CompositionPrincipes actifs
Trioxyde d'arsenic
Excipients
Hydroxyde de sodium, acide chlorhydrique, eau pour préparations injectables.
TRISENOX 2 mg/ml contient 4.14 mg de sodium par flacon.
Indications/Possibilités d’emploiTRISENOX est indiqué pour l'induction de la rémission et la consolidation chez des patients atteints de:
·Leucémie promyélocytaire aiguë (LPA) à risque faible ou intermédiaire (numération leucocytaire ≤10 x 103/μl) nouvellement diagnostiquée, en association avec l'acide tout-trans-rétinoïque (ATRA);
·Leucémie promyélocytaire aiguë (LPA) récidivante ou réfractaire (le traitement antérieur doit avoir comporté un rétinoïde et une chimiothérapie);
caractérisée par la présence d'une translocation t(15;17) et/ou la présence du gène PML/RAR-alpha (leucémie promyélocytaire/récepteur alpha de l'acide rétinoïque).
La réponse des autres sous-types de leucémie aiguë myéloblastique à TRISENOX n'a pas été étudiée.
Posologie/Mode d’emploiTRISENOX doit être administré sous la surveillance d'un médecin ayant l'expérience du traitement des leucémies aiguës. On doit suivre les instructions de surveillance décrites à la rubrique «Mises en garde et précautions». La dose recommandée est identique pour les adultes et les sujets âgés.
Leucémie promyélocytaire aiguë (LPA) à risque faible ou intermédiaire nouvellement diagnostiquée
TRISENOX est administré en association avec l'acide tout-trans-rétinoïque (ATRA). Consulter à ce sujet l'information professionnelle de la préparation d'ATRA utilisée.
Traitement d'induction
TRISENOX doit être administré par voie intraveineuse à la dose de 0,15 mg/kg/jour jusqu'à l'obtention d'une rémission complète. Si une rémission complète n'est pas atteinte en l'espace de 60 jours, le traitement doit être abandonné.
L'acide tout-trans-rétinoïque est administré à la dose de 45 mg/m2 par jour jusqu'à l'obtention d'une rémission complète. Si une rémission complète n'est pas atteinte en l'espace de 60 jours, le traitement doit être abandonné.
Traitement de consolidation
TRISENOX doit être administré par voie intraveineuse à la dose de 0,15 mg/kg/jour à raison de 5 jours par semaine. Le traitement doit comprendre au total 4 cycles composés chacun de 4 semaines de traitement suivies de 4 semaines sans traitement.
L'acide tout-trans-rétinoïque est administré à la dose de 45 mg/m2 par jour pour une durée de 7 cycles de traitement composés chacun de 2 semaines de traitement suivies de 2 semaines sans traitement.
Leucémie promyélocytaire aiguë (LPA) récidivante/réfractaire
Traitement d'induction
0,15 mg/kg/jour en perfusion intraveineuse jusqu'à obtention d'une rémission complète (<5 % de blastes dans la moelle osseuse, pas de cellules leucémiques détectables).
Si une rémission complète n'est pas atteinte en l'espace de 50 jours, le traitement par TRISENOX doit être abandonné.
Traitement de consolidation
Le traitement de consolidation doit commencer 3 à 4 semaines après la fin du traitement d'induction.
TRISENOX doit être administré à la dose de 0,15 mg/kg/jour pendant 25 jours. A cet effet, TRISENOX est administré à raison de 5 jours par semaine, suivi par 2 jours d'interruption, et ce pendant 5 semaines.
Suspension, modification et réinitiation du traitement
Le traitement par TRISENOX doit être interrompu temporairement si une toxicité de grade 3 ou plus (critères de toxicité courants du National Cancer Institute, CTCAE, version 4.03 du 14.06.2010) est observée avant la fin prévue du traitement. Le traitement ne pourra être repris qu'après résolution de l'effet toxique ou après retour à l'état initial de l'anomalie ayant provoqué l'interruption.
Dans ce cas, le traitement devra reprendre à 50% de la dose quotidienne précédente. Si l'effet toxique ne se reproduit pas dans les 7 jours suivant la reprise du traitement à la dose réduite, la dose quotidienne pourra repasser à 100 % de la dose originale.
Chez les patients présentant une récurrence de la toxicité, la posologie sera réduite à un niveau égal à 75% - 50% de la dose originale. Le traitement par TRISENOX sera abandonné chez les patients qui ne tolèrent pas cette posologie.
Pour des valeurs d'ECG et d'électrolytes anormales et pour l'hépatotoxicité, voir rubrique «Mises en garde et précautions».
Les instructions fournies dans l'information professionnelle de l'ATRA doivent être respectées et les ajustements posologiques recommandés doivent être appliqués.
Patients présentant des troubles de la fonction hépatique
Etant donné l'absence de données sur tous les types d'insuffisance hépatique et vu que des effets indésirables hépatotoxiques peuvent se produire sous TRISENOX, la prudence est de rigueur lors de l'utilisation de TRISENOX chez les patients insuffisants hépatiques (voir la rubrique «Effets indésirables»).
Patients présentant des troubles de la fonction rénale
Etant donné l'absence de données sur tous les types d'insuffisance rénale, la prudence est de rigueur lors de l'utilisation de TRISENOX chez les patients insuffisants rénaux.
Utilisation chez les patients âgés
On ne dispose que de peu de données cliniques sur l'emploi de TRISENOX chez les patients âgés. Par conséquent, une prudence particulière est de rigueur dans cette population.
Enfants et adolescents
Les données disponibles pour les enfants de 5 à 18 ans ne sont que limitées (voir rubrique «Propriétés/Effets»). La dose recommandée chez l'enfant est de 0,15 mg/kg/jour en perfusion intraveineuse. Aucune donnée n'est disponible chez les enfants de moins de 5 ans.
Mode d'administration
TRISENOX doit être administré en perfusion intraveineuse de 1 à 2 heures. La durée de la perfusion peut être portée à 4 heures en cas de réactions vasomotrices. Aucun cathéter veineux central n'est nécessaire. Les patients doivent être hospitalisés au début du traitement en raison des symptômes de la maladie et afin d'assurer une surveillance adéquate.
Pour les instructions concernant la préparation du médicament, voir «Remarques particulières».
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients.
Mises en garde et précautionsLes patients cliniquement instables atteints de LPA sont particulièrement à risque et nécessiteront un contrôle plus fréquent du profil électrolytique et de la glycémie, ainsi que des bilans hématologique, hépatique, rénal et de coagulation plus fréquents.
Syndrome d'activation des leucocytes (Syndrome de différenciation LPA)
Dans les études cliniques, 27 % des patients atteints de LPA récidivante/réfractaire et traités par TRISENOX sans administration préventive de corticostéroïdes ont présenté des symptômes analogues à ceux du syndrome de l'acide rétinoïque-LPA (RA-APL), ou syndrome de différenciation LPA.
