CompositionPrincipes actifs
Dulaglutide.
Excipients
Citrate de sodium dihydraté, acide citrique, mannitol, polysorbate 80, eau pour préparations injectables, q.s. pro 0,5 ml.
Teneur totale en sodium: 0,32 mg / 0,5 ml.
Indications/Possibilités d’emploiTrulicity est indiqué dans le traitement du diabète sucré de type 2 chez des patients âgés de 10 ans et plus chez qui le diabète n'est pas suffisamment contrôlé par un régime alimentaire et de l'exercice physique:
·en monothérapie, en cas de contre-indication ou d'intolérance à la metformine.
·en association avec d'autres médicaments hypoglycémiants (voir la section «Efficacité clinique» pour les résultats sur les associations de Trulicity avec d'autres hypoglycémiants étudiés dans les essais cliniques).
Trulicity est indiqué dans la prévention d'évènements cardiovasculaires chez des patients adultes atteints de diabète sucré de type 2 et d'une affection cardiovasculaire déjà manifeste ou subclinique (voir la section «Efficacité clinique»).
Posologie/Mode d’emploiAdultes
La dose recommandée est de 0,75 mg une fois par semaine. Si l'effet est insuffisant et que le traitement est bien toléré, la dose peut être augmentée à 1,5 mg une fois par semaine.
Pour un contrôle supplémentaire de la glycémie
·la dose de 1,5 mg peut être augmentée à 3 mg une fois par semaine au bout de 4 semaines au minimum.
·la dose de 3 mg peut être augmentée à 4,5 mg une fois par semaine au bout de 4 semaines au minimum.
La dose maximale est de 4,5 mg une fois par semaine.
(Voir «Mises en garde et précautions» et «Effets indésirables»).
Enfants et adolescents
La dose initiale pour les enfants et les adolescents âgés de 10 et plus est de 0,75 mg une fois par semaine.
Au besoin, la dose peut être augmentée à 1,5 mg une fois par semaine au bout de 4 semaines au minimum. La dose maximale est de 1,5 mg une fois par semaine.
Traitement d'association
La dose peut être administrée à toute heure de la journée, indépendemment des repas.
Lorsque Trulicity est ajouté à un traitement en cours par la metformine et/ou la pioglitazone, la dose de metformine et/ou de pioglitazone peut être conservée. Lorsque Trulicity est ajouté à un traitement en cours par la metformine et/ou un inhibiteur du SGLT-2 (co-transporteur du sodium-glucose de type 2), la dose de metformine et/ou d'inhibiteur du SGLT-2 peut être conservée. Lorsque Trulicity est ajouté à un traitement en cours par une sulfonylurée ou l'insuline, une diminution de la dose de sulfonylurée ou d'insuline devrait être envisagée afin de réduire le risque d'hypoglycémie.
L'utilisation de Trulicity ne nécessite pas d'autosurveillance additionnelle de la glycémie par le patient. Une autosurveillance de la glycémie par le patient peut toutefois s'avérer nécessaire pour ajuster la dose de la sulfonylurée ou de l'insuline. Ceci s'applique en particulier au début du traitement par Trulicity et lors d'une réduction de la dose d'insuline. Il est recommandé de réduire la dose d'insuline par étapes.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Dose retardée
En cas d'oubli, la dose doit être administrée le plus rapidement possible si le délai avant la date de la prochaine dose est d'au moins 3 jours (72 heures). Si la dose suivante est prévue dans moins de 3 jours (72 heures), la dose omise ne doit pas être administrée et la dose suivante doit être administrée le jour normalement prévu. Dans tous les cas, le patient peut ensuite reprendre son rythme hebdomadaire d'administration habituel.
En cas de besoin, le jour de la semaine prévu pour l'injection hebdomadaire peut être changé, dans la mesure où la dernière dose a été administrée au moins 3 jours (72 heures) auparavant.
Populations particulières
Patients âgés (>65 ans)
Aucun ajustement de la dose n'est requis en fonction de l'âge.
Insuffisance rénale
Aucun ajustement de la dose n'est requis chez les patients atteints d'insuffisance rénale. L'expérience clinique chez les patients présentant une insuffisance rénale terminale (eGFR<15 ml/min/1,73m2) est limitée (voir «Pharmacocinétique»).
Insuffisance hépatique
Aucun ajustement de la dose n'est requis chez les patients atteints d'insuffisance hépatique (voir «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l'efficacité de Trulicity chez les enfants et les adolescents de moins de 10 ans n'ont pas encore été étudiées.
Utilisation
Trulicity est administré par injection sous-cutanée dans l'abdomen, la cuisse ou le haut du bras. Trulicity ne doit pas être administré par injection intraveineuse ou intramusculaire.
Contre-indicationsHypersensibilité à la substance active ou à l'un des excipients.
Mises en garde et précautionsTrulicity ne doit pas être utilisé chez les patients atteints de diabète de type 1 ou pour le traitement d'une acidocétose diabétique. Le dulaglutide ne remplace pas l'insuline. Il existe des rapports décrivant la survenue d'une acidocétose diabétique chez des patients, dépendants de l'insuline, à la suite d'un arrêt brusque de l'insuline ou d'une rapide réduction de la dose d'insuline (voir «Posologie/Mode d'emploi»).
Maladies gastrointestinales sévères
L'utilisation d'agonistes du récepteur du GLP-1 peut être associée à des effets indésirables gastro-intestinaux, notamment à des nausées, des vomissements et de la diarrhée (voir «Effets indésirables»). Ces événements peuvent entraîner une déshydratation, ce qui peut provoquer une détérioration de la fonction rénale, y compris une insuffisance rénale aiguë. Trulicity ralentit la vidange gastrique. Une aspiration pulmonaire a été rapportée chez des patients qui recevaient des agonistes du récepteur GLP-1 à longue durée d'action et qui étaient soumis à une anesthésie générale ou à une sédation profonde. Ceci doit être pris en compte lors d'interventions de ce type. Trulicity n'a pas été étudié chez les patients atteints de maladie gastro-intestinale sévère, y compris de gastroparésie sévère, et n'est donc pas recommandé chez ces patients. Des événements en lien avec une altération de la vidange gastrique, y compris gastroparésie sévère, ont été rapportés. Soumettez les patients à une surveillance et envisagez une adaptation de la dose ou un arrêt du médicament s'ils développent de graves symptômes gastro-intestinaux pendant le traitement.
Pancréatite
Des cas de pancréatite ont été rapportés lors de l'utilisation d'agonistes du récepteur du GLP-1, y compris du dulaglutide. Les patients doivent être informés des symptômes caractéristiques de la pancréatite aigüe: fortes douleurs abdominales persistantes. En cas de suspicion de pancréatite, le dulaglutide et les autres médicaments potentiellement déclencheurs doivent être interrompus jusqu'à la fin des investigations. Si le diagnostic de pancréatite est confirmé, le traitement par le dulaglutide doit être durablement arrêté.
Le dulaglutide est associé à une élévation modérée des enzymes pancréatiques (lipase et/ou amylase pancréatique) de 11 à 21 % par rapport à la valeur initiale. À elle seule, une élévation des enzymes pancréatiques sans autres symptômes d'une pancréatite aiguë n'évoque pas une pancréatite aiguë.
Des patients avec une anamnèse de pancréatite aiguë ou chronique n'ont pas été traités dans les essais cliniques. Le risque de pancréatite de ces patients ne peut pas être évalué. Une prudence particulière est par conséquent de mise chez ces patients.
Hypoglycémie
Les patients traités par Trulicity en association avec des sulfonylurées ou une insuline peuvent présenter un risque accru d'hypoglycémie. Le risque d'hypoglycémie peut être diminué par une réduction de la dose de la sulfonylurée ou de l'insuline.
Ce médicament contient moins que 1 mmol de sodium (23 mg) par unité de dose, il est donc quasiment «sans sodium».
InteractionsLe dulaglutide retarde la vidange gastrique et peut avoir de ce fait une influence sur l'absorption des médicaments administrés par voie orale. Cela doit notamment être pris en compte lorsque des médicaments dont la fenêtre thérapeutique est étroite sont administrés en même temps que le dulaglutide car une libération accrue due à un temps de séjour gastrique prolongé peut augmenter légèrement l'exposition à ces médicaments.
Effet de Trulicity sur d'autres médicaments
Paracétamol
Après une première dose de 1 et 3 mg de dulaglutide, la Cmax du paracétamol a été réduite respectivement de 36 et 50 %; le tmax médian a été plus long (de respectivement 3 et 4 heures). Après la co-administration avec une dose allant jusqu'à 3 mg de dulaglutide à l'état d'équilibre (steady-state), aucune différence statistiquement significative de l'AUC(0-24), de la Cmax ou du tmax du paracétamol n'a été observée. Aucun ajustement de la dose de paracétamol n'est donc nécessaire lorsqu'il est administré avec le dulaglutide.
Atorvastatine
La co-administration (à jeun) de dulaglutide 1,5 mg et d'atorvastatine a réduit la Cmax et l'AUC(0-∞) de l'atorvastatine et de son principal métabolite l'ohydroxyatorvastatine, jusqu'à 70 % et 21 % . Le t1/2 moyen de l'atorvastatine et de l'ohydroxyatorvastatine a été prolongé respectivement de 17 % et 41 % après l'administration de dulaglutide. Ces observations ne sont pas considérées comme significatives d'un point de vue clinique. Aucun ajustement de la dose d'atorvastatine n'est donc nécessaire lorsqu'elle est administrée avec le dulaglutide.
Digoxine
Après la co-administration (à jeun) de digoxine à l'état d'équilibre (steady-state) avec 2 doses consécutives de dulaglutide 1,5 mg, l'exposition globale (AUCτ) et le tmax de la digoxine sont restés inchangés alors que la Cmax a diminué jusqu'à 22 %. Ces changements ne devraient pas avoir de conséquences cliniques. Aucun ajustement de la dose de digoxine n'est donc nécessaire lorsqu'elle est administrée avec le dulaglutide.
