CompositionPrincipes actifs
siltuximab (anticorps monoclonal chimérique (humain/murin) de type immunoglobuline G1κ (IgG1κ), produit à partir d'une lignée de cellules d'ovaire de hamster chinois (Chinese Hamster Ovary, CHO).
Excipients
L-histidine, monohydrate monohydrochloride de L-histidine, polysorbate 80, saccharose.
Indications/Possibilités d’emploiSylvant est indiqué pour le traitement des patients adultes atteints de la maladie de Castleman (MC) multicentrique, non-infectés par le VIH (virus de l'immunodéficience humaine) et le HHV-8 (virus d'herpès humain 8).
Posologie/Mode d’emploiLa perfusion intraveineuse (i.v.) de Sylvant doit être administrée par des professionnels de santé qualifiés.
Pour obtenir des instructions complètes sur la perfusion intraveineuse de Sylvant, voir «Précautions pour l'utilisation, la manipulation et l'élimination».
Sylvant 11 mg/kg est administré par perfusion intraveineuse d'une heure, toutes les 3 semaines jusqu'à échec du traitement.
Au cours des 12 premiers mois, des analyses sanguines doivent être effectuées avant chaque dose de traitement par Sylvant, puis tous les 3 cycles par la suite. Si les critères indiqués dans le Tableau 1 ne sont pas satisfaits avant la réalisation de la perfusion, le prescripteur doit envisager de retarder le traitement. La réduction posologique n'est pas recommandée.
Tableau 1: Critères de traitement
|
Paramètres biologique
|
Exigences avant la première administration de Sylvant
|
Critères de traitement répété
| |
Nombre absolu de neutrophiles
|
≥1,0× 109/l
|
≥1,0× 109/l
| |
Numération plaquettaire
|
≥75× 109/l
|
≥50× 109/l
| |
Hémoglobinea
|
<170 g/l
|
<170 g/l
|
a Sylvant peut augmenter le taux d'hémoglobine chez les patients atteints de MC multicentrique.
Le traitement par Sylvant doit être interrompu si le patient présente une infection sévère ou une toxicité non-hématologique sévère. Après guérison, le traitement peut être repris avec la même dose.
Si le patient développe une réaction sévère liée à la perfusion, une réaction anaphylactique, une réaction allergique sévère ou un syndrome de libération des cytokines en relation avec la perfusion, Sylvant doit être arrêté.
L'arrêt du traitement doit être envisagé si plus de 2 doses ont été différées en raison de toxicités liées au traitement au cours des 48 premières semaines.
Instructions spéciales pour la posologie
Patients souffrant de maladies hépatiques
Aucune étude formelle sur les propriétés pharmacologiques de Sylvant n'a été menée chez les patients souffrant de troubles hépatiques (voir «Propriétés pharmacocinétiques – Troubles hépatiques»).
Patients souffrant de maladies rénales
Aucune étude formelle sur les propriétés pharmacocinétiques de Sylvant n'a été menée chez les patients souffrant de troubles rénaux (voir «Propriétés pharmacocinétiques – Troubles rénaux»).
Patients âgés
Dans les études cliniques, aucune différence liée à l'âge n'a été observée dans les propriétés pharmacocinétiques (PC) ou dans le profil de sécurité. Il n'est pas nécessaire d'ajuster la dose (voir «Propriétés pharmacocinétiques – Patients âgés»).
Pédiatrie
La sécurité et l'efficacité de Sylvant chez les patients pédiatriques n'ont pour l'instant pas encore été prouvées.
Contre-indicationsHypersensibilité sévère au principe actif ou à l'un des autres excipients.
Mises en garde et précautionsDe graves infections survenant simultanément
Infections, y compris des infections localisées, doivent être traitées avant l'administration de Sylvant. Des infections graves, notamment pneumonie et septicémie, ont été observées au cours d'études cliniques (voir «Effets indésirables»).
Sylvant peut masquer les signes et symptômes d'une inflammation aiguë, et peut, entre autres, réprimer la fièvre et l'augmentation des protéines de phase aiguë, comme p.ex. la protéine C réactive (CRP). Par conséquent, les prescripteurs doivent surveiller attentivement les patients recevant le traitement afin de détecter d'éventuelles infections graves.
Vaccinations
Les vaccins vivants atténués ne doivent pas être administrés simultanément ou au cours des 4 semaines précédant le traitement par Sylvant, dans la mesure où la sécurité d'emploi clinique n'a jusqu'à présent pas été démontrée et l'inhibition de l'IL-6 peut compromettre la réponse immunitaire normale à de nouveaux antigènes.
Paramètres lipidiques
Des élévations des triglycérides et du cholestérol (paramètres lipidiques) ont été observées chez des patients traités par Sylvant (voir «Effets indésirables»). Les patients avec des paramètres lipidiques élevés doivent être traités conformément aux directives cliniques pour la prise en charge de l'hyperlipidémie.
Réactions liées à la perfusion et hypersensibilité
Des réactions liées à la perfusion légères à sévères peuvent survenir pendant et après la perfusion intraveineuse de Sylvant.
Le traitement par Sylvant doit être interrompu chez les patients qui ont développé de sévères réactions d'hypersensibilité liées à la perfusion (p.ex. anaphylaxie) pendant ou après la perfusion. Le traitement de réactions sévères liées à la perfusion doit être fondé sur les signes et symptômes de la réaction. Un personnel qualifié et les médicaments adaptés doivent être disponibles pour traiter une éventuelle anaphylaxie (voir «Effets indésirables»).
En cas de réactions à la perfusion légères à modérées, après disparition des réactions, la reprise de la perfusion à une vitesse inférieure et l'administration d'antihistaminiques, acétaminophènes et corticostéroïdes peuvent être envisagées. Le traitement par Sylvant doit être interrompu chez les patients qui ne tolèrent pas la perfusion après ces interventions.