Ce syndrome est caractérisé par une fièvre, une dyspnée, une prise de poids, des infiltrats pulmonaires et des épanchements pleuraux ou péricardiques, avec ou sans hyperleucocytose, et peut avoir une issue fatale.
Pour prévenir la survenue d'un syndrome de différenciation LPA pendant le traitement d'induction chez les patients atteints de LPA, on peut administrer de la prednisone (0,5 mg/kg/jour pendant toute la durée du traitement d'induction) à partir du premier jour du traitement d'induction.
Même après l'administration préventive de prednisone à la dose de 0,5 mg/kg/jour, 15 (19 %) des patients atteints de LPA nouvellement diagnostiquée et traités par TRISENOX en association avec l'acide tout-trans-rétinoïque (ATRA) ont développé un syndrome de différenciation LPA. Chez 5 (6%) des patients traités par TRISENOX en association avec l'acide tout-trans-rétinoïque (ATRA), le syndrome de différenciation a été jugé sévère.
Dès les premiers signes évoquant ce syndrome (fièvre inexpliquée, dyspnée et/ou prise de poids, constats anormaux à l'auscultation thoracique ou à la radiographie), le traitement par TRISENOX et ATRA doit être temporairement interrompu et une corticothérapie à haute dose (dexaméthasone 10 mg deux fois par jour par voie intraveineuse) doit immédiatement être instaurée indépendamment de la numération leucocytaire et poursuivie 3 jours ou plus, jusqu'à la régression des symptômes. Un traitement diurétique complémentaire est également recommandé s'il est cliniquement justifié/nécessaire.
Chez la majorité des patients, l'arrêt définitif du traitement par TRISENOX n'est pas nécessaire pendant le traitement du syndrome de différenciation LPA.
Après la régression des signes et symptômes, le traitement par TRISENOX et ou l'ATRA peut être repris en utilisant pendant les 7 premiers jours 50% de la dose précédente. En l'absence d'aggravation dans le sens de la toxicité subie antérieurement, le traitement par TRISENOX et/ou l'ATRA peut ensuite être poursuivi à la dose complète. Si cela entraîne une réapparition des symptômes, il faut revenir à la dose réduite précédente de TRISENOX et/ou d'ATRA.
Il est recommandé de ne pas administrer de chimiothérapie parallèlement à la corticothérapie, étant donné qu'on ne dispose d'aucune expérience concernant l'administration conjointe de corticostéroïdes et d'une chimiothérapie lors du traitement du syndrome d'activation des leucocytes induit par TRISENOX.
Anomalies de l'électrocardiogramme (ECG):
Le trioxyde d'arsenic peut occasionner une prolongation de l'intervalle QT et un bloc auriculo-ventriculaire complet. La prolongation de l'intervalle QT peut aboutir à une arythmie ventriculaire de type torsades de pointes, qui peut être fatale.
Tout traitement antérieur à base d'anthracyclines peut accroître le risque de prolongation de l'intervalle QT. Le risque de torsades de pointes est en corrélation avec le degré de prolongation de l'intervalle QT, avec l'administration concomitante de produits prolongeant
l'intervalle QT (tels que les antiarythmiques de classe Ia et III (ex.: quinidine, amiodarone, sotalol, dofétilide), les antipsychotiques (ex.: thioridazine), les antidépresseurs (ex.: amitriptyline), certains macrolides (ex.: érythromycine), certains antihistaminiques, certains antibiotiques de la famille des quinolones et autres médicaments connus pour prolonger l'intervalle QT, avec des antécédents de torsades de pointes, avec une prolongation préexistante de l'intervalle QT, avec une insuffisance cardiaque, avec l'administration de diurétiques éliminant le potassium, d'amphotéricine B ou avec une autre affection entraînant une hypokaliémie ou une hypomagnésémie.
Dans les études cliniques, 16 sur 40 (40%) des patients atteints de LPA récidivante/réfractaire et traités par TRISENOX ont subi au moins une prolongation de l'intervalle QT corrigé (QTc) supérieure à 500 msec.
La prolongation de l'intervalle QTc a été observée 1 à 5 semaines après la perfusion de TRISENOX, avec retour à la valeur initiale au terme de la 8ème semaine suivant la perfusion de TRISENOX. Une patiente (recevant plusieurs médicaments concomitants, dont l'amphotéricine B) a présenté un phénomène de torsades de pointes asymptomatique pendant le traitement d'induction d'une rechute de LPA par le trioxyde d'arsenic.
Dans les études cliniques, 12 sur 77 (15,6%) des patients avec une LPA nouvellement diagnostiquée ont présenté un allongement du QTc sous traitement par le trioxyde d'arsenic en association avec l'ATRA (voir la section «Patients atteints de LPA nouvellement diagnostiquée à risque non élevé» dans la rubrique «Propriétés/Effets»). Chez un patient atteint d'une LPA nouvellement diagnostiquée, le traitement d'induction a été interrompu en raison d'une prolongation importante de l'intervalle QTc et d'anomalies électrolytiques le troisième jour du traitement d'induction.
Recommandations de contrôle de l'ECG et du profil électrolytique
Avant de commencer un traitement par TRISENOX, un ECG doit être pratiqué, ainsi qu'un dosage sérique des électrolytes et de la créatinine.
Les anomalies électrolytiques préexistantes seront corrigées, les traitements connus pour prolonger l'intervalle QT seront interrompus si possible, et les patients présentant des facteurs de risque de prolongation de QTc ou des facteurs de risque de torsades de pointes devront faire l'objet d'une surveillance.
Pendant le traitement par TRISENOX, on veillera à maintenir constamment la kaliémie à plus de 4 mEq/l et la magnésémie à plus de 1,8 mg/dl. Les patients dont l'intervalle QT atteint une valeur absolue > 500 msec doivent être réévalués et une action immédiate sera entreprise pour corriger les éventuels facteurs de risque concomitants; l'avis d'un spécialiste devrait également être sollicité si possible et le rapport bénéfices/risques doit être comparé entre la poursuite et l'interruption du traitement par TRISENOX.
En cas de syncope ou d'irrégularités du rythme cardiaque, le patient devra être hospitalisé et surveillé en continu, un dosage sérique des électrolytes sera pratiqué et le traitement par TRISENOX sera suspendu jusqu'à ce que l'intervalle QTc repasse sous 460 msec, que les anomalies électrolytiques soient corrigées et que la syncope et les irrégularités du rythme cardiaque disparaissent. Après le rétablissement, le traitement doit être repris à une dose égale à 50% de la dose quotidienne précédente. En l'absence d'une réapparition de l'allongement du QTc dans les 7 jours suivant la reprise du traitement à la dose réduite, TRISENOX peut être administré la deuxième semaine à la dose de 0,11 mg/kg/jour. Si le patient ne présente toujours pas d'allongement du QTc, la dose quotidienne initiale peut être reprise à 100%. On ne dispose pas d'informations sur l'influence de TRISENOX sur l'intervalle QTc pendant la perfusion.
