CompositionPrincipe actif:
Inhibiteur de l'alpha1-protéinase humaine.
Excipients
Chlorure de sodium, dihydrogénophosphate de sodium monohydraté, mannitol, hydroxyde de sodium et acide chlorhydrique (correspondant à 1,9 mg/ml de sodium ou environ 1,6 mmol de sodium par bouteille de 1000 mg et environ 6,5 mmol de sodium par bouteille de 4000 mg).
Solvant: eau pour préparations injectables.
La poudre est de couleur blanche à crème. Le solvant est une solution claire et incolore.
La solution reconstituée présente une osmolalité d'environ 279 mOsmol/kg et un pH de 7,0.
Indications/Possibilités d’emploiRespreeza est indiqué comme traitement d'entretien/de maintien chez les adultes présentant un déficit sévère en inhibiteur de l'alpha1-protéinase (phénotypes (Z, Z), (Z, null), (null, null) ou (S, Z)) et une maladie pulmonaire cliniquement avérée (volume expiratoire maximal seconde (VEMS) ou capacité de diffusion (DLCO) pronostiqués à <70% de la valeur nominale). Respreeza ralentit la destruction sous-jacente du tissu pulmonaire, qui conduit à l'emphysème. Les données cliniques sont limitées à la tomographie spiralée de densitométrie TDM assistée par ordinateur (CT).
Posologie/Mode d’emploiPosologie pour les adultes et les enfants
Le traitement doit être mis en place et surveillé par un médecin spécialiste expérimenté dans le traitement du déficit en inhibiteur de l'alpha1-protéinase.
Les perfusions suivantes peuvent être administrées par une personne de confiance ou par le patient lui-même (voir rubrique «Mises en garde et précautions»).
Chaque fois que Respreeza est administré au patient, il est fortement recommandé de documenter le nom et la désignation du lot de la préparation afin d'établir un lien entre le patient et le lot de produit si nécessaire.
Posologie
Après reconstitution, Respreeza doit être administré exclusivement sous forme de perfusion intraveineuse.
Sauf indication contraire, la dose recommandée de Respreeza est de 60 mg/kg de poids corporel (PC), administrée une fois par semaine pour obtenir la réponse clinique souhaitée et le taux sérique d'inhibiteur de l'alpha1-protéinase désiré. La dose peut être corrigée en fonction de ces deux paramètres. Il est possible d'administrer des doses allant jusqu'à 120 mg/kg PC.
Patients âgés
La sécurité et l'efficacité de Respreeza chez les patients âgés (65 ans ou plus) n'ont pas été établies dans des études cliniques spécifiques. Les expériences acquises après la mise sur le marché n'indiquent toutefois pas de différences chez les patients âgés. Aucune adaptation posologique n'est recommandée.
Patients insuffisants rénaux ou hépatiques
Aucune étude particulière n'a été effectuée chez des patients insuffisants rénaux ou hépatiques. Aucun schéma posologique différent n'est recommandé chez ces patients.
Enfants et adolescents
La sécurité et l'efficacité de Respreeza chez l'enfant et l'adolescent (de moins de 18 ans) n'ont pas été étudiées jusqu'à présent. On ne dispose d'aucune donnée.
Mode d'administration
Administration intraveineuse.
Le produit doit être reconstituée avec de l'eau pour préparations injectables et administré avec un set de perfusion intraveineuse (voir «Instructions pour la manipulation» dans la rubrique «Remarques particulières»).
La solution reconstituée doit être administrée par voie intraveineuse avec un tube de perfusion séparé à un débit de perfusion d'environ 0,08 ml/kg PC/min. Ce débit de perfusion peut être adapté en fonction de la réponse clinique et du bien-être du patient. La durée de perfusion de la dose recommandée de 60 mg/kg de PC est d'environ 15 minutes.
Un tube de perfusion Respreeza est réservé à un emploi unique.
D'autres instructions concernant l'administration de la solution reconstituée se trouvent dans la rubrique «Remarques particulières».
Contre-indicationsHypersensibilité au Respreeza, au principe actif ou à l'un des excipients de Respreeza (voir rubrique «Composition»).
Patients souffrant d'un déficit en IgA et présentant des anticorps anti-IgA, en raison du risque d'hypersensibilité sévère et de réactions anaphylactiques.
Cœur pulmonaire décompensé.
Mises en garde et précautionsLe tabagisme est un facteur de risque important dans le développement d'un emphysème. Respreeza est par conséquent destiné exclusivement aux non-fumeurs confirmés (>6 mois). Les patients doivent également éviter d'autres facteurs de risque de maladies pulmonaires (par exemple tabagisme passif, aérocontaminants).