Tumeurs malignes
Les médicaments immunomodulateurs peuvent augmenter le risque de tumeurs malignes. Compte tenu de l'expérience limitée avec le siltuximab, insuffisante pour l'évaluation de l'incidence d'affections malignes, les données disponibles ne suggèrent pas d'augmentation du risque de tumeur maligne.
Perforation gastro-intestinale
Des cas de perforation gastro-intestinale (GI) ont été rapportés au cours d'études cliniques réalisées avec le siltuximab, mais en dehors de celles réalisées sur la MC multicentrique. Le siltuximab doit être utilisé avec précaution chez les patients présentant un risque accru de perforation GI. Les patients présentant des symptômes pouvant être associés à une perforation GI ou évocateurs de cette dernière doivent être examinés immédiatement.
Insuffisance hépatique
Au cours d'études cliniques, des augmentations légères à modérées, transitoires ou intermittentes, des transaminases hépatiques ou d'autres tests de la fonction hépatique tels que la bilirubine ont été rapportées après le traitement par Sylvant. Les patients présentant une insuffisance hépatique connue ainsi que les patients présentant des taux élevés de transaminases ou de bilirubine doivent être surveillés au cours du traitement par Sylvant.
InteractionsAucune étude formelle sur les interactions de Sylvant avec d'autres médicaments n'a été menée. Dans les études non-cliniques il est confirmé que l'interleukine 6 (IL-6) diminue l'activité du cytochrome P450 (CYP450). La liaison de l'IL-6 bioactive au siltuximab peut entraîner une augmentation du métabolisme des substrats CYP450, dans la mesure où l'activité de l'enzyme CYP450 revient à la normale. Par conséquent, l'administration simultanée de Sylvant avec des substrats du CYP450 à index thérapeutique étroit peut potentiellement changer les effets thérapeutiques et la toxicité de des médicaments en raison des variations des voies de signal du CYP450. Lors du début ou de l'arrêt du traitement par Sylvant chez des patients traités par des médicaments concomitants, à savoir des médicaments substrats du CYP450 et présentant un index thérapeutique étroit, il est recommandé de surveiller l'effet (p.ex. warfarine) ou la concentration du médicament (p.ex. ciclosporine ou théophylline). Il est indispensable d'ajuster la dose des médicaments concomitants en conséquence. L'effet de Sylvant sur l'activité de l'enzyme du CYP450 peut persister pendant plusieurs semaines après l'interruption du traitement. En outre, les prescripteurs doivent également prendre des précautions lorsque Sylvant est administré simultanément avec des médicaments substrats de CYP3A4 pour lesquels une atténuation de l'efficacité (p.ex. contraceptif oral) ne serait pas souhaitable.
Grossesse, allaitementGrossesse
Il n'existe pas de donnée sur l'utilisation de Sylvant chez la femme enceinte. Les études menées sur les singes n'ont montré aucune toxicité maternelle ou fœtale après administration du médicament par voie intraveineuse (voir «Données précliniques»). L'on ignore encore si siltuximab peut présenter un risque pour le fœtus après avoir été administré à la femme enceinte ou affecter la capacité reproductive de la femme. Sylvant doit être administré aux femmes enceintes uniquement si le bénéfice est nettement supérieur au risque encouru. Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant le traitement et jusqu'à 3 mois après l'arrêt du traitement.
En outre, les prescripteurs doivent également prendre des précautions lorsque Sylvant est administré simultanément avec des médicaments substrats de CYP3A4 pour lesquels une atténuation de l'efficacité (p.ex. des contraceptifs oraux, ne serait pas souhaitable (voir «Interactions»).
Comme avec les autres anticorps de type immunoglobuline G, le siltuximab traverse la barrière placentaire, comme cela a été mis en évidence au cours des études chez le singe. Par conséquent, les enfants nés de femmes traitées par siltuximab peuvent présenter des risques majorés d'infection et des précautions doivent être prises en cas d'administration de vaccins vivants chez ces enfants (voir «Données précliniques»).
Allaitement
On ne sait pas si le siltuximab ou ses métabolites passent dans le lait maternel humain. Parce qu'un grand nombre de médicaments et d'immunoglobulines passent dans le lait humain, et en raison d'éventuels effets indésirables liés à l'utilisation de Sylvant qui peuvent survenir chez les enfants allaités, il convient donc de décider d'arrêter l'allaitement ou d'arrêter de prendre le médicament, au regard du bénéfice du médicament pour la mère. On peut éventuellement envisager une interruption du traitement par le siltuximab ou une interruption de l'allaitement pendant la durée du traitement par le siltuximab en tenant compte de la demi-vie du siltuximab.
Fertilité
Les effets du siltuximab sur la fertilité n'ont jusqu'à maintenant pas été évalués chez des patients. Chez les singes à qui l'on a administré du siltuximab par voie intraveineuse, aucune modification histopathologique des tissus reproducteurs n'a été constatée (voir «Données précliniques»). Chez les souris à qui le médicament a été administré par voie sous-cutanée, aucune conséquence sur la fertilité des souris mâles ou femelles n'a été observée.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude n'a été menée pour étudier les effets sur l'aptitude à conduire des véhicules et à utiliser des machines. On ne sait pas si Sylvant peut avoir des conséquences sur la motricité.
Effets indésirablesLes données de l'ensemble des patients traités avec Sylvant en monothérapie (n=370) forment la base globale de l'évaluation de la sécurité. Le tableau 2 présente les fréquences d'apparition d'effets indésirables identifiés chez les 87 patients atteints de MC multicentrique (étude 1, étude 2 et étude 3) qui ont reçu la posologie recommandée de 11 mg/kg toutes les 3 semaines.
