Propriétés/EffetsCode ATC
L01EX09
Mécanisme d'action
Le nintédanib est un inhibiteur de tyrosines kinases qui appartient au type des petites molécules. Les tyrosine kinases inhibées comprennent les récepteurs du facteur de croissance dérivé des plaquettes (platelet-derived growth factor receptors, PDGFR) α et β, les récepteurs du facteur de croissance des fibroblastes (fibroblast growth factor receptors, FGFR) 1 à 3 et les récepteurs du facteur de croissance de l'endothélium vasculaire (vascular endothelial growth factor receptors, VEGFR) 1 à 3. Le nintédanib se fixe de manière compétitive au site de liaison de l'adénosine triphosphate (ATP) de ces récepteurs et bloque ainsi la signalisation intracellulaire qui est décisive pour la prolifération, la migration et la transformation des fibroblastes, et donc pour les mécanismes pathologiques de la FPI. À part cela, le nintédanib inhibe aussi les kinases Flt-3, Lck, Lyn et Src.
Pharmacodynamique
L'activation des cascades de signalisation induites par le FGFR et le PDGFR est un facteur décisif pour la prolifération et la migration des fibroblastes/myofibroblastes pulmonaires, qui sont les cellules caractéristiques de la pathologie de fibrose pulmonaire idiopathique. La signification potentielle de l'inhibition du VEGFR pour la pathologie de FPI n'a pas encore été totalement élucidée. On suppose que le nintédanib, en se liant au site de liaison d'adénosine triphosphate (ATP) du domaine intracellulaire des récepteurs à activité tyrosine kinase, inhibe à l'échelle moléculaire les cascades de signalisation du FGFR et du PDGFR impliquées dans la prolifération et la migration des fibroblastes pulmonaires, et interfère ainsi avec l'activation croisée (par autophosphorylation du récepteur homodimère).
In vitro, des concentrations faiblement nanomolaires de nintédanib suffisent pour inhiber les récepteurs cibles. Sur des fibroblastes pulmonaires de patients atteints de FPI, la prolifération stimulée par PDGF, FGF et VEGF a été inhibée par le nintédanib aux CE50 de 11 nmol/l, 5,5 nmol/l et moins de 1 nmol/l. À des concentrations de 100 à 1000 nmol/l, le nintédanib a également inhibé la migration de fibroblastes stimulée par PDGF, FGF et VEGF ainsi que la transformation des fibroblastes en myofibroblastes induite par TGF-β2. On suppose que l'activité anti-inflammatoire du nintédanib limite en outre la stimulation fibrotique en réduisant les médiateurs profibrotiques tels que l'IL-1β et l'IL-6. On ignore encore dans quelle mesure l'activité anti-angiogénique du nintédanib participe au mécanisme d'action du médicament dans le traitement des fibroses pulmonaires. Dans des études in vivo, le nintédanib a fait preuve d'une forte activité anti-fibrotique et anti-inflammatoire.
Efficacité clinique
Fibrose pulmonaire idiopathique (FPI)
L'efficacité clinique du nintédanib a été étudiée chez 1231 patients atteints de FPI dans une étude de phase II (TOMORROW) et deux études de phase III (INPULSIS-1 et INPULSIS-2). Il s'agissait d'études randomisées, en double aveugle, avec contrôle contre placebo, dans lesquelles un traitement de 52 semaines par Ofev (150 mg deux fois par jour) a été évalué versus placebo.
Les études INPULSIS-1 et INPULSIS-2 étaient de conception identique, proche de la conception de l'étude TOMORROW. Les patients ont été randomisés en proportions de 3:2 (1:1 dans l'étude TOMORROW) et assignés pour 52 semaines à un traitement par Ofev (150 mg deux fois par jour) ou par un placebo (également deux fois par jour). L'étude TOMORROW comprenait des groupes de traitement supplémentaires (50 mg par jour, 50 mg deux fois par jour et 100 mg deux fois par jour) qui ne doivent pas être discutés ici.
Le critère primaire était défini comme le déclin annuel de la capacité vitale forcée (CVF). La variation du score total au questionnaire SGRQ (Saint George's Respiratory Questionnaire) entre le début de l'étude et la semaine 52 ainsi que le temps écoulé jusqu'à la première exacerbation aiguë de la FPI étaient des critères secondaires importants des études INPULSIS et des critères secondaires de l'étude TOMORROW.
Déclin annuel de la CVF
Le déclin annuel de la CVF (en ml) était significativement plus faible chez les patients sous nintédanib que chez les patients sous placebo. L'effet du traitement était concordant entre les 3 études. Le tableau 1 présente les résultats des études individuelles et les résultats de l'analyse cumulée des études INPULSIS.
