l'efficacité des doses faibles et élevées de sécukinumab était comparable, bien que l'efficacité de la dose élevée ait été supérieure chez les patients dont le poids corporel était ≥50 kg. Les profils de sécurité des faibles doses et des doses élevées étaient comparables.
Les résultats concernant l'efficacité à la semaine 12 sont présentés dans le tableau 5.
Tableau 5: Résumé de la réponse clinique chez les enfants et adolescents atteints de psoriasis sévère à la semaine 12*
|
Critère de réponse
|
Comparaison des traitements
|
«Test»
|
«Témoin»
|
Rapport des risques
|
| |
«Test» vs «Témoin»
|
n/m** (%)
|
n/m** (%)
|
Évaluateur (IC à 95%)
|
valeur p
| |
à la semaine 12***
| |
PASI 75
|
Sécukinumab à faible dose vs placebo
|
32/40 (80,0)
|
6/41 (14,6)
|
25,78 (7,08; 114,66)
|
< 0,0001
| |
|
Sécukinumab à dose élevée vs placebo
|
31/40 (77,5)
|
6/41 (14,6)
|
22,65 (6,31; 98,93)
|
< 0,0001
| |
IGA 0/1
|
Sécukinumab à faible dose vs placebo
|
28/40 (70,0)
|
2/41 (4,9)
|
51,77 (10,02; 538,64)
|
< 0,0001
| |
|
Sécukinumab à dose élevée vs placebo
|
24/40 (60,0)
|
2/41 (4,9)
|
32,52 (6,48; 329,52)
|
< 0,0001
| |
PASI 90
|
Sécukinumab à faible dose vs placebo
|
29/40 (72,5)
|
1/41 (2,4)
|
133,67 (16,83; 6395,22)
|
< 0,0001
| |
|
Sécukinumab à dose élevée vs placebo
|
27/40 (67,5)
|
1/41 (2,4)
|
102,86 (13,22; 4850,13)
|
< 0,0001
| |
* En cas de données manquantes, une imputation en tant que non répondeur a été effectuée
** n = nombre de répondeurs, m = nombre de patients évaluables
*** fenêtre de temps prolongée pour les visites à la semaine 12
Le rapport des risques, l'intervalle de confiance à 95% et la valeur p proviennent d'un modèle de régression exact utilisant le groupe de traitement, la catégorie de poids corporel à l'inclusion et la catégorie d'âge comme facteurs.
|
Une proportion plus élevée de patients pédiatriques traités par le sécukinumab a signalé une amélioration de la qualité de vie liée à la santé, mesurée par un score CDLQI de 0 ou 1, par rapport au placebo à la semaine 12.
Psoriasis en plaques modéré à sévère
Une étude de phase III multicentrique, en ouvert, à deux bras et en parallèle a été menée chez 84 patients pédiatriques âgés de 6 à < 18 ans atteints de psoriasis en plaques modéré à sévère (défini par un score PASI ≥12, un score IGA mod 2011 ≥3 et une surface corporelle affectée ≥10%) et pour lesquels un traitement systémique était envisageable.
Les patients ont été randomisés pour recevoir un traitement par le sécukinumab aux semaines 0, 1, 2, 3 et 4, suivi de la même dose toutes les 4 semaines, de la manière suivante:
·Faible dose de sécukinumab (75 mg pour un poids corporel < 50 kg ou 150 mg pour un poids corporel ≥50 kg),
·Dose élevée de sécukinumab (75 mg pour un poids corporel < 25 kg, 150 mg pour un poids corporel ≥25 kg et < 50 kg ou 300 mg pour un poids corporel ≥50 kg).
Les co-critères d'évaluation principaux étaient la proportion de patients ayant obtenu une amélioration d'au moins 75% du score PASI (réponse PASI 75) ou un score IGA mod 2011 «sans symptômes» ou «presque sans symptômes» (0 ou 1) avec une amélioration d'au moins 2 points entre l'inclusion et la semaine 12.
L'efficacité du sécukinumab, tant à faible dose qu'à dose élevée, a été comparable et a montré une amélioration cliniquement pertinente pour les co-critères d'évaluation principaux, en comparaison historique par rapport au placebo.
L'efficacité a été évaluée, pour tous les patients, pendant au moins 24 semaines après la première administration. L'efficacité (définie par la réponse PASI 75 et la réponse IGA mod 2011 de «sans symptôme» ou «presque sans symptôme» [0 ou 1]) a été observée dès la semaine 2. La proportion de patients ayant obtenu une réponse PASI 75 et une réponse IGA mod 2011 «sans symptôme» ou «presque sans symptôme» (0 ou 1) a augmenté tout au long de la période de 24 semaines. Des améliorations du PASI 90 et du PASI 100 ont également été observées à la semaine 12 et ont augmenté tout au long de la période de 24 semaines.
Après la semaine 12, l'efficacité des doses faibles et élevées de sécukinumab était comparable. Les profils de sécurité des faibles doses et des doses élevées étaient également comparables.
Les résultats concernant l'efficacité aux semaines 12 et 24 sont présentés dans le tableau 6.
Tableau 6: Résumé de la réponse clinique en cas de psoriasis pédiatrique modéré à sévère aux semaines 12* et 24*
|
|
Semaine 12
|
Semaine 24
| |
|
Sécukinumab
faible dose
|
Sécukinumab
dose élevée
|
Sécukinumab
faible dose
|
Sécukinumab
dose élevée
| |
Nombre de patients
|
42
|
42
|
42
|
42
| |
Réponse PASI 75 n (%)
|
39 (92,9%)
|
39 (92,9%)
|
40 (95,2%)
|
40 (95,2%)
| |
Réponse IGA mod 2011 «sans symptôme» ou «presque sans symptôme» n (%)
|
33 (78,6%)
|
35 (83,3%)
|
37 (88,1%)
|
39 (92,9%)
| |
Réponse PASI 90 n (%)
|
29 (69,0%)
|
32 (76,2%)
|
37 (88,1%)
|
37 (88,1%)
| |
Réponse PASI 100 n (%)
|
25 (59,5%)
|
23 (54,8%)
|
28 (66,7%)
|
28 (66,7%)
| |
* En cas de données manquantes, une imputation en tant que non répondeur a été effectuée.
|
Flexibilité posologique en cas de psoriasis en plaques
Dans une étude multicentrique, randomisée et en double aveugle menée chez 331 patients, l'efficacité, la sécurité et la tolérance de Cosentyx 300 mg, utilisé toutes les 4 semaines, ont été comparées à celles de Cosentyx 300 mg, utilisé toutes les 2 semaines, chez des patients adultes présentant un poids corporel ≥90 kg et un psoriasis en plaques modéré à sévère. Les patients ont été randomisés selon un rapport 1:1 comme suit:
·Sécukinumab 300 mg aux semaines 0, 1, 2, 3 et 4, suivi de la même dose toutes les 2 semaines jusqu'à la semaine 52 (n = 165).
·Sécukinumab 300 mg aux semaines 0, 1, 2, 3 et 4, suivi de la même dose toutes les 4 semaines jusqu'à la semaine 16 (n = 166).
·Les patients qui avaient été randomisés dans le groupe sécukinumab 300 mg toutes les 4 semaines et avaient atteints une réponse PASI 90 à la semaine 16 ont continué à recevoir le même schéma posologique jusqu'à la semaine 52. Les patients qui avaient été randomisés dans le groupe Cosentyx 300 mg toutes les 4 semaines et qui n'avaient pas atteint une réponse PASI 90 à la semaine 16 ont continué à recevoir le même schéma posologique ou sont passés à Cosentyx 300 mg toutes les 2 semaines pour la période allant jusqu'à la semaine 52.
