CompositionPrincipes actifs
Pasireotidum (ut Pasireotidi Pamoas)
Excipients
Flacon avec principe actif: Poly(D,L-lactidecoglycolide) (50-60:40-50), Poly(D,L-lactidecoglycolide) (50:50)
Seringue préremplie avec solvant: mannitol, carmellose sodique correspondant à 1,5 mg de sodium, poloxamère 188, eau pour solution injectable.
Indications/Possibilités d’emploi-Traitement des patients acromégales chez lesquels un traitement chirurgical ou par radiothérapie est inadapté ou inefficace et/ou d’autres traitements médicamenteux n’ont pas donné les résultats souhaités.
-Traitement de la maladie de Cushing, lorsque toutes les autres options thérapeutiques non médicamenteuses sont épuisées.
Posologie/Mode d’emploiSignifor LAR ne doit être administré que par un professionnel de santé expérimenté.
Acromégalie
La dose initiale recommandée de Signifor LAR est de 40 mg toutes les 4 semaines.
Chez les patients dont le taux de GH et/ou d’IGF-1 n’est pas entièrement contrôlé au bout de 3 mois de traitement avec Signifor LAR 40 mg, la dose peut être augmentée à 60 mg maximum.
Maladie de Cushing
Utilisation chez les patients qui n'ont pas été traités antérieurement avec le pasiréotide
La dose initiale recommandée de Signifor LAR pour le traitement de la maladie de Cushing est de 10 mg toutes les 4 semaines (28 jours). En fonction de la réponse clinique et de la tolérance, la dose peut être augmentée par palier de 10 mg jusqu’à 40 mg tous les 28 jours au maximum.
Conversion depuis le pasiréotide injecté par voie sous-cutanée
Aucune donnée n'est disponible concernant le passage du pasiréotide injecté par voie sous-cutanée au Signifor LAR ou vice versa. Signifor (s.c.) et Signifor LAR n'étant pas bioéquivalents, aucune recommandation posologique ne peut être donnée (voir «Mises en garde et précautions»). La dose maximale dans ce cas est aussi de 40 mg tous les 28 jours.
Ajustement de la posologie du fait d’effets indésirables / d’interactions
Acromégalie : La gestion des effets indésirables (supposés) ou d’une réponse exagérée au traitement (IGF-1 < limite inférieure à la normale) peut nécessiter une réduction de la dose de Signifor LAR. La dose peut être réduite par paliers de 20 mg de manière provisoire ou permanente.
Morbus Cushing : En cas d'apparition d'effets indésirables ou de signes d'hypocortisolisme, une réduction de la dose, une interruption du traitement ou l'arrêt du traitement par Signifor LAR peut être nécessaire.
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la dose n’est nécessaire chez les patients présentant une insuffisance hépatique légère (Child Pugh A). Signifor LAR est contre-indiqué chez les patients présentant une insuffisance hépatique sévère (Child Pugh C).
Insuffisance hépatique modérée (Child Pugh B):
Acromégalie: la dose initiale recommandée est de 20 mg toutes les 4 semaines. La dose maximale est de 40 mg toutes les 4 semaines (voir «Pharmacocinétique»).
Maladie de Cushing: la dose initiale recommandée est de 10 mg toutes les 4 semaines. La dose maximale est de 20 mg toutes les 4 semaines (voir «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la dose n’est nécessaire chez les patients présentant une insuffisance rénale (cf. «Pharmacocinétique»).
Patients âgés
Les données concernant l’utilisation de Signifor LAR chez les patients de plus de 65 ans sont limitées. Aucune donnée n’indique cependant qu’un ajustement posologique est nécessaire chez les patients âgés.
Enfants et adolescents
La sécurité d’emploi et l’efficacité n’ont pas été évaluées chez l’enfant et l’adolescent.
Prise retardée
Si une dose de Signifor LAR est oubliée, l’injection oubliée doit être administrée le plus rapidement possible. La dose suivante devra être programmée 4 semaines après cette injection (le jour de la semaine prévu pour l’injection pourra donc changer le cas échéant).
Mode d’administration
Signifor LAR doit être administré par injection intramusculaire profonde.
La suspension de Signifor LAR doit être préparée juste avant son administration.
En cas d’injections intramusculaires répétées, le point d’injection doit être alterné entre le muscle glutéal gauche et droit (cf. «Remarques particulières» / «Remarques concernant la manipulation»).
Contre-indicationsHypersensibilité au principe actif ou à l’un des excipients.
Insuffisance hépatique sévère (Child Pugh C).
Mises en garde et précautionsL'initiation et la surveillance du traitement par Signifor ne doivent être effectuées que par des médecins ayant de l'expérience dans le traitement de ces pathologies.
Pour le traitement de la maladie de Cushing, le pasiréotide est disponible en Signifor® pour l'injection sous-cutanée deux fois par jour (en doses individuelles de 300-900 µg) et en Signifor LAR pour l'injection intramusculaire une fois par mois (en doses de 10-40 mg). Aucune donnée n'est disponible concernant le passage d'une forme galénique à l'autre. Si le traitement est passé de pasiréotide s.c. à pasiréotide i.m. (ou inversement), le patient doit faire l'objet d'un suivi particulièrement attentif au cours des 2-3 premiers mois portant sur sa réponse au traitement et une éventuelle détérioration du métabolisme du glucose.
Hypocortisolisme
Le traitement par Signifor LAR entraîne une suppression rapide de la sécrétion d'ACTH (hormone adrénocorticotrope). Cette suppression rapide de l’ACTH peut entraîner un hypocortisolisme passager dont les symptômes sont la faiblesse, la fatigue, l’anorexie, des nausées, des vomissements, l’hypotension, l’hyponatrémie, l’hypoglycémie, ou encore une insuffisance surrénalienne aigüe. Des cas d'hypocortisolisme ont été rapportés dans des études cliniques chez des patients atteints de la maladie de Cushing, notamment pendant les deux premiers mois du traitement. Il est donc recommandé de surveiller les patients régulièrement et de les informer des symptômes éventuels. En cas d'hypocortisolisme, il peut s’avérer nécessaire de réduire la dose, d’interrompre le traitement par Signifor LAR et/ou d’administrer temporairement un traitement de substitution par glucocorticoïdes.
Métabolisme du glucose
Des modifications de la régulation du glucose sont attendues au cours du traitement par le pasiréotide. Une augmentation de la glycémie à jeun, des hyperglycémies et une hausse de l’HbA1c, ainsi que, moins fréquemment, des hypoglycémies, ont été observées au cours des études cliniques, aussi bien dans des cas de maladie de Cushing que de l’acromégalie, ce qui a entraîné l’arrêt de l’étude chez jusqu’à 5% des patients. La survenue d’une hyperglycémie est corrélée à une diminution de la sécrétion d’insuline et des hormones incrétines (c.-à-d. glucagon-like peptide-1 [GLP-1] et polypeptide insulinotrope dépendant du glucose [GIP]). Le degré du trouble de la régulation du glucose est plus prononcé en présence d’un état prédiabétique ou d’un diabète sucré manifeste. Une augmentation de la glycémie à jeun et du HbA1c a été observée, notamment durant les premiers mois du traitement, après quoi l’augmentation a cessé.
Le statut glycémique (glycémie à jeun et HbA1c) doit être évalué avant l’instauration du traitement et surveillé régulièrement au cours du traitement. L’auto-mesure de la glycémie et et/ou le dosage de la glycémie à jeun doivent être effectués toutes les semaines pendant les 2-3 premiers mois du traitement, puis périodiquement lorsque cela est cliniquement justifié et toutes les semaines pendant les quatre à six premières semaines suivant une augmentation de dose.