Un ECG sera effectué deux fois par semaine, et plus fréquemment pour les patients cliniquement instables, pendant les phases d'induction et de consolidation.
Hépatotoxicité (grade 3 ou plus)
Dans les études cliniques, 43 sur 77 (63,2%) des patients atteints d'une LPA nouvellement diagnostiquée à risque faible ou intermédiaire ont subi des effets hépatotoxiques de grade 3 ou 4 sous traitement d'induction ou de consolidation par TRISENOX en association avec l'ATRA. Les effets toxiques ont toutefois régressé après l'arrêt temporaire de TRISENOX, de l'ATRA ou des deux (voir les rubriques «Propriétés/Effets» et «Effets indésirables»). Si une hépatotoxicité de grade 3 ou plus selon les critères de toxicité courants du National Cancer Institute est observée à un moment donné, le traitement par TRISENOX doit être interrompu avant sa fin prévue. Après régression des taux de bilirubine et/ou d'ASAT et/ou de phosphatase alcaline à une valeur inférieure à 4 fois la limite supérieure de la normale, le traitement par TRISENOX doit être repris à une dose égale à 50% de la dose précédente pendant les 7 premiers jours. En l'absence d'aggravation de la toxicité antérieure, le traitement par TRISENOX peut ensuite être poursuivi à la dose complète. Si l'hépatotoxicité réapparaît, le traitement par TRISENOX doit être définitivement abandonné.
Analyses de laboratoire
Le profil électrolytique, la glycémie, ainsi que les bilans hématologique, hépatique, rénal et de coagulation doivent être surveillés au moins deux fois par semaine, et plus fréquemment pour les patients cliniquement instables, pendant la phase d'induction et au moins une fois par semaine pendant la phase de consolidation.
Hyperleucocytose
Certains patients atteints de LPA récidivante/réfractaire ont développé une hyperleucocytose (≥10 x 103/μl) sous TRISENOX. Aucune corrélation n'est trouvée entre la numération leucocytaire initiale et le développement d'une hyperleucocytose ou le taux maximal de leucocytes constaté.
L'hyperleucocytose n'a jamais été traitée par une chimiothérapie complémentaire; elle a régressé au cours de la poursuite du traitement par TRISENOX. À l'exception d'un patient chez lequel on a trouvé 22 x 103 leucocytes/μl pendant le traitement de consolidation, le taux de leucocytes était plus bas pendant la phase de consolidation que dans la phase d'induction, avec des valeurs <10 x 103/μl.
Les études cliniques auprès de patients atteints de LPA récidivante/réfractaire montrent qu'une hyperleucocytose se développe en phase d'induction chez ~10% des patients, dont ~3% avec une sévérité de grade 3 ou 4. Les taux de leucocytes ont cependant baissé ou se sont normalisés avant l'obtention d'une rémission médullaire, si bien qu'il n'a pas été nécessaire de recourir à une chimiothérapie cytotoxique ou à une leucophérèse.
Dans les études cliniques, une hyperleucocytose a été observée dans le cadre du traitement d'induction chez 35 sur 74 (47%) des patients atteints de LPA nouvellement diagnostiquée à risque faible ou intermédiaire. Tous ces cas ont cependant été traités avec succès par l'hydroxycarbamide.
Les patients atteints de LPA nouvellement diagnostiquée ou de LPA récidivante/réfractaire qui développent une hyperleucocytose persistante après le début du traitement doivent recevoir de l'hydroxycarbamide. L'administration d'hydroxycarbamide doit être poursuivie à une dose permettant de maintenir la numération leucocytaire à ≤10 x 103/μl, la dose étant ensuite diminuée progressivement.
Tableau 1
|
Recommandation pour le début du traitement par l'hydroxycarbamide
| |
Taux de leucocytes
|
Hydroxycarbamide
| |
10–50 x 103/μl
|
500 mg quatre fois par jour
| |
>50 x 103/μl
|
1'000 mg quatre fois par jour
|
Développement de nouvelles tumeurs primaires
Le trioxyde d'arsenic, principe actif de TRISENOX, est cancérogène chez l'homme. Les patients doivent être surveillés quant au développement de nouvelles tumeurs primaires.
Encéphalopathie
Des cas d'encéphalopathie ont été signalés sous trioxyde d'arsenic. Une encéphalopathie de Wernicke a été rapportée après le traitement par le trioxyde d'arsenic chez des patients qui présentaient une carence en vitamine B1. Les patients présentant un risque de carence en vitamine B1 doivent être surveillés étroitement quant aux signes et symptômes d'une encéphalopathie après l'instauration du traitement par le trioxyde d'arsenic. Dans certains cas, une supplémentation en vitamine B1 a fait régresser la maladie.
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsIl n'existe aucune étude formelle des interactions pharmacocinétiques entre TRISENOX et les autres produits thérapeutiques.
Médicaments connus pour causer un allongement de l'intervalle QT/QTc, une hypokaliémie ou une hypomagnésémie
Une prolongation de l'intervalle QT/QTc est attendue sous traitement par TRISENOX, et des cas de torsades de pointes et de bloc cardiaque complet ont été décrits.
Les patients qui reçoivent ou ont reçu des médicaments connus pour provoquer une hypokaliémie ou une hypomagnésémie, comme les diurétiques ou l'amphotéricine B, peuvent avoir un risque supérieur de torsades de pointes. La prudence s'impose lorsque TRISENOX est coadministré avec des médicaments connus pour prolonger l'intervalle QT/QTc, comme les macrolides, le neuroleptique thioridazine ou les composés connus pour provoquer une hypokaliémie ou une hypomagnésémie.
Médicaments connus pour provoquer des effets hépatotoxiques
Des effets hépatotoxiques peuvent apparaître sous TRISENOX. La prudence est de rigueur lors de l'utilisation de TRISENOX en association avec d'autres médicaments qui ont des effets hépatotoxiques avérés (voir la rubrique «Mises en garde et précautions»).
Autres médicaments antileucémiques
L'influence de TRISENOX sur l'efficacité d'autres antileucémiques est inconnue.
Grossesse, allaitementEn raison du risque génotoxique des arsénieux (voir rubrique «Données précliniques»), les femmes en âge de procréer doivent utiliser un moyen de contraception efficace au cours et jusqu'à 6 mois après la fin du traitement par TRISENOX.
Les hommes doivent utiliser un moyen de contraception efficace au cours et jusqu'à 3 mois après la fin du traitement par TRISENOX, et être informés qu'ils ne doivent pas procréer.