Tableau 1: Déclin annuel de la CVF (ml) dans les études TOMORROW, INPULSIS-1 et INPULSIS-2 ainsi que dans l'analyse cumulée des études INPULSIS – population traitée2
|
|
TOMORROW
|
INPULSIS-1
|
INPULSIS-2
|
INPULSIS-1 et INPULSIS-2
Données réunies
| |
|
Placebo
|
Ofev 150 mg deux fois par jour
|
Placebo
|
Ofev 150 mg deux fois par jour
|
Placebo
|
Ofev 150 mg deux fois par jour
|
Placebo
|
Ofev 150 mg deux fois par jour
| |
Nombre de patients inclus à l'analyse
|
83
|
84
|
204
|
309
|
219
|
329
|
423
|
638
| |
Taux 1,2 (ET) de déclin sur 52 semaines
|
-190 (36)
|
-60
(39)
|
−239,9 (18,71)
|
−114,7 (15,33)
|
−207,3 (19,31)
|
−113,6 (15,73)
|
−223,5 (13,45)
|
−113,6
(10,98)
| |
Comparaison vs placebo
| |
Différence1
|
|
131
|
|
125,3
|
|
93,7
|
|
109,9
| |
IC à 95 %
|
|
(27;
235)
|
|
(77,7;
172,8)
|
|
(44,8;
142,7)
|
|
(75,9; 144,0)
| |
Valeur p
|
|
0,01363
|
|
<0,0001
|
|
0,0002
|
|
<0,0001
| |
1
Calcul sur la base d'un modèle de régression à coefficient aléatoire.
2 Population randomisée pour l'étude TOMORROW; population traitée pour l'étude INPULSIS-1, l'étude INPULSIS-2 et les données réunies des études INPULSIS
3 Valeur p nominale.
|
Toutes les analyses de sensibilité prédéfinies ont confirmé la robustesse de l'effet du nintédanib en termes de réduction du taux annuel de déclin de la CVF. À part cela, des effets comparables sur d'autres critères de la fonction pulmonaire – par exemple la variation de la CVF à 52 semaines versus valeur initiale et l'analyse des répondeurs en termes de CVF – ont été observés, confirmant les effets ralentissants du nintédanib sur la progression de la maladie. La figure 1 présente l'évolution de la CVF en fonction du temps par rapport aux valeurs initiales pour les deux groupes de traitement (sur la base de l'analyse cumulée des études INPULSIS-1 et INPULSIS-2).
Figure 1: Variation moyenne observée (ETM) de la CVF versus valeur initiale (ml) en fonction du temps – études INPULSIS-1 et INPULSIS-2 (données réunies)
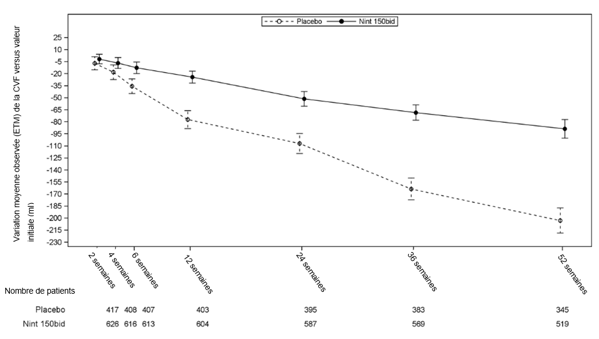
bid = deux fois par jour, ETM = erreur type de la moyenne
Variation du score SGRQ total à 52 semaines versus valeur initiale
Le score total au SGRQ (St. George's Respiratory Questionnaire) est une mesure de la qualité de vie en fonction de la santé (HRQoL). Il a été analysé à 52 semaines. Dans l'étude TOMORROW, la variation moyenne estimée du score SGRQ total entre le début de l'étude (valeur initiale) et la semaine 52 était de 5,46 sous placebo, indiquant une détérioration de la qualité de vie liée à la santé, tandis qu'elle était de -0,66 sous nintédanib, indiquant une stabilité de la qualité de vie liée à la santé. La différence moyenne estimée entre le nintédanib et le placebo était de -6,12 (IC à 95 %: -10,57; -1,67; p = 0,0071).
Dans l'étude INPULSIS-2, l'augmentation du score SGRQ total par rapport à la valeur initiale était plus prononcée chez les patients sous placebo que chez ceux traités par le nintédanib 150 mg deux fois par jour. La détérioration de la HRQoL était plus légère dans le groupe sous nintédanib. La différence entre les deux groupes de traitement était statistiquement significative (-2,69; IC à 95 %: -4,95; -0,43; p = 0,0197).