La glycémie à jeun et le HbA1c reculent en général après l’arrêt du pasiréotide, mais peuvent rester élevés par rapport aux valeurs initiales. Le taux de glycémie à jeun doit être surveillé jusqu’à trois semaines et l’HbA1c jusqu’à trois mois après la fin du traitement.
En cas de survenue d’une hyperglycémie, il est indiqué d’instaurer ou d’ajuster rapidement un traitement de l’hyperglycémie en utilisant des incrétines, des sécrétagogues de l’insuline et/ou de l’insuline. Si l’hyperglycémie ne peut être contrôlée malgré un traitement médical approprié, la dose de pasiréotide doit être réduite ou le traitement suspendu.
Après la mise sur le marché, des cas d’acidocétose ont été rapportés pendant le traitement avec le pasiréotide, que les patients aient ou non présenté un diabète avant le début du traitement. Il existait dans certains cas des facteurs de prédisposition tels que des maladies aiguës, des infections, des maladies du pancréas (par ex. tumeurs malignes du pancréas ou opérations du pancréas) ou abus d’alcool. Tous les patients présentant des symptômes qui suggèrent une acidose métabolique sévère doivent être examinés pour rechercher une acidocétose. Ces symptômes sont souvent aspécifiques et comprennent, par exemple, une soif excessive, une perte d’appétit, des douleurs abdominales, des nausées, des vomissements, une dyspnée, une fatigue inhabituelle ou un épuisement et un état confusionnel. Le patient doit être informé du risque (et en particulier de l’augmentation du risque liée, par exemple, à une consommation excessive d’alcool ou à un jeûne prolongé) et des symptômes possibles de l’acidocétose. Il doit également être informé que, si de tels symptômes surviennent, il est nécessaire de consulter sans tarder un médecin.
Chez les patients ayant un mauvais contrôle glycémique (défini comme un taux d’HbA1c >8% sous traitement antidiabétique), la surveillance et la prise en charge du diabète doivent être intensifiées avant et pendant le traitement par Signifor LAR.
Dans les deux études pivot sur l’acromégalie, la fréquence et la gravité des effets indésirables liés à l’hyperglycémie étaient supérieures sous pasiréotide administré par voie intramusculaire que sous le comparateur actif. Les patients acromégales ayant développé une hyperglycémie ont en général semblé répondre aux traitements antidiabétiques.
Paramètres hépatiques
De légères élévations temporaires des aminotransférases sont fréquemment observées au cours du traitement par le pasiréotide. Une augmentation concomitante de l’ALAT (alanine aminotransférase) supérieure à 3× LSN (limite supérieure de la normale) et des taux de bilirubine supérieurs à 2× LSN a été observée dans quelques cas.
Il est donc recommandé de contrôler la fonction hépatique avant le traitement par Signifor LAR, durant les 2 à 3 premières semaines de traitement, puis tous les mois pendant 3 mois. Ensuite, la fonction hépatique doit être vérifiée chaque fois que cela semble cliniquement indiqué.
Les patients présentant une augmentation des transaminases doivent faire l’objet d’un deuxième contrôle de la fonction hépatique pour confirmer cette augmentation. La fonction hépatique de ces mêmes patients – le cas échéant, même après l’arrêt du traitement par Signifor LAR, doit être régulièrement contrôlée jusqu’au rétablissement des valeurs pré-thérapeutiques.
Le traitement par Signifor LAR doit être interrompu dans les cas suivants:
survenue d’un ictère ou d’autres signes évocateurs d’une altération cliniquement significative de la fonction hépatique
élévation durable des taux d’aspartate aminotransférase (ASAT) ou d’ALAT à ≥5× LSN
élévation des taux d’ALAT ou d’ASAT à ≥3× LSN et augmentation simultanée de la bilirubine à ≥2× LSN.
Le traitement ne doit pas être repris si un lien entre les troubles de la fonction hépatique et le traitement par Signifor LAR est soupçonné.
Événements indésirables au niveau de la vésicule biliaire
La cholélithiase est un effet indésirable connu du traitement à long terme par des analogues de la somatostatine. Elle a été fréquemment observée au cours des études cliniques réalisées avec le pasiréotide. Il est donc recommandé de pratiquer des échographies de la vésicule biliaire avant le traitement par Signifor LAR, puis tous les 6 à 12 mois. La survenue d’une cholélithiase chez les patients traités par Signifor LAR est en grande partie asymptomatique; les concrétions symptomatiques doivent être traitées conformément à la pratique clinique courante. Des cas de cholangite, résultant, dans la plupart des cas, d’une complication d’une cholélithiase, ont été rapportés sous traitement avec Signifor LAR après la mise sur le marché.
Événements cardiovasculaires
Des bradycardies ont été observées au cours du traitement par le pasiréotide. Une surveillance étroite est recommandée chez les patients présentant une cardiopathie et/ou des facteurs de risque de bradycardie, dont antécédents de bradycardie cliniquement significative, bloc AV de type Mobitz II, insuffisance cardiaque (classe III ou IV de la NYHA), status post-infarctus du myocarde, angine de poitrine instable, antécédents de tachycardie ventriculaire ou fibrillation ventriculaire. Un ajustement de la dose de médicaments tels que bêtabloquants, inhibiteurs de canaux calciques ou substances contrôlant l’équilibre électrolytique peut s’avérer nécessaire.
Deux études menées chez des volontaires sains ont révélé que le pasiréotide allonge l’intervalle QT à l’ECG (voir également Section «Propriétés/Effets»). La signification clinique de cet allongement n'est pas connue. Un allongement de > 500 ms du QTcF a été mesuré chez 2 patients sur 201. Ces épisodes étaient sporadiques, ne sont survenus qu'une seule fois et sont restés sans conséquence clinique. Aucun épisode de torsade de pointes n’a été observé, ni dans ces études, ni dans des études cliniques réalisées sur des patients atteints de la maladie de Cushing ou acromégaliques.
Concernant l’allongement de l’intervalle QT, les études de phase III chez des patients acromégales n’ont indiqué aucune différence cliniquement significative entre Signifor LAR et les analogues de la somatostatine utilisés comme comparateurs actifs. Tous les événements relatifs à l’allongement QT étaient spontanément réversibles, sans aucune intervention thérapeutique.
Le pasiréotide doit être utilisé avec prudence chez les patients présentant un risque accru d’allongement de l’intervalle QT, par exemple en cas de :
syndrome du QT long congénital
cardiopathie cliniquement pertinente, telle qu’infarctus du myocarde récent, insuffisance cardiaque congestive, angine de poitrine instable ou bradycardie cliniquement pertinente
patients utilisant des antiarythmiques ou d’autres substances connues pour entraîner un allongement du QT
hypokaliémie et/ou hypomagnésémie
Il est recommandé de réaliser un ECG initial avant de débuter le traitement par Signifor LAR. 21 jours après le début du traitement, et à chaque indication clinique, la surveillance d’une éventuelle influence sur l’intervalle QT est recommandée.
Une hypokaliémie ou une hypomagnésémie doit être corrigée avant le traitement par Signifor LAR et le taux de sodium et de magnésium doit être régulièrement surveillé au cours du traitement.
Hormones hypophysaires
Une carence en hormones hypophysaires est une conséquence fréquente des opérations transsphénoïdales et surtout après une radiothérapie hypophysaire. Les patients ainsi traités antérieurement et atteints de la maladie de Cushing ou acromégales peuvent donc présenter une carence en une ou plusieurs hormones hypophysaires. Les effets pharmacologiques du pasiréotide imitant l’action de la somatostatine, l’inhibition d’hormones hypophysaires autres que ACTH et GH/IGF-1 ne peut pas être exclue. Une surveillance de la fonction hypophysaire (par ex. TSH/T4 libre, ACTH, GH/IGF-1) doit donc être réalisée avant le début du traitement par Signifor LAR et régulièrement au cours du traitement par Signifor LAR, tel que cliniquement indiqué.