Antihypertenseurs
La co-administration (à jeun) de plusieurs doses de dulaglutide 1,5 mg avec du lisinopril à l'état d'équilibre n'a donné lieu à aucun changement cliniquement significatif de l'AUC ou de la Cmax du lisinopril. Un retard statistiquement significatif du tmax du lisinopril d'environ 1 heure a été observé au jour 3 et au jour 24 de l'étude. L'administration concomitante d'une dose unique de dulaglutide 1,5 mg avec le métoprolol a donné lieu à une augmentation de l'AUC et de la Cmax du métoprolol respectivement de 19 % et 32 %. Bien que le tmax du métoprolol ait été retardé d'une heure, ce changement n'était pas statistiquement significatif. Les changements n'ont pas été considérés comme cliniquement significatifs; aucun ajustement de la dose de lisinopril ou de métoprolol n'est donc nécessaire lorsqu'ils sont administrés avec le dulaglutide.
Warfarine
Après co-administration de dulaglutide (1,5 mg) et d'une dose unique de 10 mg de warfarine, l'exposition des isomères S et R de la warfarine et la Cmax de la R-warfarine n'ont pas été modifiées; la Cmax de la S- warfarine a diminué de 22 %. Le tmax de la S-warfarine a été retardé de 4 heures et le tmax de la R-warfarine de 6 heures. Ces changements ne sont pas considérés comme significatifs d'un point de vue clinique. Aucun ajustement de la dose de warfarine n'est donc nécessaire lorsqu'elle est administrée avec le dulaglutide. Il n'a pas été réalisé d'études d'interaction avec la phenprocoumone et l'acénocoumarol.
Contraceptifs oraux
La co-administration de dulaglutide (1,5 mg) avec un contraceptif oral (norgestimate 0,18 mg/éthinylestradiol 0,025 mg) n'a pas affecté l'AUC globale de la norelgestromine et de l'éthinylestradiol. Des réductions statistiquement significatives de la Cmax de 26 % et 13 % et des retards du tmax de 2 heures et 0,3 heures ont été observées respectivement pour la norelgestromine et l'éthinylestradiol. Ces observations ne sont pas considérées comme significatives d'un point de vue clinique. Aucun ajustement de la dose de contraceptif oral n'est donc nécessaire lorsqu'il est administré avec le dulaglutide.
Metformine
Après co-administration de plusieurs doses de dulaglutide 1,5 mg avec de la metformine (formulation à libération immédiate) à l'état d'équilibre (steady-state), l'AUCτ de la metformine a augmenté jusqu'à 15 % et la Cmax a diminué jusqu'à 12 %, sans changement du tmax. Ces changements correspondent au retard de la vidange gastrique provoqué par le dulaglutide et restent compris dans la variabilité pharmacocinétique de la metformine; ils ne sont donc pas considérés comme significatifs d'un point de vue clinique. Aucun ajustement de la dose de la metformine à libération immédiate n'est donc nécessaire lorsqu'elle est administrée avec le dulaglutide.
Sitagliptine
L'exposition à la sitagliptine n'a pas été affectée par l'administration concomitante d'une dose unique de dulaglutide 1,5 mg. Après l'administration concomitante de 2 doses consécutives de dulaglutide 1,5 mg, l'AUC(0-τ) et la Cmax de la sitagliptine ont diminué respectivement d'environ 7,4 % et 23,1 %. Le tmax de la sitagliptine a été allongé d'environ 0,5 heure après l'administration concomitante de dulaglutide par rapport à l'administration de sitagliptine seule.
La sitagliptine peut produire une inhibition allant jusqu'à 80 % de la DPP-4 sur une période de 24 heures. L'administration concomitante de dulaglutide 1,5 mg et de sitagliptine a entraîné des augmentations respectives de l'AUC(0-τ) et de la Cmax du dulaglutide d'environ 38 % et 27 %; le tmax médian a été allongé d'environ 24 heures. Par conséquent, le dulaglutide présente un niveau de protection élevé contre l'inactivation par la DPP-4 (voir «Efficacité clinique»).
Effet d'autres médicaments sur Trulicity
Immunoglobulines administrées par voie intraveineuse
L'effet de l'administration intraveineuse d'immunoglobulines sur la pharmacocinétique et la pharmacodynamique du dulaglutide n'a pas été étudié. On ne peut donc pas exclure une réduction de la durée de l'effet du dulaglutide lors de l'administration intraveineuse concomitante d'immunoglobulines.
Grossesse, allaitementGrossesse
Il n'existe pas de données suffisantes concernant l'utilisation du dulaglutide pendant la grossesse. Les études chez l'animal ont mis en évidence une toxicité sur la reproduction (voir «Données précliniques»). Le risque possible chez l'être humain n'est pas connu. Par conséquent, l'utilisation du dulaglutide n'est pas recommandée pendant la grossesse.
Allaitement
On ignore si le dulaglutide est excrété dans le lait maternel. On ne peut exclure un risque pour le nouveau-né ou l'enfant. Des jeunes rats traités avec le dulaglutide pendant la gestation et l'allaitement ont présenté une réduction de poids et des déficits mnésiques (voir «Données précliniques»). Le dulaglutide ne doit pas être utilisé pendant l'allaitement ou l'allaitement doit être arrêté.
Fertilité
Aucune étude n'a été menée sur l'effet du dulaglutide sur la fertilité chez l'être humain.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude n'a été menée sur l'aptitude à la conduite et la capacité d'utiliser des machines. Les patients doivent être informés des précautions à prendre pour éviter une hypoglycémie lors de la conduite de véhicules ou l'utilisation de machines lorsque Trulicity est utilisé en association avec une sulfonylurée ou une insuline.
Effets indésirablesRésumé du profil de sécurité
Dans les études initiales de phases 2 et 3 terminées pour l'enregistrement du dulaglutide 0,75 mg et 1,5 mg, 4006 patients ont reçu Trulicity seul ou en association avec d'autres antidiabétiques. Les effets indésirables les plus fréquemment rapportés dans les essais cliniques et après la mise sur le marché ont été de nature gastro-intestinale, incluant nausées, vomissements et diarrhées. En général, ces effets ont été d'intensité légère à modérée.
Les résultats de l'étude à long terme cardiovasculaire menée chez 4949 patients qui ont reçu Trulicity et ont été observés sur une période médiane de 5,4 ans ont corroboré ces résultats.
Liste des effets indésirables
Les effets indésirables suivants ont été identifiés à partir des évaluations pendant toute la durée des études cliniques de phase 2 et de phase 3, de l'étude cardiovasculaire et des annonces d'effets indésirables qui ont suivi la mise sur le marché. Ils sont présentés selon la terminologie MedDRA par classe de système d'organe et par ordre décroissant de fréquence (très fréquent: ≥1/10; fréquent: ≥1/100; < 1/10; occasionnel: ≥1/1000, < 1/100; rare: ≥1/10'000, < 1/1000; très rare: < 1/10'000). Dans chaque groupe, les effets indésirables sont classés par ordre décroissant de fréquence.
La fréquence des événements a été calculée à partir de leur incidence dans les études d'enregistrement de phase 2 et de phase 3.
Affections du système immunitaire
Rare: réactions anaphylactiques.
Affections gastro-intestinales
Très fréquent: nausées (12,9 % avec 0,75 mg et 21,2 % avec 1,5 mg), diarrhée (10,7 % avec 0,75 mg et 13,7 % avec 1,5 mg), vomissements (11,5 % avec 1,5 mg de dulaglutide), douleurs abdominales (10,2 % avec 1,5 mg de dulaglutide).
Fréquent: vomissements (avec 0,75 mg de dulaglutide), diminution de l'appétit, dyspepsie, constipation, douleurs abdominales (avec 0,75 mg de dulaglutide), flatulence, météorisme, reflux gastro-œsophagien, éructation.
Rare: retard de la vidange gastrique, pancréatite aiguë.
Affections hépatobiliaires
Occasionnel: cholélithiase, cholécystite.
Troubles du métabolisme et de la nutrition
Très fréquent:
Hypoglycémie* en cas d'utilisation concomitante avec une sulfonylurée ou de l'insuline:
·avec la metformine plus glimépiride (39,0 % avec 0,75 mg et 40,3 % avec 1,5 mg de dulaglutide).
·avec le glimépiride (11,3 % avec 1,5 mg de dulaglutide).
·avec l'insuline prandiale (85,3 % avec 0,75 mg et 80 % avec 1,5 mg de dulaglutide).
·avec l'insuline basale (35,3 % avec 1,5 mg de dulaglutide).
Hypoglycémie* sans utilisation concomitante d'une sulfonylurée ou d'insuline:
·avec la metformine (10,9 % avec 1,5 mg de dulaglutide).
Fréquent:
Hypoglycémie* sans utilisation concomitante d'une sulfonylurée ou d'insuline:
·en monothérapie.
·avec la metformine plus pioglitazone.
·avec la metformine (avec 0,75 mg de dulaglutide).
Troubles généraux et anomalies au site d'administration
Fréquent: fatigue.
Occasionnel: réactions au site d'injection$.
Investigations
Fréquent: tachycardie sinusale, bloc auriculo-ventriculaire (bloc AV) de 1er degré.
*Hypoglycémie symptomatique documentée avec glucose sanguin ≤3,9 mmol/l.
$Dans des études pédiatriques, celles-ci ont été fréquemment observées; 3,9 % (2 patients) dans le groupe du dulaglutide 0,75 mg, 3,8 % (2 patients) dans le groupe du dulaglutide 1,5 mg et 2 % (1 patient) dans le groupe du placebo. Tous les événements ont été légers à moyennement graves.
Description de certains effets indésirables
Hypoglycémie
Lorsque Trulicity a été utilisé en monothérapie ou en association avec de la metformine seule ou de la metformine et de la pioglitazone, les taux d'hypoglycémies symptomatiques documentées ont été de 0,14 à 0,18 événement/patient/an sous Trulicity 0,75 mg et de 0,19 à 0,62 événement/patient/an sous Trulicity 1,5 mg; aucun épisode d'hypoglycémie sévère n'a été rapporté. Lorsque Trulicity 1,5 mg a été utilisé avec une sulfonylurée seule, les taux ont été de 0,90 événement/patient/an. Lorsque Trulicity a été utilisé en association avec une sulfonylurée plus metformine, les taux d'hypoglycémies symptomatiques documentées ont été de 1,67 événement/patient/an avec les deux dosages.