Le débit de perfusion recommandé, indiqué dans la rubrique «Posologie/Mode d'emploi», doit être respecté. L'état clinique du patient, y compris les signes vitaux, doit être étroitement surveillé pendant toute la durée des premières perfusions. S'il apparaît une réaction indésirable, susceptible d'être liée à l'administration de Respreeza, la perfusion doit être ralentie ou arrêtée, selon l'état clinique du patient. Si les symptômes disparaissent rapidement après l'arrêt de la perfusion, celle-ci peut être reprise à un débit plus faible, confortable pour le patient.
Hypersensibilité
Des réactions d'hypersensibilité peuvent apparaître, y compris chez des patients ayant toléré des traitements précédents par l'inhibiteur d'alpha1-protéinase humain.
La suspicion de réactions allergiques ou anaphylactiques peut nécessiter un arrêt immédiat de la perfusion, en fonction de la nature et de la gravité de la réaction. En cas de choc, un traitement médical d'urgence doit être administré.
Auto-administration/traitement à domicile
Les données concernant l'auto-administration/le traitement à domicile avec ce médicament sont limitées.
Les risques potentiels associés à l'auto-administration/au traitement à domicile sont liés à la manipulation et l'administration de la préparation ainsi qu'à la prise en charge des effets indésirables, notamment des réactions d'hypersensibilité. Les patients doivent être informés des premiers signes de réactions d'hypersensibilité.
La décision concernant un traitement à domicile ou l'administration de la perfusion par le patient lui-même revient au médecin traitant (voir également la rubrique «Mises en garde et précautions»). Ce dernier doit s'assurer qu'une formation appropriée est fournie au patient (notamment en ce qui concerne la reconstitution, l'utilisation du dispositif de transfert et du filtre, l'assemblage du système de perfusion, les techniques de perfusion, la tenue d'un journal de traitement ainsi que l'identification des effets indésirables et les mesures qui doivent être prises en cas d'apparition de tels effets) et que le bon usage du traitement est contrôlé à intervalles réguliers.
Sécurité par rapport aux agents pathogènes transmissibles
Respreeza est préparé à partir de plasma humain. Les mesures standard de prévention des infections qui pourraient résulter de l'emploi de médicaments préparés à partir de plasma ou de sang humain comprennent la sélection des donneurs, le contrôle des donneurs individuels et du pool de plasma à la recherche de marqueurs spécifiques d'infection ainsi que l'inclusion d'étapes efficaces de fabrication pour l'inactivation/l'élimination des virus. La possibilité d'une transmission d'agents pathogènes infectieux ne peut cependant pas être totalement exclue lors de l'administration de médicaments qui ont été préparés à partir de plasma ou de sang humain. Cela s'applique également aux virus et autres agents pathogènes inconnus jusqu'ici ou émergents.
Les mesures de sécurité prises sont considérées comme efficaces contre des virus enveloppés tels que le virus de l'immunodéficience humaine (VIH), le virus de l'hépatite B (VHB) et le virus de l'hépatite C (VHC) et contre des virus non enveloppés tels que le virus de l'hépatite A (VHA) et le parvovirus B19.
Une vaccination (hépatites A et B) appropriée est à envisager chez des patients recevant d'une façon régulière/répétée des inhibiteurs de protéinase provenant de plasma humain.
Teneur en sodium
Respreeza contient environ 37 mg de sodium par flacon de 1000 mg et 149 mg de sodium par flacon de 4000 mg, correspondant à 1,9%, et respectivement 7,4% de l'apport maximal quotidien en sodium recommandé par l'OMS pour un adulte ayant un régime alimentaire de 2 g d'apport quotidien en sodium.
InteractionsAucune étude d'interactions n'a été effectuée.
Grossesse, allaitementGrossesse
Aucune étude sur la reproduction avec Respreeza n'a été effectuée sur l'animal. La sécurité d'emploi de Respreeza chez la femme enceinte n'a pas été établie dans des études cliniques contrôlées. Étant donné que l'inhibiteur de l'alpha1-protéinase est une protéine humaine endogène, un effet nocif de Respreeza sur le fœtus est considéré comme peu probable. Respreeza doit cependant être administré avec précaution à des femmes enceintes.
Allaitement
On ignore si Respreeza et ses métabolites sont excrétés dans le lait maternel. L'excrétion de l'inhibiteur de l'alpha1-protéinase humaine dans le lait maternel n'a pas été étudiée chez l'animal. Des précautions sont à prendre pendant la période d'allaitement. Le cas échéant, il convient de soupeser soit de poursuivre/d'interrompre l'allaitement, soit de poursuivre/d'interrompre le traitement avec Respreeza en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement par l'inhibiteur de l'alpha1-protéinase humaine pour la femme.
Fertilité
Aucune étude de fertilité avec Respreeza n'a été effectuée sur l'animal. L'effet sur la fertilité humaine n'a pas été examiné dans des études cliniques contrôlées. Étant donné que l'inhibiteur de l'alpha1-protéinase est une protéine humaine endogène, aucun effet nocif sur la fertilité ne devrait être attendu.