Fertilité
Les études expérimentales sur les animaux ont révélé une incidence du pasiréotide administré par voie sous-cutanée sur la fertilité des femelles (voir «Données précliniques»). L’effet du pasiréotide sur la fertilité humaine est inconnu. Lors du traitement de femmes en âge de procréer, l’éventualité d’une diminution de la fertilité féminine doit cependant être prise en considération.
D’un autre côté, la réduction et/ou la normalisation du taux de cortisol sérique et du taux d’hormone GH («Growth hormone», hormone de croissance) et de la concentration d’IGF-1 («Insulin-like growth factor», facteur de croissance apparenté à l’insuline) obtenue par le traitement peut entraîner une amélioration de la fertilité.
Le cas échéant, il peut être demandé aux patientes en âge de procréer de prendre des mesures contraceptives adaptées pendant le traitement par le pasiréotide (cf. «Grossesse/Allaitement»).
Ce médicament contient moins de 1 mmol de sodium (23 mg) par unité de dosage, c’est-à-dire qu’il est pratiquement «sans sodium».
InteractionsInteractions pharmacocinétiques
Le pasiréotide est modérément lié aux protéines et métaboliquement stable. Il n’est ni un substrat ni un inhibiteur ni un inducteur d’enzymes importantes de type CYP450 dans le foie.
Le pasiréotide semble être un substrat du transporteur d’efflux de la glycoprotéine P (P-gp). Toutefois, le pasiréotide n’est ni un inhibiteur ni un inducteur de la P-gp. Le pasiréotide n’est ni un substrat du transporteur d’efflux BCRP (breast cancer resistance protein, protéine de résistance dans le cancer du sein) ni des transporteurs d’influx OCT1 (organic cation transporter 1, transporteur de cations organiques) et OATP («organic aniontransporting polypeptide», polypeptide transporteur d’anions organiques) 1B1, 1B3 ou 2B1. À des concentrations cliniquement significatives, on ne s’attend pas à ce que le pasiréotide inhibe l’UGT1A1 (uridine diphosphate glucuronosyltransferase 1A1), les transporteurs d’influx OAT1 ou OAT3, OATP 1B1 ou 1B3, OCT1 ou OCT2, ou les transporteurs d’efflux P-gp, BCRP, MRP2 (multidrug resistance protein 2, protéine 2 de résistance multimédicaments) et BSEP (bile salt export pump, pompe d’exportation des sels biliaires).
Sur la base de ces données in vitro, le potentiel d’interactions transmises par la liaison de protéines, le métabolisme et/ou les transporteurs entre le pasiréotide et d’autres médicaments administrés en concomitance est faible in vivo.
Interactions pharmacodynamiques
La prudence est recommandée en cas d’administration concomitante de Signifor et de médicaments antiarythmiques ou de principes actifs qui pourraient allonger l’intervalle QT (cf. «Mise en garde et précautions»).
Effet de SIGNIFOR LAR sur d’autres médicaments
Quelques rares données publiées suggèrent que les analogues de la somatostatine pourraient, par inhibition de la sécrétion de l’hormone de croissance, réduire de manière indirecte la clairance métabolique de substances métabolisées par les enzymes du CYP450. Sur la base des données disponibles, l’éventualité d’un tel effet indirect du pasiréotide ne peut pas être exclue. La prudence est recommandée en cas d’administration concomitante de pasiréotide et de médicaments ayant un faible indice thérapeutique et métabolisés essentiellement par l’intermédiaire du CYP3A4.
Chez le chien, le pasiréotide a entraîné une diminution du taux sanguin de cyclosporine par diminution de l’absorption intestinale de cyclosporine. On ignore si une telle interaction se produit aussi chez l’être humain. Un ajustement de la dose de cyclosporine peut donc s’avérer nécessaire en cas d’administration concomitante de pasiréotide et de cyclosporine.
La biodisponibilité de la bromocriptine peut être augmentée en cas d’administration concomitante de bromocriptine et d’analogues de la somatostatine. On ne peut exclure l’éventualité d’un tel effet du pasiréotide.
Effet d’autres médicaments sur SIGNIFOR LAR
L’influence de l’inhibiteur P-gp sur la pharmacocinétique du pasiréotide, administré sous forme de de Signifor en injection s.c., a été évaluée lors de l’administration concomitante de vérapamil. Aucune modification du taux ou de l’étendue de la biodisponibilité du pasiréotide n’a été constatée.
Grossesse, AllaitementGrossesse
Il n’existe aucune étude adéquate et bien contrôlée chez la femme enceinte. Les expérimentations réalisées chez les animaux en administration s.c. ont montré une toxicité pour la reproduction (cf. «Données précliniques»). Comme le risque potentiel n’est pas connu chez l’être humain, Signifor LAR ne doit être utilisé pendant la grossesse qu’en cas de nécessité absolue.
Allaitement
On ignore si le pasiréotide passe dans le lait maternel humain. Des études chez les rats femelles sur le pasiréotide en administration s.c. ont montré un passage du pasiréotide dans le lait. Un risque pour l’enfant allaité ne pouvant être exclu, les femmes ne doivent pas utiliser Signifor LAR pendant qu’elles allaitent.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n’a été menée. Lors de l’utilisation du pasiréotide, des effets indésirables tels que vertige, ainsi que de rares cas d’hypoglycémies, susceptibles d’altérer l’aptitude à la conduite, ont été rapportés. Il faudrait par conséquent savoir comment le patient réagit à Signifor LAR avant qu’il ne conduise un véhicule ou n’utilise des machines.
Effets indésirablesLe profil de sécurité est largement similaire dans le traitement de la maladie de Cushing et de l'acromégalie et comparable à celui du pasiréotide administré par voie sous-cutanée.
Les effets indésirables suivants ont été rapportés dans les études cliniques réalisées avec le pasiréotide et sont répertoriés par classe de systèmes d’organes MedDRA selon les catégories suivantes: très fréquents (≥1/10) ; fréquents (≥1/100 et <1/10) ; occasionnels (≥1/1 000 et <1/100), fréquence inconnue (principalement basés sur des rapports spontanés de pharmacovigilance, sans que la fréquence exacte puisse être estimée).
Acromégalie
L’évaluation de sécurité se base sur 491 patients acromégales ayant reçu le pasiréotide dans les études de phase I à III (419 patients ont reçu Signifor LAR et 72 Signifor s.c.).
La sécurité de Signifor LAR chez les patients acromégales a été évaluée dans deux études menées en aveugle avec contrôle par substance active. Dans l’étude menée chez les patients non traités par médicament au préalable et dont l’intervention chirurgicale avait échoué ou n’était pas indiquée, les effets indésirables les plus fréquents qui ont été rapportés dans les bras Signifor LAR et Sandostatine LAR étaient : diarrhée (33,1% vs 40,6%), cholélithiase (30,9% vs 36,7%), hyperglycémie (28,1% vs 7,2%) et diabète sucré (19,7% vs 3,9%). Les effets indésirables de grade 3 ou 4 selon les critères de toxicité (CTC, Common Toxicity Criteria), qui ont été rapportés chez plus de 2% des patients dans le bras Signifor LAR ou Sandostatine LAR, étaient diabète sucré (4,5% vs 0%), diarrhée (0,6% vs 2,8%) et hyperglycémie (2,2% vs 0,6%). Des troubles du métabolisme du glucose ont entraîné l’arrêt prématuré de l’étude chez presque 3% des patients.