Lorsque Trulicity 1,5 mg a été utilisé en association avec une insuline basale, le taux a été de 3.,8 événements/patient/an. Lorsque Trulicity a été utilisé en association avec une insuline prandiale, les taux d'hypoglycémies symptomatiques documentées ont été de 35,66 événements/patient/an sous dulaglutide 0,75 mg et de 31,06 événements/patient/an sous dulaglutide 1,5 mg.
Les taux d'hypoglycémies sévères ont été respectivement de 0 et 0,01 événement/patient/an pour le dulaglutide 0,75 mg et 1,5 mg en association avec une sulfonylurée plus metformine. Il n'y a pas eu d'épisode d'hypoglycémie sévère avec une sulfonylurée seule. Les taux d'hypoglycémie sévère ont été de 0,01 événement/patient/an avec le dulaglutide 1,5 mg en association avec l'insuline glargine et respectivement de 0,05 et 0,06 événement/patient/an pour le dulaglutide 0,75 mg et 1,5 mg en association avec une insuline prandiale.
Dans une étude de phase 3 au cours de laquelle le dulaglutide 1,5 mg, 3 mg ou 4,5 mg a été utilisé en association avec la metformine, les incidences d'une hypoglycémie symptomatique documentée jusqu'à la semaine 52 se sont élevées respectivement à 3,1 %, 2,4 % et 3,1 %, et les taux ont été respectivement de 0,07, 0,05 et 0,07 événements/patient/année. Un épisode d'hypoglycémie sévère en rapport avec le dulaglutide 1,5 mg et 4,5 mg a été rapporté.
Effets indésirables gastro-intestinaux
Les effets indésirables gastro-intestinaux (nausée, vomissements, diarrhée et autres) ont été généralement d'intensité légère à modérée. Ils ont été principalement rapportés au cours des 2 premières semaines de traitement puis ils ont rapidement diminué au cours des 4 semaines suivantes. Le taux est ensuite resté relativement stable pendant le reste de la durée du traitement.
Ces effets indésirables étaient dépendants de la dose: sous 0,75 mg, 34,5 % des patients des études ont présenté ≥1 effet indésirable gastro-intestinal, sous 1,5 mg la proportion était de 43,9 %.
Dans une étude de phase 3 portant sur les dosages de 1,5 mg, 3 mg et 4,5 mg de dulaglutide, les rapports combinés d'effets indésirables gastro-intestinaux survenus jusqu'à la semaine 52 mentionnaient la nausée (respectivement 14,2 %, 16,1 % et 17,3 %), la diarrhée (respectivement 7,7 %, 12,0 % et 11,6 %) et les vomissements (respectivement 6,4 %, 9,1 % et 10,1 %).
Lors des études de pharmacologie clinique réalisées chez des patients atteints de diabète de type 2 sur une durée allant jusqu'à 6 semaines, la majorité des effets indésirables gastro-intestinaux ont été observés au cours des 2 à 3 premiers jours après la dose initiale et ont diminué avec les doses suivantes.
Cholécystite
Dans l'étude d'outcome cardiovasculaire menée chez des patients adultes souffrant de diabète de type 2 et d'une maladie cardiovasculaire établie (CVD) ou présentant plusieurs facteurs de risque cardiovasculaire, avec un temps d'observation moyen de 5,4 ans (voir sous «Efficacité clinique», Étude d'outcome cardiovasculaire), une cholécystite a été rapportée comme événement grave chez respectivement 0,5 % et 0,3 % des patients sous Trulicity ou placebo.
Dans les études sur le contrôle glycémique menée chez des patients qui recevaient du dulaglutide, l'incidence de la cholécystite a été de 0,1 % (occasionnelle).
Réactions au site d'injection
Des effets indésirables au site d'injection, à médiation immunitaire potentielle (par ex. éruption cutanée, érythème), ont été signalés chez 0,7 % des patients qui recevaient Trulicity; les événements ont été généralement d'intensité légère.
Immunogénicité
Dans les études d'enregistrement, des anticorps anti-dulaglutide sont apparus chez 1,6 % des patients pendant le traitement au dulaglutide. Les patients développant des anticorps anti-dulaglutide présentaient généralement des titres faibles. Les données des études de phase III n'ont pas montré d'impact négatif des anticorps antidulaglutide sur l'efficacité (HbA1c).
Hypersensibilité
Lors des études d'enregistrement de phases 2 et 3, des événements d'hypersensibilité systémique (par ex. urticaire, angio-œdème) ont été rapportés chez 0,5 % des patients traités par le dulaglutide. Aucun des patients ayant présenté une hypersensibilité systémique n'a développé des anticorps dirigés contre le dulaglutide.
Augmentation de la fréquence cardiaque
Trulicity 0,75 mg et 1,5 mg sont associés à des augmentations dose-dépendantes de la fréquence cardiaque, de 2 à 4 battements par minute (bpm) et une incidence de tachycardies sinusales avec augmentation de la fréquence cardiaque ≥15 bpm de 1,3 % et 1,4 % respectivement.
Dans une étude de phase 3 portant sur les dosages de 1,5 mg, 3 mg et 4,5 mg de dulaglutide, l'incidence d'une tachycardie sinusale avec une augmentation simultanée de ≥15 battements par minute (beats per minute = bpm) a été respectivement de 2,6 %, 1,9 % et 2,6 % par rapport à la valeur initiale. Des augmentations moyennes de la fréquence cardiaque de 1 à 4 bpm ont été observées.
Bloc auriculo-ventriculaire de premier degré / allongement de l'intervalle PR
Trulicity est associé avec de légères augmentations de l'intervalle PR de 2 à 3 msec en moyenne par rapport à la valeur à l'inclusion et une fréquence de blocs auriculo-ventriculaires de premier degré de 1,5 % et 2,4 % pour le dulaglutide 0,75 mg et 1,5 mg respectivement.
Dans une étude de phase 3 portant sur les dosages de 1,5 mg, 3 mg et 4,5 mg de dulaglutide, l'incidence d'un bloc auriculo-ventriculaire (bloc AV) de premier degré a été respectivement de 1,2 %, 3,8 % et 1,7 %. Des augmentations moyennes de l'intervalle PR de 3 à 5 ms ont été observées par rapport à la valeur initiale.
Évaluation cardio-vasculaire
Au total, 51 patients (dulaglutide: 26 sur 3885; toutes les substances de comparaison: 25 sur 2125) ont eu au moins un événement cardio-vasculaire (décès d'origine cardio-vasculaire, infarctus du myocarde non mortel, accident vasculaire cérébral non mortel ou hospitalisation en raison d'un angor instable). L'analyse statistique de ces résultats a montré que le risque cardio-vasculaire sous dulaglutide n'était pas augmenté par rapport aux traitements de référence (HR: 0,57; IC ajusté à 98,02 % [0,30, 1,10]; p=0,046).
Arrêt du traitement suite à un effet indésirable
Lors d'études de 26 semaines, la fréquence des arrêts de traitement suite à des effets indésirables a été de 2,6 % avec Trulicity 0,75 mg et de 6,1 % avec Trulicity 1,5 mg, par rapport à 3,7 % pour le placebo. Pendant toute la durée de l'étude (104 semaines au maximum), la fréquence des arrêts de traitement suite à des effets indésirables a été de 5,1 % avec Trulicity 0,75 mg et de 8,4 % avec Trulicity 1,5 mg. Les effets indésirables les plus fréquents entraînant un arrêt du traitement ont été les nausées (1,0 % avec 0,75 mg; 1,9 % avec 1,5 mg), diarrhées (0,5 % avec 0,75 mg; 0,6 % avec 1,5 mg) et vomissements (0,4 % avec 0,75 mg; 0,6 % avec 1,5 mg); ils ont été généralement rapportés au cours des 4 à 6 premières semaines.
Dans une étude de phase 3 portant sur les dosages de 1,5 mg, 3 mg et 4,5 mg de dulaglutide, l'incidence des arrêts du traitement en raison d'un effet indésirable jusqu'à la semaine 52 a été respectivement de 6,0 % (1,5 mg), 7,0 % (3 mg) et 8,5 % (4,5 mg). Les effets indésirables les plus fréquents ayant entraîné un arrêt du traitement avec le dulaglutide 1,5 mg, 3 mg ou 4,5 mg ont été la nausée (respectivement 1,3 %, 1,3 %, 1,5 %), la diarrhée (respectivement 0,2 %, 1,0 %, 1,0 %) et les vomissements (respectivement 0,0 %, 0,8 %, 1,3 %).
Dosages de dulaglutide de 3 mg et 4,5 mg
Le profil de sécurité observé chez des patients traités avec 3 mg et 4,5 mg de dulaglutide une fois par semaine concorde avec le profil de sécurité déjà décrit ci-dessus pour le dulaglutide 0,75 mg et 1,5 mg une fois par semaine.
Population pédiatrique
Le profil de sécurité chez les enfants et les adolescents âgés de 10 ans et plus qui étaient traités avec 0,75 mg et 1,5 mg de dulaglutide une fois par semaine était comparable au profil de sécurité décrit ci-dessus chez les patients adultes.
Chez les patients pédiatriques traités avec le dulaglutide, le profil immunogène concorde avec le profil décrit ci-dessus pour les patients adultes. Dans l'étude pédiatrique, 2,1 % et 4,0 % des patients traités respectivement avec le placebo et le dulaglutide ont développé pendant le traitement des anticorps dirigés contre le dulaglutide.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLes effets indésirables liés à un surdosage avec le dulaglutide observés au cours des études cliniques ont été des troubles gastro-intestinaux et une hypoglycémie. En cas de surdosage, un traitement de soutien approprié doit être mis en place en fonction des symptômes cliniques du patient.