Dans l'étude INPULSIS-1, l'augmentation du score SGRQ total à 52 semaines versus valeur initiale était similaire sous nintédanib et sous placebo (différence entre les groupes de traitement: -0,05; IC à 95 %: -2,50; 2,40; p = 0,9657). Dans l'analyse cumulée des études INPULSIS, la variation moyenne estimée du score SGRQ total à 52 semaines versus valeur initiale était plus faible sous nintédanib (3,53) que sous placebo (4,96). La différence entre les traitements était de -1,43 (IC à 95 %: -3,09; 0,23; p = 0,0923). L'influence du nintédanib sur la qualité de vie liée à la santé, évaluée à l'aide du score SGRQ total, est globalement modérée et correspond à une plus faible détérioration que sous placebo.
Temps écoulé jusqu'à la première exacerbation aiguë de la FPI
Dans les études TOMORROW et INPULSIS-2, le risque de subir une première exacerbation aiguë de la FPI au cours de la période d'observation de 52 semaines était significativement plus faible sous nintédanib que sous placebo (hazard ratio [HR]: 0,16; IC à 95 %: 0,04; 0,71; p = 0,0054) et (HR: 0,38; IC 95 %: 0,19; 0,77; p = 0,005). Dans l'étude INPULSIS-1, aucune différence n'a été constatée entre les groupes de traitement (HR: 1,15; IC à 95 %: 0,54; 2,42; p = 0,6728).
Analyse de survie
Dans l'analyse cumulée prédéfinie des données de survie issues des études INPULSIS, le taux de mortalité globale au cours des 52 semaines était numériquement plus faible sous nintédanib (5,5 %) que sous placebo (7,8 %). L'analyse du temps écoulé jusqu'au décès a révélé un HR de 0,70 (IC à 95 % 0,43; 1,12; p = 0,1399). Les résultats de tous les critères d'évaluation concernant la survie (tels que la mortalité pendant le traitement et la mortalité de cause respiratoire) ont montré systématiquement une différence numérique en faveur du nintédanib, mais sans atteindre le seuil de signification statistique.
Traitement au long cours par Ofev chez les patients atteints de FPI (INPULSIS-ON)
Une étude d'extension en ouvert portant sur Ofev a inclus 734 patients atteints de FPI. Certains patients ont été traités par Ofev pendant plus de 5 ans. Les patients qui avaient achevé la phase de traitement d'une durée de 52 semaines dans l'étude INPULSIS, ont été traités en ouvert par Ofev dans le cadre de l'étude d'extension INPULSIS-ON. La durée d'exposition médiane pour les patients traités par Ofev tant dans l'étude INPULSIS que dans l'étude INPULSIS-ON s'élevait à 44,7 mois (intervalle: 11,9-68,3). Le taux annuel ajusté du déclin de la CVF sur 192 semaines était de −135,1 (5,8) ml/an chez tous les patients traités et était cohérent avec le taux annuel de déclin de la CVF chez les patients qui avaient été traités par Ofev dans les études INPULSIS de phase III (−113,6 ml par an). Le profil d'effets indésirables d'Ofev dans l'étude INPULSIS-ON était semblable à celui observé dans les études INPULSIS de phase III.
Patients atteints de FPI présentant une altération avancée de la fonction pulmonaire (INSTAGE)
Une étude en groupes parallèles, randomisée et menée en double aveugle, portant sur l'évaluation de l'efficacité et la sécurité d'Ofev en association avec le sildénafil administré par voie orale par comparaison avec le traitement par Ofev seul chez 273 patients atteints de FPI et présentant une altération avancée de la fonction pulmonaire (DLCO < 35% de la valeur prédite) pendant 24 semaines.
Le déclin de la CVF chez les patients traités par Ofev seul coïncidait avec le déclin de la CVF chez les patients présentant des affections moins avancées qui avaient été traités par Ofev dans les études INPULSIS de phase III. L'ajout de sildénafil à Ofev n'a pas apporté de bénéfice significatif en ce qui concerne la qualité de vie par comparaison avec Ofev administré seul. Le profil de sécurité et de tolérance d'Ofev chez les patients atteints de FPI présentant une altération avancée de la fonction pulmonaire coïncidait avec les profils observés dans les études INPULSIS. Le profil d'événements indésirables de l'association d'Ofev et de sildénafil correspondait au profil de sécurité connu des différents composants, sans augmentation des événements graves ou mortels par comparaison avec Ofev administré seul.
Données complémentaires issues de l'étude de phase IV INJOURNEY associant Ofev 150 mg deux fois par jour et la pirfénidone:
Au cours d'un essai exploratoire, en ouvert, randomisé, l'administration concomitante de 150 mg de nintédanib deux fois par jour et de pirfénidone (augmentation progressive de la dose jusqu'à 801 mg trois fois par jour) a été comparée à l'administration de nintédanib 150 mg deux fois par jour seul chez un total de 105 patients pendant 12 semaines. Le critère d'évaluation principal était le pourcentage de patients présentant des évènements indésirables gastro-intestinaux entre le début de l'essai et la semaine 12.