Grossesse
Il a été démontré que le trioxyde d'arsenic possède des propriétés embryotoxiques et tératogènes dans les études chez l'animal. Il n'a pas été conduit d'étude avec TRISENOX chez la femme enceinte. Les femmes en âge de procréer doivent se soumettre à un test de grossesse avant de commencer le traitement par TRISENOX. Si ce médicament est administré pendant la grossesse ou si la patiente devient enceinte pendant la prise de ce médicament, elle doit être informée du risque potentiel pour le foetus.
Allaitement
Le trioxyde d'arsenic est excrété dans le lait maternel. En raison du risque de réactions indésirables graves causées par TRISENOX chez le nourrisson allaité, l'allaitement doit être interrompu avant et pendant toute la durée du traitement, et pendant deux semaines après la dernière dose.
Fertilité
Aucune étude clinique ou préclinique de fertilité n'a été conduite avec TRISENOX.
Effet sur l’aptitude à la conduite et l’utilisation de machinesLes effets sur l'aptitude à conduire des véhicules et à utiliser des machines n'ont pas été étudiés.
Effets indésirablesRésumé du profil de sécurité
Dans les études cliniques, des effets indésirables de grade CTC 3 ou 4 sont survenus chez 37% des patients atteints de LPA récidivante/réfractaire. Les effets indésirables les plus fréquemment rapportés ont inclus une hyperglycémie (10 à 18%), une hypokaliémie (13 à 27%), une neutropénie (8%) et une augmentation des taux d'alanine aminotransférase (ALAT, 8%). Les bilans hématologiques ont révélé une hyperleucocytose chez 50% des patients atteints de LPA récidivante/réfractaire.
Ainsi qu'attendu, ce groupe de patients atteints de LPA récidivante/réfractaire a fréquemment subi des effets indésirables sérieux (1 à 10%). Dans ce cadre, les cas de syndrome de différenciation LPA (27%), d'hyperleucocytose, d'allongement du QT (jusqu'à 40%, dont 1 avec torsade de pointes), de fibrillation/flutter auriculaire et d'hyperglycémie ont été attribués à TRISENOX, de même que différents effets indésirables sérieux tels que thrombopénie (associée à des hémorragies), infections, douleurs, diarrhée, neuropathie ou nausées.
D'une façon générale, les effets indésirables apparus au cours du traitement ont diminué avec le temps – peut-être à cause d'une amélioration de la maladie de base – chez les patients atteints de LPA récidivante/réfractaire. Les patients ont globalement mieux toléré (moins d'effets toxiques) le traitement de consolidation et d'entretien que le traitement d'induction. Cela provient probablement de la superposition entre les effets indésirables et le processus pathologique non contrôlé au début du traitement, ainsi que des nombreux médicaments nécessaires pour contrôler les symptômes et le processus pathologique.
Dans une étude de non-infériorité de phase III, multicentrique, ayant évalué l'acide tout-trans-rétinoïque (ATRA) associé à une chimiothérapie en comparaison avec l'ATRA associé au trioxyde d'arsenic chez des patients atteints de LPA nouvellement diagnostiquée à risque faible à intermédiaire (étude APL0406), des effets indésirables sévères comprenant hépatotoxicité (63%), thrombopénie (88%), neutropénie (79%), syndrome de différenciation LPA (19%) et allongement du QTc (16%) ont été observés chez des patients traités par le trioxyde d'arsenic.
Liste des effets indésirables
Les effets indésirables suivants (listés selon la classification MedDRA par classes de systèmes d'organes) ont été signalés dans le cadre d'études cliniques et/ou dans des rapports postcommercialisation chez des patients atteints de LPA récidivante/réfractaire ou de LPA nouvellement diagnostiquée. Les fréquences sont indiquées comme suit: très fréquents (≥1/10), fréquents (<1/10, ≥1/100), occasionnels (<1/100, ≥1/1000) et rares (<1/1000, ≥1/10'000).
Infections et infestations:
Très fréquents: infection (22%), pneumonie (18%), sepsis (17%).
Fréquents: zona, infection fongique, taux accru de protéine C-réactive.
Occasionnels: infection mycobactérienne, pneumonie à Pneumocystis jirovecii, infection à Herpes simplex, myocardite, gastro-entérite virale.
Tumeurs bénignes, malignes et non précisées:
Fréquents: syndrome myélodysplasique.
Occasionnels: progression d'un néoplasme malin, leucémie aiguë myéloblastique, glioblastome multiforme, myélome multiple.
Cas individuels observés hors des études (expérience pratique):
Fréquents: leucémie aiguë promyélocytaire.
Occasionnels: récidive de leucémie, mélanome malin, cancer du pancréas.
Affections hématologiques et du système lymphatique:
Très fréquents: neutropénie (44%), thrombopénie (11%).
Fréquents: anémie, pancytopénie, coagulation intravasculaire disséminée, leucocytose, insuffisance médullaire sévère.
Affections du système immunitaire:
Occasionnels: hypersensibilité.
Troubles du métabolisme et de la nutrition:
Fréquents: hypokaliémie, déshydratation, hyperglycémie, réduction de l'appétit, Hyponatrémie, hypocalcémie, hyperkaliémie, hypercalcémie, prise de poids.
Occasionnels: hypoalbuminémie, hypophosphatémie, perte de poids, taux accru de bicarbonate, taux réduit de protéines totales.
Cas individuels observés hors des études (expérience pratique):
Fréquents: hypomagnésémie, syndrome de lyse tumorale, hyperuricémie.
Occasionnels: diabète, hypermagnésémie.
Affections psychiatriques:
Fréquents: état confusionnel, insomnie.
Occasionnels: agitation.
Cas individuels observés hors des études (expérience pratique):
Fréquents: modification de l'humeur.
Occasionnels: anxiété.
Affections du système nerveux:
Fréquents: neuropathie périphérique, accident vasculaire cérébral, céphalée, crise de convulsions, sensation de vertige, encéphalopathie, léthargie, coma, hydrocéphalie, somnolence.
Occasionnels: hypoesthésie, syncope, paresthésie, trouble cognitif, modification de l'état de conscience, paralysie faciale, névralgie.
Cas individuels observés hors des études (expérience pratique):
Fréquents: tremblement, aphasie.
Occasionnels: démence, neuropathie axonale, hémorragie du système nerveux central, altération du sens gustatif, hémiparésie, polyneuropathie.
Fréquence indéterminée: encéphalopathie de Wernicke.
Affections oculaires:
Fréquents: détérioration de la vue, oedème périorbitaire.
Cas individuels observés hors des études (expérience pratique):
Occasionnels: vue double, sécrétion accrue de liquide lacrymal, hyperémie oculaire, douleurs oculaires, photophobie.
Affections de l'oreille et du labyrinthe:
Occasionnels: vertige.
Cas individuels observés hors des études (expérience pratique):
Fréquents: surdité.
Occasionnels: hyperacousie, acouphène.
Affections cardiaques:
Très fréquents: allongement de l'intervalle QT à l'ECG (33%).