Propriétés/EffetsCode ATC
A10BJ05
Mécanisme d'action
Le dulaglutide est un agoniste du récepteur du GLP-1 (Glucagon-like peptide-1) à action prolongée. La molécule comprend deux peptides identiques liés par des ponts disulfures, chacune contenant une séquence analogue à celle du GLP-1 humain modifiée, liée de manière covalente par un petit pont peptidique à la chaîne lourde (fragment Fc) d'une immunoglobuline humaine G4 (IgG4) modifiée. La partie du dulaglutide analogue du GLP-1 présente une homologie d'environ 90 % avec le GLP-1 natif humain. Le GLP-1 natif a une demi-vie de 1,5 à 2 minutes du fait de sa dégradation par la DPP-4 et de la clairance rénale. Contrairement au GLP-1 natif, le dulaglutide est protégé de la dégradation par la DPP-4 et sa grande taille ralentit son absorption et réduit sa clairance rénale. Ces caractéristiques structurelles donnent lieu à une formulation soluble à la demi-vie prolongée de 4,7 jours, ce qui permet une administration sous-cutanée hebdomadaire. De plus, la molécule de dulaglutide a été conçue pour empêcher la réponse immunitaire dépendante du récepteur Fcγ et réduire son potentiel immunogène.
Le dulaglutide présente plusieurs propriétés anti-hyperglycémiantes du GLP-1. En présence de concentrations élevées de glucose, le dulaglutide augmente l'AMP cyclique (AMPc) intracellulaire dans les cellules bêta du pancréas, ce qui entraîne la sécrétion d'insuline. Le dulaglutide inhibe la sécrétion de glucagon, qui est élevée chez les patients diabétiques de type 2. Des concentrations de glucagon plus faibles entraînent une diminution de la production de glucose hépatique. Le dulaglutide ralentit également la vidange gastrique.
Pharmacodynamique
Chez les patients atteints de diabète de type 2, le dulaglutide améliore le contrôle glycémique grâce aux effets prolongés de la diminution des concentrations de glucose à jeun, avant et après les repas dès la première administration du médicament; l'effet est maintenu pendant l'intervalle d'injection hebdomadaire.
Une étude pharmacodynamique avec Trulicity a permis d'observer, chez des patients atteints de diabète de type 2, un rétablissement de la première phase de la sécrétion d'insuline à un niveau dépassant ceux observés chez des sujets sains sous placebo et une amélioration de la deuxième phase de la sécrétion d'insuline en réponse à l'administration d'un bolus intraveineux de glucose.
Conformément à son profil pharmacocinétique, le dulaglutide a un profil pharmacodynamique adapté à une administration hebdomadaire (voir «Pharmacocinétique»).
Efficacité clinique
Contrôle glycémique
La tolérance et l'efficacité du dulaglutide ont été étudiées dans dix études contrôlées et randomisées de phase 3 incluant 8035 patients adultes atteints de diabète de type 2. Parmi eux, 1644 patients avaient 65 ans et plus, et parmi ces patients 174 avaient 75 ans et plus. Ces études ont inclus 5650 patients traités par le dulaglutide, dont 1558 traités par le dulaglutide 0,75 mg une fois par semaine, 2862 patients traités par le dulaglutide 1,5 mg une fois par semaine, 616 patients traités par le dulaglutide 3 mg une fois par semaine et 614 patients traités par le dulaglutide 4,5 mg une fois par semaine. Dans toutes les études, le dulaglutide a entrainé des améliorations cliniquement significatives du contrôle glycémique, mesurées par le taux d'hémoglobine glyquée A1c (HbA1c).
Monothérapie
Une monothérapie avec le dulaglutide a été comparée à la metformine dans une étude avec comparateur actif d'une durée de 52 semaines. Au bout de 26 semaines, le dulaglutide a été supérieur à la metformine (1500-2000 mg/jour) dans la réduction de l'HbA1c (valeur moyenne initiale 7,6 %). Un pourcentage significativement plus élevé de patients sous dulaglutide a atteint la valeur cible d'HbA1c de <7,0 % et de ≤6,5 % en comparaison avec la metformine au bout de 26 semaines. Ces effets se sont maintenus jusqu'à la 52e semaine. Le taux d'hypoglycémies symptomatiques documentées a été de 0,62 épisode/patient/an sous dulaglutide 1,5 mg, de 0,15 épisode/patient/an sous dulaglutide 0,75 mg et de 0,09 épisode/patient/an sous metformine. Des hypoglycémies sévères n'ont pas été constatées.
Tableau 1: Résultats d'une étude contrôlée de 52 semaines en monothérapie versus comparateur actif, comparant deux dosages de dulaglutide à la metformine
|
|
HbA1c
à l'inclusion
|
Changement moyen de l'HbA1c
|
Patients atteignant une valeur cible d'HbA1c
|
Changement de la glycémie à jeun
|
Changement du poids corporel
| |
|
(%)
|
(%)
|
<7,0 % (%)
|
≤6,5 % (%)
|
(mmol/l)
|
(kg)
| |
26 semaines
| |
Dulaglutide 1,5 mg 1 fois par semaine
(n=269)
|
7,63
|
-0,78††
|
61,5
|
46,0
|
-1,61
|
-2,29
| |
Dulaglutide 0,75 mg 1 fois par semaine
(n=270)
|
7,58
|
-0,71††
|
62,6
|
40,0
|
-1,46
|
-1,36
| |
Metformine 1500-2000 mg/jour
(n=268)
|
7,60
|
-0,56
|
53,6
|
29,8
|
-1,34
|
-2,22
| |
52 semaines
| |
Dulaglutide 1,5 mg 1 fois par semaine
(n=269)
|
7,63
|
-0,70††
|
60,0
|
42,3
|
-1,56
|
-1,93
| |
Dulaglutide 0,75 mg 1 fois par semaine
(n=270)
|
7,58
|
-0,55†
|
53,2
|
34,7
|
-1,00
|
-1,09
| |
Metformine 1500-2000 mg/jour
(n=268)
|
7,60
|
-0,51
|
48,3
|
28,3
|
-1,15
|
-2,20
|
†† Valeur p unilatérale ajustée pour tests multiples < 0,025, pour la supériorité du dulaglutide comparé à la metformine, évaluée seulement pour l'HbA1c.
† Valeur p unilatérale ajustée pour tests multiples < 0,025, pour la non-infériorité du dulaglutide comparé à la metformine, évaluée seulement pour l'HbA1c.
Association avec la metformine
La sécurité et l'efficacité du dulaglutide ont été examinées dans une étude contrôlée avec placebo et comparateur actif (sitagliptine 100 mg, une fois par jour), d'une durée de 104 semaines, en association avec la metformine. Au bout de 26 semaines, le traitement par le dulaglutide a entrainé une plus forte réduction de l'HbA1c que le placebo. Au bout de 52 semaines, la baisse de l'HbA1c sous dulaglutide était également supérieure à celle induite par la sitagliptine, avec une proportion significativement plus élevée de patients atteignant les valeurs cibles d'HbA1c < 7,0 % et ≤6,5 %. Le traitement par le dulaglutide a en outre induit une réduction significativement plus importante du glucose plasmatique à jeun mesuré et du poids corporel, par rapport à la sitagliptine. Ces effets se sont maintenus jusqu'à la fin de l'étude (104 semaines). Le taux d'hypoglycémies symptomatiques documentées sous dulaglutide 1,5 mg, dulaglutide 0,75 mg et sitagliptine s'est élevé respectivement à 0,19, 0,18 et 0,17 épisode/patient/an. Aucune hypoglycémie sévère n'a été rapportée sous dulaglutide.
Tableau 2: Résultats d'une étude contrôlée versus placebo et comparateur actif de 104 semaines comparant deux dosages de dulaglutide à la sitagliptine
|
|
HbA1c
à l'inclusion
|
Changement moyen de l'HbA1c
|
Patients atteignant une valeur cible d'HbA1c
|
Changement de la glycémie à jeun
|
Changement du poids corporel
| |
|
(%)
|
(%)
|
<7,0 % (%)
|
≤6,5 % (%)
|
(mmol/l)
|
(kg)
| |
26 semaines
| |
Dulaglutide 1,5 mg 1 fois par semaine
(n = 304)
|
8,12
|
-1,22‡‡
|
60,9
|
46,.7
|
-2,38
|
-3,18
| |
Dulaglutide 0,75 mg 1 fois par semaine
(n=302)
|
8,19
|
-1,01‡‡
|
55,2
|
31,0
|
-1,97
|
-2,63
| |
Placebo (n= 177)
|
8,10
|
0,03
|
21,0
|
12,5
|
-0,49
|
-1,47
| |
Sitagliptine 100 mg 1 fois par jour
(n = 315)
|
8,09
|
-0,61
|
37,8
|
21,8
|
-0,97
|
-1,46
| |
52 semaines
| |
Dulaglutide 1,5 mg 1 fois par semaine
(n = 304)
|
8,12
|
-1,10††
|
57,6
|
41,7
|
-2,38
|
-3,03
| |
Dulaglutide 0,75 mg 1 fois par semaine
(n=302)
|
8,19
|
-0,87††
|
48,8
|
29,0
|
-1,63
|
-2,60
| |
Sitagliptine 100 mg 1 fois par jour
(n = 315)
|
8.,09
|
-0,39
|
33,0
|
19,2
|
-0,90
|
-1,53
| |
104 semaines
| |
Dulaglutide 1,5 mg 1 fois par semaine
(n = 304)
|
8,12
|
-0,99††
|
54,3
|
39,1
|
-1,99
|
-2,88
| |
Dulaglutide 0,75 mg 1 fois par semaine
(n=302)
|
8,19
|
-0,71††
|
44,8
|
24,2
|
-1,39
|
-2,39
| |
Sitagliptine 100 mg 1 fois par jour
(n = 315)
|
8,09
|
-0,32
|
31,1
|
14,1
|
-0,47
|
-1,75
|
†† Valeur p unilatérale ajustée pour tests multiples < 0,025, pour la supériorité du dulaglutide comparé à la sitagliptine, évaluée seulement pour l'HbA1c, aux semaines 52 et 104.