Dans l'étude 1, une étude de phase II randomisée, contrôlée versus placebo dans la MC multicentrique, 53 patients ont été randomisés dans le bras de traitement par Sylvant et ont reçu la dose recommandée de 11 mg/kg toutes les 3 semaines, et 26 patients ont été randomisés dans le bras placebo. 18 des 26 patients traités par placebo ont ensuite effectué un cross-over pour recevoir Sylvant. Dans l'étude 2, une étude de phase I, 16 des 37 patients atteints de la maladie de Castleman multicentrique ont été traités par Sylvant et ont reçu la posologie recommandée de 11 mg/kg toutes les 3 semaines. Dans l'étude 3, une étude de phase II ouverte, multicentrique, non randomisée, menée chez 60 patients atteints de MC multicentrique qui avaient été inclus auparavant dans l'étude 1 (41 patients) ou l'étude 2 (19 patients), les patients ont été traités par le siltuximab à la posologie recommandée de 11 mg/kg toutes les 3 semaines.
Tous les effets indésirables ayant un lien de causalité possible, probable ou certain avec le traitement par Sylvant à la posologie recommandée de 11 mg/kg toutes les 3 semaines sont regroupés dans le tableau 2.
Les effets indésirables les plus fréquemment identifiés (≥10%) étaient rhinopharyngite, thrombocytopénie, reflux gastro-œsophagien, fatigue, infection urinaire, hypertriglycéridémie, douleurs des extrémités, douleurs bucco-pharyngées, prurit et éruption.
Tableau 2: Effets indésirables en lien avec le siltuximab.
|
|
Placebo + BSCa
|
Siltuximab + BSCb
| |
Tous les grades (%)
|
Grade 3-4 (%)
|
Tous les grades (%)
|
Grade 3-4 (%)
| |
Affections hématologiques et du système lymphatique
| |
Thrombocytopénie
|
0,0%
|
0,0%
|
10,3%
|
2,3%
| |
Neutropénie
|
3,8%
|
0,0%
|
8,0%
|
3,4%
| |
Anémie
|
0,0%
|
0,0%
|
5,7%
|
0,0%
| |
Leucopénie
|
3,8%
|
0,0%
|
5,7%
|
0,0%
| |
Éosinophilie
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Polycythémie
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Thrombocytose
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Affections cardiaques
| |
Bloc auriculo-ventriculaire (1er degré)
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
Cardiopathie
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Insuffisance mitrale
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Épanchement péricardique
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Tachycardie sinusale
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Affections de l'oreille et du labyrinthe
| |
Vertiges
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Affections gastro-intestinales
| |
Reflux gastro-œsophagien
|
0,0%
|
0,0%
|
10,3%
|
0,0%
| |
Diarrhée
|
3,8%
|
3,8%
|
8,0%
|
0,0%
| |
Aphtes
|
0,0%
|
0,0%
|
4,6%
|
0.0%
| |
Constipation
|
0,0%
|
0,0%
|
4,6%
|
0,0%
| |
Douleurs abdominales
|
0,0%
|
0,0%
|
3,4%
|
0,0%
| |
Ulcération buccale
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
Stomatite
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
Troubles abdominaux
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Douleurs épigastriques
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Sécheresse buccale
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Dyspepsie
|
3,8%
|
0,0%
|
1,1%
|
0,0%
| |
Ulcération gingivale
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Nausées
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Douleurs dans la bouche
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Ulcération de la langue
|
3,8%
|
0,0%
|
1,1%
|
0,0%
| |
Vomissements
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Troubles généraux et anomalies au site d'administration
| |
Fatigue
|
7,7%
|
0,0%
|
10,3%
|
1,1%
| |
Œdème périphérique
|
0,0%
|
0,0%
|
5,7%
|
0,0%
| |
Sensation de malaise
|
0,0%
|
0,0%
|
4,6%
|
0,0%
| |
Pyrexie
|
0,0%
|
0,0%
|
3,4%
|
0,0%
| |
Troubles dans la poitrine
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
Œdème localisé
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
Asthénie
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Douleurs dans la poitrine
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Œdème facial
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Sensation de chaleur
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Œdème généralisé
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Extravasation au site de perfusion
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Douleurs
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Affections hépatobiliaires
| |
Anomalies de la fonction hépatique
|
3,8%
|
0,0%
|
4,6%
|
0,0%
| |
Hyperbilirubinémie
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Affections du système immunitaire
| |
Réaction anaphylactique
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Infections et infestations
| |
Rhinopharyngite
|
3,8%
|
0,0%
|
17,2%
|
0,0%
| |
Infection urinaire
|
0,0%
|
0,0%
|
10,3%
|
0,0%
| |
Infection des voies respiratoires supérieures
|
0,0%
|
0,0%
|
6,9%
|
0,0%
| |
Zona
|
0,0%
|
0,0%
|
3,4%
|
1,1%
| |
Infection des voies respiratoires inférieures
|
0,0%
|
0,0%
|
2,3%
|
1,1%
| |
Infection des voies respiratoires
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
Bronchite
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Conjonctivite
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Gastroentérite
|
3,8%
|
0,0%
|
1,1%
|
0,0%
| |
Impétigo
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Influenza
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Onychomycose
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Zona ophtalmique
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Otite moyenne chronique
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Paronychie
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Pharyngo-amygdalite
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Rhinite
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Sepsis
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Abcès dentaire
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Trachéobronchite
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Infection pulmonaire
|
3,8%
|
3,8%
|
0,0%
|
0,0%
| |
Investigations
| |
Prise de poids
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
Augmentation de l'alanine aminotransférase
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Augmentation de l'aspartate aminotransférase
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Augmentation de la créatine phosphokinase sanguine
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Augmentation de la créatinine sanguine
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Diminution du fer sanguin
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Augmentation du fer sanguin