Fréquents: insuffisance cardiaque, arythmie ventriculaire, fibrillation auriculaire, épanchement péricardique, arrêt cardiaque, tachycardie, infarctus du myocarde, syndrome du QT long, tachycardie supra-ventriculaire, bloc atrio-ventriculaire.
Occasionnels: bradycardie, cardiomyopathie, flutter auriculaire, myocardite.
Cas individuels observés hors des études (expérience pratique):
Fréquents: maladies cardiaques, anomalies à l'électrocardiogramme.
Occasionnels: dissociation atrio-ventriculaire, sclérose valvulaire aortique, cardiotoxicité, onde T inversée à l'électrocardiogramme.
Affections vasculaires:
Fréquents: hypotension, thrombose veineuse profonde des membres inférieurs, taux sanguin accru de lactate déshydrogénase, INR accru, temps de prothrombine accru, taux accru de produits de dégradation de la fibrine.
Occasionnels: hémorragie, choc.
Cas individuels observés hors des études (expérience pratique):
Fréquents: sensation de chaleur.
Occasionnels: hypertension, vascularite, temps de céphaline activée augmenté, taux accru d'éosinophiles, taux sanguin réduit de fibrinogène, taux accru de monocytes.
Affections respiratoires, thoraciques et médiastinales:
Très fréquents: syndrome de différenciation LPA (14%), dyspnée (13%).
Fréquents: épanchement pleural, insuffisance respiratoire, hypoxie, oedème pulmonaire, infiltration pulmonaire, détresse respiratoire, embolie pulmonaire, toux, syndrome de détresse respiratoire aiguë, pneumonite, pneumopathie interstitielle, affection des voies respiratoires, épistaxis.
Occasionnels: hémorragie pulmonaire, broncho-pneumopathie chronique obstructive, douleurs oro-pharyngées, sibilances, congestion des sinus, hypertension pulmonaire.
Affections gastro-intestinales:
Fréquents: nausée, diarrhée, vomissements, douleurs abdominales, hémorragie gastro-intestinale, constipation, pancréatite, ballonnement, dyspepsie, stomatite, flatulences.
Occasionnels: iléus, obstruction de l'intestin, syndrome abdominal aigu, ascite, perforation intestinale, oesophagite.
Cas individuels observés hors des études (expérience pratique):
Fréquents: hypoesthésie orale.
Occasionnels: sécheresse buccale, sécheresse des lèvres, gingivite.
Affections hépatobiliaires:
Fréquents: lésions hépatocellulaires, défaillance hépatique.
Occasionnels: cholécystite.
Affections de la peau et du tissu sous-cutané:
Fréquents: dermatite, prurit.
Occasionnels: érythème, urticaire, éruption cutanée exfoliative, hyperhidrose, affection cutanée, psoriasis.
Cas individuels observés hors des études (expérience pratique):
Fréquents: hyperpigmentation de la peau.
Occasionnels: alopécie, vésicules.
Affections musculo-squelettiques et du tissu conjonctif:
Fréquents: douleurs dorsales, arthralgie, douleurs dans une extrémité, douleurs osseuses, faiblesse musculaire, douleurs du système musculo-squelettique.
Occasionnels: douleurs de la nuque, myalgie, spasmes musculaires, douleurs au niveau des flancs.
Cas individuels observés hors des études (expérience pratique):
Fréquents: taux sanguin accru de créatine phosphokinase.
Affections du rein et des voies urinaires:
Fréquents: lésions rénales aiguës, défaillance rénale, altération de la fonction rénale.
Occasionnels: rétention urinaire, hématurie.
Cas individuels observés hors des études (expérience pratique):
Fréquents: dysurie, taux sanguin accru d'urée.
Occasionnels: néphrite tubulo-interstitielle.
Affections des organes de reproduction et du sein:
Cas individuels observés hors des études (expérience pratique):
Fréquents: saignement vaginal.
Troubles généraux et anomalies au site d'administration:
Très fréquents: fièvre (24%).
Fréquents: fatigue, douleurs thoraciques de cause non cardiaque, asthénie, oedème, frissons, décès soudain, douleurs, muqueuse enflammée, malaise, interaction médicamenteuse, syndrome de défaillance multiviscérale, taux sanguin accru de phosphatase alcaline.
Occasionnels: extravasation au site de perfusion, oedème facial, douleurs au site du cathéter, progression de la maladie.
Cas individuels observés hors des études (expérience pratique):
Fréquents: inefficacité du médicament.
Occasionnels: hématome au site du cathéter.
Actes médicaux et chirurgicaux:
Occasionnels: thoracostomie.
Lésions, intoxications et complications liées aux procédures:
Fréquents: hématome sous-dural, chute.
Occasionnels: fracture, contusion.
Cas individuels observés hors des études (expérience pratique):
Fréquents: intoxication par un métal.
Occasionnels: taux urinaire accru d'arsenic.
Description d'effets indésirables sélectionnés
Toxicité hématologique et gastro-intestinale
Des toxicités gastro-intestinales, des neutropénies de grade 3 ou 4 et des thrombopénies de grade 3 ou 4 sont survenues chez des patients atteints de LPA nouvellement diagnostiquée à risque faible à intermédiaire.
Neuropathie périphérique
Les neuropathies périphériques et les paresthésies/dysesthésies sont des effets bien connus de l'arsenic présent dans l'environnement. Deux patients ont arrêté prématurément le traitement à cause de cet effet indésirable. Un des deux a continué à recevoir TRISENOX dans le cadre de son plan de traitement suivant. Des symptômes de neuropathie se sont développés chez 44% des patients. Ils étaient d'intensité faible à modérée dans la plupart des cas et ont régressé après l'arrêt du traitement par TRISENOX.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSignes et symptômes
En cas de symptômes suggérant une toxicité aiguë grave de l'arsenic (ex.: convulsions, faiblesse musculaire et état confusionnel), il faut interrompre immédiatement le traitement par TRISENOX.
Traitement
On peut envisager l'administration d'un chélateur comme la pénicillamine à une dose quotidienne de ≤1 g par jour. La durée du traitement par pénicillamine doit être évaluée en prenant en considération les valeurs de laboratoire de l'arsenic urinaire. Chez les patients ne pouvant pas prendre de médicament par voie orale, on peut envisager l'administration de dimercaprol par voie intramusculaire à raison de 3 mg/kg toutes les 4 heures jusqu'à ce que les toxicités mettant directement la vie en péril aient régressé. Par la suite, on peut administrer de la pénicillamine à la dose de <1 g par jour. En présence d'une coagulopathie, l'administration du chélateur appelé succimer ou acide dimercaptosuccinique (selon DCI) à 10 mg/kg ou 350 mg/m2 est recommandée toutes les 8 heures pendant les 5 premiers jours, puis toutes les 12 heures pendant les 2 semaines suivantes.
