CompositionPrincipes actifs
Nintédanib (sous forme d'ésilate).
Excipients
·Contenu des capsules: triglycérides à chaîne moyenne, graisse solide, lécithine (de soja) (E322).
·Enveloppe de la capsule: gélatine, glycérol (85 %), dioxyde de titane (E171), oxyde de fer jaune (E172), oxyde de fer rouge (E172).
Indications/Possibilités d’emploiOfev est indiqué chez l'adulte pour le traitement:
·de la fibrose pulmonaire idiopathique (FPI)
·des pneumopathies interstitielles diffuses (PID) fibrosantes chroniques avec un phénotype progressif (voir rubrique «Efficacité clinique»)
·de la pneumopathie interstitielle diffuse associée à la sclérodermie systémique (PID-ScS)
Posologie/Mode d’emploiLe traitement doit être instauré par un médecin expérimenté dans le diagnostic et le traitement des maladies pour lesquelles Ofev est indiqué.
La dose recommandée d'Ofev est de 150 mg deux fois par jour, administrée à environ 12 heures d'intervalle.
Les capsules doivent être avalées intactes avec de l'eau lors d'un repas. Elles ne doivent pas être croquées.
Les capsules d'Ofev peuvent être prises avec une petite quantité (cuillère à café) d'aliment mou, tel que compote de pommes ou pudding au chocolat, froid ou à température ambiante, et doivent être immédiatement avalées sans les croquer afin qu'elles restent intactes.
Si une dose d'Ofev a été omise, le traitement sera poursuivi à la prochaine heure de prise prévue, à la dose recommandée. L'oubli d'une dose ne doit pas être compensé par l'administration d'une dose supplémentaire. La dose maximale journalière recommandée de 300 mg ne doit pas être dépassée.
Les capsules ne doivent pas être ouvertes ou écrasées. En cas de contact avec le contenu de la capsule, les mains doivent être lavées immédiatement et soigneusement.
Ajustement de la posologie
En cas d'effets indésirables d'Ofev (voir les sections «Mises en garde et précautions» et «Effets indésirables»), le traitement symptomatique (si indiqué) peut être accompagné d'une réduction de la dose ou d'une interruption transitoire de l'administration de nintédanib jusqu'à ce que l'effet indésirable en question ait disparu ou suffisamment régressé pour permettre la poursuite du traitement. Le traitement par Ofev peut être repris avec la dose complète (150 mg deux fois par jour) ou à dose réduite (100 mg deux fois par jour). Si la dose de 100 mg deux fois par jour est mal tolérée par le patient, le traitement par Ofev doit être abandonné.
En cas d'interruption du traitement à cause d'une augmentation des taux de transaminases (ASAT ou ALAT) à plus de 3 fois la limite supérieure de la normale (LSN), le traitement par Ofev peut être repris à dose réduite (100 mg deux fois par jour) après la normalisation des taux de transaminases, puis passer ensuite à la dose entière (150 mg deux fois par jour). Dans le cas d'une augmentation du taux d'ASAT ou d'ALAT à plus de 5 fois la LSN ou dans le cas d'une augmentation à plus de 3 fois la LSN en présence de constats ou symptômes indiquant une atteinte hépatique sévère, le traitement par Ofev doit être arrêté (voir «Mises en garde et précautions» et «Effets indésirables»).
Instructions posologiques particulières
Patients présentant des troubles de la fonction rénale
Moins de 1 % d'une dose unique de nintédanib est excrété par les reins (voir «Pharmacocinétique»). Aucun ajustement de la dose initiale n'est donc nécessaire chez les patients présentant une insuffisance rénale légère à modérée. La sécurité, l'efficacité et la pharmacocinétique du nintédanib n'ont pas été étudiées chez des patients atteints d'insuffisance rénale sévère (ClCr <30 ml/min).
Patients présentant des troubles de la fonction hépatique
Le nintédanib est éliminé essentiellement par voie biliaire et dans les selles (>90 %; voir «Pharmacocinétique»). L'exposition systémique est augmentée chez les patients présentant une insuffisance hépatique (Child Pugh A, Child Pugh B). Chez les patients présentant une insuffisance hépatique légère (Child Pugh A), la dose recommandée d'Ofev est de 100 mg deux fois par jour à un intervalle de 12 heures. Chez les patients présentant une insuffisance hépatique légère (Child Pugh A), une réduction de la dose ou un arrêt du traitement doivent être envisagés afin de pouvoir traiter les réactions indésirables.
La sécurité et l'efficacité du nintédanib n'ont pas été étudiées chez les patients présentant une insuffisance hépatique de grade Child-Pugh B ou C. Le traitement par Ofev n'est pas recommandé chez les patients atteints d'une insuffisance hépatique modérée (Child-Pugh B) ou sévère (Child-Pugh C) (voir les sections «Mises en garde et précautions» et «Pharmacocinétique»).
Enfants et adolescents
Le nintédanib ne doit pas être pris par les enfants et les adolescents de moins de 18 ans.
Patients âgés (≥65 ans)
Aucune différence concernant la sécurité générale ou l'efficacité n'a été observée entre les patients âgés et ceux de moins de 65 ans. Aucun ajustement posologique n'est nécessaire en fonction de l'âge du patient (voir «Pharmacocinétique»).
Contre-indicationsOfev est contre-indiqué chez les patients présentant une hypersensibilité connue au nintédanib, aux arachides, au soja ou à l'un des autres composants.
Ofev est contre-indiqué pendant la grossesse (voir «Grossesse/Allaitement» et «Données précliniques»).
Mises en garde et précautionsAffections gastro-intestinales
Diarrhée
Dans les études cliniques (voir «Efficacité clinique»), la diarrhée était l'effet indésirable gastro-intestinal le plus fréquemment rapporté.
Dans la majorité des cas, la diarrhée s'est manifestée, avec une intensité légère à modérée dans les 3 premiers mois du traitement.
Dans les études INPULSIS (patients FPI), la diarrhée a été rapportée chez 62,4% des patients sous Ofev contre 18,4% des patients sous placebo. Elle a entraîné une réduction de la dose d'Ofev chez 10,7% des patients contre aucun des patients sous placebo et l'arrêt du traitement par Ofev chez 4,4% des patients contre 0,2% des patients sous placebo.
Dans l'étude INBUILD (patients PID fibrosantes progressives), la diarrhée a été rapportée chez 66,9% des patients sous Ofev contre 23,9% des patients sous placebo. La diarrhée a entraîné une réduction de la dose d'Ofev chez 16,0% des patients contre 0,9% chez les patients sous placebo et l'arrêt du traitement par Ofev chez 5,7% des patients contre 0,3% chez les patients sous placebo.
Dans l'étude SENSCIS (patients PID-ScS), la diarrhée a été rapportée chez 75,7% des patients sous Ofev contre 31,6% des patients sous placebo.
La diarrhée a entraîné une réduction de la dose d'Ofev chez 22,2% des patients contre 1,0% chez les patients sous placebo et l'arrêt du traitement par Ofev chez 6,9% des patients contre 0,3% chez les patients sous placebo (voir rubrique «Effets indésirables»).
La diarrhée peut entraîner une déshydratation avec ou sans troubles électrolytiques, ce qui peut conduire à un dysfonctionnement rénal.
Une diarrhée doit être traitée dès les premiers signes par une réhydratation appropriée et une administration d'antidiarrhéiques tels que le lopéramide. Une réduction de la dose ou l'interruption du traitement est éventuellement nécessaire. Le traitement par Ofev peut être repris à dose réduite (100 mg deux fois par jour) ou entière (150 mg deux fois par jour). Dans le cas d'une diarrhée sévère persistant malgré un traitement symptomatique, le traitement par Ofev doit être abandonné.
Nausées et vomissements
Les nausées et vomissements sont des effets indésirables fréquemment rapportés (voir «Effets indésirables») et présentent généralement une intensité légère à modérée.
Dans les études INPULSIS (patients FPI), des nausées ont été rapportées chez 24,5% des patients sous Ofev contre 6,6% des patients sous placebo. Les nausées ont entraîné une réduction de la dose d'Ofev chez 1,7% des patients contre aucun des patients sous placebo et l'arrêt du traitement par Ofev chez 2,0% des patients contre aucun des patients sous placebo.
Dans l'étude INBUILD (patients PID fibrosantes progressives), des nausées ont été rapportées chez 28,9% des patients sous Ofev contre 9,4% des patients sous placebo. Les nausées ont entraîné une réduction de la dose d'Ofev chez 3,3% des patients contre 0,6% des patients sous placebo et l'arrêt du traitement par Ofev chez 0,3% des patients contre 0,3% des patients sous placebo.
Dans l'étude SENSCIS (patients PID-ScS), des nausées ont été rapportées chez 31,6% des patients sous Ofev contre 13,5% des patients sous placebo. Les nausées ont entraîné une réduction de la dose d'Ofev chez 2,1% des patients contre aucun des patients sous placebo et l'arrêt du traitement par Ofev chez 2,1% des patients contre aucun des patients sous placebo.
Dans les études INPULSIS (patients FPI), des vomissements ont été rapportés chez 11,6% des patients sous Ofev contre 2,6% des patients sous placebo. Les vomissements ont entraîné une réduction de la dose d'Ofev chez 1,1% des patients contre aucun des patients sous placebo et l'arrêt du traitement par Ofev chez 0,8% des patients contre aucun des patients sous placebo.
Dans l'étude INBUILD (patients PID fibrosantes progressives), des vomissements ont été rapportés chez 18,4% des patients sous Ofev contre 5,1% des patients sous placebo. Les vomissements ont entraîné une réduction de la dose d'Ofev chez 2,4% des patients contre 0,9% des patients sous placebo et l'arrêt du traitement par Ofev chez 0,9% des patients contre aucun des patients sous placebo.
Dans l'étude SENSCIS (patients PID-ScS), des vomissements ont été rapportés chez 24,7% des patients sous Ofev contre 10,4% des patients sous placebo. Les vomissements ont entraîné une réduction de la dose d'Ofev chez 2,1% des patients contre aucun des patients sous placebo et l'arrêt du traitement par Ofev chez 1,4% des patients contre 0,3% des patients sous placebo.
Les vomissements peuvent entraîner une déshydratation avec ou sans troubles électrolytiques, ce qui peut conduire à un dysfonctionnement rénal.
Si les symptômes persistent malgré des mesures de soutien adaptées (incluant un traitement antiémétique), une réduction de la dose ou une interruption du traitement peut devenir nécessaire. Le traitement peut être repris à dose réduite (100 mg deux fois par jour) ou entière (150 mg deux fois par jour). Une persistance de symptômes sévères exige un arrêt du traitement par Ofev.
Fonction hépatique
La sécurité et l'efficacité d'Ofev n'ont pas été étudiées chez les patients présentant une insuffisance hépatique modérée (Child-Pugh B) ou sévère (Child-Pugh C). L'utilisation d'Ofev n'est donc pas recommandée chez ces patients. Compte tenu de l'augmentation de l'exposition systémique, le risque de survenue d'événements indésirables peut être augmenté chez les patients atteints d'insuffisance hépatique légère (Child Pugh A). Les patients présentant une insuffisance hépatique légère (Child Pugh A) doivent être traités avec une dose d'Ofev réduite (voir «Posologie / Mode d'emploi» et «Pharmacocinétique»).