Les évènements gastro-intestinaux étaient fréquents et correspondaient au profil de sécurité connu de chaque composant. Les diarrhées, les nausées et les vomissements étaient les évènements indésirables les plus fréquemment rapportés, soit respectivement 20 patients (37,7 %) contre 16 patients (31,4 %), 22 patients (41,5 %) contre 6 patients (11,8 %) et 15 patients (28,3 %) contre 6 patients (11,8 %) traités par l'ajout de pirfénidone au nintédanib comparativement au traitement par nintédanib seul.
Les variations moyennes absolues (ET) de la CVF à la semaine 12 par rapport aux valeurs initiales étaient de −13,3 (17,4) ml chez les patients traités par nintédanib avec ajout de pirfénidone (n = 48), comparé à −40,9 (31,4) ml chez les patients traités par nintédanib seul (n = 44).
Pneumopathies interstitielles diffuses (PID) fibrosantes chroniques avec un phénotype progressif
L'efficacité clinique d'Ofev a été évaluée dans une étude de phase III randomisée, menée en double aveugle et contrôlée contre placebo (INBUILD) chez des patients atteints de PID fibrosante chronique avec un phénotype progressif.
Au total, 663 patients ont été randomisés selon un rapport 1:1 pour recevoir soit 2x150 mg d'Ofev par jour, soit le placebo pendant au moins 52 semaines.
La randomisation était stratifiée sur l'aspect de la fibrose à la tomodensitométrie à haute résolution (TDM-HR). 412 patients avec un aspect de fibrose de type pneumopathie interstitielle commune (PIC) et 251 patients avec d'autres aspects de fibrose ont été randomisés. Deux populations co-primaires ont été définies pour l'évaluation de cette étude: la population globale et la population avec un aspect de fibrose de type PIC à la TDM-HR.
Le critère d'évaluation principal était le taux annuel de déclin de la CVF (en ml) sur 52 semaines. D'autres critères d'évaluation étaient, par exemple, le délai de survenue de la première exacerbation aiguë de la PID ou du décès ou le délai de survenue du décès.
Les patients ayant un diagnostic clinique de PID fibrosante chronique étaient éligibles à l'étude s'ils présentaient une fibrose significative (>10% de lésions fibrotiques) à la TDM-HR ainsi que des signes cliniques de progression (définie comme un déclin de la CVF ≥10%, un déclin de la CVF entre 5% et 10% avec aggravation des symptômes OU aggravation à l'imagerie, ou une aggravation des symptômes ET une aggravation à l'imagerie) au cours des 24 mois précédents malgré un traitement adéquat. Les patients devaient présenter une CVF ≥45% de la valeur prédite et une DLCO comprise entre 30% et 80% de la valeur prédite.
Étaient exclus de la participation à l'étude les patients avec
·FPI, bronchoconstriction pertinente (p.ex. VEMS/CVF <0,7 avant bronchodilatation) ou hypertension pulmonaire significative
·élévation des transaminases ou de la bilirubine >1,5 LSN
·risque d'hémorragie connu, anticoagulation complète, infarctus du myocarde récent ou accident vasculaire cérébral récent
·traitement antérieur par nintédanib, pirfénidone, azathioprine, ciclosporine, mycophénolate, tacrolimus, corticoïdes oraux >20 mg/jour ou corticoïdes oraux+azathioprine-N-acétylcystéine dans les 4 semaines avant la randomisation*
·traitement antérieur par cyclophosphamide dans les 8 semaines avant la randomisation*
·traitement antérieur par rituximab dans les 6 mois avant la randomisation*
* L'utilisation des médicaments mentionnés était à nouveau autorisée au bout de 6 mois après le début de l'étude, pour autant que ceci soit cliniquement indiqué.
Les participants à l'étude étaient majoritairement caucasiens (74%) ou asiatiques (25%). 54% des patients étaient de sexe masculin, 49% n'avaient jamais fumé, l'âge moyen était de 66 ans et la CVF s'élevait en moyenne à 69% de la valeur prédite. Les PID sous-jacentes étaient des pneumopathies d'hypersensibilité (26%), des PID auto-immunes (26%), des pneumopathies interstitielles non spécifiques idiopathiques (19%), des pneumopathies interstitielles idiopathiques inclassifiables (17%) et d'autres PID (12%).
Taux annuel de déclin de la CVF (ml)
Une réduction statistiquement significative du taux de déclin de la CVF sur 52 semaines a été observée sous traitement par Ofev contre placebo. La différence constatée entre Ofev et placebo était de 107 ml (Tableau 2).