Dans l’étude menée auprès de patients pour lesquels aucun contrôle biochimique (GH ≤ 2,5 microgrammes/l et IGF-1 normalisé) n’a été obtenu avec un traitement par analogues de la somatostatine de première génération (désigné comme «patients insuffisamment contrôlés»), les effets indésirables les plus fréquemment observés avec Signifor LAR 40 mg, 60 mg et le comparateur actif étaient hyperglycémie (33,3%, 29,0% et 6,1%), diabète sucré (19,0%, 25,8% et 4,5%) et diarrhée (11,1%, 19,4 % et 1,5%). Les effets indésirables de grade 3 ou 4, qui ont été rapportés par plus de 2% des patients du groupe Signifor LAR 40 mg, 60 mg ou du groupe de contrôle par substance active, étaient hyperglycémie (11,1%, 8,1% et 0%), diabète sucré (0%, 3,2% et 0%) et douleurs abdominales (1,6%, 0%, 0%). Des troubles du métabolisme du glucose ont entraîné l’arrêt prématuré de l’étude chez presque 5% des patients.
Affections hématologiques et du système lymphatique*
Fréquent: anémie
Affections endocriniennes
Fréquent: insuffisance cortico-surrénalienne**, réduction du cortisol sérique
Troubles du métabolisme et de la nutrition
Très fréquent: hyperglycémie (28-33%), diabète sucré (19-26%)
Fréquent: altération de la tolérance au glucose, HbA1c augmentée, appétit diminué, hausse de la glycémie
Fréquence inconnue: acidocétose
Affections du système nerveux
Fréquent: vertiges, céphalées
Maladies cardiaques
Fréquent: bradycardie sinusale***, allongement de QT
Affections gastro-intestinales
Très fréquent: diarrhée (11-33%), douleurs abdominales (5-13%)
Fréquent: flatulence, nausées, vomissements, lipase augmentée, amylase sérique augmentée
Fréquence inconnue: Stéatorrhée, selles décolorées
Affections hépatobiliaires****
Très fréquent: cholélithiase (10-31%)
Fréquent: cholécystite, alanine aminotransférase augmentée, aspartate aminotransférase augmentée, gammaglutamyltransférase augmentée
Affections de la peau et du tissu sous-cutané
Très fréquent: alopécie (2-16%)
Affections musculosquelettiques et du tissu conjonctif
Fréquent: créatine phosphokinase sanguine augmentée
Troubles généraux et anomalies au site d’administration
Fréquent: réactions au site d’injection (douleurs, induration, troubles, hématome, prurit, gonflement), fatigue
* Un temps de prothrombine prolongé a été rapporté chez des patients sous Signifor atteints de la maladie de Cushing; dans les études sur l’acromégalie, les investigateurs n’ont identifié aucun lien entre ces événements et le traitement par Signifor LAR.
** L’insuffisance cortico-surrénalienne comprend l’hypocortisolisme
*** La bradycardie sinusale comprend la bradycardie
**** Des cas de cholestase ont été rapportés chez des patients sous Signifor souffrant de la maladie de Cushing, mais pas dans l’étude sur l’acromégalie et Signifor LAR.
Maladie de Cushing
Les données de sécurité rapportées ci-dessous sont basées sur un essai clinique de phase III chez n = 150 patients atteints de la maladie de Cushing ayant reçu des doses initiales de 10 mg ou 30 mg de Signifor LAR avec la possibilité d’augmenter la dose jusqu'à maximum 40 mg tous les 28 jours. La durée médiane d'exposition était de 14,8 mois (intervalle de 0,9 à 42,5 mois) pour les patients traités avec la dose initiale recommandée de 10 mg de Signifor LAR et de 12,5 mois (intervalle de 0,9 à 45,8 mois) pour les patients traités avec la dose initiale de 30 mg de Signifor LAR.
Les effets indésirables les plus fréquemment signalés (≥ 20%) soupçonnés d'être associés au traitement étaient l'hyperglycémie, la diarrhée, la cholélithiase et le diabète sucré. Des troubles du métabolisme du glucose ont entraîné l’arrêt prématuré de l’étude chez presque 5% des patients.
La fréquence et la gravité de certains effets indésirables semblaient plus élevées dans le groupe ayant reçu la dose initiale la plus élevée de 30 mg.
Affections hématologiques et du système lymphatique*
Fréquent: anémie
Occasionnel: temps de prothrombine prolongé
Affections endocriniennes
Fréquent: insuffisance cortico-surrénalienne
Troubles du métabolisme et de la nutrition
Très fréquent: hyperglycémie (47%), diabète sucré (21%)
Fréquent: diminution de l’appétit, augmentation de l’HbA1c, altération de la tolérance au glucose
Affections du système nerveux
Fréquent: céphalées, vertiges
Affections cardiaques
Fréquent: bradycardie (sinusale)
Occasionnel: allongement de QT
Affections gastro-intestinales
Très fréquent: diarrhée (32%), nausées (15%), douleurs abdominales (14%)
Fréquent: vomissements, lipase augmentée
Occasionnel: amylase augmentée
Fréquence inconnue: Stéatorrhée, selles décolorées
Affections hépatobiliaires
Très fréquent: cholélithiase (31%)
Fréquent: Gamma-GT, ALAT et ASAT augmentées; cholestase (aigüe ou chronique); cholécystite
Troubles généraux et anomalies au site d’administration
Très fréquent: fatigue (14%)
Fréquent: réactions au site d’injection (par ex. douleurs)
Effets indésirables après la commercialisation
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSignes et symptômes
En cas de surdosage, il faut s'attendre à l’apparition plus marquée des symptômes et à la modification des valeurs de laboratoire énumérés sous la rubrique «Effets indésirables». Dans les études cliniques, des doses allant jusqu'à 2,1 mg ont été administrées deux fois par jour chez des volontaires sains; dans ces cas, c’est principalement la diarrhée qui était rapportée.
Traitement
En cas de surdosage, il est recommandé d’instaurer un traitement de soutien approprié, adapté à l’état clinique du patient, jusqu’à la disparition des symptômes.
Propriétés/EffetsCode ATC
H01CB05
Mécanisme d’action
Le pasiréotide (hexapeptide cyclique) est un analogue de la somatostatine qui se lie avec une grande affinité aux récepteurs humains de la somatostatine des sous-types: SSTR 1, 2, 3 et 5.
Pharmacodynamique
Les récepteurs de la somatostatine sont exprimés dans de nombreux tissus, en particulier dans des tumeurs de l’hypophyse et les autres tumeurs neuroendocrines, dans lesquels des hormones, telles que ACTH ou l’hormone de croissance, sont sécrétées en excédent.
Acromégalie: En raison de son profil de liaison étendu concernant les récepteurs de la somatostatine, le pasiréotide possède le potentiel de stimuler aussi bien les sous-types de récepteurs sstr2 que sstr5 qui sont importants pour l’inhibition de la sécrétion de GH et d’IGF-1. Par conséquent, il est potentiellement plus efficace dans le traitement des patients acromégales en comparaison avec d’autres analogues de la somatostatine.
Maladie de Cushing: le pasiréotide lie et active les récepteurs hsst des cellules corticotropes en adénomes producteurs d’ACTH, ce qui inhibe la sécrétion de l’ACTH. Le pasiréotide montre sur ce point une forte affinité avec quatre des cinq hsst, notamment hsst5.
Pharmacodynamique de sécurité
Métabolisme du glucose
La survenue d’une hyperglycémie après administration s.c. de pasiréotide était corrélée à une diminution significative tant de la sécrétion d’insuline que des hormones incrétines (c.-à-d. glucagon-like peptide-1 [GLP-1] et polypeptide insulinotrope dépendant du glucose [GIP]). Le pasiréotide n’avait aucune influence sur la sensibilité à l’insuline. Chez les sujets sains, un traitement à base d’incrétines (agonistes du GLP-1 et inhibiteurs de la DDP-4) a montré le plus d’efficacité dans le traitement de l’hyperglycémie associée au pasiréotide.