Les critères d'évaluation principaux et les principaux critères d'évaluation secondaires étaient la proportion de patients qui avaient obtenu, à la semaine 16, une réponse PASI 90 ainsi qu'une réponse allant de «sans symptôme» à «presque sans symptôme» (0 ou 1) dans les catégories IGA mod 2011.
À la semaine 16, la proportion de patients qui avaient obtenu une réponse PASI 90 était plus élevée dans le groupe recevant le traitement toutes les 2 semaines (Q2W) que dans le groupe recevant le traitement toutes les 4 semaines (Q4W) (73,2% et 55,5%, respectivement). La différence entre les traitements était statistiquement significative (valeur p unilatérale = 0,0003).
Le groupe de patients qui n'avaient pas obtenu de réponse PASI 90 et qui étaient passés au schéma de traitement toutes les 2 semaines à la semaine 16 présentaient, à la semaine 32, une réponse PASI 90 plus élevée que dans le groupe de patients qui étaient restés au schéma de traitement toutes les 4 semaines (38,7% et 16,5%, respectivement). La différence entre les traitements était cliniquement pertinente, mais uniquement exploratoire.
Les profils de sécurité des deux schémas posologiques, Cosentyx 300 mg toutes les 4 semaines et Cosentyx 300 mg toutes les 2 semaines, étaient comparables chez les patients présentant un poids corporel ≥90 kg et correspondaient au profil de sécurité rapporté pour les patients atteints de psoriasis.
Rhumatisme psoriasique
Chez les patients adultes atteints de rhumatisme psoriasique actif, il a été démontré que Cosentyx améliore les signes et les symptômes, la fonction physique et la qualité de vie liée à la santé, et réduit en outre le taux de progression des lésions articulaires périphériques.
La sécurité et l'efficacité de Cosentyx ont été démontrées dans trois études de phase III randomisées, en double aveugle et contrôlées contre placebo menées chez 1999 patients qui présentaient un rhumatisme psoriasique actif (≥3 articulations gonflées et ≥3 articulations sensibles à la pression) malgré un traitement par anti-inflammatoires non stéroïdiens (AINS), corticostéroïdes ou médicaments antirhumatismaux modificateurs de la maladie (DMARD). Le diagnostic de RP des patients de ces études avait été posé depuis au moins cinq ans. La majorité des patients présentaient également une lésion de la peau due à un psoriasis actif ou des antécédents documentés de psoriasis. Afin d'obtenir une évaluation non biaisée de l'efficacité de Cosentyx dans le traitement du psoriasis, l'utilisation concomitante d'un traitement topique par corticostéroïdes ou d'une thérapie à base d'UV n'était pas autorisée pendant les études. Plus de 61% et 42% des patients atteints de RP présentaient respectivement une enthésite et une dactylite à l'inclusion. Le nombre de patients atteints de RP avec une atteinte axiale était trop faible pour permettre une évaluation significative.
L'efficacité et la sécurité de Cosentyx aux doses de 75 mg, 150 mg et 300 mg ont été évaluées par rapport à un placebo avec une dose initiale soit par voie intraveineuse soit par voie sous-cutanée. Dans l'étude 1 portant sur le rhumatisme psoriasique (étude PsA1) ou l'étude 2 portant sur le rhumatisme psoriasique (étude PsA2) et l'étude 3 portant sur le rhumatisme psoriasique (étude PsA3), respectivement 29%, 35% et 30% des patients avaient été précédemment traités par des médicaments anti-TNF-alpha, ce traitement ayant été interrompu soit en raison d'une absence de réponse, soit en raison d'une intolérance (patients anti-TNF-alpha-IR).
Dans l'étude PsA1 (FUTURE 1), 606 patients ont été évalués; 60,7% d'entre eux recevaient du MTX de manière concomitante. Des patients présentant tous les sous-groupes de RP ont été inclus, notamment l'arthrite polyarticulaire sans détection de nodules rhumatoïdes (76,7%), la spondylarthrite périphérique (18,5%), l'arthrite périphérique asymétrique (60,2%), l'atteinte interphalangienne distale (59,6%) et l'arthrite mutilante (7,9%). Les patients randomisés pour recevoir Cosentyx ont reçu 10 mg/kg par voie IV aux semaines 0, 2 et 4, suivis d'une dose mensuelle de 75 mg ou 150 mg SC, à partir de la semaine 8. Les patients randomisés pour recevoir un placebo et qui n'ont pas répondu au traitement sont ensuite passés au traitement par 75 mg ou 150 mg de Cosentyx SC une fois par mois à la semaine 16. Au cours de la semaine 24, les patients encore sous placebo ont été transférés vers le traitement par Cosentyx 75 mg ou 150 mg SC. Le critère d'évaluation principal était la réponse selon l'American College of Rheumatology (ACR) 20 à la semaine 24.
L'étude PsA2 (FUTURE 2) a évalué 397 patients, dont 46,6% étaient traités de manière concomitante par MTX. Des patients présentant tous les sous-groupes de RP ont été inclus, notamment l'arthrite polyarticulaire sans détection de nodules rhumatoïdes (85,9%), la spondylarthrite périphérique (21,7%), l'arthrite périphérique asymétrique (64,0%), l'atteinte interphalangienne distale (57,9%) et l'arthrite mutilante (6,3%). Les patients randomisés pour recevoir Cosentyx ont reçu 75 mg, 150 mg ou 300 mg SC aux semaines 0, 1, 2, 3 et 4, puis la même dose chaque mois. Les patients randomisés pour recevoir un placebo et qui n'avaient pas répondu au traitement à la semaine 16 sont ensuite passés à Cosentyx 150 mg ou 300 mg SC une fois par mois à la semaine 16. Au cours de la semaine 24, les patients encore sous placebo ont été transférés vers le traitement par Cosentyx 150 mg ou 300 mg SC. Le critère d'évaluation principal était la réponse ACR 20 à la semaine 24.
L'étude PsA3 (FUTURE 5) a évalué 996 patients, dont 50,1% étaient traités de manière concomitante par MTX. Des patients présentant tous les sous-groupes de RP ont été inclus, notamment l'arthrite polyarticulaire sans détection de nodules rhumatoïdes (78,7%), la spondylarthrite périphérique (19,8%), l'arthrite périphérique asymétrique (65,0%), l'atteinte interphalangienne distale (56,7%) et l'arthrite mutilante (6,8%). Les patients ont été randomisés dans les groupes suivants: Cosentyx 150 mg, Cosentyx 300 mg ou placebo, respectivement par voie SC aux semaines 0, 1, 2, 3 et 4, puis la même dose chaque mois, ou Cosentyx 150 mg une fois par mois sans dose de charge initiale. Les patients initialement randomisés pour recevoir un placebo et n'ayant pas répondu au traitement à la semaine 16 sont ensuite passés à un traitement par Cosentyx (150 mg ou 300 mg SC) une fois par mois à la semaine 16. Au cours de la semaine 24, les patients encore sous placebo ont été transférés vers le traitement par Cosentyx (150 mg ou 300 mg) une fois par mois. Le critère d'évaluation principal était la réponse ACR 20 à la semaine 16 et le principal critère d'évaluation secondaire était la différence du score total de Sharp modifié (mTSS) à la semaine 24 par rapport à l'inclusion.
