Propriétés/EffetsCode ATC L01XX27
Mécanisme d'action
Le mécanisme d'action de TRISENOX n'est pas complètement élucidé. Le trioxyde d'arsenic induit in vitro des altérations morphologiques et des fragmentations de l'acide désoxyribonucléique (ADN) caractéristiques de l'apoptose des cellules NB4 humaines de la leucémie promyélocytaire. Le trioxyde d'arsenic provoque également la lésion ou la dégradation de la protéine de fusion PML/RAR-alpha.
Pharmacodynamique
Voir chapitre Efficacité clinique.
Efficacité clinique
Patients atteints de LPA nouvellement diagnostiquée à risque non élevé:
TRISENOX a été évalué dans une étude de non-infériorité de phase III, randomisée et contrôlée, auprès de 77 patients atteints de LPA nouvellement diagnostiquée à risque faible à intermédiaire. Cette étude a évalué l'efficacité et la sécurité du traitement par TRISENOX en association avec l'acide tout-trans-rétinoïque (ATRA) versus ATRA en association avec une chimiothérapie (p.ex. idarubicine et mitoxantrone) (étude APL0406). Les patients inclus étaient atteints de leucémie promyélocytaire aiguë (LPA) nouvellement diagnostiquée, confirmée par détection de la translocation t(15; 17) ou du gène de fusion PML-RARα par RT-PCR ou par la structure microponctuée des corps nucléaires PML dans les cellules leucémiques. On ne dispose pas de données concernant les patients porteurs d'autres translocations telles que t(11;17) (PLZF-RARα). Les patients présentant des arythmies significatives, des anomalies à l'ECG (syndrome du QT long congénital, antécédents ou présence de tachyarythmie ventriculaire ou auriculaire significative, bradycardie cliniquement significative [<50/min], QTc >450 msec à l'ECG enregistré dans le cadre du screening, bloc de branche droit plus hémibloc antérieur gauche, bloc bifasciculaire) ou une neuropathie étaient exclus de l'étude. Les patients du groupe traité par ATRA + TRISENOX ont reçu l'ATRA par voie orale à la dose de 45 mg/m2/jour et TRISENOX par voie intraveineuse à la dose de 0,15 mg/kg/jour jusqu'à obtention d'une rémission complète. Dans le cadre du traitement de consolidation, les doses d'ATRA et de TRISENOX sont restées inchangées, mais l'ATRA a été administré sur un nombre total de 7 cycles composés chacun de 2 semaines de traitement suivies de 2 semaines sans traitement, tandis que TRISENOX a été administré sur un nombre total de 4 cycles composés chacun de 4 semaines de traitement (à raison de 5 jours par semaine) suivies de 4 semaines sans traitement.
Les résultats de 34 mois (médiane) de cette étude ont été présentés dans le travail de Lo-Coco et al. (2013). Le critère primaire de l'étude était défini comme la survie sans événement (EFS, event-free survival) 2 ans après le diagnostic. Le taux d'EFS à 2 ans était de 97% dans le groupe traité par ATRA + TRISENOX et de 86 % dans le groupe traité par ATRA + chimiothérapie (p <0,001 pour la non-infériorité). Les résultats ont suggéré post hoc une supériorité d'ATRA + TRISENOX versus ATRA + chimiothérapie. Les critères secondaires de l'étude ont inclus la rémission hématologique complète (HCR; 100% sous ATRA + TRISENOX vs 95% sous ATRA + chimiothérapie), la survie totale à 2 ans (OS; 99% sous ATRA + TRISENOX vs 91% sous ATRA + chimiothérapie), la survie sans maladie à 2 ans (DSF; 97% sous ATRA + TRISENOX vs 90% sous ATRA + chimiothérapie) et l'incidence cumulative de récidives à 2 ans (CIR; 1% sous ATRA + TRISENOX vs 6% sous ATRA + chimiothérapie).
Cette étude a reçu une extension et les résultats de la cohorte de l'étude d'extension (129 patients sous ATRA + TRISENOX) ont été publiés (Platzbecker et al. 2016). À 24 et à 50 mois, tous les 4 critères d'efficacité (EFS – survie sans événement, OS – survie globale, DFS survie sans maladie – et CIR incidence cumulative de récidives) révélaient une différence cliniquement significative entre les traitements étudiés, en faveur du traitement par ATRA + TRISENOX. Les résultats à 50 mois étaient les suivants: EFS de 97% sous ATRA + TRISENOX vs 80% sous ATRA + chimiothérapie; OS de 99% sous ATRA + TRISENOX vs 93% sous ATRA + chimiothérapie; DFS de 97% sous ATRA + TRISENOX vs 83% sous ATRA + chimiothérapie; CIR de 1,9% sous ATRA + TRISENOX vs 14% sous ATRA + chimiothérapie. Ces données ont démontré la non-infériorité du traitement par ATRA + ATO versus ATRA + chimiothérapie en termes d'EFS. Elles ont confirmé le bénéfice du traitement non seulement pour l'EFS et l'OS, mais aussi pour la DFS et la CIR.
LPA récidivante/réfractaire
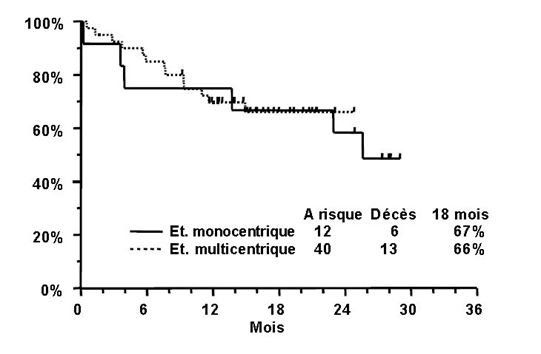
TRISENOX a été étudié chez 52 patients atteints de LPA, précédemment traités par une anthracycline et un rétinoïde, dans deux essais ouverts non comparatifs. L'un était une étude monocentrique (n=12) et l'autre une étude multicentrique effectuée dans 9 centres (n=40). Les patients de la première étude ont reçu une dose moyenne de TRISENOX de 0,16 mg/kg/jour (limites: 0,06 à 0,20 mg/kg/jour) et ceux de l'étude multicentrique une dose fixe de 0,15 mg/kg/jour. TRISENOX a été administré en perfusions intraveineuses de 1 à 2 heures.