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Augmentation du phosphore sanguin
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Augmentation de la gamma-glutamyltransférase
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Augmentation de l'hémoglobine
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Diminution du nombre de réticulocytes
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Diminution du pourcentage de réticulocytes
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Diminution de la ferritine sérique
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Augmentation de la ferritine sérique
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Diminution de la saturation de la transferrine
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Augmentation de la saturation de la transferrine
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Urobilinogène urinaire
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Perte de poids
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Diminution de la capacité totale de fixation du fer
|
3,8%
|
0,0%
|
0,0%
|
0,0%
| |
Diminution de la capacité latente de fixation du fer
|
3,8%
|
0,0%
|
0,0%
|
0,0%
| |
Troubles du métabolisme et de la nutrition
| |
Hypertriglycéridémie
|
0,0%
|
0,0%
|
11,5%
|
2,3%
| |
Hypercholestérolémie
|
0,0%
|
0,0%
|
6,9%
|
0,0%
| |
Hypokaliémie
|
0,0%
|
0,0%
|
4,6%
|
0,0%
| |
Hypomagnésémie
|
0,0%
|
0,0%
|
3,4%
|
0,0%
| |
Diminution de l'appétit
|
3,8%
|
0,0%
|
2,3%
|
0,0%
| |
Hyperuricémie
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
Anomalie des enzymes
|
7,7%
|
0,0%
|
1,1%
|
0,0%
| |
Hyperamylasémie
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Hyperphosphatémie
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Hypocalcémie
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Hyponatrémie
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Hypophosphatémie
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Hypoalbuminémie
|
3,8%
|
0,0%
|
0,0%
|
0,0%
| |
Hypoglycémie
|
3,8%
|
0,0%
|
0,0%
|
0,0%
| |
Affections musculosquelettiques et du tissu conjonctif
| |
Douleurs des extrémités
|
0,0%
|
0,0%
|
10,3%
|
0,0%
| |
Arthralgie
|
0,0%
|
0,0%
|
4,6%
|
0,0%
| |
Myalgie
|
0,0%
|
0,0%
|
3,4%
|
0,0%
| |
Douleurs osseuses
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Raideur articulaire
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Faiblesse musculaire
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Douleurs musculosquelettiques
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Raideur musculosquelettique
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Cervicalgies
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Ostéopénie
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Tumeurs bénignes, malignes et non précisées (incl. kystes et polypes)
| |
Douleurs tumorales
|
3,8%
|
0,0%
|
0,0%
|
0,0%
| |
Affections du système nerveux
| |
Céphalées
|
0,0%
|
0,0%
|
3,4%
|
0,0%
| |
Neuropathie motrice périphérique
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
Neuropathie sensitive périphérique
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
Sensation vertigineuse
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Hypoesthésie
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Somnolence
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Affections psychiatriques
| |
Hallucinations somatiques
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Dépression
|
3,8%
|
0,0%
|
0,0%
|
0,0%
| |
Affections du rein et des voies urinaires
| |
Insuffisance rénale
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Rétention urinaire
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Affections respiratoires, thoraciques et médiastinales
| |
Douleurs bucco-pharyngées
|
3,8%
|
0,0%
|
13,8%
|
0,0%
| |
Toux
|
3,8%
|
0,0%
|
3,4%
|
0,0%
| |
Dyspnée
|
0,0%
|
0,0%
|
3,4%
|
1,1%
| |
Inflammation pulmonaire
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
Épistaxis
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Toux productive
|
3,8%
|
0,0%
|
1,1%
|
0,0%
| |
Tachypnée
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Affections de la peau et du tissu sous-cutané
| |
Prurit
|
0,0%
|
0,0%
|
14,9%
|
0,0%
| |
Éruption maculo-papuleuse
|
0,0%
|
0,0%
|
12,6%
|
0,0%
| |
Éruption
|
0,0%
|
0,0%
|
4,6%
|
0,0%
| |
Sécheresse cutanée
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
Érythème
|
3,8%
|
0,0%
|
2,3%
|
0,0%
| |
Lupus érythémateux cutané (LEC)
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Dermatite acnéiforme
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Dermatite allergique
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Exanthème médicamenteux
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Eczéma
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Hyperhidrose
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Sueurs nocturnes
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Prurit généralisé
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Éruption érythémateuse
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Éruption généralisée
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Éruption prurigineuse
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Hyperpigmentation cutanée
|
0,0%
|
0,0%
|
1,1%
|
0,0%
| |
Ulcère cutané
|
0,0%
|
0,0%
|
1,1%
|
1,1%
| |
Affections vasculaires
| |
Hypertension
|
0,0%
|
0,0%
|
3,4%
|
0,0%
| |
Hypotension
|
0,0%
|
0,0%
|
3,4%
|
1,1%
| |
Bouffées de chaleur
|
0,0%
|
0,0%
|
2,3%
|
0,0%
| |
a
Tous les patients atteints de la maladie de Castleman (MC) multicentrique traités par le placebo (N = 26).
b Tous les patients atteints de la maladie de Castleman (MC) multicentrique traités par Sylvant à la posologie recommandée (y compris les patients de l'étude croisée (N = 87).
BSC = Best Supportive Care (soins de soutien optimaux)
|
Réactions liées à la perfusion et hypersensibilité
Au cours des études cliniques, Sylvant a été associé, chez 5,1% des patients traités en monothérapie par Sylvant, à une réaction liée à la perfusion ou à une réaction d'hypersensibilité (réaction sévère chez 0,8%).
Chez les patients atteints de MC multicentrique traités à long terme par le siltuximab à la posologie recommandée de 11 mg/kg toutes les 3 semaines, des réactions liées à la perfusion ou des réactions d'hypersensibilité sont survenues avec une fréquence de 6,3% (réactions sévères: 1,3%).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucun cas de surdosage n'a été rapporté. La dose répétée de 15 mg/kg toutes les 3 semaines a été effectuée sans effet indésirable supplémentaire.