Des cas de dommages du foie liés à la prise de médicaments ont été observés dans le cadre du traitement par nintédanib. Lors de l'observation du marché, des cas non sévères et sévères de dommages du foie liés à la prise de médicaments ont été signalés, y compris des dommages du foie sévères à issue fatale.
La plupart des événements hépatiques indésirables sont survenus au cours des trois premiers mois de traitement. Par conséquent, les transaminases hépatiques et les concentrations de bilirubine doivent être contrôlées au début du traitement par Ofev, à intervalles réguliers au cours des trois premiers mois du traitement puis périodiquement ou lorsque cliniquement indiqué.
Les augmentations des valeurs d'enzymes hépatiques (ALAT, ASAT, PAL, gamma glutamyl-transférase (GGT)) et de la valeur de bilirubine étaient réversibles dans la plupart des cas après réduction de la dose ou interruption du traitement. Des valeurs accrues d'ALAT et/ou ASAT qui ont augmenté à au moins plus de 3 fois la limite supérieure normale (LSN) ont été observées chez jusqu'à respectivement 5,0%, 7,8% et 4,9% des patients sous Ofev dans les études INPULSIS (patients FPI), INBUILD (patients PID fibrosantes progressives) et SENSCIS (patients PID-ScS), tandis que chez les patients sous placebo, cela concernait moins de 2% des patients au total.
En présence de taux accrus de transaminases (ASAT ou ALAT) à plus de 3 fois la limite supérieure de la normale (LSN), on interrompra l'administration d'Ofev ou réduira la dose à 100 mg deux fois par jour. En même temps, le patient sera étroitement surveillé et les autres causes possibles d'une augmentation des taux d'enzymes hépatiques seront explorées. Après normalisation des taux de transaminases, le traitement par Ofev peut être repris à dose réduite (100 mg deux fois par jour), puis poursuivi à la dose entière (150 mg deux fois par jour). Si l'augmentation des valeurs hépatiques est accompagnée de signes cliniques ou symptômes d'une atteinte hépatique tels qu'un ictère, ou si les taux de transaminases (ASAT ou ALAT) augmentent à plus de 5 fois la LSN, le traitement par Ofev doit être définitivement abandonné (voir «Posologie/Mode d'emploi»).
Les patients à faible poids corporel (< 65 kg), les Asiatiques et les femmes présentent un risque plus élevé d'augmentations des taux d'enzymes hépatiques.
L'exposition au nintédanib a augmenté de façon linéaire avec l'âge des patients. Ceci pourrait également entraîner une hausse du risque d'élévations des enzymes hépatiques (voir «Pharmacocinétique»).
Chez les patients présentant ces facteurs de risque, une surveillance étroite est recommandée.
Fonction rénale
Des cas d'atteinte / insuffisance rénale, dont certains conduisant à la mort, ont été rapportés avec l'utilisation de nintédanib (voir «Effets indésirables»).
Les patients doivent être surveillés pendant le traitement par nintédanib, avec une attention particulière en cas d'existence de facteurs de risque d'atteinte/insuffisance rénale. En cas d'atteinte/insuffisance rénale, une adaptation du traitement doit être envisagée (voir «Ajustement de la posologie»).
Hémorragies
En raison de son mécanisme d'action (inhibition du récepteur du facteur de croissance de l'endothélium vasculaire (vascular endothelial growth factor receptor) VEGFR)), Ofev peut être associé à un risque hémorragique accru.
Dans les études cliniques sur le médicament Ofev, la fréquence des événements hémorragiques chez les patients sous Ofev était légèrement plus élevée ou comparable entre les groupes de traitement (10,3% versus 7,8% dans le groupe placebo dans les études INPULSIS (patients FPI); 11,1% versus 12,7% dans le groupe placebo dans l'étude INBUILD (patients PID fibrosantes progressives), 11,1% versus 8,3% dans le groupe placebo dans l'étude SENSCIS (patients PID-ScS)).
Les saignements les plus fréquemment rapportés étaient une épistaxis sans gravité.
La fréquence des événements hémorragiques graves était faible dans les 2 groupes de traitement (Ofev 1,3% contre placebo 1,4% dans INPULSIS (patients FPI); Ofev 0,9% contre placebo 1,5% dans INBUILD (patients PID fibrosantes progressives); Ofev 1,4% contre placebo 0,7% dans SENSCIS (patients PID-ScS)).
Les études cliniques n'ont pas inclus de patients présentant un risque hémorragique connu (par exemple coagulopathie congénitale ou traitement anticoagulant à dose entière). Des saignements ont été rapportés après la commercialisation, dont certains sévères et mortels (y compris chez des patients avec ou sans traitement par anticoagulants ou autres médicaments pouvant provoquer des saignements). Les patients présentant des risques supplémentaires ne doivent donc être traités par Ofev que si le bénéfice attendu prédomine par rapport au risque potentiel.
Événements thromboemboliques artériels
Les patients ayant récemment subi un infarctus du myocarde ou un accident vasculaire cérébral étaient exclus d'une participation aux études cliniques.
Dans les études cliniques, les événements thromboemboliques artériels ont été rapportés à une faible fréquence (Ofev 2,5% contre placebo 0,7% dans les études INPULSIS (patients FPI); Ofev 0,9% contre placebo 0,9% dans INBUILD (patients PID fibrosantes progressives); Ofev 0,7% contre placebo 0,7% dans SENSCIS (patients PID-ScS)). Dans les études INPULSIS, des infarctus du myocarde ont été décrits chez un pourcentage plus élevé de patients dans le groupe Ofev (1,6%) que dans le groupe placebo, alors que les événements indésirables évoquant une cardiopathie ischémique étaient du même ordre dans le groupe Ofev et dans le groupe placebo. Dans l'étude INBUILD et l'étude SENSCIS, des infarctus du myocarde ont été observés à une faible fréquence: Ofev 0,9% contre placebo 0,9% dans INBUILD; Ofev 0% contre placebo 0,7% dans SCENSIS.
La prudence est de rigueur chez les patients à risque cardiovasculaire accru (par exemple patients présentant une cardiopathie coronarienne connue). Une interruption du traitement doit être envisagée chez les patients développant des signes ou symptômes d'ischémie aiguë du myocarde.
Anévrismes et dissections artérielles
L'utilisation d'inhibiteurs des voies du VEGF chez les patients souffrant ou non d'hypertension peut favoriser la formation d'anévrismes et/ou de dissections artérielles. Avant l'instauration d'Ofev, ce risque doit être soigneusement pris en considération chez les patients présentant des facteurs de risque tels que l'hypertension ou des antécédents d'anévrisme.
Thromboembolies veineuses
Aucune augmentation du risque de thromboembolies veineuses n'a été observée sous nintédanib dans les études cliniques. Le mécanisme d'action du nintédanib pourrait être associé à un risque accru d'événements thromboemboliques.
Perforations gastro-intestinales et colite ischémique
Les patients sous nintédanib peuvent avoir un risque accru de perforation gastro-intestinale en raison du mécanisme d'action du médicament. Dans les études INPULSIS (patients FPI), une perforation gastro-intestinale a été rapportée chez 0,3 % des patients sous Ofev et chez aucun (0) des patients sous placebo. Dans l'étude SENSCIS (patients PID-ScS) et dans l'étude INBUILD (patients PID fibrosantes progressives), aucun cas de perforation gastro-intestinale n'a été rapporté dans les deux groupes de traitement.
Après l'introduction sur le marché, des cas de perforations gastro-intestinales et de colite ischémique, dont certains d'issue fatale, ont été signalés. Une prudence particulière est recommandée chez les patients récemment soumis à une intervention chirurgicale abdominale, en cas de perforation récente d'un organe creux, d'ulcères peptiques dans les antécédents, de diverticulose ou en cas d'utilisation concomitante de corticostéroïdes ou d'AINS. En cas de survenue d'une perforation gastro-intestinale ou d'une colite ischémique, le traitement par Ofev doit être arrêté définitivement.
Les patients à risque connu de perforation gastro-intestinale ou de colite ischémique ne doivent être traités par Ofev que si le bénéfice attendu prédomine par rapport au risque potentiel.
Protéinurie d'ordre néphrotique et microangiopathie thrombotique
Très peu de cas de protéinurie néphrotique, avec ou sans atteinte de la fonction rénale, ont été rapportés après la mise sur le marché. Dans certains cas, les résultats histologiques correspondaient à une microangiopathie glomérulaire avec ou sans thrombus rénal. Après l'arrêt du traitement par Ofev, les symptômes étaient réversibles, avec une protéinurie résiduelle dans certains cas. L'interruption du traitement doit être envisagée chez les patients qui développent des signes ou des symptômes de syndrome néphrotique.
Les inhibiteurs de la voie du VEGF ont été associés à des microangiopathies thrombotiques (MAT), dont très peu de cas ont été rapportés avec le nintédanib. Si des signes biologiques ou cliniques de MAT surviennent chez un patient recevant du nintédanib, le traitement par nintédanib doit être arrêté et une évaluation approfondie de la MAT doit être réalisée.
Syndrome d'encéphalopathie postérieure réversible (SEPR)
Très peu de cas de syndrome d'encéphalopathie postérieure réversible (SEPR) ont été rapportés après la mise sur le marché.
Le SEPR est un syndrome neurologique pouvant se manifester par des céphalées, des troubles visuels, des convulsions, une léthargie, une confusion et autres troubles neurologiques.
Une hypertension légère à sévère peut survenir. Un examen d'imagerie par résonance magnétique est nécessaire afin de confirmer le diagnostic de SEPR.
En cas de suspicion de SEPR, le traitement par le nintédanib doit être arrêté.
On ignore comment évaluer la sécurité en cas de reprise du traitement par le nintédanib chez les patients ayant des antécédents de SEPR.
Troubles de la cicatrisation des plaies
Aucune augmentation de l'incidence des troubles de la cicatrisation n'a été observée dans les études cliniques. Le mécanisme d'action du nintédanib peut perturber la cicatrisation des plaies. Aucune étude n'a été effectuée spécifiquement sur l'impact du nintédanib sur la cicatrisation des plaies. Le traitement par Ofev ne doit donc pas être commencé – ou repris après une interruption due à une intervention chirurgicale – avant que la cicatrisation des plaies soit jugée cliniquement satisfaisante.
Lécithine du soja
Les capsules molles Ofev contiennent de la lécithine du soja (voir «Contre-indications»).
InteractionsLes études effectuées pour identifier les interactions n'ont inclus que des adultes.
Influence d'autres agents actifs sur le nintédanib
Glycoprotéine P (P-gp) et inhibiteurs/inducteurs du CYP3A4
Le nintédanib est un substrat de la P-gp (voir «Pharmacocinétique») et en plus faible mesure un substrat du CYP3A4. Une co-administration de kétoconazole – inhibiteur puissant de la P-gp et inhibiteur du CYP3A4 – a fait augmenter l'AUC du nintédanib d'un facteur 1,61 et la Cmax d'un facteur 1,83.