L'étude PsA4 (FUTURE 3) a évalué 414 patients, dont 47,6% étaient traités de manière concomitante par MTX. Les patients randomisés dans le groupe Cosentyx ont reçu 150 mg ou 300 mg SC aux semaines 0, 1, 2, 3 et 4, puis la même dose chaque mois. Les patients randomisés pour recevoir un placebo et qui n'avaient pas répondu au traitement à la semaine 16 sont ensuite passés à Cosentyx 150 mg ou 300 mg SC une fois par mois à la semaine 16. Au cours de la semaine 24, les patients encore sous placebo ont été transférés vers le traitement par Cosentyx 150 mg ou 300 mg SC. Le critère d'évaluation principal était la réponse ACR 20 à la semaine 24.
Réponse clinique
Signes et symptômes
Le traitement par Cosentyx a entraîné une amélioration significative de l'ampleur de l'activité de la maladie aux semaines 16, 24 et 52 par rapport au placebo. Ces mesures comprenaient la réponse des symptômes articulaires en termes d'ACR 20, ACR 50, ACR 70, la réponse des symptômes cutanés (Psoriasis Area and Severity Index, PASI) 75, PASI 90, ainsi que d'autres scores d'activité de la maladie et d'état de santé (Disease Activity Score, DAS28-CRP, Short Form Health Survey - Physical Component Summary; SF-36 PCS, Health Assessment Questionnaire - Disability Index, HAQ-DI) (voir tableau 7).
Tableau 7: Réponse clinique dans les études PsA2 et PsA3 aux semaines 16, 24 et 52
|
|
PsA2
|
PsA3
| |
Placebo
|
150 mg1
|
300 mg1
|
Placebo
|
150 mg1
|
300 mg1
| |
Nombre de patients randomisés
|
98
|
100
|
100
|
332
|
220
|
222
| |
Réponse ACR 20 n (%)
| |
Semaine 16
|
18
(18,4%)
|
60
(60,0%***)
|
57
(57,0%***)
|
91◊
(27,4%)
|
122◊
(55,5%***)
|
139◊
(62,6%***)
| |
Semaine 24
|
15◊
(15,3%)
|
51◊
(51,0%***)
|
54◊
(54,0%***)
|
78
(23,5%)
|
117
(53,2%***)
|
141
(63,5%***)
| |
Semaine 52
|
-
|
64
(64,0%)
|
64
(64,0%)
|
NA
|
NA
|
NA
| |
Réponse ACR 50 n (%)
| |
Semaine 16
|
6
(6,1%)
|
37
(37,0%***)
|
35
(35,0%***)
|
27
(8,1%)
|
79
(35,9%***)
|
88
(39,6%***)
| |
Semaine 52
|
-
|
39
(39,0%)
|
44
(44,0%)
|
NA
|
NA
|
NA
| |
Réponse ACR 70 n (%)
| |
Semaine 16
|
2
(2,0%)
|
17
(17,0%**)
|
15
(15,0%**)
|
14
(4,2%)
|
40
(18,2%***)
|
45
(20,3%***)
| |
Semaine 52
|
-
|
20
(20,0%)
|
24
(24,0%)
|
NA
|
NA
|
NA
| |
DAS28-CRP
| |
Semaine 16
|
-0,50
|
-1,45***
|
-1,51***
|
-0,63
|
-1,29***
|
-1,49***
| |
Semaine 52
|
-
|
-1,69
|
-1,78
|
NA
|
NA
|
NA
| |
Réponse PASI 75 n (%)
| |
Semaine 16
|
3
(7,0%)
|
33
(56,9%***)
|
27
(65,9%***)
|
20
(12,3%)
|
75
(60,0%***)
|
77
(70,0%***)
| |
Semaine 52
|
-
|
33
(56,9%)
|
30
(73,2%)
|
-
|
-
|
-
| |
Réponse PASI 90 n (%)
| |
Semaine 16
|
3
(7,0%)
|
22
(37,9%***)
|
18
(43,9%***)
|
15
(9,3%)
|
46
(36,8%***)
|
59
(53,6%***)
| |
Semaine 52
|
-
|
25
(43,1%)
|
23
(56,1%)
|
-
|
-
|
-
| |
Disparition de la dactylite n (%)†
| |
Semaine 16
|
10
(37%)
|
21
(65,6%*)
|
26
(56,5%)
|
40
(32,3%)
|
46
(57,5%***)
|
54
(65,9%***)
| |
Semaine 52
|
-
|
21
(65,6%)
|
32
(69,6%)
|
NA
|
NA
|
NA
| |
Disparition de l'enthésite n (%)‡
| |
Semaine 16
|
17
(26,2%)
|
32
(50,0%**)
|
32
(57,1%***)
|
68
(35,4%)
|
77
(54,6%***)
|
78
(55,7%***)
| |
Semaine 52
|
-
|
31
(48,4%)
|
30
(53,6%)
|
NA
|
NA
|
NA
| |
* p < 0,05, ** p < 0,01, *** p < 0,001; par rapport au placebo
Toutes les valeurs p sont présentées sans correction pour les tests multiples.
Les patients dont les critères d'évaluation binaires étaient manquants ont été enregistrés comme non répondeurs («non-responder imputation»).
NA: non disponible (Not Available); ACR: American College of Rheumatology; PASI: Psoriasis Area and Severity Index; DAS: score d'activité de la maladie (Disease Activity Score); BSA: surface corporelle (Body Surface Area, BSA)
◊ Critère d'évaluation principal
1 Cosentyx 150 mg ou 300 mg SC aux semaines 0, 1, 2, 3 et 4, puis la même dose chaque mois.
† Pour les patients présentant une dactylite à l'inclusion (n = 27, 32 et 46 respectivement dans PsA2 et n = 124, 80 et 82 respectivement dans PsA3), la disparition complète de la dactylite a été évaluée dans le sous-groupe de patients présentant une dactylite à l'inclusion et elle est exprimée en pourcentage de patients avec une valeur de l'indice de dactylite de Leeds (LDI) égale à zéro.
‡ Pour les patients présentant une enthésite à l
'inclusion (n = 65, 64 et 56 respectivement dans PsA2 et n = 192, 141 et 140 respectivement dans PsA3), la disparition complète de l'enthésite a été évaluée dans le sous-groupe de patients présentant une enthésite à l'inclusion et elle est exprimée en pourcentage de patients avec une valeur de l'indice d'enthésite de Leeds (LEI) égale à zéro.
|
L'effet de Cosentyx est apparu à la semaine 2. Une différence statistiquement significative pour l'ACR 20 par rapport au placebo a été obtenue à la semaine 3. Dans l'étude PsA2, l'efficacité a été maintenue jusqu'à la semaine 104 (64,4% et 69,4% pour 150 mg et 300 mg, respectivement).
À la semaine 16, les patients traités par Cosentyx ont présenté des améliorations significatives des signes et symptômes, notamment une réponse significativement plus élevée en termes d'ACR 20 (60,0% et 57,0% pour 150 mg et 300 mg, respectivement) par rapport au placebo (18,4%).