Électrophysiologie cardiaque
L’effet de Signifor administré par voie s.c. sur l’intervalle QT a été évalué dans deux études sur le QT. Dans les deux études, un effet du pasiréotide sur l’intervalle QT a été observé. Dans l’une des études réalisées avec une dose de 1950 µg deux fois par jour, l’allongement du QTcF moyen maximal ajusté en fonction du placebo était de 17,5 ms (IC à 90% : 15,53; 19,38). Dans l’autre étude réalisée à des doses de 600 µg deux fois par jour et de 1950 µg deux fois par jour, l’allongement du QTcI moyen ajusté en fonction du placebo était de 13,19 ms (IC à 90% : 11,38; 15,01) et 16,12 ms respectivement (IC à 90% : 14,30 ; 17,95 ms). Une réduction de la fréquence cardiaque est survenue aux deux posologies, avec un écart maximal par rapport au placebo après 1 heure pour le pasiréotide 600 µg deux fois par jour (-10,39 bpm) et après 30 minutes pour le pasiréotide 1950 µg deux fois par jour (-14,91 bpm).
Les concentrations maximales prédites pour la dose maximale de Signifor LAR de 40 mg chez les patients atteints de la maladie de Cushing et présentant une fonction hépatique normale et de 20 mg chez les patients présentant une insuffisance hépatique de gravité modérée étaient de 14 ng/ml et 11,7 ng/ml. Ces deux valeurs sont inférieures aux concentrations maximales observées dans les deux études précitées.
Les concentrations maximales prédites pour la dose maximale de Signifor LAR de 60 mg chez les patients acromégales avec fonction hépatique normale et de 40 mg chez les patients acromégales avec une insuffisance hépatique modérée de 25,8 ng/ml et 28,8 ng/ml sont similaires aux concentrations maximales observées sous Signifor s.c. 600 microgrammes deux fois par jour (24,3 ng/ml) et se situent en-dessous de la concentration maximale observée de 1 950 microgrammes deux fois par jour (80,6 ng/ml).
L’allongement de l’intervalle QT pendant l’administration de pasiréotide n’est pas déterminé par un effet du canal potassique hERG. La restitution cardiaque, c.-à-d. la capacité du cœur à se reposer après chaque battement précédent, a été examinée au cours d’un ECG continu de 24 heures, pour déterminer l’action du pasiréotide sur la susceptibilité aux arythmies. Le pasiréotide a amélioré de manière significative tous les paramètres de restitution en présence d’un allongement QT. Cela indique que l’allongement QT généré par le pasiréotide n’est potentiellement pas associé à une augmentation du risque pro-arythmique. Une autre analyse quantitative de la morphologie de l’onde T n’a montré aucune modification qui pourrait indiquer une atteinte à l’hétérogénéité spatiale de la repolarisation du cœur pendant le traitement par pasiréotide.
Efficacité clinique
Acromégalie
L’efficacité et la sécurité de Signifor LAR chez des patients atteints d’acromégalie active ont été évaluées dans deux études menées en aveugle avec contrôle par substance active. Le principal critère d’efficacité était défini comme la comparaison du pourcentage de patients qui avaient atteint un contrôle biochimique (défini comme taux GH moyen < 2,5 microgrammes/l et normalisation du taux IGF-1 ajusté au sexe et à l’âge).
Une étude a inclus 358 patients pour lesquels une adénectomie avait échoué ou une opération n’était pas indiquée (contre-indications, non approbation de l’opération). Le principal critère d’efficacité a été analysé au bout de 12 mois. Le pourcentage de patients ayant atteint un contrôle biochimique était de 31,3% et 19,2% pour le pasiréotide LAR et l’octréotide LAR (p = 0,007). Un contrôle biochimique a été rapidement atteint dans les deux bras de l’étude (c.-à-d. au mois 3).
Le pourcentage de patients présentant une diminution du volume de la tumeur de plus de 20 % au mois 12 était de 80,8% pour le pasiréotide en intramusculaire et de 77,4% pour l’octréotide LAR. La détermination de la qualité de vie au mois 12 a montré, en comparaison avec la valeur initiale, des améliorations statistiquement significatives du score de qualité de vie physique et psychologique ainsi que du score AcroQol aussi bien sous Signifor LAR que sous octréotide.
Pendant la phase d’extension, 74 patients ont continué d’être traités par pasiréotide intramusculaire et 46 patients par Octréotide LAR. Au mois 25, 48,6% des patients (36/74) du groupe pasiréotide intramusculaire et 45,7% (21/46) du groupe Octréotide LAR ont atteint un contrôle biochimique. Le pourcentage de patients qui avaient atteint au même moment un taux GH moyen inférieur à 2,5 microgrammes/l et une normalisation du taux de l’IGF-1 était comparable dans les deux bras de traitement. Pendant la phase d’extension, le volume de la tumeur avait continuellement réduit et l’amélioration des symptômes de l’acromégalie restait comparable entre les deux bras de traitement. Les scores AcroQol étaient plus élevés dans le bras pasiréotide intramusculaire que dans le bras Octréotide LAR pendant toute la durée de la phase d’extension.
Dans une étude réalisée auprès de patients acromégales préalablement traités, insuffisamment contrôlés (définis comme patients ayant une concentration GH moyenne sur un profil 5 points sur une durée de 2 heures >2,5 microgrammes/l et un taux de l’IGF-1 ajusté au sexe et à l’âge 1,3 fois supérieur à la LSN), [les patients ont dû être traités pendant au moins 6 mois avant la randomisation avec la dose maximale indiquée de Sandostatine LAR (30 mg) ou de Lanréotide ATG (120 mg)], soit pasiréotide 40 mg (n=65) ou 60 mg (n=65) soit le médicament précédent (n=68) a été administré. Deux tiers des patients avaient déjà subi une opération. La valeur initiale moyenne du GH était de 17,6 microgrammes/l, 12,1 microgrammes/l et 9,5 microgrammes/l dans le groupe recevant 40 mg, celui recevant 60 mg et le groupe de contrôle actif. Les valeurs IGF-1 moyennes se situaient au début de l’étude à 2,6, 2,8 et 2,9 x LSN.
Le pourcentage de patients ayant atteint un contrôle biochimique à la semaine 24 se montait à 15,4% (p = 0,006) ou 20,0% (p <0,0001) pour pasiréotide intramusculaire 40 mg ou 60 mg en comparaison avec 0% dans le bras de contrôle (Octréotide LAR, Lanréotide ATG).
Le pourcentage de patients qui ont montré une réduction ou une absence de modification du volume de la tumeur hypophysaire à la semaine 24, était de 81,0% et 70,3% sous pasiréotide intramusculaire 40 mg ou 60 mg et 50,0% dans le groupe de contrôle. Par ailleurs, un plus grand pourcentage de patients a obtenu sous pasiréotide intramusculaire (18,5% et 10,8% pour 40 mg et 60 mg) une réduction du volume de la tumeur d’au moins 25% en comparaison avec le comparateur actif (1,5%). À la semaine 24, des améliorations du score de la qualité de vie physique et psychologique ainsi que du score global AcroQol ont été identifiées aussi bien dans le groupe pasiréotide intramusculaire à 40 mg que dans celui à 60 mg. Dans le groupe de traitement par pasiréotide 40 mg intramusculaire, par rapport aux valeurs initiales, ces modifications ont été statistiquement significatives dans le sous-score AcroQol de la qualité de vie physique. Dans le groupe recevant le pasiréotide 60 mg intramusculaire, ces modifications ont été statistiquement significatives aussi bien dans les sous-scores pour la qualité de vie physique et psychologique que pour le score global. Dans le groupe Octréotide LAR et le groupe Lanréotide ATG, aucune modification statistiquement significative n’a été constatée par rapport aux valeurs initiales. L’amélioration moyenne par rapport à la valeur initiale était plus grande pour tous les scores dans le groupe recevant le pasiréotide 60 mg en intramusculaire. Toutefois, les différences entre les modifications à la semaine 24 en comparaison avec la valeur initiale n’étaient pas statistiquement significatives entre les groupes de traitement
Maladie de Cushing
L'efficacité et l'innocuité de Signifor LAR ont été étudiées dans une étude de phase III multicentrique, en double aveugle et randomisée 1:1 chez n = 150 patients atteints de la maladie de Cushing persistante ou récidivante après une résection d'adénome et chez des patients pour lesquels une chirurgie de l'hypophyse n'était pas envisageable. Les patients inclus présentaient un cortisol urinaire libre moyen de 24 heures (CLUm) entre ≥ 1,5 et ≤ 5x LSN. Les patients ont été traités pendant 12 mois et ont initialement reçu 10 mg ou 30 mg de Signifor LAR i.m. tous les 28 jours.