Chez un patient traité par Ofev, la co-administration d'un inhibiteur puissant de la P-gp (p.ex. kétoconazole ou érythromycine) peut faire augmenter l'exposition au nintédanib. Une surveillance étroite de la tolérance du nintédanib est alors recommandée. Les effets indésirables peuvent exiger une interruption du traitement par Ofev, une réduction de la dose ou un arrêt d'administration d'Ofev (voir «Posologie/Mode d'emploi»).
Une co-administration de rifampicine (inducteur puissant de la P-gp et inducteur du CYP3A4) a fait diminuer l'AUC du nintédanib de 50,3 % et sa Cmax de 60,3 % par rapport à l'administration de nintédanib seul.
Les inducteurs puissants de la P-gp (p.ex. rifampicine, carbamazépine, phénytoïne et millepertuis) peuvent faire diminuer l'exposition au nintédanib. Le choix d'une co-médication alternative dont le potentiel d'induction de la P-gp est faible ou inexistant doit être envisagé.
Aliments
Ofev doit être pris avec un repas pour améliorer la biodisponibilité (voir «Pharmacocinétique»).
pH
La solubilité du nintédanib dépend fortement du pH et baisse lors d'un pH >3. Dans les études cliniques, la co-administration de médicaments causant une augmentation du pH intragastrique n'a pas influencé les valeurs de Ctrough du nintédanib pris avec un repas. Lors d'une prise à jeun, la co-administration de médicaments induisant une augmentation du pH intragastrique pourrait réduire la biodisponibilité du nintédanib. Le nintédanib doit donc être pris avec des aliments.
Tabagisme
Le tabagisme est associé à une diminution de l'exposition au nintédanib, susceptible d'altérer son efficacité. Les patients doivent être incités à arrêter de fumer avant de commencer le traitement par Ofev ou à réduire leur consommation de tabac pendant le traitement par Ofev.
D'après les études in vitro, le nintédanib n'est pas un substrat des transporteurs OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, BCRP ou MRP-2. Des études in vitro ont révélé que le nintédanib est un substrat du transporteur OCT-1. On estime que ces observations n'ont guère de signification clinique.
Influence du nintédanib sur d'autres agents actifs
Transporteurs
De faibles effets inhibiteurs sur les transporteurs OCT-1, BCRP et P-gp ont été observés in vitro. La signification clinique de ces effets n'a pas été étudiée, mais elle est considérée comme faible.
D'après les études in vitro, le nintédanib n'est pas un inhibiteur des transporteurs OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 ou MRP-2.
Il est apparu que le métabolite BIBF 1202 était in vitro un faible inhibiteur des transporteurs OATP-1B1, OATP-1B3, OATP-2B1, OCT-1 et BCRP. La signification clinique de ces effets n'a pas été étudiée, mais elle est considérée comme faible.
Enzymes du cytochrome (CYP)
La biodégradation du nintédanib ne s'effectue qu'en faible mesure par voie des enzymes du CYP. Le nintédanib et ses métabolites – le groupement acide libre BIBF 1202 et le BIBF 1202 glucuronidé – n'ont pas eu d'effet inhibiteur ou inducteur sur les enzymes du CYP dans les études précliniques (voir «Pharmacocinétique»). On estime donc que la probabilité d'interactions médicamenteuses dues à une influence du nintédanib sur la voie métabolique du CYP est faible.
Administration en association avec d'autres médicaments
Traitement concomitant par pirfénidone
Les données issues d'une étude spécifique de pharmacocinétique n'ont pas mis en évidence d'interaction pharmacocinétique significative entre le nintédanib et la pirfénidone lorsque ces principes actifs sont administrés en association.
Traitement concomitant par bosentan
Dans une étude spéciale de pharmacocinétique, le traitement concomitant par Ofev associé à du bosentan a été étudié sur des sujets volontaires sains. Les participants à l'étude ont reçu une dose unique de 150 mg d'Ofev avant et après une administration répétée de 125 mg de bosentan deux fois par jour à l'état d'équilibre. Les rapports ajustés des valeurs moyennes géométriques (intervalle de confiance (IC) à 90%) étaient respectivement de 103% (86% - 124%) et 99% (91% - 107%) pour la Cmax et l'AUC0-tz du nintédanib (n=13), ce qui indique que l'administration concomitante du nintédanib avec du bosentan n'a pas modifié la pharmacocinétique du nintédanib.
Traitement concomitant par des contraceptifs hormonaux oraux
L'administration concomitante de nintédanib et de contraceptifs hormonaux oraux n'a pas eu d'influence notable sur la pharmacocinétique des contraceptifs hormonaux oraux.
Dans une étude spécifique de PK, des patientes atteintes de PID-ScS ont reçu une dose unique d'une association de 30 µg d'éthinylestradiol et de 150 µg de lévonorgestrel avant et après l'administration de 150 mg de nintédanib deux fois par jour pendant au moins 10 jours. Les rapports ajustés des moyennes géométriques (IC à 90%) s'élevaient respectivement à 117% (108%-127%; Cmax) et 101% (93%-111%; AUC0-tz) pour l'éthinylestradiol et à 101% (90%-113%; Cmax) et 96% (91%-102%; AUC0-tz) pour le lévonorgestrel (n=15), ce qui indique que l'administration concomitante de nintédanib n'a pas d'effets notables sur les taux plasmatiques d'éthinylestradiol et de lévonorgestrel.
Grossesse, allaitementGrossesse
On ne dispose pas de données sur l'utilisation d'Ofev chez la femme enceinte, mais les expérimentations animales précliniques ont révélé une toxicité du médicament sur la reproduction (voir «Données précliniques»). Vu que le nintédanib peut nuire également au fœtus humain, il ne doit pas être utilisé pendant la grossesse.
Un test de grossesse doit être effectué avant le début du traitement par Ofev et être répété si nécessaire pendant le traitement.
Il faut informer les patientes qu'elles devront prévenir leur médecin ou leur pharmacien si elles constatent le début d'une grossesse au cours de leur traitement par Ofev.
Si une patiente est enceinte pendant le traitement par Ofev, le traitement doit être arrêté et la patiente doit être informée des risques potentiels pour le fœtus.
Contraception
Le nintédanib peut nuire au fœtus (voir la rubrique «Données précliniques»). Les patientes susceptibles de procréer qui sont traitées par Ofev doivent être averties de la nécessité d'éviter une grossesse pendant le traitement par Ofev et qu'au début du traitement, pendant le traitement et jusqu'à 3 mois au moins après la dernière dose d'Ofev, des méthodes contraceptives très efficaces doivent être utilisées. Le nintédanib n'a pas d'influence notable sur les taux plasmatiques d'éthinylestradiol et de lévonorgestrel (voir «Interactions»). L'efficacité des contraceptifs hormonaux oraux peut être diminuée en cas de vomissements et/ou de diarrhée ainsi que dans toute autre situation entravant l'absorption. Il convient de recommander aux femmes qui prennent des contraceptifs hormonaux oraux et chez lesquelles une telle situation se produit, d'utiliser une autre méthode contraceptive très fiable.
Allaitement
On ne dispose pas de données concernant le passage du nintédanib et de ses métabolites dans le lait maternel.
L'excrétion de faibles quantités de nintédanib et de ses métabolites (≤0,5 % de la dose administrée) dans le lait des rates allaitantes a été constatée dans des études précliniques.
Un risque pour le nouveau-né / l'enfant ne peut pas être exclu. L'allaitement doit être interrompu pendant le traitement par Ofev.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Les patients doivent être instruits d'être prudents en conduisant un véhicule ou en utilisant une machine au cours du traitement par Ofev.
Effets indésirablesLa sécurité d'Ofev a été évaluée dans des études cliniques auprès de plus de 1000 patients atteints de FPI, dont plus de 200 ont reçu Ofev plus de 2 ans. Chez des patients atteints de FPI, Ofev a été évalué dans trois études de 52 semaines avec randomisation, double aveugle et contrôle versus placebo. Dans l'étude de phase II (TOMORROW) et les études de phase III (INPULSIS-1 et INPULSIS-2), 723 patients atteints de FPI ont reçu Ofev à la dose de 150 mg deux fois par jour et 508 patients ont reçu un placebo. La durée médiane d'exposition a été de 10 mois chez les patients traités par Ofev et de 11 mois chez les patients sous placebo. Les participants aux études étaient âgés de 42 à 89 ans (âge médian: 67 ans). La majorité des patients étaient de sexe masculin (79 %) et de type caucasoïde (60 %).
Ofev a été en outre évalué dans une étude de phase III, randomisée, menée en double aveugle et contrôlée contre placebo (INBUILD), dans laquelle le traitement par Ofev 150 mg deux fois par jour (n=332) a été comparé avec le placebo chez 663 patients atteints d'autres PID fibrosantes chroniques avec un phénotype progressif. Alors que les patients ont été traités pendant au moins 52 semaines, le traitement a duré jusqu'à 27 mois chez certains patients. La durée médiane d'exposition était de 16 mois chez les patients sous Ofev et de 17 mois chez les patients sous placebo. L'âge des participants à l'étude allait de 27 à 87 ans (âge médian: 67 ans). La plupart des patients étaient de sexe masculin (54%). Les patients étaient majoritairement caucasiens (74%) ou asiatiques (25%).
Ofev a été évalué dans une étude de phase III avec randomisation, double aveugle et contrôle versus placebo (SENSCIS) dans laquelle 576 patients atteints de PID-ScS ont reçu deux fois par jour 150 mg d'Ofev (n=288) ou le placebo (n=288). Tandis que les patients ont été traités durant au moins 52 semaines, le traitement de certains patients a duré jusqu'à 100 semaines. La durée d'exposition médiane était de 15 mois pour les patients sous Ofev et 16 mois pour les patients sous placebo. L'âge des participants allait de 20 à 79 ans (âge médian: 55 ans). La plupart des patients étaient de sexe féminin (75 %). Les patients étaient majoritairement caucasiens (67 %), asiatiques (25 %) ou avaient la peau noire (6 %). 49 % des patients ont reçu un traitement stable par mycophénolate au début de l'étude.
Les effets indésirables le plus souvent décrits en rapport avec l'administration d'Ofev étaient des diarrhées, nausées et vomissements, des douleurs abdominales, une perte d'appétit, une perte de poids, des hémorragies, des taux accrus d'enzymes hépatiques et une éruption cutanée.
Le profil de sécurité était similaire chez les patients traités sous Ofev, et ce indépendamment du fait qu'ils aient reçu du mycophénolate au début de l'étude ou non.
La section «Mises en garde et précautions» décrit des effets indésirables sélectionnés et la marche à suivre correspondante.
Les catégories de fréquence des effets indésirables sont définies selon la convention suivante: «très fréquents» (≥1/10), «fréquents» (< 1/10, ≥1/100), «occasionnels» (< 1/100, ≥1/1000), «rares» (< 1/1000, ≥1/10'000), «très rares» (< 1/10.000).
Fibrose pulmonaire idiopathique (FPI)
Affections hématologiques et du système lymphatique
Occasionnels: thrombocytopénie.
Troubles du métabolisme et de la nutrition
Fréquents: perte d'appétit, perte de poids.
Affections du système nerveux
Fréquents: céphalées.