Tableau 2. Taux annuel de déclin de la CVF (ml, critère d'évaluation principal de l'étude INBUILD)
|
|
Population totale
|
Sous-population
de type PIC
|
Sous-population
Autres aspects de fibrose à la TDM-HR
| |
|
Ofev
|
Placebo
|
Ofev
|
Placebo
|
Ofev
|
Placebo
| |
Nombre de patients évalués
|
332
|
331
|
206
|
206
|
126
|
125
| |
Tauxa (ES) de déclin sur 52 semaines
|
-80,8
|
-187,8
|
-82,9
|
-211,1
|
-79,0
|
-154,2
| |
Comparaison contre placebo
Différencea
|
107,0
|
128,2
|
75,2*
| |
IC à 95%
|
(65,4, 148,5)
|
(70,8, 185,6)
|
(15,5, 135,0)*
| |
Valeur p
|
< 0,0001
|
< 0,0001
|
| |
* Une comparaison reposant sur la sous-population avec d'autres aspects de fibrose à la TDM-HR n'a pas été reprise dans la procédure de tests multiples. Les valeurs présentées ici servent à des fins descriptives.
a Basé sur un modèle de régression avec coefficients aléatoires et les effets de catégories fixes du traitement, l'aspect TDM-HR, les effets continus fixes du temps, la valeur initiale de la CVF (ml), y compris l'interaction entre le traitement et le temps et l'interaction entre la valeur initiale et le temps.
|
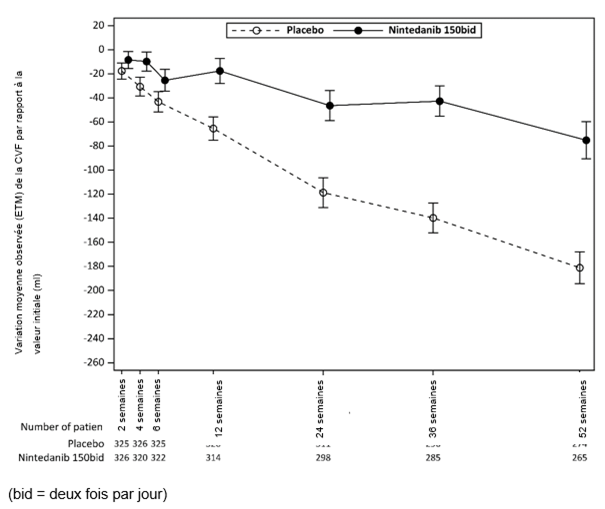
La Figure 2 montre l'évolution de la variation de la CVF dans le temps par rapport à la valeur initiale, dans les groupes de traitement.
Figure 2: Variation moyenne observée (ETM) de la CVF par rapport à la valeur initiale (ml) sur 52 semaines
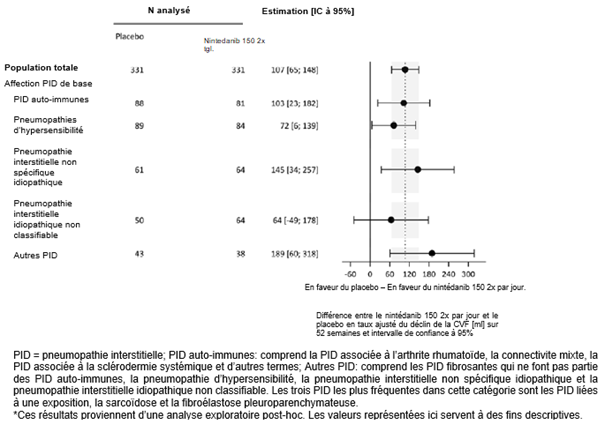
Une analyse exploratoire post-hoc réalisée selon le diagnostic de PID a été effectuée (Figure 3). Dans l'ensemble, un effet thérapeutique cohérent a été mis en évidence au sein des différents diagnostics de PID.
Figure 3: Taux annuel de déclin de la CVF (ml) sur 52 semaines reposant sur le diagnostic de base de PID dans l'étude 5*
Déclin de la CVF en pourcentage
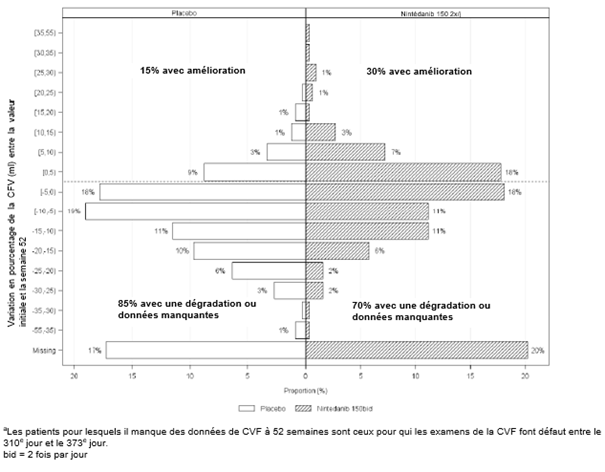
La Figure 4 représente la variation en pourcentage de la CVF (ml) de la valeur initiale à la semaine 52. La dégradation de la fonction pulmonaire était moins importante chez la majorité des patients sous Ofev que chez les patients sous placebo.