Propriétés/EffetsCode ATC
L04AC11
Mécanisme d'action
Le siltuximab est un anticorps monoclonal chimérique homme/souris qui forme des complexes stables de plus haute affinité avec les formes bioactives solubles de l'IL-6 humaine. Le siltuximab empêche la liaison de l'IL-6 humaine aux récepteurs solubles et membranaires de l'IL-6 (IL-6R), inhibant ainsi la formation du complexe de signalisation hexamérique avec la gp130 à la surface cellulaire. L'IL-6 est une cytokine pro-inflammatoire produite par différents types de cellules, notamment les lymphocytes T et B, d'autres types de lymphocytes, les monocytes, les macrophages et les fibroblastes, ainsi que les cellules malignes. Il a été déterminé que l'IL-6 participait à différents processus physiologiques normaux, tels que l'induction de la sécrétion d'immunoglobulines, l'initiation de la synthèse des protéines hépatiques de la phase aiguë et la stimulation de la prolifération et de la différenciation des cellules précurseurs hématopoïétiques. L'hyperproduction d'IL-6, au cours des maladies inflammatoires chroniques et des affections malignes, a été liée à l'anémie et à la cachexie, et il a été supposé qu'elle jouait un rôle central dans la prolifération des plasmocytes et les manifestations systémiques observées chez les patients atteints de la maladie de Castleman multicentrique.
Pharmacodynamique
In-vitro, le siltuximab a inhibé de manière dose-dépendante la croissance d'une lignée cellulaire de plasmocytome murin dépendant de l'IL-6 en réponse à l'IL-6 humaine. Dans des cultures de cellules d'hépatome humain, la production stimulée par l'IL-6 de la protéine sérique de la phase aiguë, l'amyloïde A, a été inhibée de manière dose-dépendante par le siltuximab. De même, dans des cultures de cellules humaines de lymphome de Burkitt, la production d'immunoglobulines M (IgM) en réponse à l'IL-6 a été inhibée de manière dose-dépendante par le siltuximab.
Biomarqueurs
L'IL-6 stimule au cours de la phase aiguë l'expression de la CRP. Le mécanisme d'action du siltuximab est la neutralisation de la bioactivité de l'IL-6, qui peut être mesurée indirectement par la suppression de la CRP. Le traitement par Sylvant de la MC multicentrique entraîne une diminution rapide et prolongée des concentrations sériques de CRP. Le dosage des concentrations d'IL-6 dans le sérum ou le plasma au cours du traitement ne doit pas être utilisé comme un marqueur pharmacodynamique, dans la mesure où les complexes entre l'anticorps neutralisé par le siltuximab et l'IL-6 interfèrent avec les méthodes de quantification immunologiques actuelles de l'IL-6.
Efficacité clinique
Étude 1
Une étude de phase 2 internationale, randomisée (2:1), en double aveugle, contrôlée versus placebo a été menée pour évaluer l'efficacité et la sécurité de Sylvant (11 mg/kg toutes les 3 semaines) par rapport au placebo en association avec les soins de soutien optimaux (Best Supportive Care, BSC) chez des patients atteints de MC multicentrique. Le traitement a été poursuivi jusqu'à un échec thérapeutique (défini par une progression de la maladie basée sur l'augmentation des symptômes, une progression radiologique ou une détérioration de l'indice de performance) ou une toxicité inacceptable. Au total, 79 patients atteints de MC multicentrique symptomatique ont été randomisés et traités. L'âge moyen était de 47 ans (intervalle de 20 à 74 ans) dans le bras sous Sylvant et de 48 ans (intervalle de 27 à 78 ans) dans le bras sous placebo. Un nombre plus important de patients masculins a été inclus dans le bras sous placebo (85% dans le groupe sous placebo contre 56% dans le groupe sous Sylvant). L'indice de performance ECOG (0/1/2) au début du traitement a été respectivement de 42%/45%/13% dans le bras sous Sylvant et de 39%/62%/0% dans le bras sous placebo. Au début de la thérapie, 55% des patients dans le bras sous Sylvant et 65% des patients dans le bras sous placebo avaient reçu des traitements systémiques antérieurs pour la MC multicentrique et 30% des patients dans le bras sous Sylvant et 31% dans le bras sous placebo utilisaient des corticoïdes. Les sous-types histologiques ont été similaires dans les deux bras de traitement, avec 33% de sous-type hyalino-vasculaire, 23% de sous-type plasmocytaire et 44% de sous-type mixte. Les paramètres biologiques relatifs à la maladie sont présentés dans le tableau 3. La vitesse de sédimentation de la CRP et des érythrocytes (ESR) a montré une grande diversité dans les deux bras de traitement.
Tableau 3: Paramètres biologiques relatifs à la maladie
|
|
Sylvant + BSC*
|
Placebo + BSC
| |
Patients appartenant à la population en intention de traiter
|
53
|
26
| |
Moyenne en hémoglobine (g/l) (écart-type)
|
115,8 (24,70)
|
130,0 (25,70)
| |
Moyenne des plaquettes (109/l) (écart-type)
|
323,2 (156,58)
|
302,6 (123,54)
| |
Moyenne de l'albumine (g/dl) (écart-type)
|
3,5 (0,76)
|
3,6 (0,46)
| |
Moyenne de l'ESR (mm/h) (écart-type)
|
68,3 (48,66)
|
34,6 (35,06)
| |
Moyenne de la CRP (mg/l) (écart-type)
|
43,2 (53,63)
|
24,8 (34,53)
| |
Moyenne du fibrinogène (µmol/l) (écart-type)
|
16,9 (7,52)
|
15,3 (7,48)
|
* Best Supportive Care (soins de soutien optimaux).
Le critère d'évaluation principal de l'étude était la réponse tumorale et symptomatique durable, définie comme une réponse tumorale examinée de manière indépendante et une résolution complète ou une stabilisation des symptômes de MC multicentrique collectés de manière prospective, pendant au moins 18 semaines sans échec thérapeutique.
L'étude 1 a démontré une amélioration statistiquement significative du taux de réponse tumorale et symptomatique examinée de manière indépendante dans le bras sous Sylvant, par rapport au bras sous placebo (34% vs 0%, intervalle de confiance à 95%: 11,1; 54,8; p=0,0012). Les analyses de sensibilité ont également soutenu l'analyse du critère d'évaluation principal qui a présenté un taux de réponse tumorale et symptomatique durable évaluée par l'investigateur significativement plus élevé de 45% chez les patients traités par Sylvant par rapport à 0% des patients traités par le placebo (p <0,0001). Le taux de réponse tumorale global a été évalué de manière indépendante et par l'investigateur, sur la base des critères de Cheson modifiés.