‡‡ Valeur p unilatérale ajustée pour tests multiples < 0,001, pour la supériorité du dulaglutide comparé au placebo, évaluée seulement pour l'HbA1c.
Association avec la metformine et une sulfonylurée
Dans une étude avec comparateur actif d'une durée de 78 semaines, le dulaglutide a été comparé à l'insuline glargine, tous deux en association avec la metformine et une sulfonylurée (glimépiride). Au bout de 52 semaines, le dulaglutide a été supérieur à l'insuline glargine pour abaisser l'HbA1c; cette supériorité s'est maintenue jusqu'à la fin des 78 semaines. Cet effet s'est accompagné d'un pourcentage significativement plus élevé de patients qui atteignaient des valeurs cibles d'HbA1c < 7,0 % et ≤6,5 % au bout de 52 (contrôle principal) et 78 semaines (contrôle final).
Durant les 52 premières semaines, les patients sous dulaglutide ont perdu en moyenne 1,87 kg alors que les patients sous insuline glargine ont gagné 1,44 kg; cet effet s'est maintenu jusqu'à la 78e semaine. Le taux d'hypoglycémies symptomatiques documentées a été de 1,67 épisode/patient/an sous dulaglutide 1,5 mg, de 1,67 sous dulaglutide 0,75 mg et de 3,02 épisodes/patient/an sous insuline glargine. Deux cas d'hypoglycémies sévères ont été observés sous dulaglutide 1,5 mg, aucun cas sous dulaglutide 0,75 mg et deux cas sous insuline glargine. Les taux (épisodes/patient/an) d'hypoglycémies nocturnes ont été de 1,8 sous insuline glargine, de 0,59 sous dulaglutide 0,75 mg et de 0,74 sous dulaglutide 1,5 mg.
Tableau 3: Résultats d'une étude contrôlée de 78 semaines avec comparateur actif comparant deux dosages de dulaglutide à l'insuline glargine
|
|
HbA1c
à l'inclusion
|
Changement moyen de l'HbA1c
|
Patients atteignant une valeur cible d'HbA1c
|
Changement de la glycémie à jeun
|
Changement du poids corporel
| |
|
(%)
|
(%)
|
<7,0 % (%)
|
≤6,5 % (%)
|
(mmol/l)
|
(kg)
| |
52 semaines
| |
Dulaglutide 1,5 mg 1 fois par semaine
(n = 273)
|
8,18
|
-1,08††
|
53,2
|
27,0
|
-1,50
|
-1,87
| |
Dulaglutide 0,75 mg 1 fois par semaine
(n=272)
|
8,13
|
-0,76†
|
37,1
|
22,5
|
-0,87
|
-1,33
| |
Insuline glargine+ 1 fois par jour
(n = 262)
|
8,10
|
-0,63
|
30,9
|
13,5
|
-1,76
|
1,44
| |
78 semaines
| |
Dulaglutide 1,5 mg 1 fois par semaine
(n = 273)
|
8,18
|
-0,90††
|
49,0
|
28,1
|
-1,10
|
-1,96
| |
Dulaglutide 0,75 mg 1 fois par semaine
(n=272)
|
8,13
|
-0,62†
|
34,1
|
22,1
|
-0,58
|
-1,54
| |
Insuline glargine+
1 fois par jour
(n = 262)
|
8,10
|
-0,59
|
30,5
|
16,6
|
-1,58
|
1,28
|
†† Valeur p unilatérale ajustée pour tests multiples < 0,025, pour la supériorité du dulaglutide comparé à l'insuline glargine, évaluée seulement pour l'HbA1c.
† Valeur p unilatérale ajustée pour tests multiples < 0,025, pour la non-infériorité du dulaglutide comparé à l'insuline glargine, évaluée seulement pour l'HbA1c.
+ Les doses d'insuline glargine ont été ajustées en utilisant un algorithme avec une glycémie à jeun cible < 5,6 mmol/l.
Association avec une sulfonylurée
La sécurité et l'efficacité du dulaglutide en association avec la sulfonylurée glimépiride ont été étudiées dans une étude contrôlée avec placebo d'une durée de 24 semaines. Le traitement par le dulaglutide 1,5 mg a entraîné une réduction statistiquement significative de l'HbA1c par rapport au placebo au bout de 24 semaines (différence HbA1c LSM [95% CI] de-1,27 [-1,57, -0,97]). Avec le dulaglutide 1,5 mg, le pourcentage de patients ayant atteint les taux cibles d'HbA1c < 7,0 % et ≤6,5 % au bout de 24 semaines a été significativement plus élevé qu'avec le placebo.
Les taux d'hypoglycémie symptomatique documentée avec le dulaglutide 1,5 mg et avec placebo ont été respectivement de 0,90 et 0,04 épisode/patient/an. Aucun cas d'hypoglycémie sévère n'a été observé avec le duaglutide ni avec le placebo.
Tableau 4: Résultats d'une étude de 24 semaines contrôlée versus placebo comparant le dulaglutide en association avec le glimépiride
|
|
HbA1c à l'inclusion
|
Changement moyen de l'HbA1c
|
Patients atteignant une valeur cible d'HbA1c
|
Changement de la glycémie à jeun
|
Changement du poids corporel
| |
|
(%)
|
(%)
|
<7,0 % (%)
|
≤6,5 % (%)
|
(mmol/l)
|
(kg)
| |
24 semaines
| |
Dulaglutide 1,5 mg une fois par semaine (n=239)
|
8,39
|
-1,38‡‡
|
55,3‡‡
|
40,0**
|
-1,70‡‡
|
-0,91
| |
Placebo (n=60)
|
8,39
|
-0,11
|
18,9
|
9,4
|
0,16
|
-0,24
|
‡‡ p < 0,001 pour la supériorité du dulaglutide comparé au placebo, avec une erreur globale de type I contrôlée
** p < 0,001 pour la comparaison du groupe dulaglutide au placebo
Association avec un inhibiteur du SGLT-2 avec ou sans metformine
Une étude de 24 semaines à trois bras contrôlée par placebo a évalué l'efficacité du dulaglutide (1,5 mg et 0,75 mg une fois par semaine) chez des patients déjà traités avec un inhibiteur du SGLT-2 (avec ou sans metformine) et n'ayant pas un contrôle adéquat de la glycémie (HbA1c ≥7,0% - ≤9,5 %). Le critère d'évaluation principal était la diminution du taux d'HbA1c entre le début de l'étude et la semaine 24 (ΔHbA1c). Comparé au contrôle placebo, l'effet hypoglycémiant (ΔHbA1c) était statistiquement significativement supérieur dans les deux bras du dulaglutide (différence moyenne [IC à 95 %] pour 1,5 mg -0,82 % [-1,00, -0,64] et 0,75 mg -0,69% [-0,86, -0,51]). De plus, dans les deux bras du dulaglutide (1,5 mg et 0,75 mg), une proportion significativement supérieure au contrôle placebo a atteint la valeur cible de HbA1c <7,0% (71,5% et 61,8% contre 32,5%, p <0,001).
Association avec la metformine et la pioglitazone
Dans une étude contrôlée avec placebo et comparateur actif (exénatide deux fois par jour), tous deux en association avec la metformine et la pioglitazone, le dulaglutide a démontré sa supériorité pour améliorer l'HbA1c par rapport au placebo au bout de 26 semaines. En outre, le dulaglutide a entraîné une réduction de l'HbA1c supérieure à celle induite par l'exénatide au bout de 26 et 52 semaines. Cet effet s'est accompagné d'une baisse significativement plus importante de la glycémie à jeun et un pourcentage plus important de patients ont atteint des valeurs cibles d'HbA1c < 7,0 % et ≤ à 6,5 % au bout de 26 et 52 semaines. La perte de poids a été comparable sous dulaglutide et sous exénatide. Les taux d'hypoglycémie symptomatique documentés avec le dulaglutide 1,5 mg et 0,75 mg et l'exénatide deux fois par jour ont été respectivement de 0,19, 0,14 et 0,75 épisode/patient/an. Aucun cas d'hypoglycémie sévère n'a été observé sous dulaglutide et deux cas d'hypoglycémie sévère ont été observés avec l'exénatide deux fois par jour.
Tableau 5: Résultats d'une étude contrôlée de 52 semaines avec comparateur actif comparant deux dosages de dulaglutide à l'exénatide
|
|
HbA1c
à l'inclusion
|
Changement moyen de l'HbA1c
|
Patients atteignant une valeur cible d'HbA1c
|
Changement de la glycémie à jeun
|
Changement du poids corporel
| |
|
(%)
|
(%)
|
<7,0 % (%)
|
≤6,5 % (%)
|
(mmol/l)
|
(kg)
| |
26 semaines
| |
Dulaglutide 1,5 mg 1 fois par semaine
(n = 279)
|
8,10
|
-1,51‡‡,††
|
78,2
|
62,7
|
-2,36
|
-1,30
| |
Dulaglutide 0,75 mg 1 fois par semaine
(n=280)
|
8,05
|
-1,30‡‡,††
|
65.8
|
53,2
|
-1,90
|
0,20
| |
Placebo (n = 141)
|
8,06
|
-0,46
|
42,9
|
24,4
|
-0.26
|
1,24
| |
Exénatide+ 10 µg 2 fois par jour
(n = 276)
|
8,07
|
-0,99
|
52,3
|
38,0
|
-1,35
|
-1,07
| |
52 semaines
| |
Dulaglutide 1.5 mg 1 fois par semaine
(n = 279)
|
8,10
|
-1,36††
|
70,8
|
57,2
|
-2,04
|
-1,10
| |
Dulaglutide 0.75 mg 1 fois par semaine
(n=280)
|
8,05
|
-1,07††
|
59,1
|
48,3
|
-1,58
|
0,44
| |
Exénatide+ 10 µg 2 fois par jour
(n = 276)
|
8,07
|
-0,80
|
49,2
|
34,6
|
-1,03
|
-0,80
|
†† Valeur p unilatérale ajustée pour tests multiples < 0,025, pour la supériorité du dulaglutide comparé l'exénatide, évaluée seulement pour l'HbA1c
‡‡ Valeur p unilatérale ajustée pour tests multiples < 0,001, pour la supériorité du dulaglutide comparé au placebo, évaluée seulement pour l'HbA1c
+ Dosage de l'exénatide: 5 µg 2 fois par jour pendant les 4 premières semaines, puis 10 µg 2 fois par jour
En association avec l'insuline basale titrée, avec ou sans metformine
Dans une étude contrôlée avec placebo de 28 semaines, le dulaglutide 1,5 mg a été comparé au placebo en association avec de l'insuline glargine basale titrée (88 % avec et 12 % sans metformine) pour étudier l'effet sur le contrôle glycémique et la sécurité. Pour optimiser la dose d'insuline glargine, les deux groupes étaient titrés pour cibler une glycémie à jeun < 5,6 mmol/l. La dose moyenne initiale d'insuline glargine était de 37 unités/jour pour les patients recevant le placebo et de 41 unités/jour pour les patients recevant le dulaglutide 1,5 mg. Les doses initiales d'insuline glargine chez les patients ayant une HbA1c < 8,0 % étaient réduites de 20 %. A la fin des 28 semaines de traitement, la dose était de 65 unités/jour pour les patients recevant le placebo et de 51 unités/jour pour les patients recevant le dulaglutide 1,5 mg. A la 28e semaine, le traitement par Trulicity 1,5 mg une fois par semaine a entraîné une réduction statistiquement significative de l'HbA1c par rapport au placebo (différence HbA1c LSM [95% CI] de -0,77 [-0,97, -0,56]) et à un pourcentage significativement plus élevé de patients ayant atteint les taux cibles d'HbA1c < 7.0 % et ≤6.5 %.