Fréquence inconnue: syndrome d'encéphalopathie postérieure réversible (SEPR).
Maladies cardiaques
Occasionnels: Infarctus du myocarde.
Affections vasculaires
Fréquents: hémorragiesa.
Occasionnels: hypertensionb.
Fréquence non connue: anévrismes et dissections artérielles.
Affections gastro-intestinales
Très fréquents: diarrhées (53,6%), nausées (19,1%), douleurs abdominalesc (10,2%).
Fréquents: vomissements.
Occasionnels: pancréatites, perforations gastro-intestinales, colites.
Fréquence non connue: colites ischémiques (observées après l'introduction sur le marché).
Affections hépatobiliaires
Très fréquents: augmentation des enzymes hépatiques (10,5%).
Fréquents: augmentation de l'alanine aminotransférase (ALAT), augmentation de l'aspartate aminotransférase (ASAT), augmentation de la gamma-glutamyltransférase (GGT).
Occasionnels: augmentation du taux sanguin des phosphatases alcalines (PAL), hyperbilirubinémie, atteinte hépatique d'origine médicamenteuse.
Affections de la peau et du tissu sous-cutané
Fréquents: éruption cutanée.
Occasionnels: prurit, alopécie.
Affections du rein et des voies urinaires
Occasionnels: protéinurie.
Fréquence non connue: insuffisance rénale (voir «Mises en garde et précautions»).
a Des évènements hémorragiques sévères et non sévères, dont certains mortels, correspondant à l'expérience acquise pendant les études cliniques, ont été observés après la mise sur le marché. Les évènements hémorragiques les plus fréquents comprenaient des saignements rectaux/hématochézie, des saignements de nez et des hématomes. Les événements mortels concernaient des saignements gastro-intestinaux, intracrâniens et pulmonaires ainsi que des CIVD.
b Incluant: hypertension, tension artérielle accrue, poussée hypertensive et cardiopathie hypertensive.
c Incluant: douleurs abdominales, douleurs épigastriques, douleurs pelviennes, douleurs gastro-intestinales, sensation douloureuse de pression abdominale.
Autres pneumopathies interstitielles diffuses (PID) fibrosantes chroniques avec un phénotype progressif
Affections hématologiques et du système lymphatique
Occasionnels: thrombocytopénie.
Troubles du métabolisme et de la nutrition
Très fréquents: perte d'appétit (11,1%).
Fréquents: perte de poids.
Affections du système nerveux
Fréquents: céphalées.
Fréquence inconnue: syndrome d'encéphalopathie postérieure réversible (SEPR).
Maladies cardiaques
Occasionnels: Infarctus du myocarde.
Affections vasculaires
Fréquents: hypertension, hémorragies.
Fréquence non connue: anévrismes et dissections artérielles.
Affections gastro-intestinales
Très fréquents: diarrhées (59%), nausées (24%), douleurs abdominales (11%), vomissements (12%).
Occasionnels: pancréatites, colites.
Fréquence non connue: perforations gastro-intestinales, colites ischémiques (observées après l'introduction sur le marché).
Affections hépatobiliaires
Très fréquents: augmentation des enzymes hépatiques (18,4%), augmentation de l'alanine aminotransférase (ALAT) (10,8%).
Fréquents: augmentation de l'aspartate aminotransférase (ASAT), augmentation de la gamma-glutamyl transférase (GGT), augmentation du taux sanguin des phosphatases alcalines (PAL), atteinte hépatique d'origine médicamenteuse.
Occasionnels: hyperbilirubinémie
Affections de la peau et du tissu sous-cutané
Fréquents: éruption cutanée.
Occasionnels: prurit, alopécie.
Affections du rein et des voies urinaires
Occasionnels: protéinurie.
Fréquence non connue: insuffisance rénale (voir «Mises en garde et précautions»).
Pneumopathie interstitielle associée à la sclérodermie systémique (PID-ScS)
Affections hématologiques et du système lymphatique
Occasionnels: thrombocytopénie.
Troubles du métabolisme et de la nutrition
Fréquents: perte d'appétit, perte de poids.
Affections du système nerveux
Fréquents: céphalées.
Fréquence inconnue: syndrome d'encéphalopathie postérieure réversible (SEPR).
Maladies cardiaques
Fréquence non connue: Infarctus du myocarde.
Affections vasculaires
Fréquents: hypertension, hémorragies.
Fréquence non connue: anévrismes et dissections artérielles.
Affections gastro-intestinales
Très fréquents: diarrhées (68,4%), nausées (24,7%), douleurs abdominales (11,8%), vomissements (17,7%).
Occasionnels: colites.
Fréquence non connue: pancréatites, perforations gastro-intestinales, colites ischémiques (observées après l'introduction sur le marché).
Affections hépatobiliaires
Très fréquents: augmentation des enzymes hépatiques (11,1%).
Fréquents: augmentation de l'alanine aminotransférase (ALAT), augmentation de l'aspartate aminotransférase (ASAT), augmentation de la gamma-glutamyltransférase (GGT).
Occasionnels: atteinte hépatique d'origine médicamenteuse.
Fréquence non connue: hyperbilirubinémie.
Affections de la peau et du tissu sous-cutané
Occasionnels: éruption cutanée, prurit.
Fréquence non connue: alopécie.
Affections du rein et des voies urinaires
Occasionnels: insuffisance rénale (voir «Mises en garde et précautions»).
Fréquence non connue: protéinurie.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageIl n'existe aucun antidote spécifique et aucun traitement spécifique d'un surdosage d'Ofev. La plus forte dose unique de nintédanib administrée dans une étude de phase I était de 450 mg une fois par jour. À part cela, 2 patients ont reçu une surdose ne dépassant pas 600 mg deux fois par jour pendant une période allant jusqu'à 8 jours. Les effets indésirables observés étaient en accord avec le profil de sécurité connu du nintédanib, comprenant des taux accrus d'enzymes hépatiques et des symptômes gastro-intestinaux. Les deux patients se sont rétablis de ces effets indésirables.
Dans le cadre des études INPULSIS (patients FPI), un patient reçu accidentellement une dose de 600 mg par jour pendant 21 jours au total. Un effet indésirable sans gravité (rhinopharyngite) est apparu et a régressé au cours de la période de surdosage. Aucun autre événement décrit ne s'est produit.
Lors d'un surdosage, le traitement doit être interrompu et des mesures générales de soutien adaptées à la situation clinique doivent être initiées.
Propriétés/EffetsCode ATC
L01EX09
Mécanisme d'action
Le nintédanib est un inhibiteur de tyrosines kinases qui appartient au type des petites molécules. Les tyrosine kinases inhibées comprennent les récepteurs du facteur de croissance dérivé des plaquettes (platelet-derived growth factor receptors, PDGFR) α et β, les récepteurs du facteur de croissance des fibroblastes (fibroblast growth factor receptors, FGFR) 1 à 3 et les récepteurs du facteur de croissance de l'endothélium vasculaire (vascular endothelial growth factor receptors, VEGFR) 1 à 3. Le nintédanib se fixe de manière compétitive au site de liaison de l'adénosine triphosphate (ATP) de ces récepteurs et bloque ainsi la signalisation intracellulaire qui est décisive pour la prolifération, la migration et la transformation des fibroblastes, et donc pour les mécanismes pathologiques de la FPI. À part cela, le nintédanib inhibe aussi les kinases Flt-3, Lck, Lyn et Src.
Pharmacodynamique
L'activation des cascades de signalisation induites par le FGFR et le PDGFR est un facteur décisif pour la prolifération et la migration des fibroblastes/myofibroblastes pulmonaires, qui sont les cellules caractéristiques de la pathologie de fibrose pulmonaire idiopathique. La signification potentielle de l'inhibition du VEGFR pour la pathologie de FPI n'a pas encore été totalement élucidée. On suppose que le nintédanib, en se liant au site de liaison d'adénosine triphosphate (ATP) du domaine intracellulaire des récepteurs à activité tyrosine kinase, inhibe à l'échelle moléculaire les cascades de signalisation du FGFR et du PDGFR impliquées dans la prolifération et la migration des fibroblastes pulmonaires, et interfère ainsi avec l'activation croisée (par autophosphorylation du récepteur homodimère).
In vitro, des concentrations faiblement nanomolaires de nintédanib suffisent pour inhiber les récepteurs cibles. Sur des fibroblastes pulmonaires de patients atteints de FPI, la prolifération stimulée par PDGF, FGF et VEGF a été inhibée par le nintédanib aux CE50 de 11 nmol/l, 5,5 nmol/l et moins de 1 nmol/l. À des concentrations de 100 à 1000 nmol/l, le nintédanib a également inhibé la migration de fibroblastes stimulée par PDGF, FGF et VEGF ainsi que la transformation des fibroblastes en myofibroblastes induite par TGF-β2. On suppose que l'activité anti-inflammatoire du nintédanib limite en outre la stimulation fibrotique en réduisant les médiateurs profibrotiques tels que l'IL-1β et l'IL-6. On ignore encore dans quelle mesure l'activité anti-angiogénique du nintédanib participe au mécanisme d'action du médicament dans le traitement des fibroses pulmonaires. Dans des études in vivo, le nintédanib a fait preuve d'une forte activité anti-fibrotique et anti-inflammatoire.
Efficacité clinique
Fibrose pulmonaire idiopathique (FPI)
L'efficacité clinique du nintédanib a été étudiée chez 1231 patients atteints de FPI dans une étude de phase II (TOMORROW) et deux études de phase III (INPULSIS-1 et INPULSIS-2). Il s'agissait d'études randomisées, en double aveugle, avec contrôle contre placebo, dans lesquelles un traitement de 52 semaines par Ofev (150 mg deux fois par jour) a été évalué versus placebo.
Les études INPULSIS-1 et INPULSIS-2 étaient de conception identique, proche de la conception de l'étude TOMORROW. Les patients ont été randomisés en proportions de 3:2 (1:1 dans l'étude TOMORROW) et assignés pour 52 semaines à un traitement par Ofev (150 mg deux fois par jour) ou par un placebo (également deux fois par jour). L'étude TOMORROW comprenait des groupes de traitement supplémentaires (50 mg par jour, 50 mg deux fois par jour et 100 mg deux fois par jour) qui ne doivent pas être discutés ici.
Le critère primaire était défini comme le déclin annuel de la capacité vitale forcée (CVF). La variation du score total au questionnaire SGRQ (Saint George's Respiratory Questionnaire) entre le début de l'étude et la semaine 52 ainsi que le temps écoulé jusqu'à la première exacerbation aiguë de la FPI étaient des critères secondaires importants des études INPULSIS et des critères secondaires de l'étude TOMORROW.
Déclin annuel de la CVF
Le déclin annuel de la CVF (en ml) était significativement plus faible chez les patients sous nintédanib que chez les patients sous placebo. L'effet du traitement était concordant entre les 3 études. Le tableau 1 présente les résultats des études individuelles et les résultats de l'analyse cumulée des études INPULSIS.