Après quatre mois de traitement, les patients ayant un CLU moyen ≤ 1,5x LSN ont continué à recevoir la dose à laquelle ils avaient été randomisés. Pour les patients ayant un CLU moyen > 1,5x LSN après quatre mois de traitement, la dose a été augmentée respectivement de 10 mg à 30 mg ou de 30 mg à 40 mg. Si le CLU était toujours > 1x LSN, une nouvelle augmentation de dose pouvait se produire après 7 et 9 mois, la dose maximale étant fixée à 40 mg. En cas d'intolérance, la dose pouvait être ajustée à tout moment vers le bas, la dose minimale possible étant de 5 mg.
Le critère d'évaluation primaire de l'efficacité était la proportion de patients pour lesquels les valeurs moyennes de CLU s'étaient normalisées après 7 mois de traitement (c.-à-d. CLU ≤ ULN; = «répondant»), indépendamment d'une augmentation possible de la dose au mois 4. Les patients sortis avant le mois 4 ont été classés comme non-répondants.
Le principal critère d'évaluation secondaire était la proportion de patients répondants CLUm après 7 mois sans que leur dose n’ait été augmentée jusqu'à ce moment-là. Les autres paramètres secondaires étaient les changements par rapport aux valeurs de référence dans le test CLU sur 24 heures, l'ACTH plasmatique, les taux de cortisol sérique, les symptômes cliniques de la maladie de Cushing et la qualité de vie liée à la santé (selon SF-12v2 et CushingQoL). Le critère d'efficacité a été défini comme suit: le taux de réponse sous la limite de l'intervalle de confiance à 95% devait être > 15 %. Ce critère a été atteint dans les deux groupes. Dans le groupe 10 mg, le taux de réponse après 7 mois était de 41, 9% (IC à 95%: 30,5-53,9%), et dans le groupe 30 mg, il était de 40,8% (IC à 95%: 29,7-52,7%). La réduction du CLU s'est maintenue pendant toute la durée du traitement. Deux strates ont été distinguées sur la base de la CLU moyenne de référence: Chez les patients présentant un CLU compris entre 1,5 et 2x LSN, le taux de réponse dans les deux groupes était de 52% (IC à 95%: 31-72%). Chez les patients présentant un CLU > 2x LSN par contre, le taux de réponse était de 37 % dans le groupe recevant 10 mg (IC à 95%: 23-52%) et de 35% dans le groupe recevant 30 mg (IC à 95%: 22-50 %).
La dose a été augmentée au mois 4 pour 31 des 74 patients du groupe recevant 10 mg et pour 28 des 76 patients du groupe recevant 30 mg. Dans le principal critère d'évaluation secondaire (c.-à-d. la proportion de répondants sans augmentation de dose antérieure), 28,4% (IC à 95%: 18,5-40,1%) des patients ont répondu après 7 mois dans le groupe recevant 10 mg, et 31,6% (IC à 95%: 21,4-43,3%) dans le groupe recevant 30 mg.
Après 7 mois, une réduction cliniquement pertinente de la pression artérielle systolique et diastolique en position couchée et une réduction du poids corporel ont été observées dans les deux groupes. Les changements dans les deux paramètres avaient tendance à être plus marqués chez les patients classés dans la catégorie des répondants CLUm. La même tendance a été observée après 12 mois.
Après 7 mois, la majorité des patients montrait une amélioration ou une stabilisation des symptômes cliniques par rapport aux résultats initiaux. La rougeur du visage s'est améliorée chez 43,5% des patients, et un bon tiers des patients ont montré une amélioration des coussinets adipeux supraclaviculaires et dorsaux. Il y a également eu une amélioration de la qualité de vie, quoique non significative sur le plan statistique.
PharmacocinétiqueAbsorption
Le plateau de concentration après administration de pasiréotide sous forme de Signifor LAR est atteint au bout de quatre mois. Après des doses mensuelles répétées de Signifor LAR, le pasiréotide présente une exposition pharmacocinétique approximativement proportionnelle à la dose.
Distribution
Le pasiréotide a un volume de distribution (Vz/F) > 100 l et il est présent principalement dans le plasma (91%). La liaison aux protéines plasmatiques est modérée (88%) et est indépendante de la concentration.
Le pasiréotide est vraisemblablement un substrat de la P-gp, le passage dans le liquide céphalo-rachidien n’a pas été évalué.
Métabolisme
Le pasiréotide s’est avéré avoir une stabilité métabolique très élevée dans les microsomes hépatiques et rénaux humains.
Élimination
Le pasiréotide est éliminé essentiellement par les voies biliaires sous forme inchangée et seulement dans une faible mesure par les reins. La clairance est de 4,5-8,5 l/h et la demi-vie d’élimination est d’environ 16 jours. 55,9 ± 6,63% du pasiréotide radiomarqué ont été retrouvés pendant les 10 premiers jours après l’injection, dont 48,3 ± 8,16% de la radioactivité ont été mesurés dans les fèces et 7,63 ± 2,03% dans les urines.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Chez des patients atteints d’une insuffisance hépatique modérée à sévère (Child-Pugh B et C) et ayant reçu une dose unique de Signifor s.c., des expositions significativement élevées ont été observées en comparaison aux sujets présentant une fonction hépatique normale. Dans les groupes présentant une insuffisance hépatique modérée à sévère, l’AUCinf a augmenté de 60% et 79%, la Cmax de 67% et 69% et la CL/F a diminué de 37% et 44% par rapport au groupe de contrôle.
Troubles de la fonction rénale
Dans une étude menée chez des patients souffrant d’une insuffisance rénale légère, modérée ou sévère, ainsi que d’une insuffisance rénale terminale, la pharmacocinétique du pasiréotide après administration d’une dose unique de 0,9 mg par voie sous-cutanée n’était pas significativement différente de celle de sujets ayant une fonction rénale normale.
Patients âgés
Des analyses de cinétique des populations indiquent que la pharmacocinétique du pasiréotide n’est pas notablement influencée par l’âge. On ne dispose toutefois d’aucune étude pharmacocinétique spécifique menée chez des patients de ≥65 ans.
Enfants et adolescents
La pharmacocinétique du pasiréotide n’a pas été étudiée dans ce groupe d’âge.
Sexe, ethnicité et poids corporel
Les analyses pharmacocinétiques de population suggèrent que le sexe, le poids corporel et l’appartenance ethnique, n’ont pas d’influence cliniquement pertinente sur les paramètres pharmacocinétiques de Signifor LAR.