Figure 4: Histogramme de la variation en pourcentage de la CVF (ml) de la valeur initiale à la semaine 52 après traitement avec incréments (%) ou décréments par paliers de 5 (étude 5)a
Délai de survenue de la première exacerbation aiguë de la PID ou du décès
Le risque d'une première exacerbation aiguë de la PID ou de décès a diminué dans le groupe Ofev par rapport au groupe placebo sur 52 semaines (HR Ofev contre placebo: 0,80; IC à 95%: 0,48; 1,34) ou sur toute la durée de l'étude (HR Ofev contre placebo: 0,67; IC à 95%: 0,46; 0,98), ce qui correspond à une réduction de 33% du risque d'une première exacerbation aiguë de la PID ou de décès chez les patients sous Ofev par comparaison avec le placebo.
Analyse de la survie
Dans l'étude INBUILD, les données de survie sous Ofev par comparaison avec le placebo ont été évaluées afin d'étayer le critère d'évaluation principal (CFV). La mortalité toutes causes confondues a été évaluée sur toute la durée de l'étude et sur la période de suivi disponible, indépendamment de la cause du décès et de la poursuite ou non du traitement. L'analyse n'a pas montré de différence statistiquement significative sur 52 semaines (HR Ofev contre placebo 0,94, IC à 95% 0,47, 1,86), et sur toute la durée de l'étude (HR Ofev contre placebo 0,78, IC 95% 0,5, 1,21).
Pneumopathie interstitielle associée à la sclérodermie systémique (PID-ScS)
L'efficacité clinique du nintédanib a été évaluée dans une étude de phase III randomisée, en double aveugle et contrôlée versus placebo (SENSCIS) chez des patients atteints de PID-ScS.
En tout, 580 patients avec une proportion de 1:1 ont été randomisés pour recevoir soit 150 mg d'Ofev deux fois par jour ou le placebo pendant au moins 52 semaines. La randomisation a été stratifiée selon le statut des anticorps anti-topoisomérase (ATA). Certains patients ont poursuivi le traitement en aveugle durant jusqu'à 100 semaines.
Le diagnostic des patients atteints de PID-ScS reposait sur les critères de classification pour la sclérodermie systémique (ScS) de l'American College of Rheumatology / de la European League Against Rheumatism lorsque la survenue de la maladie (premier symptôme non Raynaud) remontait à moins de 7 ans et en présence d'une fibrose pulmonaire d'au moins 10%, constatée sur la base d'une image obtenue par tomodensitométrie haute résolution effectuée au cours des 12 mois précédents.
Chez les patients, la CVF devait atteindre au moins 40 % de la valeur estimée, tandis que la DLCO devait être entre 30 et 89 % de la valeur estimée. Les patients atteints d'importantes obstructions des voies respiratoires (c'est-à-dire VEMS/CVF avant la bronchodilatation inférieur à 0,7) ou ayant reçu ou allant recevoir une transplantation de cellules souches hématopoïétiques ne pouvaient pas participer à l'étude. D'autres critères d'exclusion étaient des valeurs ALAT, ASAT ou de bilirubine élevées (>1,5xLNS), un risque accru d'hémorragies (y compris avec anticoagulation thérapeutique), un infarctus du myocarde ou un accident vasculaire cérébral récent, une hypertension pulmonaire considérable, des ulcérations à au moins trois bouts de doigt, un antécédent de nécrose sévère du doigt avec hospitalisation nécessaire ainsi qu'un antécédent de crise rénale sclérodermique. Les patients qui recevaient d'autres médicaments à l'étude, qui avaient déjà été traités par nintédanib ou pirfénidone et avaient reçu de l'azathioprine 8 semaines avant la randomisation ou du cyclophosphamide ou de la cyclosporine A 6 mois avant la randomisation ont également été exclus. Un traitement stable par mycophénolate ou méthotrexate ainsi que par prednisone ≤10 mg/jour ou une dose équivalente était autorisé.
Le critère d'évaluation primaire était le déclin annuel de la capacité vitale forcée (CVF) sur 52 semaines.
Les critères secondaires importants étaient la variation absolue du Rodnan Skin Score modifié (mRSS) à la semaine 52 par rapport à la valeur initiale, la variation absolue du score total au Saint George's Respiratory Questionnaire (SGRQ) à la semaine 52 par rapport à la valeur initiale ainsi que la mortalité pendant toute l'étude.