Les taux d'hypoglycémie symptomatique documentée ont été de 3,38 épisodes/patient/an avec le dulaglutide 1,5 mg comparé à 4,38 épisodes/patient/an avec le placebo. Une hypoglycémie sévère a été rapportée chez un patient avec le dulaglutide 1,5 mg en association à l'insuline glargine, et aucune n'a été rapportée avec le placebo.
Tableau 6: Résultats d'une étude de 28 semaines comparant le dulaglutide au placebo en association avec de l'insuline glargine titrée
|
|
HbA1c
à l'inclusion
|
Changement moyen de l'HbA1c
|
Patients atteignant une valeur cible d'HbA1c
|
Changement de la glycémie à jeun
|
Changement du poids corporel
| |
|
(%)
|
(%)
|
<7,0 % (%)
|
≤6,5 % (%)
|
(mmol/l)
|
(kg)
| |
28 semaines
| |
Dulaglutide 1,5 mg une fois par semaine (n=150)
|
8,41
|
-1,44‡‡
|
66,7‡‡
|
50,0**
|
-2,48‡‡
|
-1,91‡‡
| |
Placebo une fois par semaine (n=150)
|
8,32
|
-0,67
|
33,3
|
16,7
|
-1,55
|
0,50
|
‡‡ p < 0,001 pour la supériorité du dulaglutide comparé au placebo, avec une erreur globale de type I contrôlée
** p < 0,001 pour la comparaison du groupe dulaglutide au placebo
Association avec l'insuline prandiale, avec ou sans metformine
Dans cette étude, des patients traités par 1 ou 2 injections quotidiennes d'insuline avant l'entrée dans l'étude ont arrêté leur traitement d'insuline préalable et ont été randomisés pour recevoir soit le dulaglutide une fois par semaine 1,5 mg ou 0,75 mg, soit de l'insuline glargine une fois par jour, en association avec de l'insuline prandiale lispro trois fois par jour, avec ou sans metformine. Au bout de 26 semaines, la baisse d'HbA1c sous dulaglutide a été supérieure à celle observée sous l'insuline glargine. Cet effet était maintenu à la fin des 52 semaines de l'étude. Cet effet s'est accompagné d'une perte de poids significative par rapport à l'insuline glargine, avec laquelle une prise pondérale a été observée au cours de l'étude. De même, un pourcentage plus important de patients ont atteint des valeurs cibles d'HbA1c < 7,0 % et ≤6,5 % au bout de 26 semaines et <7,0 % au bout de 52 semaines dans le groupe dulaglutide que dans le groupe insuline glargine. Le taux d'hypoglycémies symptomatiques documentées a été de 31,06 épisodes/patient/an sous dulaglutide 1,5 mg, de 35,66 épisodes/patient/an sous dulaglutide 0,75 mg et de 40,95 épisodes/patient/an sous insuline glargine. Sous dulaglutide 1,5 mg, 10 patients ont rapporté des hypoglycémies sévères, sous dulaglutide il y a eu 7 patients et 15 sous insuline glargine.
Les taux (épisodes/patient/an) d'hypoglycémies nocturnes ont été de 7,99 sous insuline glargine, de 3,94 sous dulaglutide 0,75 mg et de 3,61 sous dulaglutide 1,5 mg.
Tableau 7: Résultats d'une étude contrôlée de 52 semaines avec comparateur actif comparant deux dosages de dulaglutide à l'insuline glargine
|
|
HbA1c
à l'inclusion
|
Changement moyen de l'HbA1c
|
Patients atteignant une valeur cible d'HbA1c
|
Changement de la glycémie à jeun
|
Changement du poids corporel
| |
|
(%)
|
(%)
|
<7,0 % (%)
|
≤6,5 % (%)
|
(mmol/l)
|
(kg)
| |
26 semaines
| |
Dulaglutide 1,5 mg 1 fois par semaine
(n = 295)
|
8,46
|
-1,64††
|
67,6
|
48,0
|
-0,27
|
-0,87
| |
Dulaglutide 0,75 mg 1 fois par semaine
(n=293)
|
8,40
|
-1,59††
|
69,0
|
43,0
|
0,22
|
0,18
| |
Insuline glargine+
1 fois par jour
(n = 296)
|
8,53
|
-1,41
|
56,8
|
37,5
|
-1,58
|
2,33
| |
52 semaines
| |
Dulaglutide 1,5 mg 1 fois par semaine
(n = 295)
|
8,46
|
-1,48††
|
58,5
|
36,7
|
0,08
|
-0,35
| |
Dulaglutide 0,75 mg 1 fois par semaine
(n=293)
|
8,40
|
-1,42††
|
56,3
|
34,7
|
0,41
|
0,86
| |
Insuline glargine+
1 fois par jour
(n = 296)
|
8,53
|
-1,23
|
49,3
|
30,4
|
-1,01
|
2,89
|
†† Valeur p unilatérale ajustée pour tests multiples < 0,025, pour la supériorité du dulaglutide comparé l'insuline glargine, évaluée seulement pour l'HbA1c
+ Les doses d'insuline glargine ont été ajustées en utilisant un algorithme avec une valeur cible de glycémie à jeun de 4,0-5,5 mmol/l.
Étude d'outcome cardiovasculaire
L'étude à long terme sur l'outcome cardiovasculaire sous dulaglutide était une étude clinique en double aveugle, contrôlée avec placebo. Des patients atteints de diabète de type 2 ont reçu, en plus du traitement standard pour leur diabète de type 2, un traitement de dulaglutide 1,5 mg (4949) ou un placebo (4952). La durée médiane d'observation de l'étude a été de 5,4 ans.
L'âge moyen était de 66,2 ans, l'IMC moyen s'est élevé à 32,3 kg / m² et 46,3% des patients étaient de sexe féminin.
L'étude a inclus des patients dans les trois catégories de risque suivantes: 2892 (29,2%) patients ≥50 ans souffrant d'affection cardiovasculaire avérée (par ex. infarctus du myocarde), 2965 (29,9%) patients ≥55 ans présentant une affection cardiovasculaire subclinique (par ex. mise en évidence d'une ischémie cardiaque ou >50% de sténose coronarienne) et 3725 (37,6%) patients ≥60 ans qui présentaient ≥2 facteurs de risque cardiovasculaire (par ex. tabagisme ou hypertension artérielle).
La valeur initiale moyenne de l'HbA1c s'est élevée à 7,2%. La majorité des patients avaient une valeur initiale d'HbA1c située entre 6,0% et 8,9% (du 10e au 90e percentile). La durée moyenne du diabète était de 10,5 ans. Le bras de traitement avec le dulaglutide incluait des patients ≥65 ans (n = 2619; 52,9%) et ≥75 ans (n = 484; 9,7%) et des patients atteints d'insuffisance rénale légère (n = 2435; 50,2%), modérée (n = 1031; 21,3%) ou sévère (n = 50; 1,1%).
Le critère primaire a été le temps écoulé entre la randomisation et la première survenue d'un évènement cardiovasculaire sévère (MACE): mort cardiovasculaire, infarctus du myocarde non mortel ou accident vasculaire cérébral non mortel. Le statut au moment del'objectif primaire ou le statut vital à la fin de l'étude a été disponible pour 99,7% des participants qui avaient été randomisés pour recevoir le dulaglutide ou le placebo. Le dulaglutide était supérieur au placebo dans la prévention des MACE (Figure 1). Les patients traités avec le dulaglutide avaient un taux de MACE plus bas que ceux traités avec le placebo [HR: 0,88; IC à 95% (0,79, 0,99]). Chacune des composantes du MACE a contribué à la réduction du MACE, comme cela est illustré dans la Figure 2. L'efficacité du dulaglutide a été cohérente sur divers sous-groupes, notamment ceux formés en fonction de l'HbA1c de départ, du sexe, de la durée du diabète, de l'âge et de l'eGFR.
L'analyse par sous-groupes en fonction des catégories de risque cardiovasculaire a donné les résultats suivants: les patients ≥50 ans atteints d'une affection cardiovasculaire avérée [18,3% des patients sous dulaglutide avec MACE vs. 20,7% des patients sous placebo, HR: 0,87; IC à 95% (0,74, 1,03)], patients ≥55 ans présentant une affection cardiovasculaire subclinique [10,2% vs. 12,3%, HR: 0,81; IC à 95% (0,65, 1,01)] et patients ≥60 ans qui présentaient ≥2 facteurs de risque cardiovasculaire [8,9% vs. 9,1%, HR: 0,96; IC à 95% CI (0,78, 1,19).