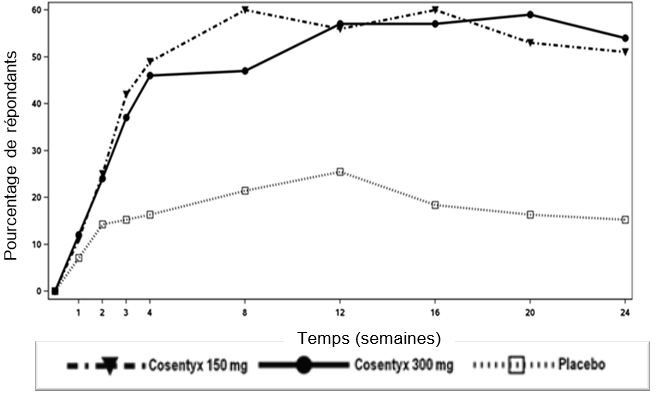
Le pourcentage de patients ayant présenté une réponse ACR 20 par visite est présenté à la figure 1
Figure 1: Réponse ACR 20 dans l'étude PsA2 au cours du temps jusqu'à la semaine 24
Pour les critères d’évaluation principaux et les critères d’évaluation secondaires importants, une réponse similaire a été observée chez les patients atteints de RP, qu’ils reçoivent ou non du MTX en même temps.
Les patients non traités antérieurement par un anti-TNF-alpha, mais également les patients anti-TNFalpha-IR qui ont reçu Cosentyx ont présenté une réponse ACR 20 significativement plus élevée aux semaines 16 et 24 par rapport au placebo, la réponse étant numériquement plus élevée dans le groupe non antérieurement traité par anti-TNF-alpha (non traité par anti-TNF-alpha dans PsA2: 64% et 58% pour 150 mg et 300 mg, respectivement, par rapport à 15,9% pour le placebo; anti-TNFalpha-IR: 30% et 46% pour 150 mg et 300 mg, respectivement, par rapport à 14,3% pour le placebo).
Les patients anti-TNF-alpha-IR qui ont été traités par une dose de 300 mg ont présenté un taux de réponse plus élevé selon l'ACR20 (p < 0,05) par rapport aux patients sous placebo et ont montré un bénéfice cliniquement significatif par rapport à la dose de 150 mg pour l'ACR 50, le PASI 75, le PASI 90, l'HAQ-DI, la dactylite et l'enthésite.
Dans PsA2, la proportion de patients ayant obtenu une réponse modifiée selon les critères de réponse au RP (PsARC) était plus élevée à la semaine 24 dans le groupe de patients traités par Cosentyx (59,0% et 61,0% pour 150 mg et 300 mg, respectivement) que dans le groupe de patients traités par placebo (26,5%).
Les résultats des composantes des critères ACR de réponse sont présentés dans le tableau 8.
Tableau 8: Différence des moyennes des composantes de l'ACR par rapport à l'inclusion dans l'étude PsA2 à la semaine 24
|
|
Placebo
(N = 98)
|
150 mg
(N = 100)
|
300 mg
(N = 100)
| |
Nombre d'articulations gonflées
| |
Inclusion
|
12,1
|
11,9
|
11,2
| |
Différence
|
-5,14
|
-6,32
|
-7,28*
| |
Nombre d'articulations sensibles à la pression
| |
Inclusion
|
23,4
|
24,1
|
20,2
| |
Différence
|
-4,28
|
-11,42***
|
-10,84**
| |
Évaluation de la douleur par le patient
| |
Inclusion
|
55,4
|
58,9
|
57,7
| |
Différence
|
-11,71
|
-23,39**
|
-22,35**
| |
Évaluation globale par le patient
| |
Inclusion
|
57,6
|
62,0
|
60,7
| |
Différence
|
-10,14
|
-25,78***
|
-26,70***
| |
Évaluation globale par le médecin
| |
Inclusion
|
55,0
|
56,7
|
55,0
| |
Différence
|
-25,23
|
-32,97*
|
-38,52***
| |
Indice de handicap (HAQ)
| |
Inclusion
|
1,1684
|
1,2200
|
1,2828
| |
Différence
|
-0,31
|
-0,48*
|
-0,56**
| |
CRP (mg/dl)
| |
Inclusion
|
7,71
|
14,15
|
10,69
| |
hsCRP, (rapport post-inclusion/inclusion)
|
0,75
|
0,55*
|
0,55*
| |
* p < 0,05, ** p < 0,01, *** p < 0,001 basé sur une valeur p nominale mais non ajustée
|
Dans l'étude PsA1, les patients traités par Cosentyx ont présenté des signes et symptômes de RP significativement améliorés à la semaine 24, avec une réponse similaire à celle de l'étude PsA2. L'efficacité a été maintenue jusqu'à la semaine 104.
Réponse radiographique
Dans l'étude PsA3, les atteintes structurales ont été évaluées par radiographie et exprimées sous la forme du score total de Sharp modifié (mTSS) et de ses composantes, le score d'érosion (ES) et le score de pincement articulaire (JSN). Des radiographies des mains, des poignets et des pieds ont été réalisées à l'inclusion, à la semaine 16 et/ou à la semaine 24, et évaluées indépendamment par au moins deux évaluateurs, qui étaient en situation d'aveugle quant au groupe de traitement et au numéro de la visite.
Le traitement par Cosentyx 150 mg ou 300 mg a permis de réduire significativement, par rapport au traitement par placebo, le taux de progression des lésions articulaires périphériques, évalué par la variation du mTSS à la semaine 24 par rapport à l'inclusion (tableau 8).
Le pourcentage de patients sans progression de la maladie (définie par une modification du mTSS ≤0,5) entre la randomisation et la semaine 24 était respectivement de 79,8%, 88,0% et 73,6% pour Cosentyx 150 mg, 300 mg et le placebo. Une inhibition des atteintes structurales a été constatée indépendamment de l'utilisation concomitante éventuelle de MTX ou du statut TNF.
Le traitement par Cosentyx 150 mg a permis de réduire significativement le taux de progression des lésions articulaires périphériques jusqu'à la semaine 24 par rapport au traitement par placebo, d'après la variation du mTSS par rapport à l'inclusion (voir tableau 9). L'inhibition des atteintes structurales s'est maintenue sous traitement par Cosentyx jusqu'à la semaine 52.
Tableau 9: Modification du score total de Sharp modifié dans les études PsA3 et PsA1
|
|
PsA3
| |
|
Placebo
n = 296
|
150 mg1
n = 213
|
300 mg1
n = 217
| |
Score total
| |
Inclusion
|
15,0
|
13,6
|
12,9
| |
(ET)
|
(38,2)
|
(25,9)
|
(23,7)
| |
Modification moyenne à la semaine 24
|
0,5
|
0,17*
|
0,08*
| |
* p < 0,05, sur la base de la valeur p nominale non corrigée pour les tests multiples.
1Cosentyx 150 mg ou 300 mg SC aux semaines 0, 1, 2, 3 et 4, puis la même dose chaque mois.
|
Manifestations axiales dans le RP
Une étude randomisée, en double aveugle et contrôlée par placebo (MAXIMISE) a évalué l'efficacité du sécukinumab chez 485 patients atteints de RP avec des manifestations axiales, qui n'avaient pas été prétraités par des médicaments biologiques et qui ne répondaient pas suffisamment aux anti-inflammatoires non stéroïdiens (AINS). La variable principale, une amélioration d'au moins 20% des critères ASAS 20 (Assessment of SpondyloArthritis International Society, ASAS) à la semaine 12 a été atteinte (voir tableau 10).
Tableau 10: Réponse clinique dans l'étude MAXIMISE à la semaine 12
|
|
Placebo
(n = 164)
|
150 mg
(n = 157)
|
300 mg
(n = 164)
| |
Réponse ASAS 20, % (IC à 95%)
|
31,2 (24,6; 38,7)
|
66,3 (58,4; 73,3)*
|
62,9 (55,2; 70,0)*
| |
* p < 0,0001; par rapport au placebo en utilisant l'imputation multiple.