Tableau 1: Déclin annuel de la CVF (ml) dans les études TOMORROW, INPULSIS-1 et INPULSIS-2 ainsi que dans l'analyse cumulée des études INPULSIS – population traitée2
|
|
TOMORROW
|
INPULSIS-1
|
INPULSIS-2
|
INPULSIS-1 et INPULSIS-2
Données réunies
| |
|
Placebo
|
Ofev 150 mg deux fois par jour
|
Placebo
|
Ofev 150 mg deux fois par jour
|
Placebo
|
Ofev 150 mg deux fois par jour
|
Placebo
|
Ofev 150 mg deux fois par jour
| |
Nombre de patients inclus à l'analyse
|
83
|
84
|
204
|
309
|
219
|
329
|
423
|
638
| |
Taux 1,2 (ET) de déclin sur 52 semaines
|
-190 (36)
|
-60
(39)
|
−239,9 (18,71)
|
−114,7 (15,33)
|
−207,3 (19,31)
|
−113,6 (15,73)
|
−223,5 (13,45)
|
−113,6
(10,98)
| |
Comparaison vs placebo
| |
Différence1
|
|
131
|
|
125,3
|
|
93,7
|
|
109,9
| |
IC à 95 %
|
|
(27;
235)
|
|
(77,7;
172,8)
|
|
(44,8;
142,7)
|
|
(75,9; 144,0)
| |
Valeur p
|
|
0,01363
|
|
<0,0001
|
|
0,0002
|
|
<0,0001
| |
1
Calcul sur la base d'un modèle de régression à coefficient aléatoire.
2 Population randomisée pour l'étude TOMORROW; population traitée pour l'étude INPULSIS-1, l'étude INPULSIS-2 et les données réunies des études INPULSIS
3 Valeur p nominale.
|
Toutes les analyses de sensibilité prédéfinies ont confirmé la robustesse de l'effet du nintédanib en termes de réduction du taux annuel de déclin de la CVF. À part cela, des effets comparables sur d'autres critères de la fonction pulmonaire – par exemple la variation de la CVF à 52 semaines versus valeur initiale et l'analyse des répondeurs en termes de CVF – ont été observés, confirmant les effets ralentissants du nintédanib sur la progression de la maladie. La figure 1 présente l'évolution de la CVF en fonction du temps par rapport aux valeurs initiales pour les deux groupes de traitement (sur la base de l'analyse cumulée des études INPULSIS-1 et INPULSIS-2).
Figure 1: Variation moyenne observée (ETM) de la CVF versus valeur initiale (ml) en fonction du temps – études INPULSIS-1 et INPULSIS-2 (données réunies)
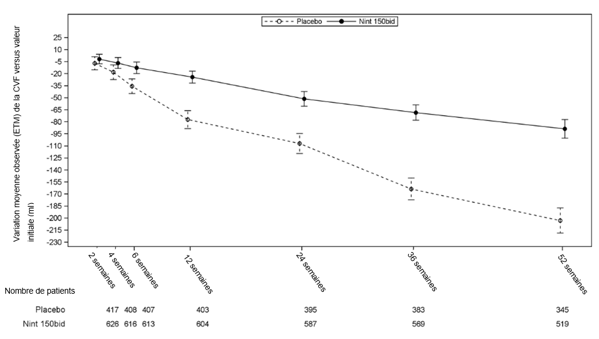
bid = deux fois par jour, ETM = erreur type de la moyenne
Variation du score SGRQ total à 52 semaines versus valeur initiale
Le score total au SGRQ (St. George's Respiratory Questionnaire) est une mesure de la qualité de vie en fonction de la santé (HRQoL). Il a été analysé à 52 semaines. Dans l'étude TOMORROW, la variation moyenne estimée du score SGRQ total entre le début de l'étude (valeur initiale) et la semaine 52 était de 5,46 sous placebo, indiquant une détérioration de la qualité de vie liée à la santé, tandis qu'elle était de -0,66 sous nintédanib, indiquant une stabilité de la qualité de vie liée à la santé. La différence moyenne estimée entre le nintédanib et le placebo était de -6,12 (IC à 95 %: -10,57; -1,67; p = 0,0071).
Dans l'étude INPULSIS-2, l'augmentation du score SGRQ total par rapport à la valeur initiale était plus prononcée chez les patients sous placebo que chez ceux traités par le nintédanib 150 mg deux fois par jour. La détérioration de la HRQoL était plus légère dans le groupe sous nintédanib. La différence entre les deux groupes de traitement était statistiquement significative (-2,69; IC à 95 %: -4,95; -0,43; p = 0,0197).
Dans l'étude INPULSIS-1, l'augmentation du score SGRQ total à 52 semaines versus valeur initiale était similaire sous nintédanib et sous placebo (différence entre les groupes de traitement: -0,05; IC à 95 %: -2,50; 2,40; p = 0,9657). Dans l'analyse cumulée des études INPULSIS, la variation moyenne estimée du score SGRQ total à 52 semaines versus valeur initiale était plus faible sous nintédanib (3,53) que sous placebo (4,96). La différence entre les traitements était de -1,43 (IC à 95 %: -3,09; 0,23; p = 0,0923). L'influence du nintédanib sur la qualité de vie liée à la santé, évaluée à l'aide du score SGRQ total, est globalement modérée et correspond à une plus faible détérioration que sous placebo.
Temps écoulé jusqu'à la première exacerbation aiguë de la FPI
Dans les études TOMORROW et INPULSIS-2, le risque de subir une première exacerbation aiguë de la FPI au cours de la période d'observation de 52 semaines était significativement plus faible sous nintédanib que sous placebo (hazard ratio [HR]: 0,16; IC à 95 %: 0,04; 0,71; p = 0,0054) et (HR: 0,38; IC 95 %: 0,19; 0,77; p = 0,005). Dans l'étude INPULSIS-1, aucune différence n'a été constatée entre les groupes de traitement (HR: 1,15; IC à 95 %: 0,54; 2,42; p = 0,6728).
Analyse de survie
Dans l'analyse cumulée prédéfinie des données de survie issues des études INPULSIS, le taux de mortalité globale au cours des 52 semaines était numériquement plus faible sous nintédanib (5,5 %) que sous placebo (7,8 %). L'analyse du temps écoulé jusqu'au décès a révélé un HR de 0,70 (IC à 95 % 0,43; 1,12; p = 0,1399). Les résultats de tous les critères d'évaluation concernant la survie (tels que la mortalité pendant le traitement et la mortalité de cause respiratoire) ont montré systématiquement une différence numérique en faveur du nintédanib, mais sans atteindre le seuil de signification statistique.
Traitement au long cours par Ofev chez les patients atteints de FPI (INPULSIS-ON)
Une étude d'extension en ouvert portant sur Ofev a inclus 734 patients atteints de FPI. Certains patients ont été traités par Ofev pendant plus de 5 ans. Les patients qui avaient achevé la phase de traitement d'une durée de 52 semaines dans l'étude INPULSIS, ont été traités en ouvert par Ofev dans le cadre de l'étude d'extension INPULSIS-ON. La durée d'exposition médiane pour les patients traités par Ofev tant dans l'étude INPULSIS que dans l'étude INPULSIS-ON s'élevait à 44,7 mois (intervalle: 11,9-68,3). Le taux annuel ajusté du déclin de la CVF sur 192 semaines était de −135,1 (5,8) ml/an chez tous les patients traités et était cohérent avec le taux annuel de déclin de la CVF chez les patients qui avaient été traités par Ofev dans les études INPULSIS de phase III (−113,6 ml par an). Le profil d'effets indésirables d'Ofev dans l'étude INPULSIS-ON était semblable à celui observé dans les études INPULSIS de phase III.
Patients atteints de FPI présentant une altération avancée de la fonction pulmonaire (INSTAGE)
Une étude en groupes parallèles, randomisée et menée en double aveugle, portant sur l'évaluation de l'efficacité et la sécurité d'Ofev en association avec le sildénafil administré par voie orale par comparaison avec le traitement par Ofev seul chez 273 patients atteints de FPI et présentant une altération avancée de la fonction pulmonaire (DLCO < 35% de la valeur prédite) pendant 24 semaines.
Le déclin de la CVF chez les patients traités par Ofev seul coïncidait avec le déclin de la CVF chez les patients présentant des affections moins avancées qui avaient été traités par Ofev dans les études INPULSIS de phase III. L'ajout de sildénafil à Ofev n'a pas apporté de bénéfice significatif en ce qui concerne la qualité de vie par comparaison avec Ofev administré seul. Le profil de sécurité et de tolérance d'Ofev chez les patients atteints de FPI présentant une altération avancée de la fonction pulmonaire coïncidait avec les profils observés dans les études INPULSIS. Le profil d'événements indésirables de l'association d'Ofev et de sildénafil correspondait au profil de sécurité connu des différents composants, sans augmentation des événements graves ou mortels par comparaison avec Ofev administré seul.
Données complémentaires issues de l'étude de phase IV INJOURNEY associant Ofev 150 mg deux fois par jour et la pirfénidone:
Au cours d'un essai exploratoire, en ouvert, randomisé, l'administration concomitante de 150 mg de nintédanib deux fois par jour et de pirfénidone (augmentation progressive de la dose jusqu'à 801 mg trois fois par jour) a été comparée à l'administration de nintédanib 150 mg deux fois par jour seul chez un total de 105 patients pendant 12 semaines. Le critère d'évaluation principal était le pourcentage de patients présentant des évènements indésirables gastro-intestinaux entre le début de l'essai et la semaine 12.
Les évènements gastro-intestinaux étaient fréquents et correspondaient au profil de sécurité connu de chaque composant. Les diarrhées, les nausées et les vomissements étaient les évènements indésirables les plus fréquemment rapportés, soit respectivement 20 patients (37,7 %) contre 16 patients (31,4 %), 22 patients (41,5 %) contre 6 patients (11,8 %) et 15 patients (28,3 %) contre 6 patients (11,8 %) traités par l'ajout de pirfénidone au nintédanib comparativement au traitement par nintédanib seul.
Les variations moyennes absolues (ET) de la CVF à la semaine 12 par rapport aux valeurs initiales étaient de −13,3 (17,4) ml chez les patients traités par nintédanib avec ajout de pirfénidone (n = 48), comparé à −40,9 (31,4) ml chez les patients traités par nintédanib seul (n = 44).
Pneumopathies interstitielles diffuses (PID) fibrosantes chroniques avec un phénotype progressif
L'efficacité clinique d'Ofev a été évaluée dans une étude de phase III randomisée, menée en double aveugle et contrôlée contre placebo (INBUILD) chez des patients atteints de PID fibrosante chronique avec un phénotype progressif.
Au total, 663 patients ont été randomisés selon un rapport 1:1 pour recevoir soit 2x150 mg d'Ofev par jour, soit le placebo pendant au moins 52 semaines.
La randomisation était stratifiée sur l'aspect de la fibrose à la tomodensitométrie à haute résolution (TDM-HR). 412 patients avec un aspect de fibrose de type pneumopathie interstitielle commune (PIC) et 251 patients avec d'autres aspects de fibrose ont été randomisés. Deux populations co-primaires ont été définies pour l'évaluation de cette étude: la population globale et la population avec un aspect de fibrose de type PIC à la TDM-HR.
Le critère d'évaluation principal était le taux annuel de déclin de la CVF (en ml) sur 52 semaines. D'autres critères d'évaluation étaient, par exemple, le délai de survenue de la première exacerbation aiguë de la PID ou du décès ou le délai de survenue du décès.