75,2 % des patients étaient de sexe féminin. Les patients étaient majoritairement de type caucasien (67 %), asiatique (25 %) ou avaient la peau noire (6 %). L'âge moyen (écart type [ET], min.-max.) s'élevait à 54,0 (12,2, 20-79) ans. En tout, 51,9 % des patients souffraient d'une sclérose systémique cutanée diffuse (ScS) et 48,1 % d'une ScS cutanée limitée. La durée moyenne (ET) depuis la première apparition d'un symptôme non Raynaud était de 3,49 (1,7) ans. 49,0 % des patients ont reçu un traitement stable par mycophénolate (46,5% mycophénolate mofétil, 1,9% mycophénolate de sodium, 0,5% acide mycophénolique) et la profil de sécurité observé chez les patients sous mycophénolate ou non était similaire.
Déclin annuel de la CVF
Le déclin annuel de la CVF (en ml) sur 52 semaines a nettement diminué de 41,0 ml chez les patients sous Ofev par rapport au placebo (voir Tableau 3), ce qui correspond à un effet relatif du traitement de 43,8 %.
Tableau 3: Déclin annuel de la CVF (ml) sur 52 semaines
|
|
Placebo
|
Ofev
150 mg deux fois par jour
| |
Nombre de patients évalués
|
288
|
287
| |
Taux1 (ET) du déclin sur 52 semaines
|
-93,3 (13,5)
|
-52,4 (13,8)
| |
Comparaison versus placebo
| |
Différence1
|
|
41,0
| |
IC à 95 %
|
|
(2,9, 79,0)
| |
Valeur p
|
|
<0,05
| |
1
Sur la base d'une régression avec des coefficients aléatoires, ajustés pour le traitement, le sexe, la taille, l'âge, le statut ATA, la valeur initiale de CVF, CVF*la durée et traitement*la durée
|
Une analyse exploratoire des données jusqu'à 100 semaines (durée de traitement max. dans l'étude SENSCIS) a montré que l'efficacité, avec un traitement par Ofev, sur le ralentissement de la progression de la PID-ScS durait plus de 52 semaines.
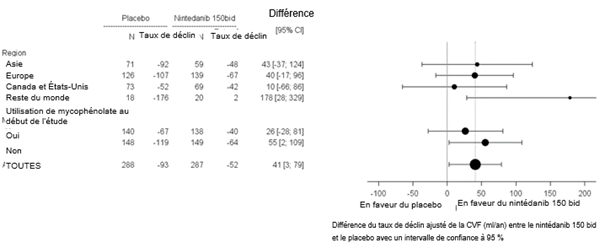
Dans deux analyses d'efficacité de sous-groupes prédéfinis, la différence moyenne de traitement relative au déclin de la CVF à la semaine 52 chez les patients selon la région et l'utilisation du mycophénolate a été étudiée (voir Figure 5).
Figure 5: Analyse de sous-groupes de la différence moyenne de traitement relative au déclin de la CVF (ml) à la semaine 52 selon la région et l'utilisation du mycophénolate (SENSCIS)
Variation en pourcentage de la capacité vitale forcée à partir du début de l'étude
La Figure 6 représente pour l'étude SENSCIS la variation en pourcentage de la CVF en ml à la semaine 52 par rapport à la valeur initiale. La dégradation de la fonction pulmonaire était moins importante chez la majorité des patients sous Ofev que chez les patients sous placebo.
Figure 6: Histogramme de la variation en pourcentage de la CVF (ml) à la semaine 52 par rapport à la valeur initiale selon le traitement et augmentation ou diminution en pourcentage de 5 (SENSCIS)a
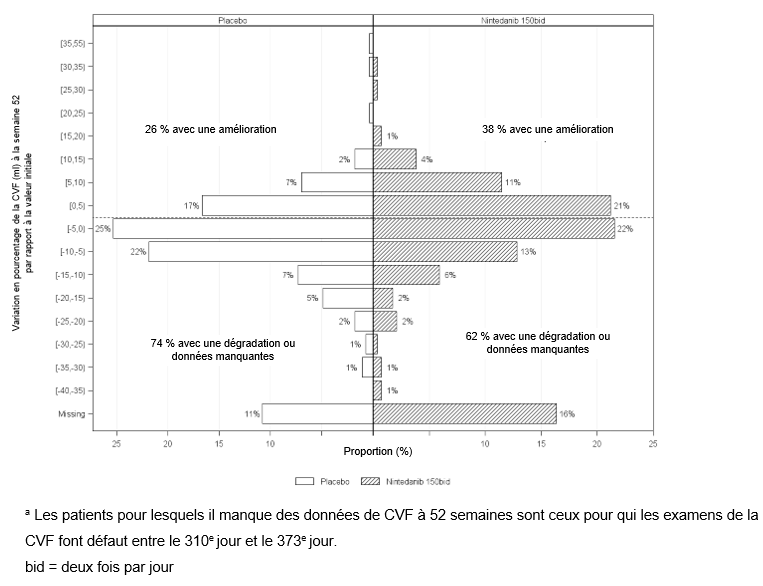
Figure 7: Variation moyenne observée de la CVF par rapport à la valeur initiale (ml) sur 52 semaines