Les principaux résultats d'efficacité de l'étude 1 sont présentés dans le tableau 4.
Tableau 4: Critères d'efficacité de l'étude 1
|
Critères d'efficacité:
|
Sylvant + BSC
|
Placebo + BSC
|
valeur pa
| |
Critère d'efficacité principal
| |
Réponse tumorale et symptomatique durable (examen indépendant)
|
18/53 (34,0%)
|
0/26 (0%)
|
0,0012
| |
Critères d'efficacité secondaires
| |
Meilleure réponse tumorale (examen indépendant)
|
20/53 (37,7%)
|
1/26 (3,8%)
|
0,0022
| |
Meilleure réponse tumorale (évaluation de l'investigateur)
|
27/53 (50,9%)
|
0/26 (0%)
|
<0,0001
| |
Délai avant un échec thérapeutique
|
Non atteint
|
134 jours
|
0,0084; RR 0,418
| |
Augmentation de l'hémoglobine >15 g/l à la semaine 13/population évaluable pour la réponse sur l'hémoglobine
|
19/31 (61,3%)
|
0/11 (0%)
|
0,0002
| |
Durée de la réponse tumorale et symptomatique (jours) – examen indépendant; médiane (min, max)
|
340 (55, 676)b
|
Non applicablec
|
| |
Réponse symptomatique complète durabled
|
13/53 (24,5%)
|
0/26 (0%)
|
0,0037
| |
Durée de la réponse symptomatique complète durable (jours), médiane (minimum, maximum)
|
472 (169, 762)e
|
Non applicablec
|
|
a Ajustée pour l'utilisation des corticoïdes lors de la randomisation.
b Au moment de l'analyse principale, les données de 19 répondeurs tumoraux et symptomatiques sur 20 ont été censurées à cause d'une réponse en cours.
c Non applicable, il n'y a eu aucun répondeur dans le bras placebo, par conséquent la durée n'est pas applicable.
d Une réponse symptomatique complète est définie par une réduction de 100% du score global initial des symptômes de MC multicentrique, maintenue pendant au moins 18 semaines avant échec thérapeutique.
e Les données de 11 répondeurs symptomatiques complets sur 13 ont été censurées à cause d'une réponse en cours.
Les signes et les symptômes liés à la MC multicentrique ont été collectés de manière prospective. Un score global pour l'ensemble des symptômes (désigné par le terme de Score global des symptômes liés à la MC multicentrique) a été constitué par la somme des grades de sévérité (grade NCI-CTCAE) des signes et des symptômes liés à la MC multicentrique [symptômes généraux liés à la MC multicentrique (fatigue, malaise, hyperhidrose, sueurs nocturnes, fièvre, perte de poids, anorexie, douleur tumorale, dyspnée et prurit), phénomènes auto-immuns, rétention liquidienne, neuropathie et troubles cutanés]. Le pourcentage de changement par rapport aux valeurs initiales du score des signes et des symptômes liés à la MC multicentrique et du score global des symptômes liés à la MC multicentrique à chaque cycle a été calculé. Une réponse symptomatique complète a été définie par une réduction de 100% par rapport aux valeurs initiales du score global des symptômes liés à la MC multicentrique, maintenue pendant au moins 18 semaines avant échec thérapeutique.
La réponse sur l'hémoglobine a été définie comme un changement lors de l'inclusion à ≥15 g/l à la Semaine 13. La valeur moyenne de l'hémoglobine à chaque cycle au cours de la période de traitement en aveugle est représentée dans le schéma 1.
Schéma 1: Valeur moyenne de l'hémoglobine à chaque cycle au cours de la période de traitement en aveugle
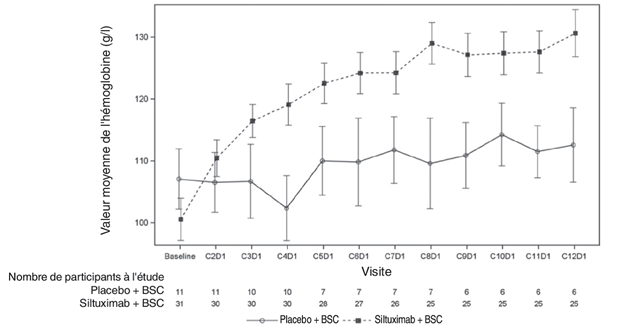
Le taux de survie d'un an a augmenté de 100% dans le bras Sylvant et de 92% dans le bras placebo.
Analyses en sous-groupes:
Les analyses des critères d'évaluation principaux et secondaires dans différents sous-groupes définis selon l'âge (<65 ans et ≥65 ans); l'ethnie (origine caucasienne et non caucasienne); la région (Amérique du Nord, EMEA [Europe, Moyen-Orient, Afrique] et Asie-Pacifique); l'utilisation initiale de corticoïdes (oui et non); l'utilisation de traitements antérieurs (oui et non); et l'histologie de la MC multicentrique (plasmocytaire et mixte) ont montré de façon cohérente un effet thérapeutique en faveur du bras Sylvant, sauf pour le sous-groupe hyalino-vasculaire. Un effet thérapeutique cohérent en faveur des patients traités par Sylvant a été mis en évidence dans le sous-groupe hyalino-vasculaire pour tous les critères secondaires majeurs.
Étude 2
En plus de l'étude 1, des données d'efficacité chez des patients atteints de la maladie de Castleman (MC) issus de l'étude de phase I à bras unique (étude 2) sont disponibles. Dans cette étude, 37 patients atteints de MC ont été traités par Sylvant. 35 de ces patients étaient atteints de MC multicentrique. Au total, 16 patients atteints de MC multicentrique ont reçu 11 mg/kg toutes les 3 semaines. Les données démographiques des patients et les caractéristiques de la maladie des patients ayant reçu 11 mg/kg toutes les 3 semaines étaient similaires à celles de l'étude 1. L'âge moyen était de 51 ans (21 à 76 ans) et 50% des patients étaient des hommes. L'indice de performance ECOG (0/1/2) au début du traitement était de 6%/69%/25%. Soixante-neuf pour cent (69%) des patients avaient reçu des traitements systémiques antérieurs pour la MC multicentrique. Les sous-types histologiques étaient de 44% pour le sous-type hyalino-vasculaire, 50% pour le sous-type plasmocytaire et 6% pour le sous-type mixte. Ce taux d'hémoglobine moyen (écart-type) s'élevait à 125 (23) g/l.