Pour les patients présentant un surdosage aigu et grave à l'arsenic, il est nécessaire d'envisager une dialyse.
Propriétés/EffetsCode ATC L01XX27
Mécanisme d'action
Le mécanisme d'action de TRISENOX n'est pas complètement élucidé. Le trioxyde d'arsenic induit in vitro des altérations morphologiques et des fragmentations de l'acide désoxyribonucléique (ADN) caractéristiques de l'apoptose des cellules NB4 humaines de la leucémie promyélocytaire. Le trioxyde d'arsenic provoque également la lésion ou la dégradation de la protéine de fusion PML/RAR-alpha.
Pharmacodynamique
Voir chapitre Efficacité clinique.
Efficacité clinique
Patients atteints de LPA nouvellement diagnostiquée à risque non élevé:
TRISENOX a été évalué dans une étude de non-infériorité de phase III, randomisée et contrôlée, auprès de 77 patients atteints de LPA nouvellement diagnostiquée à risque faible à intermédiaire. Cette étude a évalué l'efficacité et la sécurité du traitement par TRISENOX en association avec l'acide tout-trans-rétinoïque (ATRA) versus ATRA en association avec une chimiothérapie (p.ex. idarubicine et mitoxantrone) (étude APL0406). Les patients inclus étaient atteints de leucémie promyélocytaire aiguë (LPA) nouvellement diagnostiquée, confirmée par détection de la translocation t(15; 17) ou du gène de fusion PML-RARα par RT-PCR ou par la structure microponctuée des corps nucléaires PML dans les cellules leucémiques. On ne dispose pas de données concernant les patients porteurs d'autres translocations telles que t(11;17) (PLZF-RARα). Les patients présentant des arythmies significatives, des anomalies à l'ECG (syndrome du QT long congénital, antécédents ou présence de tachyarythmie ventriculaire ou auriculaire significative, bradycardie cliniquement significative [<50/min], QTc >450 msec à l'ECG enregistré dans le cadre du screening, bloc de branche droit plus hémibloc antérieur gauche, bloc bifasciculaire) ou une neuropathie étaient exclus de l'étude. Les patients du groupe traité par ATRA + TRISENOX ont reçu l'ATRA par voie orale à la dose de 45 mg/m2/jour et TRISENOX par voie intraveineuse à la dose de 0,15 mg/kg/jour jusqu'à obtention d'une rémission complète. Dans le cadre du traitement de consolidation, les doses d'ATRA et de TRISENOX sont restées inchangées, mais l'ATRA a été administré sur un nombre total de 7 cycles composés chacun de 2 semaines de traitement suivies de 2 semaines sans traitement, tandis que TRISENOX a été administré sur un nombre total de 4 cycles composés chacun de 4 semaines de traitement (à raison de 5 jours par semaine) suivies de 4 semaines sans traitement.
Les résultats de 34 mois (médiane) de cette étude ont été présentés dans le travail de Lo-Coco et al. (2013). Le critère primaire de l'étude était défini comme la survie sans événement (EFS, event-free survival) 2 ans après le diagnostic. Le taux d'EFS à 2 ans était de 97% dans le groupe traité par ATRA + TRISENOX et de 86 % dans le groupe traité par ATRA + chimiothérapie (p <0,001 pour la non-infériorité). Les résultats ont suggéré post hoc une supériorité d'ATRA + TRISENOX versus ATRA + chimiothérapie. Les critères secondaires de l'étude ont inclus la rémission hématologique complète (HCR; 100% sous ATRA + TRISENOX vs 95% sous ATRA + chimiothérapie), la survie totale à 2 ans (OS; 99% sous ATRA + TRISENOX vs 91% sous ATRA + chimiothérapie), la survie sans maladie à 2 ans (DSF; 97% sous ATRA + TRISENOX vs 90% sous ATRA + chimiothérapie) et l'incidence cumulative de récidives à 2 ans (CIR; 1% sous ATRA + TRISENOX vs 6% sous ATRA + chimiothérapie).
Cette étude a reçu une extension et les résultats de la cohorte de l'étude d'extension (129 patients sous ATRA + TRISENOX) ont été publiés (Platzbecker et al. 2016). À 24 et à 50 mois, tous les 4 critères d'efficacité (EFS – survie sans événement, OS – survie globale, DFS survie sans maladie – et CIR incidence cumulative de récidives) révélaient une différence cliniquement significative entre les traitements étudiés, en faveur du traitement par ATRA + TRISENOX. Les résultats à 50 mois étaient les suivants: EFS de 97% sous ATRA + TRISENOX vs 80% sous ATRA + chimiothérapie; OS de 99% sous ATRA + TRISENOX vs 93% sous ATRA + chimiothérapie; DFS de 97% sous ATRA + TRISENOX vs 83% sous ATRA + chimiothérapie; CIR de 1,9% sous ATRA + TRISENOX vs 14% sous ATRA + chimiothérapie. Ces données ont démontré la non-infériorité du traitement par ATRA + ATO versus ATRA + chimiothérapie en termes d'EFS. Elles ont confirmé le bénéfice du traitement non seulement pour l'EFS et l'OS, mais aussi pour la DFS et la CIR.
LPA récidivante/réfractaire
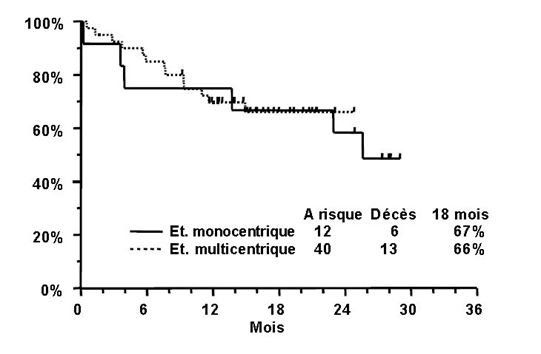
TRISENOX a été étudié chez 52 patients atteints de LPA, précédemment traités par une anthracycline et un rétinoïde, dans deux essais ouverts non comparatifs. L'un était une étude monocentrique (n=12) et l'autre une étude multicentrique effectuée dans 9 centres (n=40). Les patients de la première étude ont reçu une dose moyenne de TRISENOX de 0,16 mg/kg/jour (limites: 0,06 à 0,20 mg/kg/jour) et ceux de l'étude multicentrique une dose fixe de 0,15 mg/kg/jour. TRISENOX a été administré en perfusions intraveineuses de 1 à 2 heures.
Le traitement a été administré jusqu'à disparition complète des cellules leucémiques de la moelle osseuse ou pendant 60 jours au maximum. Les patients obtenant une rémission complète ont reçu un traitement de consolidation par TRISENOX consistant en 25 doses supplémentaires sur une période de 5 semaines. Le traitement de consolidation a commencé 6 semaines (limites: 3 à 8 semaines) après le traitement d'induction dans l'étude monocentrique et 4 semaines (limites: 3 à 6 semaines) après le traitement d'induction dans l'étude multicentrique. Par définition, la rémission complète (RC) était caractérisée par l'absence de cellules leucémiques visibles dans la moelle osseuse et par la régénération au niveau périphérique des thrombocytes et des leucocytes.