ASAS: critères de l'Assessment of SpondyloArthritis International Society.
|
Une amélioration de la réponse ASAS 20 pour les deux doses de sécukinumab a été observée à la semaine 4 et s'est maintenue jusqu'à 52 semaines.
Fonction physique et qualité de vie liée à la santé
Dans l'étude PsA2 et l'étude PsA3, les patients traités par Cosentyx 150 mg et 300 mg ont présenté une amélioration de la fonction physique à la semaine 24 et à la semaine 16, respectivement, par rapport aux patients traités par placebo, mesurée par le Health Assessment Questionnaire - Disability Index (HAQ-DI). Les améliorations des résultats aux questionnaires HAQ-DI ont été observées indépendamment de toute exposition antérieure aux anti-TNF-alpha. Des résultats comparables ont été observés dans l'étude PsA1.
Les patients traités par Cosentyx ont rapporté des améliorations visibles de la qualité de vie liée à la santé mesurée par le Short Form (36) Health Survey - Physical Component Summary (SF-36 PCS) (p < 0,001). Il y a également eu des améliorations statistiquement significatives du score FACIT-F (Functional Assessment of Chronic Illness Therapy - Fatigue) pour 150 mg et 300 mg par rapport au placebo à la semaine 24 (p < 0,01), et ces améliorations ont été maintenues dans l'étude PsA2 jusqu'à la semaine 104.
Escalade de dose à 300 mg chez les patients dont la réponse à 150 mg n'est pas satisfaisante.
Les études PsA1, PsA2 et PsA4 ont fourni des données évaluables concernant l'escalade de dose de 150 mg à 300 mg chez 159 patients dont la réponse était non satisfaisante après 92 à 156 semaines. Cela correspond à 21,1% des patients restant dans les études. Après l'escalade de la dose de sécukinumab de 150 mg à 300 mg, une amélioration des taux de réponse ACR et PASI et une réduction de la proportion de patients non répondeurs (< ACR 20; < PASI 75) ont été observées.
Spondylarthrite axiale (axSpA)
Spondylarthrite ankylosante (maladie de Bechterew)
La sécurité et l'efficacité de Cosentyx ont été démontrées dans deux études de phase III randomisées, en double aveugle et contrôlées contre placebo, menées chez 590 patients qui présentaient une spondylarthrite ankylosante (SA) active avec une activité de la maladie définie au moyen de l'indice Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 malgré un traitement par anti-inflammatoires non stéroïdiens (AINS), corticostéroïdes ou médicaments antirhumatismaux modificateurs de la maladie (DMARD). Le diagnostic de SA avait été posé en moyenne depuis 2,7 à 5,8 ans chez les patients de ces études. Les patients présentant une ankylose complète étaient exclus.
Pour les deux études, le critère d'évaluation principal était une amélioration d'au moins 20% selon les critères de l'Assessment of SpondyloArthritis International Society (réponse ASAS 20) à la semaine 16.
Dans l'étude 1 portant sur la spondylarthrite ankylosante (étude AS1) ou l'étude 2 portant sur la spondylarthrite ankylosante (étude AS2), respectivement 27,0% et 38,8% des patients avaient été précédemment traités par des médicaments anti-TNF-alpha, ce traitement ayant été interrompu soit en raison d'une absence de réponse, soit en raison d'une intolérance (patients anti-TNF-alpha-IR).
L'étude AS1 (MEASURE 1) a évalué 371 patients; 14,8%, 33,4% et 13,5% d'entre eux ont reçu respectivement du MTX, de la sulfasalazine et des corticostéroïdes de manière concomitante. Les patients randomisés dans le groupe Cosentyx ont reçu 10 mg/kg par voie IV aux semaines 0, 2 et 4, suivis de 75 mg ou 150 mg SC une fois par mois. Les patients randomisés pour recevoir un placebo et qui n'avaient pas répondu au traitement à la semaine 16, ainsi que tous les patients encore sous placebo à la semaine 24, sont passés au traitement par Cosentyx 75 mg ou 150 mg SC une fois par mois.
L'étude AS2 (MEASURE 2) a évalué 219 patients, dont 11,9%, 14,2% et 8,2% étaient respectivement traités de manière concomitante par MTX, sulfasalazine et corticostéroïdes. Les patients randomisés dans le groupe Cosentyx ont reçu 75 mg ou 150 mg SC aux semaines 0, 1, 2, 3 et 4, puis la même dose chaque mois. Les patients randomisés pour recevoir un placebo à l'inclusion ont été re-randomisés à la semaine 16 pour recevoir Cosentyx SC tous les mois (soit 75 mg, soit 150 mg).
Réponse clinique
Signes et symptômes
Dans l'étude AS2, le traitement par Cosentyx 150 mg est celui qui a entraîné la plus nette amélioration des mesures de l'activité de la maladie par rapport au placebo à la semaine 16 (voir tableau 11).
Tableau 11: Réponse clinique dans l'étude AS2 à la semaine 16
|
Résultat (valeur p par rapport au placebo)
|
Placebo
(n = 74)
|
75 mg
(n = 73)
|
150 mg
(n = 72)
| |
Efficacité à la semaine 16
| |
Réponse ASAS 20, %
|
28,4
|
41,1
|
61,1***
| |
Réponse ASAS 40, %
|
10,8
|
26,0
|
36,1***
| |
hsCRP, (rapport post-inclusion/inclusion)#
|
1,13
|
0,61
|
0,55***
| |
ASAS 5/6, %
|
8,1
|
34,2
|
43,1***
| |
BASDAI, variation moyenne des LS par rapport à la valeur à l'inclusion†
|
–0,85
|
–1,92
|
–2,19***
| |
ASAS, rémission partielle, %
|
4,1
|
15,1
|
13,9
| |
BASDAI 50, %
|
10,8
|
24,7*
|
30,6**
| |
ASDAS-CRP «major improvement»ˆ
|
4,1
|
15,1*
|
25,0***
| |
* p < 0,05; ** p < 0,01; *** p < 0,001 par rapport au placebo
Toutes les valeurs p ont été ajustées pour la multiplicité des tests basés sur une hiérarchie prédéfinie, à l'exception du BASDAI 50 et de l'ASDAS-CRP.
Les patients qui n'ont pas répondu ont été comptabilisés pour le critère d'évaluation binaire manquant.
ASAS: critères de l'Assessment of SpondyloArthritis International Society; BASDAI: indice d'activité de la spondylarthrite ankylosante de Bath; hsCRP: protéine C-réactive hautement sensible; ASDA: score d'activité de la spondylarthrite ankylosante; LS: moindres carrés (least square)
# les valeurs moyennes de la hsCRP à l'inclusion sont respectivement 8,30, 5,30 et 8,40
† Les valeurs moyennes du BASDAI à l'inclusion sont respectivement de 6,78, 6,57 et 6,59
ˆ ASDAS-CRP «major improvement» défini comme amélioration ≥2 unités
|
À la semaine 16, le traitement par Cosentyx 150 mg a entraîné des améliorations cliniquement pertinentes par rapport à l'inclusion pour chaque composante des critères de réponse ASAS 20 (évaluation globale par le patient, douleur rachidienne totale, BASFI et inflammation du rachis).