Les données cliniques observées dans l'étude 1 ont été soutenues par l'étude 2. La durée moyenne du traitement par Sylvant s'élevait à 1278 jours et le nombre moyen des administrations de Sylvant s'élevait à 51 patients sous Sylvant. Chez les 16 patients atteints de MC multicentrique traités à la posologie de 11 mg/kg toutes les 3 semaines, le taux de réponse tumorale global déterminé par un examen indépendant a été de 43,8% avec 6,3% de réponse complète. Toutes les réponses tumorales ont été durables pendant >18 semaines. Pour les patients ayant une valeur de l'hémoglobine inférieure au seuil de la valeur normale, le taux de réponse de l'hémoglobine s'élevait à 50% à la semaine 13. Le taux de survie à un an des patients traités par Sylvant s'élevait à 100%.
Étude 3
Dans cette étude de phase II ouverte, multicentrique et non randomisée, la sécurité et l'efficacité d'un traitement prolongé par le siltuximab ont été évaluées chez 60 patients atteints de MC multicentrique qui avaient été inclus auparavant dans l'étude 1 (41 patients) ou l'étude 2 (19 patients). La durée médiane du traitement par le siltuximab a été de 5,52 ans (intervalle: de 0,8 à 10,8 ans); plus de 50% des patients ont été traités pendant ≥5 ans par le siltuximab. Après un suivi médian de 6 ans, aucun des 60 patients n'était décédé et le maintien du contrôle de la maladie a été observé chez 58 des 60 patients.
PharmacocinétiqueAbsorption
Après la première administration de siltuximab (posologies comprises entre 0,9 et 15 mg/kg), l'aire sous la courbe de la concentration par rapport au temps (ASC) et la concentration sérique maximale (Cmax) ont augmenté de manière proportionnelle à la dose.
Distribution
Après l'administration de doses répétées à la posologie recommandée, la clairance du siltuximab n'a pas varié par rapport au temps et l'accumulation systémique a été modérée (indice d'accumulation de 1,7). Les concentrations sériques ont atteint l'état d'équilibre à la sixième perfusion administrée (intervalles toutes les 3 semaines) avec des concentrations maximale et minimale moyennes de 332 ± 139 et 84 ± 66 μg/ml.
Immunogénicité
Comme avec toutes les protéines thérapeutiques, il existe un risque de développement d'anticorps contre le médicament (immunogénicité). L'immunogénicité du Siltuximab a été évaluée avec différentes méthodes: un dosage immunoenzymatique (EIA) relatif à l'antigène-anticorps et un immunodosage à base d'électro-chimioluminescence (ECLIA).
Au cours des études cliniques, incluant des études en monothérapie et en association, 4 des 432 (0,9%) patients évaluables ont été testés positifs aux anticorps anti-siltuximab. Des analyses d'immunogénicité complémentaires ont été réalisées pour tous les échantillons positifs des 4 patients présentant des anticorps anti-siltuximab détectables.
Aucun de ces patients n'avait d'anticorps neutralisants. Aucune preuve de modification de la sécurité ou de l'efficacité n'a été mise en évidence chez les patients ayant développé des anticorps dirigés contre le siltuximab.
Métabolisme
Pas applicable.
Élimination
Dans les études cliniques, la clairance du siltuximab a été comprise entre 4,03 et 4,59 ml/jour/kg et la demi-vie moyenne entre 17,7 et 20,6 jours, à la dose de 11 mg/kg.
Cinétique pour certains groupes de patients
Des analyses pharmacocinétiques de population ont été effectuées à travers l'utilisation des données de 378 patients présentant différents types de pathologies ayant reçu le siltuximab en monothérapie à des posologies comprises entre 0,9 et 15 mg/kg. Les effets de différentes co-variables sur les propriétés pharmacocinétiques du siltuximab ont été évalués au cours des analyses.
La clairance du siltuximab a augmenté parallèlement au poids corporel. Cependant, aucun ajustement posologique n'est nécessaire par rapport au poids corporel, dans la mesure où l'administration est effectuée sur la base d'une dose en mg/kg. Les facteurs suivants n'ont eu aucun effet clinique sur la clairance du siltuximab: sexe, âge et ethnicité. L'effet du statut des anticorps anti-siltuximab n'a pas été examiné, dans la mesure où le nombre de patients positifs aux anticorps anti-siltuximab était trop faible.
Pédiatrie
La sécurité et l'efficacité du siltuximab n'ont pas été établies chez les patients pédiatriques.
Patients âgés
Les propriétés PK de population du siltuximab ont été analysées afin d'évaluer les effets des caractéristiques démographiques. Les résultats n'ont montré aucune différence significative au niveau des propriétés PK du siltuximab chez les patients âgés de plus de 65 ans comparé aux patients âgés de 65 ans ou moins.
Dysfonctionnement rénal
Aucune étude formelle n'a été menée sur les effets d'une atteinte de la fonction rénale sur la pharmacocinétique du siltuximab. Aucun effet significatif sur les propriétés PK du siltuximab n'a été observé chez les patients présentant une clairance de la créatinine initiale de 12 ml/min ou plus. Quatre patients avec une insuffisance rénale sévère (clairance de la créatinine comprise entre 12 et 30 ml/min) ont été inclus dans l'ensemble des données.