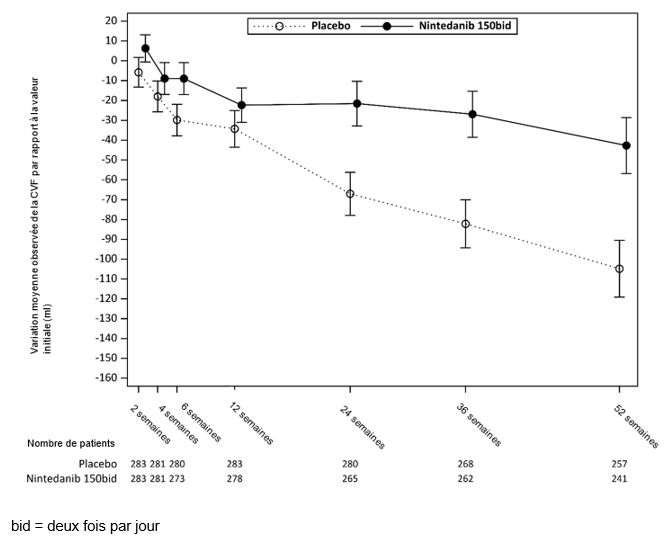
Tableau 4 Déclin annuel de la CVF (% de la valeur théorique) sur 52 semaines
|
|
Placebo
|
Ofev
150 mg deux fois par jour
| |
Nombre de patients évalués
|
288
|
287
| |
Taux1 (ET) de déclin sur 52 semaines
|
-2,6 (0,4)
|
-1,4 (0,4)
| |
Compraison versus placebo
| |
Différence1
|
|
1,15
| |
IC à 95%
|
|
(0,09, 2,21)
| |
Valeur p
|
|
<0,05
| |
1
Sur la base d'une régression avec des coefficients aléatoires et les effets de catégories fixes du traitement, le statut ATA, les effets continus fixes du temps, la valeur initiale de la CVF [% de la valeur théorique] y compris l'interaction entre le traitement et le temps et l'interaction entre la valeur initiale et le temps. L'intercept et le temps étaient donnés de manière spécifique au patient en tant qu'effet aléatoire. La variance d'erreur de mesure (within-patient error) inhérente à un patient a été modélisée au moyen d'une matrice de variance-covariance non structurée. La variabilité inter-individuelle a été modélisée au moyen d'une matrice de variance-covariance des composantes de variance.
|
Variation du score de Rodnan Skin modifié (mRSS) à 52 semaines par rapport à la valeur initiale
La variation absolue moyenne ajustée du mRSS à 52 semaines par rapport à la valeur initiale était similaire entre le groupe sous Ofev (-2,17 (IC à 95% -2,69, -1,65)) et le groupe sous placebo (-1,96 (IC à 95% -2,48, -1,45)). La différence moyenne ajustée entre les groupes de traitement s'élevait à -0,21 (IC à 95% -0,94, 0,53; p = 0,5785).
Variation du score total au Saint George's Respiratory Questionnaire (SGRQ) à 52 semaines par rapport à la valeur initiale
La variation absolue moyenne ajustée du score total de SGRD à 52 semaines par rapport à la valeur initiale était similaire entre le groupe sous Ofev (0,81 (IC à 95% -0,92, 2,55)) et le groupe sous placebo (-0,88 (IC à 95% -2,58, 0,82)). La différence moyenne ajustée entre les groupes de traitement s'élevait à 1,69 (IC à 95% -0,73, 4,12; p = 0,1711).
Analyse de survie
Sur toute l'étude, la mortalité entre le groupe Ofev (N = 10; 3,5 %) et le groupe sous placebo (N = 9; 3,1 %) était similaire. Sur toute l'étude, l'analyse du temps jusqu'au décès a donné un HR de 1,16 (IC à 95% 0,47, 2,84; p = 0,7535).
Effet sur l'intervalle QT
Dans une étude spécifique auprès de patients atteints d'un cancer du rein à cellules claires, des mesures du QT/QTc ont révélé que ni l'administration d'une dose orale unique de 200 mg de nintédanib, ni l'administration de doses orales répétées de 200 mg de nintédanib deux fois par jour pendant 15 jours n'a prolongé l'intervalle QTcF. Aucune étude approfondie complète de l'intervalle QT n'a été effectuée sur le nintédanib.
Études auprès de la population pédiatrique
Le nintédanib ne doit pas être utilisé chez la population pédiatriques atteinte de pneumopathies interstitielles diffuses (PID) fibrosantes chroniques.
|