Les patients ayant un diagnostic clinique de PID fibrosante chronique étaient éligibles à l'étude s'ils présentaient une fibrose significative (>10% de lésions fibrotiques) à la TDM-HR ainsi que des signes cliniques de progression (définie comme un déclin de la CVF ≥10%, un déclin de la CVF entre 5% et 10% avec aggravation des symptômes OU aggravation à l'imagerie, ou une aggravation des symptômes ET une aggravation à l'imagerie) au cours des 24 mois précédents malgré un traitement adéquat. Les patients devaient présenter une CVF ≥45% de la valeur prédite et une DLCO comprise entre 30% et 80% de la valeur prédite.
Étaient exclus de la participation à l'étude les patients avec
·FPI, bronchoconstriction pertinente (p.ex. VEMS/CVF <0,7 avant bronchodilatation) ou hypertension pulmonaire significative
·élévation des transaminases ou de la bilirubine >1,5 LSN
·risque d'hémorragie connu, anticoagulation complète, infarctus du myocarde récent ou accident vasculaire cérébral récent
·traitement antérieur par nintédanib, pirfénidone, azathioprine, ciclosporine, mycophénolate, tacrolimus, corticoïdes oraux >20 mg/jour ou corticoïdes oraux+azathioprine-N-acétylcystéine dans les 4 semaines avant la randomisation*
·traitement antérieur par cyclophosphamide dans les 8 semaines avant la randomisation*
·traitement antérieur par rituximab dans les 6 mois avant la randomisation*
* L'utilisation des médicaments mentionnés était à nouveau autorisée au bout de 6 mois après le début de l'étude, pour autant que ceci soit cliniquement indiqué.
Les participants à l'étude étaient majoritairement caucasiens (74%) ou asiatiques (25%). 54% des patients étaient de sexe masculin, 49% n'avaient jamais fumé, l'âge moyen était de 66 ans et la CVF s'élevait en moyenne à 69% de la valeur prédite. Les PID sous-jacentes étaient des pneumopathies d'hypersensibilité (26%), des PID auto-immunes (26%), des pneumopathies interstitielles non spécifiques idiopathiques (19%), des pneumopathies interstitielles idiopathiques inclassifiables (17%) et d'autres PID (12%).
Taux annuel de déclin de la CVF (ml)
Une réduction statistiquement significative du taux de déclin de la CVF sur 52 semaines a été observée sous traitement par Ofev contre placebo. La différence constatée entre Ofev et placebo était de 107 ml (Tableau 2).
Tableau 2. Taux annuel de déclin de la CVF (ml, critère d'évaluation principal de l'étude INBUILD)
|
|
Population totale
|
Sous-population
de type PIC
|
Sous-population
Autres aspects de fibrose à la TDM-HR
| |
|
Ofev
|
Placebo
|
Ofev
|
Placebo
|
Ofev
|
Placebo
| |
Nombre de patients évalués
|
332
|
331
|
206
|
206
|
126
|
125
| |
Tauxa (ES) de déclin sur 52 semaines
|
-80,8
|
-187,8
|
-82,9
|
-211,1
|
-79,0
|
-154,2
| |
Comparaison contre placebo
Différencea
|
107,0
|
128,2
|
75,2*
| |
IC à 95%
|
(65,4, 148,5)
|
(70,8, 185,6)
|
(15,5, 135,0)*
| |
Valeur p
|
< 0,0001
|
< 0,0001
|
| |
* Une comparaison reposant sur la sous-population avec d'autres aspects de fibrose à la TDM-HR n'a pas été reprise dans la procédure de tests multiples. Les valeurs présentées ici servent à des fins descriptives.
a Basé sur un modèle de régression avec coefficients aléatoires et les effets de catégories fixes du traitement, l'aspect TDM-HR, les effets continus fixes du temps, la valeur initiale de la CVF (ml), y compris l'interaction entre le traitement et le temps et l'interaction entre la valeur initiale et le temps.
|
La Figure 2 montre l'évolution de la variation de la CVF dans le temps par rapport à la valeur initiale, dans les groupes de traitement.
Figure 2: Variation moyenne observée (ETM) de la CVF par rapport à la valeur initiale (ml) sur 52 semaines
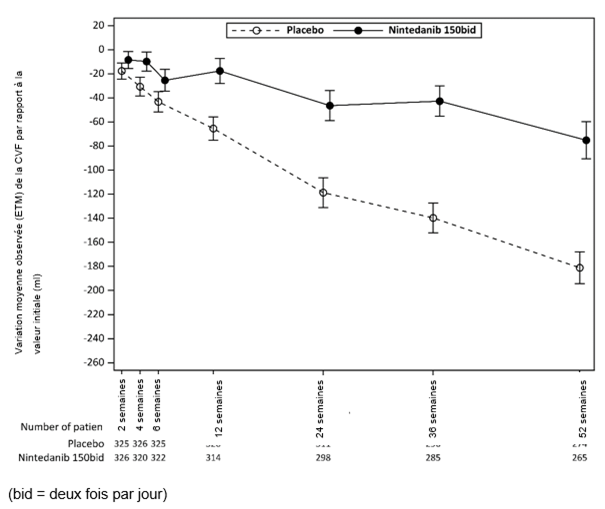
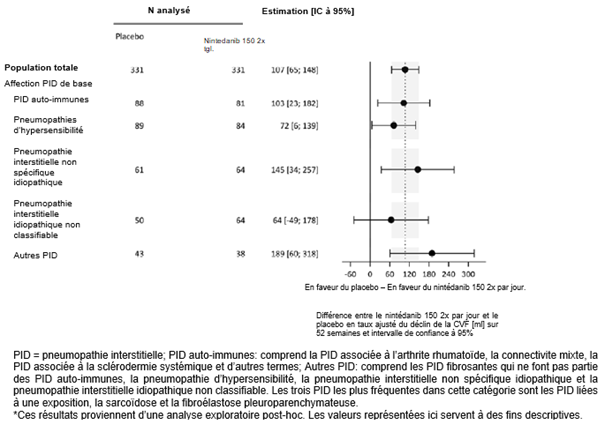
Une analyse exploratoire post-hoc réalisée selon le diagnostic de PID a été effectuée (Figure 3). Dans l'ensemble, un effet thérapeutique cohérent a été mis en évidence au sein des différents diagnostics de PID.
Figure 3: Taux annuel de déclin de la CVF (ml) sur 52 semaines reposant sur le diagnostic de base de PID dans l'étude 5*
Déclin de la CVF en pourcentage
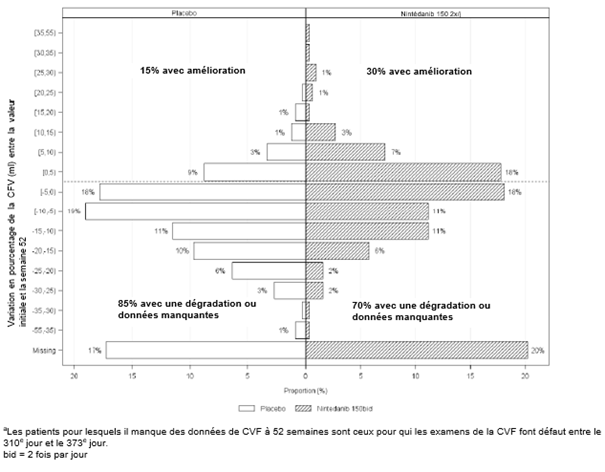
La Figure 4 représente la variation en pourcentage de la CVF (ml) de la valeur initiale à la semaine 52. La dégradation de la fonction pulmonaire était moins importante chez la majorité des patients sous Ofev que chez les patients sous placebo.
Figure 4: Histogramme de la variation en pourcentage de la CVF (ml) de la valeur initiale à la semaine 52 après traitement avec incréments (%) ou décréments par paliers de 5 (étude 5)a
Délai de survenue de la première exacerbation aiguë de la PID ou du décès
Le risque d'une première exacerbation aiguë de la PID ou de décès a diminué dans le groupe Ofev par rapport au groupe placebo sur 52 semaines (HR Ofev contre placebo: 0,80; IC à 95%: 0,48; 1,34) ou sur toute la durée de l'étude (HR Ofev contre placebo: 0,67; IC à 95%: 0,46; 0,98), ce qui correspond à une réduction de 33% du risque d'une première exacerbation aiguë de la PID ou de décès chez les patients sous Ofev par comparaison avec le placebo.
Analyse de la survie
Dans l'étude INBUILD, les données de survie sous Ofev par comparaison avec le placebo ont été évaluées afin d'étayer le critère d'évaluation principal (CFV). La mortalité toutes causes confondues a été évaluée sur toute la durée de l'étude et sur la période de suivi disponible, indépendamment de la cause du décès et de la poursuite ou non du traitement. L'analyse n'a pas montré de différence statistiquement significative sur 52 semaines (HR Ofev contre placebo 0,94, IC à 95% 0,47, 1,86), et sur toute la durée de l'étude (HR Ofev contre placebo 0,78, IC 95% 0,5, 1,21).
Pneumopathie interstitielle associée à la sclérodermie systémique (PID-ScS)
L'efficacité clinique du nintédanib a été évaluée dans une étude de phase III randomisée, en double aveugle et contrôlée versus placebo (SENSCIS) chez des patients atteints de PID-ScS.
En tout, 580 patients avec une proportion de 1:1 ont été randomisés pour recevoir soit 150 mg d'Ofev deux fois par jour ou le placebo pendant au moins 52 semaines. La randomisation a été stratifiée selon le statut des anticorps anti-topoisomérase (ATA). Certains patients ont poursuivi le traitement en aveugle durant jusqu'à 100 semaines.
Le diagnostic des patients atteints de PID-ScS reposait sur les critères de classification pour la sclérodermie systémique (ScS) de l'American College of Rheumatology / de la European League Against Rheumatism lorsque la survenue de la maladie (premier symptôme non Raynaud) remontait à moins de 7 ans et en présence d'une fibrose pulmonaire d'au moins 10%, constatée sur la base d'une image obtenue par tomodensitométrie haute résolution effectuée au cours des 12 mois précédents.
Chez les patients, la CVF devait atteindre au moins 40 % de la valeur estimée, tandis que la DLCO devait être entre 30 et 89 % de la valeur estimée. Les patients atteints d'importantes obstructions des voies respiratoires (c'est-à-dire VEMS/CVF avant la bronchodilatation inférieur à 0,7) ou ayant reçu ou allant recevoir une transplantation de cellules souches hématopoïétiques ne pouvaient pas participer à l'étude. D'autres critères d'exclusion étaient des valeurs ALAT, ASAT ou de bilirubine élevées (>1,5xLNS), un risque accru d'hémorragies (y compris avec anticoagulation thérapeutique), un infarctus du myocarde ou un accident vasculaire cérébral récent, une hypertension pulmonaire considérable, des ulcérations à au moins trois bouts de doigt, un antécédent de nécrose sévère du doigt avec hospitalisation nécessaire ainsi qu'un antécédent de crise rénale sclérodermique. Les patients qui recevaient d'autres médicaments à l'étude, qui avaient déjà été traités par nintédanib ou pirfénidone et avaient reçu de l'azathioprine 8 semaines avant la randomisation ou du cyclophosphamide ou de la cyclosporine A 6 mois avant la randomisation ont également été exclus. Un traitement stable par mycophénolate ou méthotrexate ainsi que par prednisone ≤10 mg/jour ou une dose équivalente était autorisé.