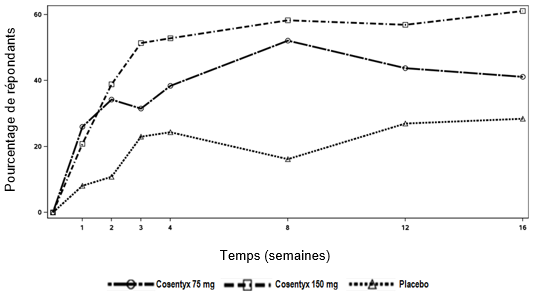
Dans l'étude AS2, le traitement par Cosentyx 150 mg a entraîné une amélioration significative de l'ASAS 20 dès 1 semaine après le début du traitement par rapport au placebo. Le pourcentage de patients ayant présenté une réponse ASAS 20 par visite est présenté à la figure 2.
Figure 2: Réponse ASAS 20 dans l'étude AS2 jusqu'à la semaine 16
La réponse ASAS 20 était supérieure à celle du placebo à la semaine 16, tant chez les patients n'ayant jamais reçu d'anti-TNF-alpha auparavant (68,2% contre 31,1%; p < 0,05) que chez les patients anti-TNF-alpha-IR (50,0% contre 24,1%; p < 0,05) avec Cosentyx 150 mg. La réponse ASAS 20 en termes de statut HLA B27 a été analysée dans l'étude AS2. 57/72 (79%) patients randomisés pour recevoir 150 mg et 58/74 (78%) patients randomisés pour recevoir un placebo étaient HLA B27 positifs. Pour 3 patients du groupe 150 mg et 5 patients du groupe placebo, le statut HLA B27 n'était pas connu.
Parmi les patients dont les mesures ASAS 20 étaient disponibles, l'ASAS 20 pour la cohorte de 150 mg à la semaine 16 était de 37/54 (68,5%) pour les patients HLA B27 positifs et de 5/9 (55,6%) pour les patients HLA B27 négatifs. Pour le placebo, l'ASAS 20 à la semaine 16 était de 18/51 (35,3%) pour les patients HLA B27 positifs et de 3/8 (37,5%) pour les patients HLA B27 négatifs.
Dans les deux études, la réponse s'est maintenue dans un ordre de grandeur comparable sous Cosentyx/- SensoReady jusqu'à la semaine 52. Sur les 72 patients randomisés pour recevoir Cosentyx 150 mg dans l'étude AS2, 61 (84,7%) étaient toujours sous traitement à la semaine 52. Respectivement 45 et 35 patients présentaient une réponse ASAS 20/40.
Mobilité de la colonne vertébrale:
La mobilité de la colonne vertébrale a été évaluée à l'aide de l'index BASMI (Bath Ankylosing Spondylitis Metrology Index) jusqu'à la semaine 52. Dans l'étude AS2 (150 mg) et l'étude AS1 (75 mg et 150 mg), des améliorations numériques ont été mises en évidence dans chacune des composantes de l'index BASMI pour les patients traités par Cosentyx par rapport aux patients traités par placebo aux semaines 4, 8, 12 et 16 (à l'exception de la flexion lombaire latérale aux semaines 4, 8 et 12 chez les patients traités par une dose de 75 mg après la dose initiale intraveineuse).
Fonction physique et qualité de vie liée à la santé
Dans les études AS1 et AS2, les patients traités par Cosentyx 150 mg ont montré une amélioration de la qualité de vie liée à la santé, mesurée par le questionnaire ASQoL (Ankylosing Spondylitis Quality of Life) (p < 0,001) et le questionnaire SF-36 Physical Component Summary (SF-36 PCS) (p < 0,001). Les patients traités par Cosentyx 150 mg ont également montré des améliorations significatives par rapport au placebo en ce qui concerne les critères d'évaluation exploratoires concernant la fonction physique mesurée par l'indice BASFI (indice fonctionnel de la spondylarthrite ankylosante de Bath [Bath Ankylosing Spondylitis Functional Index]), la douleur mesurée à l'aide de l'échelle de dorsalgie globale et nocturne (Total and Nocturnal Back Pain) et l'épuisement mesuré à l'aide de l'échelle FACIT-F (Functional Assessment of Chronic Illness Therapy - Fatigue). Toutes ces améliorations des fonctions corporelles ont été maintenues jusqu'à la semaine 52.
Spondylarthrite axiale non radiographique (nr-axSpA)
La sécurité et l'efficacité du sécukinumab ont été évaluées dans le cadre d'une étude de phase III randomisée, en double aveugle et contrôlée par placebo, menée chez 555 patients atteints de spondylarthrite axiale non radiographique (nr-axSpA) active qui répondaient aux critères de classification de l'ASAS (Assessment of SpondyloArthritis international Society) pour la spondyloarthrite axiale (axSpA) sans preuve radiographique de modifications des articulations sacro-iliaques, qui auraient répondu aux critères de New York modifiés pour la spondylarthrite ankylosante (AS). Les patients participant à l'étude présentaient une activité de la maladie définie par le Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4, un degré de douleur sur l'échelle visuelle analogique (EVA) pour les dorsalgies totales ≥40 (sur une échelle de 0 à 100 mm), malgré un traitement actuel ou antérieur par des anti-inflammatoires non stéroïdiens (AINS), un taux élevé de protéine C-réactive (CRP) et/ou des signes de sacro-iliite à l'imagerie par résonance magnétique (IRM). Chez les patients de cette étude, le diagnostic d'axSpA avait été posé en moyenne 2,1 à 3,0 ans auparavant et 54% des participants à l'étude étaient des femmes.
Dans l'étude nr-axSpA 1, 9,7% des patients avaient déjà été traités auparavant par un inhibiteur du TNF-α, qui avait été arrêté soit en raison d'un manque d'efficacité, soit en raison d'une intolérance (patients anti-TNFα-IR).
L'étude nr-axSpA 1 (PREVENT) a évalué 555 patients, dont 9,9% et 14,8% recevaient simultanément du MTX ou de la sulfasalazine, respectivement. Au cours de la phase en double aveugle, les patients ont reçu soit un placebo, soit le sécukinumab pendant 52 semaines. Les patients randomisés pour recevoir du sécukinumab ont reçu 150 mg par voie sous-cutanée aux semaines 0, 1, 2, 3 et 4, puis la même dose à intervalles mensuels ou une injection mensuelle de 150 mg de sécukinumab. Le critère d'évaluation principal consistait en une amélioration d'au moins 40% selon les critères de l'Assessment of SpondyloArthritis International Society (réponse ASAS 40) à la semaine 16 chez les patients naïfs de TNFα.
Réponse clinique
Signes et symptômes:
Dans l'étude nr-axSpA 1, le traitement par 150 mg de sécukinumab a entraîné à la semaine 16 une amélioration plus significative des paramètres d'activité de la maladie par rapport au placebo. Ces paramètres comprennent la réponse ASAS 40, ASAS 5/6, BASDAI, BASDAI 50, la CRP haute sensibilité (hsCRP), la réponse ASAS 20 et la rémission partielle ASAS par rapport au placebo à la semaine 16 (tableau 12).
Tableau 12: Réponse clinique dans l'étude nr-axSpA 1 à la semaine 16
|
Résultat (valeur p par rapport au placebo)
|
Placebo
|
150 mg1
| |
Nombre de patients naïfs d'anti-TNFα randomisés
|
171
|
164
| |
Réponse ASAS 40, %
|
29,2%
|
41,5%*
| |
Nombre total de patients randomisés
|
186
|
185
| |
Réponse ASAS 40, %
|
28,0%
|
40,0%*
| |
ASAS 5/6, %
|
23,7%
|
40,0%**
| |
BASDAI, variation moyenne des LS par rapport à l'inclusion
|
-1,46
|
-2,35**
| |
BASDAI 50, %
|
21,0%
|
37,3%**
| |
hsCRP, (rapport post-inclusion/inclusion)
|
0,91
|
0,64**
| |
Réponse ASAS 20, %
|
45,7%
|
56,8%*
| |
Rémission partielle ASAS, %
|
7,0%
|
21,6%**
| |
* p < 0,05; ** p < 0,001 par rapport au placebo
Toutes les valeurs p ont été soumises à un ajustement pour les tests multiples sur la base d'une hiérarchie prédéfinie. En l'absence de données pour un critère d'évaluation binaire, une imputation en tant que non répondeur a été effectuée.