Effet sur l’aptitude à la conduite et l’utilisation de machinesUne sensation de vertige peut apparaître après l'administration de Respreeza (voir rubrique «Effets indésirables»). Respreeza peut donc avoir un léger effet sur l'aptitude à la conduite ou à l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
Dans de très rares cas, des réactions d'hypersensibilité ou des réactions allergiques ont été observées au cours du traitement. Dans les cas les plus sérieux, des réactions allergiques peuvent conduire à des réactions anaphylactiques graves, même lorsque le patient n'a pas présenté de réactions d'hypersensibilité lors d'administrations précédentes (voir rubrique «Mises en garde et précautions»).
Effets indésirables sous forme de liste
Les effets indésirables du médicament (EIM), qui ont été déterminés lors de six études cliniques menées sur 221 patients et d'après les expériences acquises après la mise sur le marché, sont indiqués dans la liste ci-dessous conformément aux classes d'organes de la terminologie MedDRA (SOC, classe de système d'organes et terme préféré). La fréquence par perfusion a été jugée selon les définitions suivantes: occasionnel (≥1/1000 à <1/100), rare (≥1/10'000 à <1/1000), très rare (<1/10'000).
À l'intérieur de chaque groupe de fréquence, les EIM sont indiqués par ordre décroissant de gravité.
Affections hématologiques et du système lymphatique:
Fréquences indéterminées: Douleurs au niveau des ganglions lymphatiques.
Affections du système immunitaire:
Occasionnels: Réactions d'hypersensibilité (y compris tachycardie, hypotonie, confusion, syncope, hypoxie et œdème du pharynx).
Très rares: Réactions anaphylactiques.
Affections du système nerveux:
Fréquents: Sensation de vertige, céphalées.
Occasionnels: Paresthésie.
Très rares: Hypoesthésie.
Affections oculaires:
Fréquences indéterminées: Gonflement des yeux.
Affections vasculaires:
Peu fréquents: Bouffées de chaleur.
Affections respiratoires, thoraciques et médiastinales
Fréquents: Dyspnée.
Affections gastro-intestinales:
Fréquents: Nausées
Fréquences indéterminées: Gonflement des lèvres.
Affections de la peau et du tissu sous-cutané:
Occasionnels: Urticaire, éruptions cutanées (y compris exfoliantes et généralisées).
Très rares: Hyperhidrose, prurit.
Fréquences indéterminées: Œdème du visage.
Troubles généraux et anomalies au site d'administration:
Occasionnels: Asthénie, réactions au niveau du site de perfusion (y compris hématome au site de perfusion).
Très rares: Douleurs thoraciques, frissons, fièvre.
Effets indésirables du médicament après commercialisation
Des évaluations fiables de la fréquence de ces réactions exclusivement constatées après la mise sur le marché ou la détermination d'une relation causale avec l'exposition au produit ne sont pas possibles, car les effets secondaires sont rapportés volontairement et par des populations de patients de taille inconnue. La fréquence de ces EIM est par conséquent considérée comme «Fréquence indéterminée (ne pouvant pas être évaluée à l'aide des données disponibles)».
Informations sur la sécurité vis-à-vis des virus: voir rubrique «Mises en garde et précautions».
Le signalement des effets indésirables présumés des médicaments après l'autorisation de mise sur le marché est d'une grande importance. Il permet de surveiller en permanence le rapport bénéfice/risque du médicament. Les professionnels de santé sont invités à signaler tout soupçon d'effet indésirable nouveau ou grave via le portail en ligne ElViS (système de vigilance électronique). Des informations à ce sujet sont disponibles sur le site www.swissmedic.ch.
SurdosageLes conséquences d'un surdosage sont inconnues.
En cas de surdosage, le patient doit être étroitement surveillé pour détecter d'éventuels EIM. En même temps, des mesures de soutien requises doivent être disponibles.
Propriétés/EffetsCode ATC
B02AB02
Classe pharmaco-thérapeutique: antihémorrhagique, inhibiteur de protéinase
Mécanisme d'action
L'inhibiteur de l'alpha1-protéinase est considéré comme le plus important inhibiteur anti-protéase pour l'inhibition de l'élastase des neutrophiles dans les voies respiratoires inférieures. Chez les personnes saines, la quantité d'inhibiteur de l'alpha1-protéinase produite empêche une protéolyse excessive du tissu pulmonaire par inactivation de l'EN produite par les polynucléaires neutrophiles activés. Les facteurs qui stimulent la production de polynucléaires neutrophiles et leur activation dans le poumon, comme par exemple les infections des voies respiratoires et le tabagisme, augmentent à leur tour la concentration d'EN. Les patients ayant un déficit en inhibiteur de l'alpha1-protéinase endogène ne peuvent pas maintenir une défense anti-protéase suffisante et souffrent d'une protéolyse rapide des parois alvéolaires, qui peut conduire au développement d'une pneumopathie obstructive chronique cliniquement avérée au cours de la trentaine ou la quarantaine.