Le critère d'évaluation primaire était le déclin annuel de la capacité vitale forcée (CVF) sur 52 semaines.
Les critères secondaires importants étaient la variation absolue du Rodnan Skin Score modifié (mRSS) à la semaine 52 par rapport à la valeur initiale, la variation absolue du score total au Saint George's Respiratory Questionnaire (SGRQ) à la semaine 52 par rapport à la valeur initiale ainsi que la mortalité pendant toute l'étude.
75,2 % des patients étaient de sexe féminin. Les patients étaient majoritairement de type caucasien (67 %), asiatique (25 %) ou avaient la peau noire (6 %). L'âge moyen (écart type [ET], min.-max.) s'élevait à 54,0 (12,2, 20-79) ans. En tout, 51,9 % des patients souffraient d'une sclérose systémique cutanée diffuse (ScS) et 48,1 % d'une ScS cutanée limitée. La durée moyenne (ET) depuis la première apparition d'un symptôme non Raynaud était de 3,49 (1,7) ans. 49,0 % des patients ont reçu un traitement stable par mycophénolate (46,5% mycophénolate mofétil, 1,9% mycophénolate de sodium, 0,5% acide mycophénolique) et la profil de sécurité observé chez les patients sous mycophénolate ou non était similaire.
Déclin annuel de la CVF
Le déclin annuel de la CVF (en ml) sur 52 semaines a nettement diminué de 41,0 ml chez les patients sous Ofev par rapport au placebo (voir Tableau 3), ce qui correspond à un effet relatif du traitement de 43,8 %.
Tableau 3: Déclin annuel de la CVF (ml) sur 52 semaines
|
|
Placebo
|
Ofev
150 mg deux fois par jour
| |
Nombre de patients évalués
|
288
|
287
| |
Taux1 (ET) du déclin sur 52 semaines
|
-93,3 (13,5)
|
-52,4 (13,8)
| |
Comparaison versus placebo
| |
Différence1
|
|
41,0
| |
IC à 95 %
|
|
(2,9, 79,0)
| |
Valeur p
|
|
<0,05
| |
1
Sur la base d'une régression avec des coefficients aléatoires, ajustés pour le traitement, le sexe, la taille, l'âge, le statut ATA, la valeur initiale de CVF, CVF*la durée et traitement*la durée
|
Une analyse exploratoire des données jusqu'à 100 semaines (durée de traitement max. dans l'étude SENSCIS) a montré que l'efficacité, avec un traitement par Ofev, sur le ralentissement de la progression de la PID-ScS durait plus de 52 semaines.
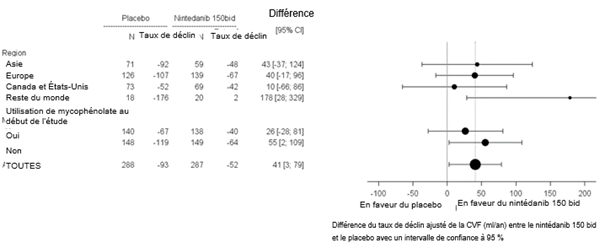
Dans deux analyses d'efficacité de sous-groupes prédéfinis, la différence moyenne de traitement relative au déclin de la CVF à la semaine 52 chez les patients selon la région et l'utilisation du mycophénolate a été étudiée (voir Figure 5).
Figure 5: Analyse de sous-groupes de la différence moyenne de traitement relative au déclin de la CVF (ml) à la semaine 52 selon la région et l'utilisation du mycophénolate (SENSCIS)
Variation en pourcentage de la capacité vitale forcée à partir du début de l'étude
La Figure 6 représente pour l'étude SENSCIS la variation en pourcentage de la CVF en ml à la semaine 52 par rapport à la valeur initiale. La dégradation de la fonction pulmonaire était moins importante chez la majorité des patients sous Ofev que chez les patients sous placebo.
Figure 6: Histogramme de la variation en pourcentage de la CVF (ml) à la semaine 52 par rapport à la valeur initiale selon le traitement et augmentation ou diminution en pourcentage de 5 (SENSCIS)a
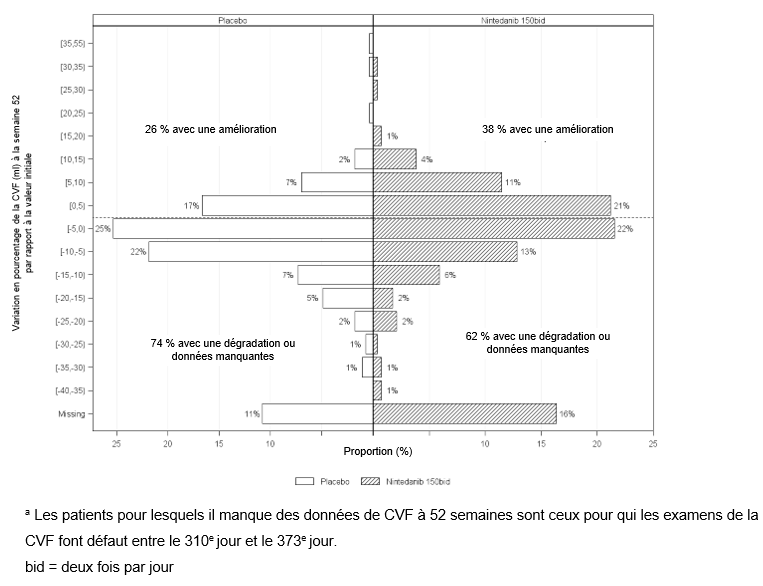
Figure 7: Variation moyenne observée de la CVF par rapport à la valeur initiale (ml) sur 52 semaines
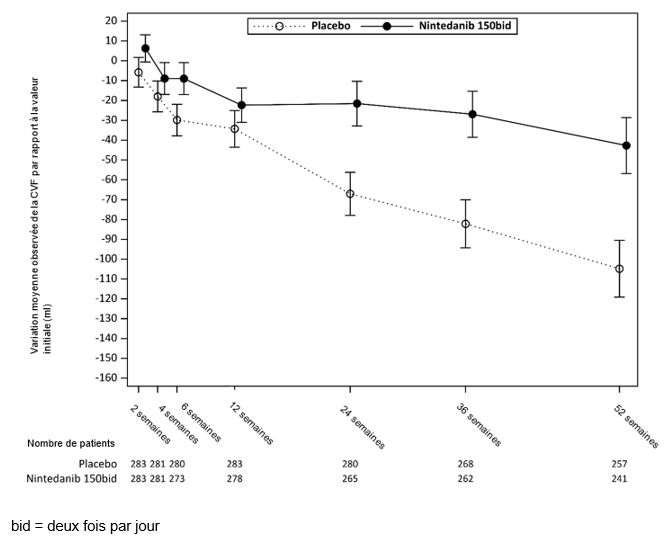
Tableau 4 Déclin annuel de la CVF (% de la valeur théorique) sur 52 semaines
|
|
Placebo
|
Ofev
150 mg deux fois par jour
| |
Nombre de patients évalués
|
288
|
287
| |
Taux1 (ET) de déclin sur 52 semaines
|
-2,6 (0,4)
|
-1,4 (0,4)
| |
Compraison versus placebo
| |
Différence1
|
|
1,15
| |
IC à 95%
|
|
(0,09, 2,21)
| |
Valeur p
|
|
<0,05
| |
1
Sur la base d'une régression avec des coefficients aléatoires et les effets de catégories fixes du traitement, le statut ATA, les effets continus fixes du temps, la valeur initiale de la CVF [% de la valeur théorique] y compris l'interaction entre le traitement et le temps et l'interaction entre la valeur initiale et le temps. L'intercept et le temps étaient donnés de manière spécifique au patient en tant qu'effet aléatoire. La variance d'erreur de mesure (within-patient error) inhérente à un patient a été modélisée au moyen d'une matrice de variance-covariance non structurée. La variabilité inter-individuelle a été modélisée au moyen d'une matrice de variance-covariance des composantes de variance.
|
Variation du score de Rodnan Skin modifié (mRSS) à 52 semaines par rapport à la valeur initiale
La variation absolue moyenne ajustée du mRSS à 52 semaines par rapport à la valeur initiale était similaire entre le groupe sous Ofev (-2,17 (IC à 95% -2,69, -1,65)) et le groupe sous placebo (-1,96 (IC à 95% -2,48, -1,45)). La différence moyenne ajustée entre les groupes de traitement s'élevait à -0,21 (IC à 95% -0,94, 0,53; p = 0,5785).
Variation du score total au Saint George's Respiratory Questionnaire (SGRQ) à 52 semaines par rapport à la valeur initiale
La variation absolue moyenne ajustée du score total de SGRD à 52 semaines par rapport à la valeur initiale était similaire entre le groupe sous Ofev (0,81 (IC à 95% -0,92, 2,55)) et le groupe sous placebo (-0,88 (IC à 95% -2,58, 0,82)). La différence moyenne ajustée entre les groupes de traitement s'élevait à 1,69 (IC à 95% -0,73, 4,12; p = 0,1711).
Analyse de survie
Sur toute l'étude, la mortalité entre le groupe Ofev (N = 10; 3,5 %) et le groupe sous placebo (N = 9; 3,1 %) était similaire. Sur toute l'étude, l'analyse du temps jusqu'au décès a donné un HR de 1,16 (IC à 95% 0,47, 2,84; p = 0,7535).
Effet sur l'intervalle QT
Dans une étude spécifique auprès de patients atteints d'un cancer du rein à cellules claires, des mesures du QT/QTc ont révélé que ni l'administration d'une dose orale unique de 200 mg de nintédanib, ni l'administration de doses orales répétées de 200 mg de nintédanib deux fois par jour pendant 15 jours n'a prolongé l'intervalle QTcF. Aucune étude approfondie complète de l'intervalle QT n'a été effectuée sur le nintédanib.
Études auprès de la population pédiatrique
Le nintédanib ne doit pas être utilisé chez la population pédiatriques atteinte de pneumopathies interstitielles diffuses (PID) fibrosantes chroniques.
PharmacocinétiqueLes propriétés pharmacocinétiques du nintédanib étaient similaires chez des volontaires sains, des patients atteints de FPI, de PID-ScS, de PID et des patients atteints de cancer.
La pharmacocinétique du nintédanib est linéaire par rapport au temps. Une proportionnalité entre la dose et l'augmentation de l'exposition au nintédanib a été démontrée à l'aide de doses croissantes (allant de 50 à 450 mg une fois par jour et de 150 à 300 mg deux fois par jour). Après l'administration de doses multiples chez des patients atteints de FPI, l'accumulation se traduisait par une AUCτ augmentée d'un facteur 1,76. Les concentrations plasmatiques à l'état d'équilibre étaient atteintes après une semaine d'administration. Les concentrations minimales de nintédanib sont restées stables pendant plus d'un an. La pharmacocinétique du nintédanib présente une variabilité interindividuelle modérée à élevée (coefficient de variation de 30 % à 70 % pour les paramètres pharmacocinétiques standard). La variabilité intra-individuelle est faible à modérée (coefficient de variation inférieur à 40 %).