Données précliniquesLes études de sécurité non cliniques qui ont été menées avec le pasiréotide administré par voie sous-cutanée ont porté sur la pharmacologie de sécurité, la toxicité après administration répétée, la génotoxicité, la carcinogénicité et la toxicité de reproduction et de développement. Par ailleurs, des études sur la tolérance et la toxicité en cas d’administration répétée de Pasiréotide LAR en injection intramusculaire ont également été réalisées. La plupart des observations notées dans les études de toxicité après administration répétée ont été réversibles et imputables à la pharmacologie du pasiréotide. Les effets constatés dans les études précliniques ont généralement été observés à des expositions comparables ou supérieures à l’exposition maximale après des doses thérapeutiques humaines.
Pharmacologie de sécurité
Dans les études de pharmacologie de sécurité (avec le pasiréotide en injection s.c.), le pasiréotide n’a pas entraîné d’effets indésirables sur les fonctions respiratoires ou cardiovasculaires. Une diminution de l’activité générale et de l’activité comportementale a été observée chez la souris à une dose de 12 mg/kg, ce qui correspond à 27 fois la dose journalière maximale recommandée pour Pasiréotide LAR par rapport à la masse corporelle.
Mutagénicité
Le pasiréotide s’est avéré non génotoxique dans deux tests de génotoxicité in vitro (test d’Ames et test d’aberration chromosomique sur des lymphocytes périphériques humains). Le pasiréotide n’a pas eu d’effets génotoxiques dans un test du micronoyau sur moelle osseuse in vivo chez le rat, à des doses atteignant jusqu’à 50 mg/kg. Ceci correspond à une dose presque 224 fois supérieure à la dose journalière maximale recommandée de pasiréotide par rapport à la masse corporelle.
Carcinogénicité
Les études de carcinogénicité chez le rat et la souris transgénique n’ont pas montré de potentiel carcinogène.
Toxicité sur la reproduction
Dans des études sur le développement embryofœtal chez le rat et le lapin, le pasiréotide en injection sous-cutanée s’est avéré non tératogène à des doses induisant une toxicité maternelle (correspondant à 10 mg/kg/jour [rat] et 5 mg/kg/jour [lapin]) qui ont conduit à une exposition (AUC0- 24 h) correspondant à 106 fois voire 30 fois la DMRH de Pasiréotide LAR. À des doses de 10 mg/kg/jour chez le rat, une fréquence accrue de résorptions précoces/totales et de déformations des membres a été notée. À des doses de 5 mg/kg/jour chez le lapin, une fréquence accrue d’avortements, de réduction du poids des fœtus et ultérieurement de modifications du squelette a été constatée. La réduction du poids des fœtus et ultérieurement un retard de formation osseuse ont été observés à une dose de 1 mg/kg/jour (exposition 4.8 fois supérieure à la DMRH pour Pasiréotide LAR). Dans une étude pré- et postnatale réalisée chez le rat, le pasiréotide n’a eu aucun effet sur le travail et la mise bas jusqu’à une dose de 10 mg/kg/jour (45 fois la DMRH pour Pasiréotide LAR par rapport à la masse corporelle).
Le pasiréotide est excrété dans le lait. Un retard dans la croissance physiologique des jeunes animaux a été observé à une dose de 2 mg/kg/jour (9 fois supérieure à la dose journalière maximale recommandée de Signifor LAR par rapport à la masse corporelle). Après le sevrage, la prise de poids chez les jeunes rats ayant arrêté le pasiréotide a été comparable à celle des animaux témoins, ce qui indique que cet effet est réversible.
Des doses de pasiréotide allant jusqu’à 10 mg/kg/jour (45 fois la dose journalière recommandée de Signifor LAR, par rapport à la masse corporelle) n’ont pas eu d’influence sur la fertilité des rats mâles. Comme attendu en raison de la pharmacologie du pasiréotide, la fertilité des rats femelles à des doses quotidiennes de 0,1 mg/kg/jour (0,5 fois la dose journalière maximale recommandée de Pasiréotide LAR par rapport à la masse corporelle) a diminué, comme l’a montré le nombre réduit de corps jaunes et de sites d’implantation. Des cycles anormaux ou une absence de cycle ont été retrouvés à des doses de 1 mg/kg/jour.
Remarques particulièresIncompatibilités
Signifor LAR poudre pour solution injectable doit être utilisée en tant que récipient unidose et ne doit pas être mélangée avec d’autres médicaments. C’est la raison pour laquelle aucune donnée de tolérance avec d’autres produits n’a été collectée.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l’emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8°C).
Ne pas congeler.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Instructions de préparation et d’injection intramusculaire de Signifor LAR
UNIQUEMENT POUR INJECTION INTRAMUSCULAIRE PROFONDE
PRUDENCE :
Il existe 2 étapes critiques pour la reconstitution de Signifor LAR. Le non-respect de ces étapes peut empêcher le médicament d’atteindre correctement le site d’action.
Le kit d’injection doit être amené à température ambiante. Sortez le kit d’injection du réfrigérateur et laissez-le au moins 30 minutes, mais en aucun cas plus de 24 heures, à température ambiante avant de le reconstituer.
Après avoir ajouté le solvant, secouez le flacon légèrement dans le sens horizontal pendant au moins 30 secondes, jusqu’à obtenir une suspension homogène.
Contenu:
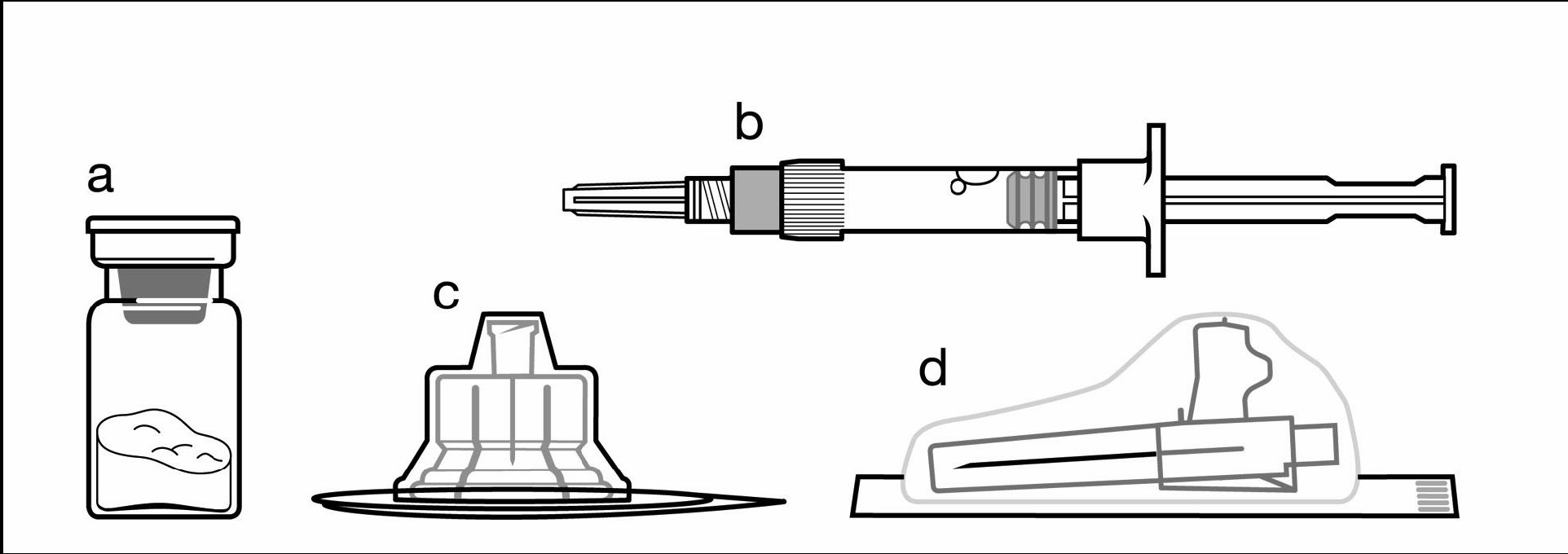
a Un flacon de Signifor LAR poudre
b Une seringue préremplie de solvant pour la reconstitution
c Un adaptateur pour le flacon en vue de la reconstitution du médicament
d Une aiguille de sécurité (20G x 1.5")
Suivez scrupuleusement les instructions ci-dessous pour garantir une reconstitution correcte de Signifor LAR avant d’effectuer l’injection intramusculaire profonde.