Dysfonctionnement hépatique
Aucune étude formelle n'a été menée sur les effets d'une atteinte de la fonction hépatique sur la pharmacocinétique du siltuximab. Aucun effet significatif sur les propriétés PK du siltuximab n'a été observé chez les patients présentant une concentration d'alanine aminotransférase initiale jusqu'à 3,7 fois la limite supérieure de la normale, une concentration initiale d'albumine comprise entre 1,5 et 5,8 g/dl et une concentration initiale de bilirubine comprise entre 1,7 et 42,8 mg/dl.
Données précliniquesCancérogénicité et mutagénicité
Les données non cliniques n'indiquent pas de risque particulier pour l'homme sur la base des études conventionnelles sur la toxicité par dose répétée, la reproduction et la toxicité pour le développement. Aucune étude de cancérogénicité et mutagénicité formelle n'a été réalisée sur le siltuximab.
Toxicologie de la reproduction et du développement
Fertilité
Le siltuximab n'a entraîné aucune toxicité sur l'appareil reproducteur chez le macaque. Chez les souris recevant un anticorps monoclonal anti-IL-6 de souris par voie sous-cutanée à une dose de 40 ou 100 mg/kg/semaine, aucun effet sur la fertilité des animaux mâles ou femelles n'a été observé.
Grossesse
Les résultats d'une étude de développement embryo-fœtal où le siltuximab était administré par voie intraveineuse à des singes femelles gestantes (jours de gestation 20 à 118) à des doses de 9,2 et 46 mg/kg/semaine, n'ont montré aucune toxicité maternelle ou fœtale. Le siltuximab a traversé la barrière placentaire au cours de la gestation et les fœtus ont été exposés au siltuximab durant leur développement. Au 140e jour de gestation (environ 25 jours avant la naissance naturelle), les concentrations sériques fœtales de siltuximab étaient similaires aux concentrations maternelles. L'examen histopathologique du tissu lymphatique des fœtus au 140e jour de gestation n'a montré aucune anormalité morphologique dans le développement du système immunitaire.
Toxicité en cas d'administrations répétées
Les études de toxicologie d'une durée de trois et six mois en rapport à des doses intraveineuses de siltuximab menées chez de jeunes macaques (à des doses de 9,2 et 46 mg/kg/semaine) n'ont montré aucun signe de toxicité. Une légère réduction de la réponse immunologique dépendante des lymphocytes T et une réduction de la taille des centres germinatifs spléniques après une immunisation par l'hémocyanine de patelle (KLH) ont été observées et considérées comme des réponses pharmacologiques à l'inhibition de l'IL-6 ne présentant pas de signification toxicologique.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, Sylvant ne doit pas être mélangé avec d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
À conserver au réfrigérateur entre 2 °C et 8 °C. Conserver à l'abri de la lumière. Tenir hors de portée des enfants
Remarques concernant la manipulation
Remarque concernant l'utilisation, la manipulation et l'élimination
Utiliser une technique aseptique
1. Calculer la dose, le volume total de solution reconstituée de Sylvant requis et le nombre de flacons nécessaires. L'aiguille recommandée pour la préparation est une aiguille de calibre 21 de 1,5 pouce (38 mm). Les poches de perfusion (250 ml) doivent contenir du dextrose à 5% et doivent être constituées de chlorure de polyvinyle (PVC), de polyoléfine (PO), de polypropylène (PP) ou de polyéthylène (PE). Des flacons en PE peuvent également être utilisés.
2. Laisser les flacons de Sylvant atteindre la température ambiante pendant environ 30 minutes. Sylvant doit rester à température ambiante pendant toute la durée de la préparation.
Afin d'obtenir un concentré de Sylvant (20 mg/ml), chaque flacon doit être reconstitué comme indiqué dans le tableau ci-dessous.
Tableau 6: instruction pour la reconstitution
|
Dosage
|
Quantité d'eau stérile pour préparations injectables nécessaire à la reconstitution
|
Concentration finale après reconstitution
| |
Flacon de 100 mg
|
5,2 ml
|
20 mg/ml
| |
Flacon de 400 mg
|
20,0 ml
|
20 mg/ml
|
Remuer doucement les flacons reconstitués en effectuant un mouvement de rotation (NE PAS AGITER, NI PASSER AU VORTEX, NI SECOUER VIGOUREUSEMENT) afin de faciliter la dissolution de la poudre. Ne pas prélever le contenu avant que toute la poudre n'ait été complètement dissoute. La poudre doit se dissoudre en moins de 60 minutes. Inspecter le concentré de Sylvant afin de contrôler qu'il ne présente pas de particules ou de décoloration. Ne pas l'utiliser en cas d'opacité visible, de particules étrangères et/ou de décoloration de la solution.
Diluer le volume total de la dose de Sylvant reconstituée dans 250 ml de dextrose stérile à 5%: veuillez retirer au préalable un volume équivalent au volume de la solution reconstituée de Sylvant de la poche de dextrose à 5%. Ajouter lentement le volume total de la solution reconstituée de Sylvant à la poche de perfusion de 250 ml. Mélanger doucement.
3. La solution reconstituée de Sylvant ne doit pas être conservée plus de deux heures avant d'être ajoutée à la poche de perfusion intraveineuse. La perfusion doit être effectuée dans un délai de 6 heures après l'ajout de la solution reconstituée dans la poche de perfusion. Administrer la solution diluée pendant une période d'une heure en utilisant des ensembles de perfusion avec tubulures de PVC, de polyuréthane (PU) ou de PE, contenant un filtre en ligne de polyéthersulfone (PESC) de 0,2 micron. Ne pas conserver la fraction de solution de perfusion non utilisée pour une utilisation ultérieure.
4. Aucune étude de compatibilité biochimique et physique n'a été menée pour évaluer la possibilité d'administrer Sylvant avec d'autres médicaments. Ne pas perfuser Sylvant simultanément dans la même tubulure de perfusion que celle utilisée pour d'autres agents.
5. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Numéro d’autorisation65183 (Swissmedic)
Présentation1 flacon de 100 mg [A]
1 flacon de 400 mg [A]
Titulaire de l’autorisationMedius AG, Muttenz
Mise à jour de l’informationJuillet 2025
|