1 Sécukinumab 150 mg SC aux semaines 0, 1, 2, 3 et 4, puis la même dose à intervalles mensuels
ASAS: critères de l'Assessment of SpondyloArthritis International Society; BASDAI: indice d'activité de la maladie de la spondylarthrite ankylosante de Bath; hsCRP: protéine C-réactive hautement sensible; LS: moindres carrés (least square)
|
Dans une analyse exploratoire, les patients qui présentaient au début de l'étude à la fois un taux de CRP anormal et des signes d'inflammation à l'IRM sont ceux qui ont le plus bénéficié du sécukinumab.
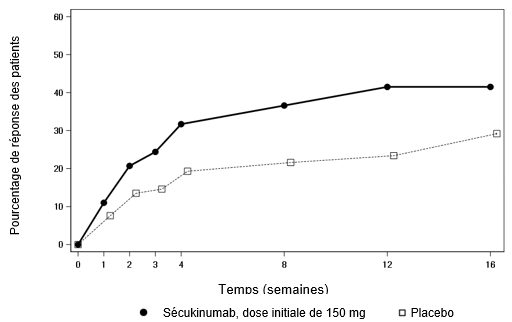
L'effet du sécukinumab 150 mg s'est manifesté dès la semaine 3 (supérieur au placebo) dans l'étude nr-axSpA 1 chez les patients naïfs d'anti-TNFα en termes de réponse ASAS 40. Le pourcentage de patients qui obtiennent une réponse ASAS 40 parmi les patients naïfs d'anti-TNFα est représenté à la figure 3 au fil des dates de visite. Les patients traités par le sécukinumab ont continué à présenter une réponse jusqu'à la semaine 52 par rapport au placebo.
Figure 3: Réponse ASAS 40 des patients naïfs d'anti-TNFα dans l'étude nr-axSpA 1 au fil du temps jusqu'à la semaine 16
Dans une analyse exploratoire portant sur 54 patients précédemment traités par anti-TNF, une réponse a été observée à la semaine 16 chez 6 patients sur 21 sous Cosentyx et chez 2 patients sur 15 sous placebo.
Fonction physique et qualité de vie liée à la santé:
Les patients traités par le sécukinumab 150 mg ont montré des améliorations statistiquement significatives par rapport aux patients traités par placebo en termes de fonction physique, telle qu'évaluée par BASFI, jusqu'à la semaine 16 (semaine 16: -1,75 contre -1,01, p < 0,01). Par rapport aux patients traités par placebo, les patients traités par le sécukinumab ont rapporté à la semaine 16 des améliorations significatives de la qualité de vie liée à la santé, évaluée à l'aide des questionnaires ASQoL (variation de la moyenne des LS: semaine 16: -3,45 contre -1,84, p < 0,001) et du score cumulé des composantes physiques du SF-36 (SF-36 PCS) (variation de la moyenne des LS: semaine 16: 5,71 contre 2,93, p < 0,001). Ces améliorations ont été maintenues jusqu'à la semaine 52.
Inhibition de l'inflammation à l'imagerie par résonance magnétique (IRM):
Les signes d'inflammation ont été évalués par IRM à l'inclusion et à la semaine 16 et exprimés sous la forme d'une modification du score d'œdème de l'articulation SI de Berlin pour les articulations sacro-iliaques, et du score ASspiMRI-a et du score de la colonne vertébrale de Berlin pour la colonne vertébrale. Une inhibition des signes d'inflammation a été observée chez les patients traités par le sécukinumab, tant au niveau de l'articulation sacro-iliaque que de la colonne vertébrale. La variation moyenne par rapport à l'inclusion du score d'œdème de l'articulation SI de Berlin était de -1,68 chez les patients traités par le sécukinumab 150 mg (n = 180) contre -0,39 chez les patients traités par placebo (n = 174) (p < 0,0001).
Arthrite juvénile idiopathique (AJI)
Arthrite liée à l'enthésite (ERA) et rhumatisme psoriasique juvénile (RPJ)
L'efficacité et la sécurité du sécukinumab ont été examinées dans une étude de phase III randomisée, basée sur les événements, contrôlée contre placebo, en double aveugle et en trois parties, menée chez 86 patients âgés de 2 ans à < 18 ans atteints d'ERA ou de RPJ actifs, qui avaient été diagnostiqués selon les critères modifiés pour la classification des AJI de l'International League of Associations for Rheumatology (ILAR). L'étude se composait d'une partie en ouvert (partie 1), suivie d'un sevrage randomisé (partie 2) et enfin d'un traitement en ouvert (partie 3). Les sous-groupes de patients atteints d'AJI au début de l'étude étaient les suivants: 60,5% ERA et 39,5% RPJ.
À l'inclusion, 65,1% des patients avaient reçu du MTX (63,5% (33 sur 52) des patients atteints d'ERA et 67,6% (23 sur 34) des patients atteints de RPJ). Les patients ont reçu une dose de 75 mg lorsqu'ils pesaient < 50 kg, ou de 150 mg lorsqu'ils pesaient ≥50 kg. Le critère d'évaluation principal était le délai jusqu'à une poussée de la maladie dans la partie 2. Une poussée de la maladie était définie comme une dégradation de ≥30% dans au moins trois des six critères de réponse ACR pour l'AJI et une amélioration de ≥30% dans pas plus d'un des six critères de réponse ACR pour l'AJI.
Dans la partie 1 en ouvert de l'étude, tous les patients ont reçu du sécukinumab jusqu'à la semaine 12. Les patients qui avaient été classés comme répondeurs en semaine 12 ont intégré la phase en double aveugle de la partie 2 et ont été randomisés selon un rapport de 1:1 soit pour poursuivre le traitement par sécukinumab, soit pour commencer un traitement par placebo. À la fin de la partie 1, 75 des 86 patients (90,4%) présentaient une réponse ACR 30 pour l'AJI et ont pris part à la partie 2.
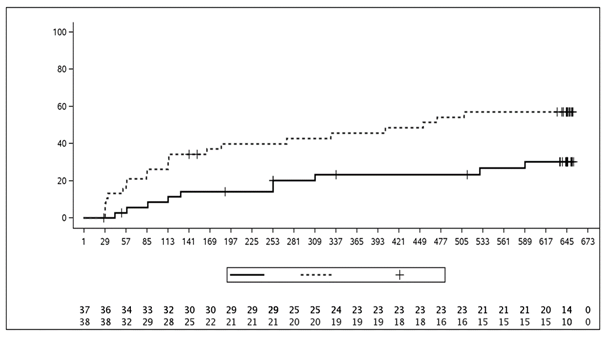
L'étude a atteint son critère d'évaluation principal en démontrant un allongement statistiquement significatif du délai jusqu'à une poussée de la maladie chez les patients traités par sécukinumab par rapport au placebo. Le risque d'une poussée de la maladie a été réduit de 72% chez les patients ayant reçu du sécukinumab par rapport aux patients ayant reçu le placebo (hazard ratio des poussées de la maladie = 0,28, IC à 95%: 0,13 à 0,63, p < 0,001) (figure 4). Dans la deuxième partie de l'étude, des poussées ont été observées chez 21 patients au total dans le groupe placebo (11 RPJ et 10 ERA) contre 10 patients dans le groupe sous sécukinumab (4 RPJ et 6 ERA). L'analyse de survie du délai jusqu'à la survenue d'une poussée de la maladie a montré que le traitement par sécukinumab a conduit, par rapport au groupe placebo de la partie 2, à un allongement du délai jusqu'à la survenue d'une poussée de la maladie, indépendamment de la prise de MTX à l'inclusion. Le risque d'une poussée de la maladie était réduit de 35% chez les sujets sous sécukinumab avec traitement simultané par MTX (hazard ratio = 0,65, IC à 95%: 0,23, 1,83) et de 92% chez les sujets sous sécukinumab sans traitement simultané par MTX (hazard ratio = 0,08, IC à 95%: 0,02, 0,32), par rapport aux sujets qui avaient été randomisés dans le groupe placebo dans la partie 2.