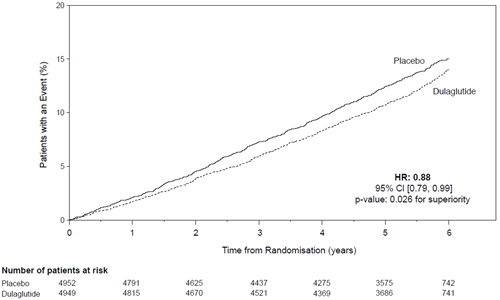
Figure 1: Diagramme selon Kaplan-Meier du temps écoulé jusqu'à la première survenue de l'objectif combiné: de mort CV, d'infarctus du myocarde non mortel ou d'accident vasculaire cérébral non mortel, dans l'étude à long terme sur l'outcome cardiovasculaire sous dulaglutide.
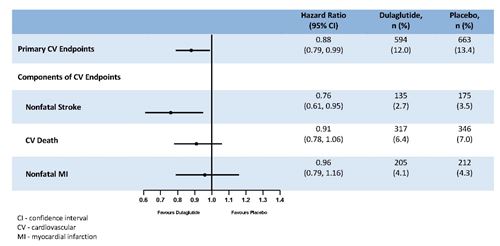
Figure 2: Représentation selon Forest de l'analyse du critère cardiovasculaire principal et de ses composantes individuelles: mort cardiovasculaire, infarctus du myocarde non mortel et accident vasculaire cérébral non mortel.
Sous dulaglutide vs. placebo ajoutés au traitement standard, une diminution significative et persistante de la valeur d'HbA1c entre le début de l'étude et le mois 60 a été observée (-0,29% vs. 0,22%; différence estimée entre les traitements – 0,51% [-0,57; -0,45]; p <0,001). Il y a eu significativement moins de patients dans le groupe dulaglutide qui ont nécessité une intervention glycémique additionnelle, par rapport au groupe sous placebo (dulaglutide: 2086 [42,2%]; placebo: 2825 [57,0%]; p <0,001).
Pression artérielle
L'effet du dulaglutide sur la pression artérielle, mesurée dans le cadre d'une surveillance ambulatoire de la pression artérielle, a été évalué dans une étude de 755 patients atteints de diabète de type 2. Le traitement avec le dulaglutide a entraîné 16 semaines après le début du traitement une réduction de la pression artérielle systolique par rapport au placebo (dose de 1,5 mg: -2,8 mmHg [97,3 %, CI -4,6, -1,0]; dose de 0,75 mg: (-1,1 mmHg [97,3 %, CI -2,8, 0,7]).
Aucune différence n'a été observée sur la pression artérielle diastolique. Des résultats similaires pour la pression artérielle systolique et diastolique ont été obtenus au bout de 26 semaines, au temps d'évaluation final de l'étude.
Traitement d'association du dulaglutide 4,5 mg, 3 mg et 1,5 mg avec la metformine
La sécurité d'emploi et l'efficacité de l'association du dulaglutide 3 mg et 4,5 mg une fois par semaine avec la metformine, en comparaison avec l'association de dulaglutide 1,5 mg une fois par semaine avec la metformine, ont été examinées dans une étude portant sur 52 semaines. Le dulaglutide 3 mg et 4,5 mg se sont avérés supérieurs au dulaglutide 1,5 mg à la semaine 36, en termes de réduction de l'HbA1c et du poids corporel. Un pourcentage plus élevé de patients a atteint les objectifs d'HbA1c de <7,0 % ou de ≤6,5 % au bout de 36 semaines avec le dulaglutide 3 mg et le dulaglutide 4,5 mg. Une réduction du poids corporel de ≥5 % par rapport à la valeur initiale a été obtenue respectivement par 31 %, 40 % et 49 % des patients sous dulaglutide 1,5 mg, 3 mg et 4,5 mg. Ces effets se sont maintenus sur 52 semaines.
Tableau 8: Résultats d'une étude avec contrôle actif comparant trois dosages de dulaglutide
|
|
HbA1c à l'inclusion
|
Changement moyen de l'HbA1c
|
Pourcentage de patients atteignant la valeur cible d'HbA1c
|
Changement de la glycémie à jeun
|
Changement du poids corporel
| |
(%)
|
(%)
|
<7,0 % (%)
|
≤6,5 % (%)
|
(mmol/l)
|
(kg)
| |
36 semaines
| |
Dulaglutide 1,5 mg 1x par semaine
(n = 612)
|
8,64
|
-1,53
|
57,0
|
38,1
|
-2,45
|
-3,1
| |
Dulaglutide 3 mg 1x par semaine
(n = 616)
|
8,63
|
-1,71#
|
64,7#
|
48,4‡‡
|
-2,66
|
-4,0#
| |
Dulaglutide 4,5 mg 1x par semaine
(n = 614)
|
8,64
|
-1,87##
|
71,5#
|
51,7‡‡
|
-2,90#
|
-4,7##
| |
52 semaines
| |
Dulaglutide 1.5 mg 1x par semaine
(n = 612)
|
8,64
|
-1,52
|
58,6
|
40,4
|
-2,39
|
-3,5
| |
Dulaglutide 3 mg 1x par semaine
(n = 616)
|
8,63
|
-1,71‡
|
65,4‡
|
49,2‡
|
-2,70‡
|
-4,3‡
| |
Dulaglutide 4,5 mg 1x par semaine
(n = 614)
|
8,64
|
-1,83‡‡
|
71,7‡‡
|
51,3‡‡
|
-2,92‡‡
|
-5,0‡‡
|
# p < 0,05, ## p < 0,001 pour la supériorité par rapport au dulaglutide 1,5 mg, valeurs de p ajustées avec le contrôle de l'erreur systématique de type 1
‡ p < 0,05, ‡‡ p < 0,001 par rapport au dulaglutide 1,5 mg
Les résultats visent l'effet sous traitement (l'analyse se base sur des modèles mixtes pour mesures répétées ou sur une régression logistique longitudinale).
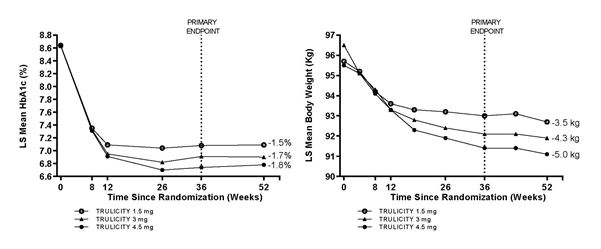
Figure 3. Variation moyenne de l'HbA1c (%) et du poids corporel (kg) entre la valeur initiale et la valeur à la semaine 52.
Les taux d'hypoglycémies symptomatiques documentées sous dulaglutide 1,5 mg, 3 mg et 4,5 mg ont été respectivement de 0,07, 0,05 et 0,07 épisode/patient/an. Un patient a rapporté une hypoglycémie sévère sous dulaglutide 1,5 mg, aucun patient sous dulaglutide 3 mg et un patient sous dulaglutide 4,5 mg.
Utilisation chez des patients atteints d'insuffisance rénale
Dans une étude de 52 semaines, le dulaglutide 1,5 mg et 0,75 mg ont été comparés à l'insuline glargine titrée en association avec l'insuline lispro prandiale pour étudier l'effet anti-hyperglycémiant chez des patients atteints de diabète de type 2 et d'insuffisance rénale chronique modérée à sévère (eGFR [selon CKD-EPI] < 60 et ≥15 ml/min/1,73m2). A l'inclusion, le eGFR global moyen était de 38 ml/min/1,73m2; 30 % des participants à l'étude avaient un eGFR < 30 ml/min/1,73m2. Le critère d'évaluation principal était le changement du taux d'HbA1c de l'inclusion jusqu'à la semaine 26, pour lequel les deux groupes dulaglutide (1,5 mg et 0,75 mg) étaient non-inférieurs par rapport au groupe insuline glargine (différence pour le changement de HbA1c [95 % CI]: -0,05 % [-0,26 %, 0,15 %] pour 1,5 mg et 0,02 % [-0,18 %, 0,22 %] pour 0,75 mg). Ceci a été confirmé par les résultats à 52 semaines de traitement.
Utilisation dans la population pédiatrique
La sécurité et l'efficacité du dulaglutide une fois par semaine (QW) ont été étudiées sur un total de 52 semaines dans une étude randomisée à 3 bras (1 [placebo]: 1 [dulaglutide 0,75 mg QW]: 1 [dulaglutide 1,5 mg QW]) portant sur un total de 154 enfants et adolescents âgés de 10 à 18 ans. Les participants à l'étude pouvaient recevoir, en plus du régime et de l'exercice physique, un prétraitement de metformine et/ou d'insuline basale. Le critère principal d'efficacité (changement de HbA1c) a été déterminé sur la période de la phase en double aveugle jusqu'à la semaine 26. Ensuite, les participants à l'étude pouvaient continuer à prendre pour 26 autres semaines un traitement ouvert de dulaglutide 0,75 mg QW. Le dulaglutide s'est avéré supérieur au contrôle de placebo en ce qui concerne le critère principal.
Il n'a pas été observé de différences statistiquement significatives ou pertinentes sur le plan clinique entre le dulaglutide et le placebo en termes de variation de l'IMC par rapport à la valeur initiale jusqu'à la semaine 26.