Les patients de l'étude monocentrique avaient rechuté après 1 à 6 traitements antérieurs et 2 patients avaient rechuté après une transplantation de cellules souches. Les patients de l'étude multicentrique avaient rechuté après 1 à 4 cycles de traitement; ici, 5 patients avaient eu antérieurement une transplantation de cellules souches. L'âge moyen des patients était de 33 ans (limites: 9 à 75 ans) dans l'étude monocentrique et de 40 ans (limites: 5 à 73 ans) dans l'étude multicentrique.
Les résultats sont résumés dans le Tableau 2:
Tableau 2
|
|
Etude monocentrique
n=12
|
Etude multicentrique
n=40
| |
Dose de TRISENOX (mg/kg/jour)
|
0,16 (0,06-0,20)
|
0,15
| |
Rémission complète (RC)
|
11 (92%)
|
34 (85%)
| |
Délai de rémission médullaire (médiane)
|
32 jours
|
35 jours
| |
Délai de RC
|
54 jours
|
59 jours
| |
Taux de survie à 18 mois
|
67%
|
66%
|
L'étude monocentrique comprenait 2 enfants (<18 ans) et tous deux ont obtenu une rémission complète (RC). L'essai multicentrique comprenait 5 enfants (< 18 ans), dont 3 ont obtenu une RC. Aucun enfant de moins de 5 ans n'a été traité.
Dans le suivi après le traitement de consolidation, 7 patients de l'étude monocentrique et 18 patients de l'étude multicentrique ont reçu un traitement d'entretien par TRISENOX.
Trois patients de l'étude monocentrique et 15 patients de l'étude multicentrique ont reçu une transplantation de cellules souches après avoir terminé le traitement par TRISENOX. La durée moyenne de la RC, selon la méthode de Kaplan-Meier, est de 14 mois pour l'étude monocentrique (elle n'a pas été atteinte pour l'étude multicentrique). À la dernière visite de contrôle, 6 patients sur 12 étaient vivants dans l'étude monocentrique, avec un suivi moyen de 28 mois (limites: 25 à 29 mois). Dans l'étude multicentrique, 27 patients sur 40 étaient vivants, avec un suivi moyen de 16 mois (limites: 9 à 25 mois). Les estimations de Kaplan-Meier de la survie sur une durée de 18 mois pour les deux études sont présentées dans la figure ci-dessous.
Le tableau 3 présente la confirmation cytogénétique de la conversion à un génotype normal et la détection par RT-PCR (Reverse Transcriptase - Polymerase Chain Reaction) de la conversion du facteur PML/RAR-α à la normale.
Analyse cytogénétique après traitement par TRISENOX:
Tableau 3
|
|
Étude pilote monocentrique
nombre de patients avec RC = 11
|
Étude multicentrique
nombre de patients avec RC = 34
| |
Cytogénétique classique [t(15;17)]
| |
Absente
|
8 (73%)
|
31 (91%)
| |
Présente
|
1 (9%)
|
0 (0%)
| |
Non évaluable
|
2 (18%)
|
3 (9%)
| |
RT-PCR pour PML/RARα
| |
Négative
|
8 (73%)
|
27 (79%)
| |
Positive
|
3 (27%)
|
4 (12%)
| |
Non évaluable
|
-
|
3 (9%)
|
Des réponses ont été observées dans toutes les tranches d'âge étudiées (limites: 6 à 75 ans). Les taux de réponses étaient similaires dans les deux sexes.
Il n'existe aucune expérience de l'effet de TRISENOX sur la variante de la LPA caractérisée par la présence des translocations chromosomiques t(11;17) et t(5;17).
Sécurité et efficacité en pédiatrie
L'expérience concernant les enfants est limitée. Dans une étude de phase I auprès de 13 enfants atteints de LPA récidivante/réfractaire (âge médian de 17 ans, fourchette de 4 à 21 ans, données publiées par Fox et al. 2008), l'ATO a été administré 5 jours par semaine par voie intraveineuse à la dose de 0,15 mg/kg en 2 heures (20 doses par cycle). 85% des patients ont atteint une rémission morphologique complète. 70 doses ont été nécessaires chez 3 patients sur 7 pour atteindre un résultat négatif de RT-PCR. À la dose recommandée de 0,15 mg/kg par administration, l'ATO a été bien toléré par les enfants et adolescents atteints de leucémie; le profil de toxicité était comparable aux données publiées sur les patients adultes.
PharmacocinétiqueAbsorption
Le degré d'exposition à l'arsenic augmente avec l'augmentation de la dose.
Les taux plasmatiques d'arsenic atteignent un pic maximum à la fin de la perfusion.
La forme inorganique, lyophilisée du trioxyde d'arsenic, mise en solution, forme immédiatement le produit d'hydrolyse: l'acide arsénieux (AsIII). AsIII est la forme pharmacologiquement active du trioxyde d'arsenic.
Distribution
Le volume de distribution (Vd) de l'AsIII est élevé (> 400 l) indiquant une distribution significative dans les tissus, avec une liaison négligeable aux protéines.
En administration quotidienne, les taux plasmatiques ont atteint leur niveau d'équilibre en 8 à 10 jours.
Le volume de distribution est aussi dépendant du poids, augmentant avec l'augmentation du poids corporel. L'arsenic s'accumule principalement dans le foie, le rein et le cœur, et dans une moindre mesure, dans le poumon, les cheveux et les ongles.
Métabolisme
Les sels trivalents d'arsenic (AsIII) sont méthylés chez l'homme et excrétés dans l'urine.
Le métabolisme du trioxyde d'arsenic implique l'oxydation de l'acide arsénieux (AsIII), la forme active du trioxyde d'arsenic, en acide arsénique (AsV) ainsi que la méthylation oxydative en acide monométhylarsonique (MMAV) et en acide diméthylarsinique (DMAV) par des méthyltransférases, essentiellement dans le foie.
Les métabolites pentavalents MMAV et DMAV sont lents à apparaître dans le plasma (environ 10-24 heures après la première administration de trioxyde d'arsenic), mais du fait de leur demi-vie longue, ils s'accumulent plus que l'AsIII lors d'administration de doses multiples.
L'étendue de l'accumulation de ces métabolites dépend du schéma posologique. L'accumulation suivant une administration de doses multiples est approximativement de 1,4 à 8 fois supérieure à celle suivant une administration en dose unique. L'AsV est présent dans le plasma uniquement à des concentrations relativement faibles.