Figure 4: Estimations de Kaplan-Meier du délai jusqu'à la poussée de la maladie dans la partie 2
Hidrosadénite suppurée
La sécurité et l'efficacité du sécukinumab ont été évaluées chez 1084 patients adultes atteints d'hidrosadénite suppurée (HS) modérée à sévère et éligibles à un traitement systémique par des médicaments biologiques, dans deux études de phase III randomisées, en double aveugle et contrôlées contre placebo. Les patients inclus dans l'étude HS 1 (SUNSHINE) ou dans l'étude HS 2 (SUNRISE) présentaient un stade I de Hurley (4,6% et 2,8% resp.), II (61,4% et 56,7% resp.) ou III (34,0% et 40,5% resp.) à l'inclusion et présentaient au moins cinq lésions inflammatoires affectant deux régions anatomiques. La proportion de patients ayant un poids corporel ≥90 kg était de 54,7% dans l'étude HS 1 et de 50,8% dans l'étude HS 2. Les patients inclus dans ces études présentaient un diagnostic d'HS modérée à sévère depuis 7,3 ans en moyenne, et 56,3% des participants à l'étude étaient des femmes. Dans l'étude HS 1 et l'étude HS 2, 82,3% et 83,6% des patients avaient été précédemment traités par des antibiotiques systémiques et 23,8% et 23,2% des patients avaient été précédemment traités par un médicament biologique qu'ils avaient arrêté, soit en raison d'une efficacité insuffisante, soit en raison d'une intolérance (patients exposés à un médicament biologique).
L'étude HS 1 a porté sur 541 patients et l'étude HS 2 sur 543 patients, dont respectivement 12,8% et 10,7% ont reçu simultanément une dose constante d'antibiotiques. Dans les deux études, les patients randomisés pour recevoir le sécukinumab ont reçu 300 mg de sécukinumab par voie sous-cutanée aux semaines 0, 1, 2, 3 et 4, puis 300 mg toutes les 2 semaines (Q2W) ou toutes les 4 semaines (Q4W). À la semaine 16, les patients randomisés pour recevoir le placebo ont été réaffectés et ont reçu 300 mg de sécukinumab à chacune des semaines 16, 17, 18, 19 et 20, puis soit 300 mg de sécukinumab Q2W, soit 300 mg de sécukinumab Q4W.
Le critère d'évaluation principal dans les deux études (étude HS 1 et étude HS 2) était la proportion de patients ayant obtenu une réponse clinique de l'hidrosadénite suppurée à la semaine 16. Celle-ci était définie par une diminution d'au moins 50% du nombre total d'abcès et de nodules inflammatoires et par l'absence d'augmentation du nombre d'abcès et/ou du nombre de fistules drainantes par rapport à l'inclusion (HiSCR50). La diminution des douleurs cutanées liées à l'HS a été évaluée en tant que critère d'évaluation secondaire à partir des données regroupées des études HS 1 et HS 2 à l'aide d'une échelle d'évaluation numérique (NRS [Numeric Rating Scale]) chez les patients présentant un score initial égal ou supérieur à 3 lors de l'inclusion.
Dans l'étude HS 1 et l'étude HS 2, une proportion significativement plus élevée de patients traités par 300 mg de sécukinumab Q2W a obtenu une réponse HiSCR50 à la semaine 16, avec une diminution significative du nombre d'abcès et de nodules inflammatoires (AN) par rapport au placebo, ce qui n'a cependant pas été le cas des patients traités par 300 mg de sécukinumab Q4W par rapport au placebo dans l'étude HS 1. Dans l'étude HS 2, une différence statistiquement significative a également été observée dans la réponse HiSCR50 et le nombre d'AN avec le traitement sécukinumab 300 mg Q4W par rapport au placebo. Dans le groupe sécukinumab 300 mg Q4W de l'étude HS 2 ainsi que dans le groupe sécukinumab 300 mg Q2W de l'étude HS 1, les poussées de la maladie ont été moins nombreuses jusqu'à la semaine 16 par rapport au placebo. Une proportion significativement plus élevée de patients traités par 300 mg de sécukinumab Q2W (données regroupées) a enregistré une diminution cliniquement significative des douleurs cutanées liées à l'HS à la semaine 16 par rapport au placebo.
Tableau 13: Réponse clinique dans l'étude HS 1 et l'étude HS 2 à la semaine 161
|
|
Étude HS 1
|
Étude HS 2
| |
|
Placebo
|
300 mg Q4W
|
300 mg Q2W
|
Placebo
|
300 mg Q4W
|
300 mg Q2W
| |
Nombre des patients randomisés
|
180
|
180
|
181
|
183
|
180
|
180
| |
Réponse HiSCR50, %
|
33,7
|
41,8
|
45,0*
|
31,2
|
46,1*
|
42,3*
| |
Nombre d'AN, %
Moyenne des LS pour la variation par rapport à l'inclusion
|
-24,3
|
-42,4
|
-46,8*
|
-22,4
|
-45,5*
|
-39,3*
| |
Poussées, %
|
29,0
|
23,2
|
15,4*
|
27,0
|
15,6*
|
20,1
| |
|
Données regroupées (étude HS 1 et étude HS 2)
| |
|
Placebo
|
300 mg Q4W
|
300 mg Q2W
| |
Nombre de patients présentant une NRS ≥3 à l'inclusion
|
251
|
252
|
266
| |
Réponse NRS 30, %
|
23,0
|
33,5
|
36,6*
| |
1
Une imputation multiple a été effectuée en cas de données manquantes
* Statistiquement significatif par rapport au placebo sur la base de la hiérarchie prédéfinie avec un alpha global = 0,05
AN: abcès et nodules inflammatoires; HiSCR: réponse clinique à l'hidrosadénite; NRS: échelle d'évaluation numérique
|
Dans les deux études, le sécukinumab a commencé à agir dès la semaine 2, son efficacité a augmenté jusqu'à la semaine 16 et s'est maintenue jusqu'à la semaine 52.
Des améliorations ont été observées pour les critères d'évaluation principaux et les critères secondaires importants chez les patients atteints d'HS, indépendamment de tout traitement antibiotique antérieur ou concomitant. La réponse HiSCR50 s'est améliorée à la semaine 16, tant chez les patients naïfs de traitement biologique que chez les patients exposés aux traitements biologiques. Des améliorations plus importantes à la semaine 16 par rapport à la valeur initiale en comparaison avec le placebo ont été mises en évidence en ce qui concerne la qualité de vie liée à la santé, mesurée avec le Dermatology Life Quality Index.
|