Tableau 9: Résultats d'une étude de 52 semaines destinée à comparer deux dosages de dulaglutide avec un placebo chez des patients pédiatriques qui étaient traités avec un régime alimentsaire et de l'exercice physique seuls, avec ou sans metformine et/ou insuline basale
|
|
HbA1c à l'inclusion
|
Changement moyen de l'HbA1c
|
Pourcentage de patients atteignant la valeur cible d'HbA1c
|
Changement de la glycémie à jeun
| |
|
(%)
|
(%)
|
<7,0 % (%)
|
≤6,5 % (%)
|
(mmol/l)
| |
26 semaines
| |
Dulaglutide combinéa (n = 103)
|
8,0
|
-0,7##
|
56,5##
|
46,7‡‡
|
-1,0#
| |
Dulaglutide 0,75 mg une fois par semaine (n = 51)
|
7,9
|
-0,5#
|
60,0##
|
48,9‡
|
-0,5#
| |
Dulaglutide 1,5 mg une fois par semaine (n = 52)
|
8,2
|
-1,0##
|
53,2##
|
44,7‡‡
|
-1,5#
| |
Placebo une fois par semaine (n = 51)
|
8,1
|
0,5
|
18,4
|
13,2
|
1,0
| |
52 semaines
| |
Dulaglutide combiné (n = 103)
|
8,0
|
-0,4
|
59,5
|
45,2
|
-0,63
| |
Dulaglutide 0,75 mg une fois par semaine (n = 51)
|
7,9
|
-0,2
|
65,0
|
55,0
|
-0,21
| |
Dulaglutide 1,5 mg une fois par semaine (n = 52)
|
8,2
|
-0,6
|
54,6
|
36,4
|
-0,95
| |
Placebo/dulaglutide 0,75 mg une fois par semaineb (n = 51)
|
8,1
|
-0,1
|
50,0
|
29,4
|
0,24
|
# p < 0,05, ## p < 0,001 pour la supériorité par rapport au placebo, valeurs p ajustées avec un contrôle général d'erreurs de type I.
‡ p < 0,05, ‡‡ p < 0,001 pour la supériorité par rapport au placebo
a Résultats combinés pour Trulicity 0,75 mg et 1,5 mg. La comparaison des deux doses combinées et individuelles était préspécifiée avec un contrôle général d'erreurs de type 1.
b Les patients qui, dans la phase initiale de 26 semaines en double aveugle, avaient été assignés à un traitement avec le placebo, ont commencé le traitement avec le dulaglutide 0,75 mg une fois par semaine dans la phase ouverte subséquente de 26 semaines.
PharmacocinétiqueAbsorption
Après une administration sous-cutanée chez des patients atteints de diabète de type 2, le dulaglutide a atteint les concentrations plasmatiques maximales au bout de 48 heures. Les expositions maximales moyenne (Cmax) et totale (AUC) ont été respectivement d'environ 114 ng/ml et 14000 ngh/ml, après plusieurs injections sous-cutanées de doses de 1,5 mg de dulaglutide chez des patients atteints de diabète de type 2. Les concentrations plasmatiques à l'état d'équilibre (steady-state) ont été atteintes 2 à 4 semaines après administration hebdomadaire de dulaglutide (1,5 mg). L'accumulation s'est élevée à environ 56% après administrations multiples. La pharmacocinétique du dulaglutide a été linéaire en fonction du temps. Dans le domaine des doses de 0,5 mg à 1,5 mg, l'exposition au dulaglutide a été moins que proportionnelle d'environ 20 %. Les expositions après administration sous-cutanée de doses uniques de dulaglutide (1,5 mg) dans l'abdomen, la cuisse ou le haut du bras ont été comparables. Selon les estimations, les biodisponibilités absolues des dosages de 3 mg et 4,5 mg sont similaires à celle d'une dose de 1,5 mg, bien qu'il n'y ait pas d'études splcifiques à ce sujet. Sur le domaine de doses de 0,75 mg à 4,5 mg, l'élévation de la concentration de dulaglutide est approximativement proportionnelle.
Distribution
Le volume central moyen de distribution pour la population examinée s'est élevé à 3,09 l, le volume de distribution périphérique moyen a été de 5,98 l.
Métabolisme
Le dulaglutide est supposé être dégradé en différents acides aminés qui le composent par les voies cataboliques générales des protéines.
Élimination
La clairance moyenne du dulaglutide obtenue pour la population examinée s'est élevée à 0,142 l/h, et la demi-vie d'élimination a été d'environ 5 jours.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
La pharmacocinétique du dulaglutide a été évaluée chez des patients atteints d'insuffisance hépatique de divers degrés de gravité. Les patients atteints d'insuffisance hépatique ont présenté des baisses statistiquement significatives de l'exposition au dulaglutide pouvant atteindre 30 % à 33 % respectivement pour la Cmax et l'AUC moyennes, par rapport aux volontaires sains. Le tmax du dulaglutide était allongé en fonction de la gravité de l'insuffisance hépatique. Néanmoins, aucune tendance n'a été observée en ce qui concerne le rapport entre l'exposition au dulaglutide et le degré de l'insuffisance hépatique. Ces effets n'ont pas été considérés comme cliniquement significatifs.
Troubles de la fonction rénale
La pharmacocinétique du dulaglutide a été examinée chez des patients atteints d'insuffisance rénale de divers degrés de gravité; elle a été généralement comparable chez les volontaires sains et les patients présentant une insuffisance rénale légère à sévère (ClCr < 30 ml/min), y compris une insuffisance rénale terminale (nécessitant une dialyse).
Patients âgés (≥65 ans)
L'âge n'a pas eu d'effet cliniquement pertinent sur les propriétés pharmacocinétiques et pharmacodynamiques du dulaglutide.
Enfants et adolescents
Une analyse pharmacocinétique de population avec le dulaglutide 0,75 mg et 1,5 mg a porté sur les données de 128 patients pédiatriques (âgés de 10 ans jusqu'à moins de 18 ans) atteints de diabète de type 2. Le profil pharmacocinétique du dulaglutide chez les patients pédiatriques était comparable à celui des adultes.
Sexe et origine ethnique
Le sexe et l'origine ethnique n'ont eu aucun effet cliniquement significatif sur les paramètres pharmacocinétiques du dulaglutide.
Poids corporel et indice de masse corporelle
Les analyses pharmacocinétiques ont mis en évidence une relation inverse statistiquement significative entre le poids corporel ou l'indice de masse corporelle (IMC) et l'exposition au dulaglutide, en dépit de l'absence d'impact cliniquement significatif du poids ou de l'IMC sur le contrôle de la glycémie.
Données précliniquesToxicité à long terme (ou toxicité en cas d'administration répétée)
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité et de toxicologie en administration répétée n'ont pas révélé de risque particulier pour l'être humain, à l'exception des aspects suivants.
Dans des études sur la pharmacologie de sécurité et la toxicité du dulaglutide chez le singe, des élévations hétérogènes de la fréquence cardiaque et de l'intervalle QT corrigé ont été observées.
Le dulaglutide a augmenté les lésions sous-jacentes du pancréas exocrine chez des rats diabétiques lors d'une exposition 30 fois supérieure à l'exposition clinique chez l'être humain. Lors d'une exposition avec une dose de 8,15 mg/kg (exposition estimée comme correspondant à environ 500 fois l'exposition clinique chez l'être humain), le dulaglutide a augmenté le nombre de cellules caliciformes du canal pancréatique chez des singes non diabétiques. Il n'y a pas eu toutefois de signes de pancréatite avec nécrose chez le rat ni chez le singe. Pour l'heure, on ne peut exclure définitivement un risque potentiel de pancréatite ou de cancer du pancréas chez l'être humain.
Mutagénicité
Le dulaglutide étant une protéine recombinante, sa génotoxicité n'a pas été étudiée.
Carcinogénicité
Lors d'une étude de carcinogénicité sur 2 ans menée chez des rats soumis à des expositions ≥3 fois l'exposition clinique humaine avec le dulaglutide 4,5 mg par semaine, le dulaglutide a entraîné des augmentations statistiquement significatives, dépendantes de la dose, de la fréquence de tumeurs à cellules C de la glande thyroïde (adénomes et carcinomes combinés). La relevance de ces résultats pour l'être humain n'est pas encore connue. Lors d'une étude de carcinogénicité d'une durée de 6 mois réalisée chez des souris transgéniques, aucune réponse tumorigène n'a été observée.
Toxicité sur la reproduction
Des études menées chez l'animal n'ont pas révélé de signes d'effets nocifs directs sur la fertilité. Lors d'expositions ≥37 fois supérieures à l'exposition clinique humaine, une augmentation de la mortalité embryo-fœtale a néanmoins été observée. Dans des études de toxicologie de la reproduction menées chez des rates et des lapines gestantes, de fortes doses de dulaglutide (correspondant à 5 à 18 fois l'exposition clinique humaine en termes d'AUC), ont diminué la croissance fœtale et/ou ont eu des effets sur le squelette, très probablement en lien avec des effets sur la mère (diminution de la prise de nourriture et perte de poids); il n'y a toutefois pas eu de signes de malformations fœtales. Des doses de dulaglutide équivalant à 7 fois l'exposition humaine, administrées à des rates pendant la gestation ou la lactation, ont entraîné un retard de croissance chez la progéniture mâle et femelle et des déficits de mémoire. Des doses de dulaglutide allant jusqu'à 7 mg/kg à partir du jour 7 jusqu'au jour 91 après la naissance (ce qui correspond, au jour 91, à 153 fois l'exposition chez des enfants pour la dose la plus élevée de 1,5 mg/semaine) n'ont pas produit de déficits de la mémoire chez des jeunes rats mâles et femelles.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8°C)
Ne pas congeler.
A conserver dans l'emballage original, afin de protéger le contenu de la lumière.
Stockage temporaire: Le stylo peut être conservé jusqu'à 14 jours non réfrigéré à une température inférieure à 30°C.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Suivre attentivement les instructions d'utilisation du stylo pré-rempli.
Trulicity ne doit pas être utilisé si la solution contient des particules ou si elle est trouble et/ou colorée.
Trulicity ne doit pas être utilisé s'il a été congelé.
Numéro d’autorisation65236 (Swissmedic).
PrésentationTrulicity 0,75 mg solution injectable en stylo pré-rempli à usage unique: 4 stylos (B)
Trulicity 1,5 mg solution injectable en stylo pré-rempli à usage unique: 4 stylos (B)
Trulicity 3 mg solution injectable en stylo pré-rempli à usage unique: 4 stylos (B)
Trulicity 4,5 mg solution injectable en stylo pré-rempli à usage unique: 4 stylos (B)
Titulaire de l’autorisationEli Lilly (Suisse) SA, 1214 Vernier/GE.
Mise à jour de l’informationJuin 2025
|