Absorption
Après l'administration orale de nintédanib en capsules molles de gélatine avec un repas, les concentrations plasmatiques maximales de nintédanib ont été atteintes en l'espace d'environ 2 à 4 heures (entre 0,5 et 8 heures). La biodisponibilité absolue d'une dose de 100 mg chez des volontaires sains était de 4,69 % (IC à 90 %: 3,62-6,08). L'absorption et la biodisponibilité sont réduites par l'influence de transporteurs (P-gp), par un métabolisme substantiel de premier passage et éventuellement par la faible solubilité du nintédanib dans un milieu de pH neutre.
Après l'ingestion de nourriture, l'exposition au nintédanib était accrue d'environ 20 % par rapport à une prise à jeun (IC: 95,3 %;152,5 %) et l'absorption était ralentie (tmax médian: 2,00 heures à jeun, 3,98 h après une prise de nourriture).
L'étude in vitro n'a mis en évidence aucune influence significative sur l'intégrité de la capsule, sur la libération du principe actif, sur les potentiels produits de dégradation et sur la quantité de principe actif contenue dans la capsule lors de l'exposition de la capsule (tous dosages confondus) à de la compote de pommes ou du pudding au chocolat pendant 15 minutes maximum; aucune influence sur l'efficacité clinique n'est donc à craindre.
Distribution
Le nintédanib présente une cinétique d'élimination au moins biphasique. Un volume de distribution élevé (Vss: 1050 l) a été observé après une administration par perfusion intraveineuse.
La liaison du nintédanib aux protéines plasmatiques humaines était élevée in vitro (97,8 %). On suppose que l'albumine sérique est la principale protéine de liaison. Le nintédanib est principalement distribué dans le plasma, avec un rapport sang-plasma de 0,869.
Métabolisme
La métabolisation du nintédanib s'effectue essentiellement par une hydrolyse due à des estérases. Il en résulte la formation du métabolite principal, le groupement acide libre BIBF 1202. Des enzymes (UGT 1A1, UGT 1A7, UGT 1A8 et UGT 1A10) transforment ensuite le BIBF 1202 en glucuronide BIBF 1202.
La biodégradation du nintédanib ne s'effectue qu'en faible mesure par voie des enzymes du CYP (surtout CYP 3A4). Dans l'étude ADME, la plus grande partie de la radioactivité retrouvée dans le plasma humain était répartie entre le nintédanib (24 %), le BIBF 1202 (32 %) et le glucuronide BIBF 1202 (30 %), tandis que les principaux métabolites CYP-dépendants étaient indétectables dans le plasma humain. In vitro, le métabolisme CYP-dépendant correspondait à environ 5 %, par rapport à environ 25 % pour l'hydrolyse de l'ester dans ces conditions expérimentales.
Élimination
La clairance plasmatique totale après une perfusion intraveineuse était élevée (CL: 1390 ml/min). L'élimination de la substance active inchangée dans les urines en l'espace de 48 h était d'environ 0,05 % après l'administration d'une dose orale et d'environ 1,4 % après l'administration d'une dose intraveineuse. La clairance rénale était de 20 ml/min.
Excrétion
Après administration orale de nintédanib radiomarqué au [14C], la voie principale d'élimination de la radioactivité liée au médicament était l'excrétion fécale/biliaire (93,4 % de la dose). La plus grande partie de la dose d'Ofev (58 % de la dose) était éliminée dans les selles sous forme de BIBF 1202; 20 % étaient éliminés en tant que substance inchangée et moins de 7 % sous forme de divers métabolites de moindre importance. La contribution de l'excrétion rénale à la clairance totale était faible (0,649 % de la dose). La dose récupérée était considérée comme totale (plus de 90 %) dans les 4 jours suivant l'administration. La demi-vie terminale du nintédanib était de 10 à 15 h.
Rapport exposition/réponse
Des analyses exploratoires pour évaluer la relation entre la pharmacocinétique et les effets indésirables, effectuées à partir des données de l'étude de phase II sur la FPI, ont montré qu'une exposition supérieure au nintédanib était en tendance associée à une augmentation des taux d'enzymes hépatiques (voir «Mises en garde et précautions»).
Cinétique pour certains groupes de patients, facteurs intrinsèques et extrinsèques
D'après les résultats d'une analyse pharmacocinétique de population portant sur des patients atteints de FPI et de cancer bronchique non à petites cellules (CBNPC) (N = 1191) et d'après des études descriptives, l'exposition au nintédanib n'était pas influencée par le sexe (après correction en fonction du poids corporel), l'insuffisance rénale légère à modérée (estimée sur la base de la clairance de la créatinine), la consommation d'alcool ou le génotype de la P-gp. L'analyse pharmacocinétique de population a révélé que l'exposition au nintédanib était modérément influencée par l'âge, le poids corporel et l'origine ethnique. Ces facteurs sont présentés plus en détail ci-dessous. Dans le contexte de la forte variabilité inter-individuelle observée pour l'exposition, les effets modérés de ces co-variables ne permettent pas de donner des recommandations d'ajustement posologique (voir «Mises en garde et précautions»).
Âge
L'exposition au nintédanib augmente de façon linéaire avec l'âge. L'AUCτ,ss d'un patient de 45 ans (5e percentile) était inférieure de 16 % et celle d'un un patient de 76 ans (95e percentile) était supérieure de 13 % à celle d'un patient âgé de 62 ans (âge médian). La tranche d'âge couverte par l'analyse allait de 29 à 85 ans et environ 5 % de la population avaient plus de 75 ans.
Aucune étude n'a été effectuée auprès d'enfants et d'adolescents.
Poids corporel
Une corrélation inverse a été observée entre le poids corporel et l'exposition au nintédanib. L'AUCτ,ss d'un patient de 50 kg (5e percentile) était supérieure de 25 % et celle d'un un patient de 100 kg (95e percentile) était inférieure de 19 % à celle d'un patient pesant 71,5 kg (poids corporel médian).
Origine ethnique
La moyenne de la population d'exposition au nintédanib était supérieure de 33 à 50 % chez les Chinois, Taïwanais et Indiens et supérieure de 16 % chez les patients japonais, et plus faible de 16 à 22 % chez les Coréens, comparé aux patients caucasoïdes (après correction en fonction du poids corporel). Les données obtenues chez les patients noirs sont très limitées mais du même ordre que celles observées chez les patients caucasoïdes.
Fumeurs
Dans l'analyse pharmacocinétique de population, l'exposition au nintédanib était plus faible de 21 % chez les fumeurs actuels que chez les ex-fumeurs et les non-fumeurs de toujours. L'ampleur de cet effet ne justifie pas un ajustement posologique.
Insuffisance hépatique
Dans une étude dédiée de Phase I en dose unique, l'exposition systémique au nintédanib évaluée par la Cmax et l'ASC a été 2,2 fois plus élevée chez les sujets présentant une insuffisance hépatique légère (Child Pugh A; Cmax : IC à 90 %: 1,3 – 3,7et ASC: 1,2 – 3,8) que chez les volontaires sains. Chez les sujets présentant une insuffisance hépatique modérée (Child Pugh B), l'exposition systémique a été augmentée de 7,6 fois selon la Cmax (IC à 90 %: 4,4 – 13,2) et 8,7 fois selon l'ASC (IC à 90 %: 5,7 – 13,1) comparativement aux volontaires sains. Les sujets présentant une insuffisance hépatique sévère (Child Pugh C) n'ont pas été étudiés.
Insuffisance rénale
La pharmacocinétique du nintédanib n'a pas été examinée dans une étude PC spécifique chez les patients présentant une insuffisance rénale. D'après les résultats de l'analyse pharmacocinétique de population, une insuffisance rénale légère (ClCr de 60 à 90 ml/min) ou modérée (ClCr de 30 à 60 ml/min) n'a pas influencé l'exposition au nintédanib. Les données disponibles sur les patients présentant une insuffisance rénale sévère (ClCr inférieure à 30 ml/min) sont limitées.
Données précliniquesToxicologie générale
Les études de toxicité après administration en dose unique chez le rat et la souris indiquent un faible potentiel de toxicité aiguë du nintédanib. Dans des études sur la toxicité en administration répétée chez le rat, les effets indésirables (tels que épaississement des plaques épiphysaires, lésions des incisives) étaient principalement liés au mécanisme d'action du nintédanib (inhibition du VEGFR-2). Des anomalies de ce type sont connues pour d'autres inhibiteurs du VEGFR-2 et peuvent être considérées comme des effets de classe.
Des diarrhées et des vomissements, associés à une moindre consommation de nourriture et à une perte de poids, ont été observés dans des études de toxicité avec administration de doses multiples chez d'autres animaux que les rongeurs.
Aucun indice d'augmentation des taux d'enzymes hépatiques n'a été trouvé chez le rat, le chien ou le singe cynomolgus. Uniquement chez le singe Rhésus, de légères élévations des enzymes hépatiques non attribuables à des effets indésirables sérieux tels que la diarrhée ont été observées.
Mutagénicité/carcinogénicité
Les études de génotoxicité n'ont révélé aucun potentiel mutagène du nintédanib.
Les études de 2 ans sur la carcinogénicité chez la souris et le rat n'ont pas mis en évidence de potentiel carcinogène du nintédanib.
Toxicité de reproduction
Chez le rat, une létalité embryo-fœtale et des effets tératogènes ont été observés à des niveaux d'exposition environ 3,6 à 7,2 fois inférieurs à l'exposition humaine observée à la dose quotidienne maximale recommandée chez l'homme (DMRH), de 150 mg deux fois par jour. Des effets sur le développement du squelette axial et des grosses artères ont également été observés à des niveaux d'exposition environ 12 à 18 fois inférieurs à la DMRH.
Chez le lapin, une létalité embryo-fœtale et des effets tératogènes ont été observés à une exposition environ 3 fois supérieure à la DMRH, tandis qu'une plus faible exposition correspondant à la DMRH de 150 mg deux fois par jour était déjà associée à des effets plus équivoques sur le développement embryo-fœtal du squelette axial et du cœur.
Une étude sur la fertilité du rat mâle et le développement embryonnaire précoce jusqu'à l'implantation n'a révélé aucune influence sur le taux de reproduction des mâles ou sur leur fertilité à des expositions environ 3 fois supérieures à la DMRH.
Chez des rates, de faibles quantités de nintédanib et/ou de ses métabolites radiomarqués ont été excrétées dans le lait (≤0,5 % de la dose administrée).
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Tenir hors de portée des enfants.
Conserver à température ambiante (15-25°C). Conserver le récipient dans son emballage d'origine pour protéger son contenu de l'humidité.
Numéro d’autorisation65330 (Swissmedic)
PrésentationOfev 100 mg: boîtes de 60 capsules [B].
Ofev 150 mg: boîtes de 60 capsules [B].
Titulaire de l’autorisationBoehringer Ingelheim (Schweiz) GmbH, Bâle.
Mise à jour de l’informationMars 2025
|