Dans des études enzymatiques menées in vitro sur microsomes hépatiques humains, il a été démontré que le trioxyde d'arsenic ne possédait pas d'activité inhibitrice sur les substrats des enzymes principaux de cytochromes P450 (tels que 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5, 4A9/11).
Les médicaments substrats de ces enzymes P450 ne sont pas censés induire une interaction avec le TRISENOX.
Élimination
Environ 15% de la dose administrée de TRISENOX est excrétée dans les urines en AsIII inchangé. Les métabolites méthylés de l'AsIII (MMAV, DMAV) sont principalement excrétés dans les urines. La concentration plasmatique de l'AsIII décline de manière biphasique à partir de la concentration du pic plasmatique avec une demi-vie moyenne d'élimination terminale de 10 à 14 heures. La clairance totale de l'AsIII pour un intervalle de dose unique de 7-32 mg (administrée à 0,15 mg/kg) est de 49 l/h; la clairance rénale est de 9 l/h.
La clairance ne dépend ni du poids corporel ni d'une dose supérieure à la marge posologique étudiée. Les demi-vies moyennes d'élimination terminale estimées des métabolites MMAV et DMAV sont respectivement de 32 h et de 70 h.
Chez les patients atteints de LPA, l'administration quotidienne de 0,15 mg/kg/jour de trioxyde d'arsenic a pratiquement quadruplé l'excrétion urinaire d'arsenic après 2 à 4 semaines, par rapport aux valeurs de référence.
Linéarité/non-linéarité
Dans l'intervalle de dose unique absolu de 7 à 32 mg (administrée à 0,15 mg/kg), la biodisponibilité (ASC) apparaît linéaire. La décroissance à partir de la concentration du pic plasmatique d'AsIII intervient de manière biphasique et est caractérisée par une phase initiale de distribution rapide suivie d'une phase terminale d'élimination lente. Après administration de 0,15 mg/kg à une fréquence journalière (n=6) ou bihebdomadaire (n=3), l'accumulation d'AsIII a été approximativement doublée par rapport à celle observée en administration unique.
Cette accumulation était légèrement supérieure à ce qui était attendu sur la base des résultats en dose unique.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Des données pharmacocinétiques chez des patients ayant une insuffisance hépatique faible à modérée avec un carcinome hépatocellulaire indiquent que l'AsIII ou l'AsV ne s'accumulent pas lorsque les perfusions sont bihebdomadaires. Sur la base des ASC doses-normalisées (par mg de dose), une augmentation de la biodisponibilité de l'AsIII, de l'AsV, du MMAV et du DMAV n'a pas été clairement constatée en cas d'altération de la fonction hépatique.
Troubles de la fonction rénale
La clairance plasmatique de l'AsIII n'a pas été altérée chez les patients présentant une insuffisance rénale faible (clairance de la créatinine de 50-80 ml/min) ou une insuffisance rénale modérée (clairance de la créatinine de 30-49 ml/min). La clairance plasmatique de l'AsIII chez les patients ayant une insuffisance rénale sévère (clairance de la créatinine < 30 ml/min) était 40% inférieure à celle de patients ayant une fonction rénale normale.
La biodisponibilité du MMAV et du DMAV semble augmenter chez les patients ayant une insuffisance rénale; la conséquence clinique en est inconnue mais aucune augmentation de la toxicité n'a été notée.
L'utilisation de TRISENOX chez les patients dialysés n'a pas été étudiée.
L'exposition systémique au MMAV (acide monométhylarsonique) et au DMAV (acide diméthylarsinique) était en tendance accrue chez les patients insuffisants rénaux. Les conséquences cliniques de cette particularité ne sont pas connues, mais aucune toxicité supplémentaire n'a été observée.
Données précliniquesDes études limitées chez l'animal de toxicité sur la reproduction avec le trioxyde d'arsenic indiquent des propriétés embryotoxiques et tératogènes (anomalies du tube neural, anophtalmie et microphtalmie) en cas d'administration de 1 à 10 fois la dose clinique recommandée (mg/m2).
Il n'a pas été conduit d'études de fertilité avec TRISENOX.
Les arsénieux induisent des aberrations chromosomiques et des transformations morphologiques dans des cellules de mammifères in vitro et in vivo. Il n'a pas été conduit d'études formelles de potentiel carcinogène avec le trioxyde d'arsenic. Cependant, le trioxyde d'arsenic et d'autres arsénieux inorganiques sont reconnus comme cancérogènes chez l'homme.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments, à l'exception de ceux mentionnés sous «Préparation et administration de la solution pour perfusion».
Stabilité
TRISENOX ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Stabilité après ouverture
La préparation diluée pour perfusion ne contient pas de conservateur. Une fois dilué dans une solution pour administration intraveineuse, la stabilité chimique et physique de TRISENOX a été démontrée pendant 24 heures à 15-30°C et pendant 72 heures à 2-8°C.
D'un point de vue microbiologique, le produit prêt à l'emploi doit être utilisé immédiatement après la dilution/reconstitution. S'il n'est pas utilisé immédiatement, les durées et conditions de conservation avant utilisation relèvent de la responsabilité de l'utilisateur et ne doivent normalement pas dépasser 24 heures à 2-8°C, à moins que la dilution/reconstitution se soit déroulée dans des conditions d'asepsie contrôlées et validées.
Remarques particulières concernant le stockage
Conserver dans l'emballage d'origine et ne pas conserver au-dessus de 30°C. Conserver hors de portée des enfants.
Remarques concernant la manipulation
Une technique aseptique doit être strictement observée durant la manipulation de TRISENOX car il ne contient aucun agent conservateur.
Préparation et administration de la solution pour perfusion
TRISENOX doit être dilué dans 100 à 250 ml de solution de glucose à 50 mg/ml (5%) ou de sérum physiologique à 0,9%, immédiatement après ouverture du flacon.
Est destiné seulement à un usage unique.
Toute fraction inutilisée de chaque flacon doit être jetée en respectant les mesures de sécurité. Les solutions non utilisées doivent être jetées.
TRISENOX ne doit pas être mélangé avec ou administré en même temps et dans la même sonde intraveineuse que d'autres médicaments.
TRISENOX doit être administré en perfusion intraveineuse de 1 à 2 heures. La durée de la perfusion peut être portée à 4 heures en cas de réactions vasomotrices. Il n'est pas nécessaire de mettre en place un cathéter veineux central.
La solution diluée doit être limpide et incolore. L'absence de particules et de coloration doit être contrôlée visuellement dans toute solution parentérale avant administration. Ne pas utiliser la solution si des particules sont visibles.
Numéro d’autorisation65178 (Swissmedic).
PrésentationTRISENOX 2 mg/ml: flacons de 6 ml: EO 10 [A]
Titulaire de l’autorisationTeva Pharma AG, Basel.
Mise à jour de l’informationJuin 2022.
Numéro de version interne: 9.1
|