Le traitement a été administré jusqu'à disparition complète des cellules leucémiques de la moelle osseuse ou pendant 60 jours au maximum. Les patients obtenant une rémission complète ont reçu un traitement de consolidation par TRISENOX consistant en 25 doses supplémentaires sur une période de 5 semaines. Le traitement de consolidation a commencé 6 semaines (limites: 3 à 8 semaines) après le traitement d'induction dans l'étude monocentrique et 4 semaines (limites: 3 à 6 semaines) après le traitement d'induction dans l'étude multicentrique. Par définition, la rémission complète (RC) était caractérisée par l'absence de cellules leucémiques visibles dans la moelle osseuse et par la régénération au niveau périphérique des thrombocytes et des leucocytes.
Les patients de l'étude monocentrique avaient rechuté après 1 à 6 traitements antérieurs et 2 patients avaient rechuté après une transplantation de cellules souches. Les patients de l'étude multicentrique avaient rechuté après 1 à 4 cycles de traitement; ici, 5 patients avaient eu antérieurement une transplantation de cellules souches. L'âge moyen des patients était de 33 ans (limites: 9 à 75 ans) dans l'étude monocentrique et de 40 ans (limites: 5 à 73 ans) dans l'étude multicentrique.
Les résultats sont résumés dans le Tableau 2:
Tableau 2
|
|
Etude monocentrique
n=12
|
Etude multicentrique
n=40
| |
Dose de TRISENOX (mg/kg/jour)
|
0,16 (0,06-0,20)
|
0,15
| |
Rémission complète (RC)
|
11 (92%)
|
34 (85%)
| |
Délai de rémission médullaire (médiane)
|
32 jours
|
35 jours
| |
Délai de RC
|
54 jours
|
59 jours
| |
Taux de survie à 18 mois
|
67%
|
66%
|
L'étude monocentrique comprenait 2 enfants (<18 ans) et tous deux ont obtenu une rémission complète (RC). L'essai multicentrique comprenait 5 enfants (< 18 ans), dont 3 ont obtenu une RC. Aucun enfant de moins de 5 ans n'a été traité.
Dans le suivi après le traitement de consolidation, 7 patients de l'étude monocentrique et 18 patients de l'étude multicentrique ont reçu un traitement d'entretien par TRISENOX.
Trois patients de l'étude monocentrique et 15 patients de l'étude multicentrique ont reçu une transplantation de cellules souches après avoir terminé le traitement par TRISENOX. La durée moyenne de la RC, selon la méthode de Kaplan-Meier, est de 14 mois pour l'étude monocentrique (elle n'a pas été atteinte pour l'étude multicentrique). À la dernière visite de contrôle, 6 patients sur 12 étaient vivants dans l'étude monocentrique, avec un suivi moyen de 28 mois (limites: 25 à 29 mois). Dans l'étude multicentrique, 27 patients sur 40 étaient vivants, avec un suivi moyen de 16 mois (limites: 9 à 25 mois). Les estimations de Kaplan-Meier de la survie sur une durée de 18 mois pour les deux études sont présentées dans la figure ci-dessous.
Le tableau 3 présente la confirmation cytogénétique de la conversion à un génotype normal et la détection par RT-PCR (Reverse Transcriptase - Polymerase Chain Reaction) de la conversion du facteur PML/RAR-α à la normale.
Analyse cytogénétique après traitement par TRISENOX:
Tableau 3
|
|
Étude pilote monocentrique
nombre de patients avec RC = 11
|
Étude multicentrique
nombre de patients avec RC = 34
| |
Cytogénétique classique [t(15;17)]
| |
Absente
|
8 (73%)
|
31 (91%)
| |
Présente
|
1 (9%)
|
0 (0%)
| |
Non évaluable
|
2 (18%)
|
3 (9%)
| |
RT-PCR pour PML/RARα
| |
Négative
|
8 (73%)
|
27 (79%)
| |
Positive
|
3 (27%)
|
4 (12%)
| |
Non évaluable
|
-
|
3 (9%)
|
Des réponses ont été observées dans toutes les tranches d'âge étudiées (limites: 6 à 75 ans). Les taux de réponses étaient similaires dans les deux sexes.
Il n'existe aucune expérience de l'effet de TRISENOX sur la variante de la LPA caractérisée par la présence des translocations chromosomiques t(11;17) et t(5;17).
Sécurité et efficacité en pédiatrie
L'expérience concernant les enfants est limitée. Dans une étude de phase I auprès de 13 enfants atteints de LPA récidivante/réfractaire (âge médian de 17 ans, fourchette de 4 à 21 ans, données publiées par Fox et al. 2008), l'ATO a été administré 5 jours par semaine par voie intraveineuse à la dose de 0,15 mg/kg en 2 heures (20 doses par cycle). 85% des patients ont atteint une rémission morphologique complète. 70 doses ont été nécessaires chez 3 patients sur 7 pour atteindre un résultat négatif de RT-PCR. À la dose recommandée de 0,15 mg/kg par administration, l'ATO a été bien toléré par les enfants et adolescents atteints de leucémie; le profil de toxicité était comparable aux données publiées sur les patients adultes.
|