La suspension Signifor LAR doit être préparée avant l’administration immédiatement avant l’injection.
Signifor LAR ne doit être administré que par un personnel médical formé.
|
Étape 1
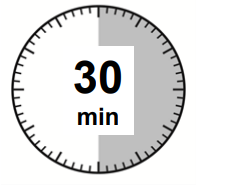
Sortez le kit d’injection Signifor LAR du réfrigérateur.
PRUDENCE: Il est extrêmement important de ne commencer la procédure de reconstitution qu’après que le kit d’injection a atteint la température ambiante. Laissez le kit au moins 30 minutes, mais en aucun cas plus de 24 heures, à température ambiante avant de le reconstituer.
Remarque: le kit d’injection peut, si nécessaire, être réfrigéré de nouveau.
|
|
| |
Étape 2
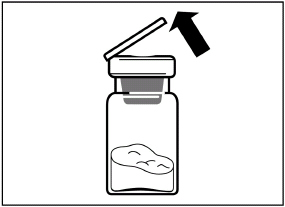
Enlevez le bouchon plastique du flacon et désinfecter le bouchon en caoutchouc avec une compresse d’alcool.
|
|
| |
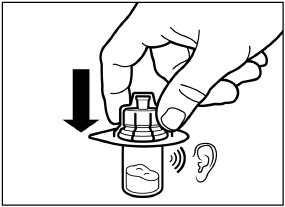
Enlevez le film du couvercle de l’emballage de l’adaptateur pour le flacon, mais ne sortez PAS l’adaptateur de l’emballage.
Tenez l’emballage de l’adaptateur et placez l’adaptateur sur le flacon. Appuyez ensuite entièrement sur l’adaptateur entièrement, jusqu’à ce qu’il s’enclenche. Ce qui sera reconnaissable par un «clic» sonore.
|
|
| |
Enlevez alors l’emballage de l’adaptateur en effectuant un mouvement vertical.
|
|

| |
Étape 3
Enlevez le capuchon de fermeture de la seringue préremplie avec le solvant et vissez la seringue sur l’adaptateur du flacon.
|
|
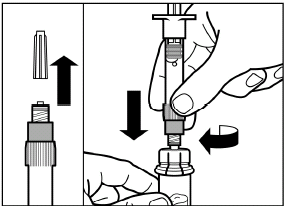
| |
Poussez avec précaution le piston entièrement vers le bas pour injecter la totalité du solvant dans le flacon.
|
|
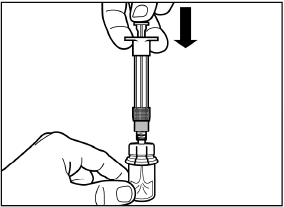
| |
Étape 4
PRUDENCE: Maintenez le piston appuyé et secouez le flacon pendant au moins 30 secondes légèrement dans le sens horizontal jusqu’à ce que la poudre soit complètement en suspension. Secouez encore une fois légèrement pendant 30 secondes si la poudre n’est pas complètement en suspension.
|
|

| |
Étape 5
Retournez la seringue et le flacon (de manière à ce que le flacon soit en haut), tirez le piston lentement en arrière et remplissez la seringue avec tout le contenu du flacon.
|
|

| |
Dévissez la seringue de l’adaptateur.
|
|
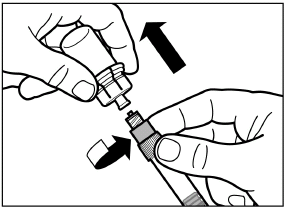
| |
|
| |
|
| |
Étape 6
Vissez l’aiguille de sécurité sur la seringue.
|
|
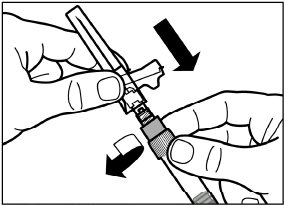
| |
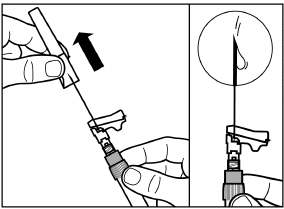
Enlevez le capuchon protecteur de l’aiguille en tirant tout droit.
Pour éviter des dépôts, secouez prudemment la seringue afin de conserver une suspension homogène.
Tapotez avec un doigt prudemment contre la seringue pour éliminer les bulles d’air et expulsez-les de la seringue.
Signifor LAR reconstitué est maintenant prêt pour une administration immédiate.
|
|
| |
Étape 7
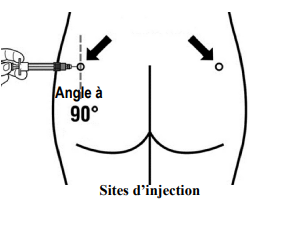
Signifor LAR ne doit être administré qu’en injection intramusculaire profonde, JAMAIS en intraveineuse.
Désinfectez le site d’injection avec une compresse d’alcool.
Piquez l’aiguille complètement dans le fessier gauche ou droit (muscle glutéal) à un angle de 90° par rapport à la surface de la peau.
Tirez lentement le piston en arrière (aspirez) pour garantir qu’aucun vaisseau sanguin n’a été piqué (si un vaisseau sanguin a été piqué, un autre site d’injection doit être choisi).
Poussez lentement le piston jusqu’à ce que la seringue soit vide. Retirez l’aiguille du site d’injection et activez la protection de l’aiguille (comme montré à l’étape 8).
|
|
| |
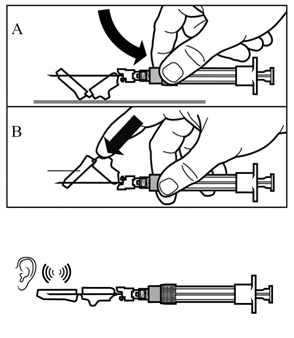
Étape 8
Activez la protection de l’aiguille conformément à l’une des deux procédures suivantes:
·Appuyez fermement la section rabattable de la protection de l’aiguille sur une surface dure (figure A)
·ou poussez la section rabattable avec votre doigt vers l’avant (figure B).
Un «clic» sonore confirme l’activation correcte.
Jetez l’aiguille immédiatement dans un récipient de sécurité.
|
|
|
Précautions spéciales d’élimination
Tout produit non utilisé ou tout produit résiduel doit être éliminé dans les règles conformément aux exigences locales en vigueur.
Numéro d’autorisation65148 (Swissmedic)
PrésentationSignifor LAR 10 mg poudre dans un flacon, 2 ml de solvant pour suspension injectable dans une seringue préremplie. [A]
Signifor LAR 20 mg poudre dans un flacon, 2 ml de solvant pour suspension injectable dans une seringue préremplie. [A]
Signifor LAR 30 mg poudre dans un flacon, 2 ml de solvant pour suspension injectable dans une seringue préremplie. [A]
Signifor LAR 40 mg poudre dans un flacon, 2 ml de solvant pour suspension injectable dans une seringue préremplie. [A]
Signifor LAR 60 mg poudre dans un flacon, 2 ml de solvant pour suspension injectable dans une seringue préremplie. [A]
Titulaire de l’autorisationRecordati AG, 6340 Baar
Mise à jour de l’informationFévrier 2025
|