Pharmacodynamique
L'administration de Respreeza augmente et maintient le taux sérique de l'inhibiteur de l'alpha1-protéinase et son taux dans le film de liquide qui tapisse l'épithélium pulmonaire (epithelial lining fluid (ELF)), de sorte que l'emphysème progresse plus lentement.
Efficacité clinique
Étude RAPID et étude RAPID d'extension
La sécurité et l'efficacité de Respreeza ont été examinées dans une étude randomisée, en double aveugle, multicentrique, contrôlée par placebo (RAPID) suivie d'une étude d'extension en ouvert de 2 ans (étude RAPID d'extension). Au total 180 patients (93 patients traités par Respreeza et 87 patients traités par un placebo) présentant un déficit en inhibiteur de l'alpha1-protéinase et des signes cliniques d'un emphysème ont été randomisés, pendant une période allant jusqu'à 24 mois, pour recevoir une dose intraveineuse hebdomadaire de 60 mg/kg PC de Respreeza ou d'un placebo. Ensuite, 140 patients furent inclus dans l'étude d'extension RAPID (76 patients précédemment traités par Respreeza et 64 précédemment traités par placebo provenant de l'étude principale RAPID) et traités par une dose intraveineuse hebdomadaire de 60 mg/kg PC de Respreeza pendant une période atteignant 24 mois.
Les données cliniques dans les deux études reposent sur une tomographie spiralée assistée par ordinateur (CT), avec laquelle était examiné l'effet de Respreeza sur la diminution de la densité pulmonaire et la progression de l'emphysème.
La vitesse de diminution annuelle de la densité pulmonaire, mesurée à l'aide des enregistrements CT à la capacité pulmonaire totale (total lung capacity (TLC)) sur 2 ans, était significativement plus faible sous Respreeza (-1,45 g/l) que sous placebo (-2,19 g/l), ce qui correspond à une réduction de 34% (p = 0,017; unilatérale). Les valeurs plus élevées de la densité pulmonaire, mesurées par CT, étaient en corrélation avec un plus grand VEMS1 (0,31; p <0,001), une plus grande DLCO (0,46; p <0,001), une plus grande capacité de charge (0,26; p=0,002) et un plus faible score d'activités sur le questionnaire respiratoire du St. George's (-0,26; p = 0,002) tout au long de l'étude.
L'analyse finale des études RAPID a montré
·qu'une vitesse de diminution de la densité pulmonaire plus faible a été maintenue chez les patients qui avaient reçu un traitement continu par Respreeza pendant 4 ans
·que dans l'étude d'extension RAPID, une vitesse de diminution de la densité pulmonaire plus faible avait été obtenue chez les patients traités précédemment par placebo dans l'étude principale RAPID;
·que chez les patients qui n'avaient pas été traités précédemment par Respreeza dans l'étude principale RAPID, les pertes de densité pulmonaire sont restées irréversibles malgré le traitement ultérieur par Respreeza dans l'étude d'extension.
Dans l'étude RAPID, Respreeza a montré un profil de sécurité et de tolérance semblable à celui du placebo. Les doses unitaires de 120 mg/kg PC de Respreeza, qui avaient été administrées à 75 patients, n'ont de même pas entraîné de problèmes particuliers en ce qui concerne la sécurité. Une posologie hebdomadaire régulière de 120 mg/kg PC n'a pas été étudiée.
PharmacocinétiqueQuatre études cliniques avec Respreeza ont été effectuées sur 89 patients (59 hommes et 30 femmes) pour évaluer l'effet de Respreeza sur le taux sérique de l'inhibiteur de l'alpha1-protéinase. Les patients étaient âgés de 29 à 68 ans (moyenne d'âge 49 ans). À la sélection, les taux sériques d'inhibiteur de l'alpha1-protéinase étaient de 3,2 à 10,1 µM (moyenne 5,6 µM).
Sur la base d'une étude de pharmacocinétique avec 13 hommes et 5 femmes présentant un déficit en inhibiteur de l'alpha1-protéinase, âgés de 36 à 66 ans, les données pharmacocinétiques suivantes ont été relevées (voir Tableau 1):
Tableau 1: paramètres pharmacocinétiques de l'inhibiteur de l'alpha1-protéinase après une dose unitaire de 60 mg/kg PC de Respreeza
|
Paramètres pharmacocinétiques
|
Moyenne
(± écart-type)*
| |
Aire sous la courbe (AUC0-∞)
|
144 (±27) µM x jour
| |
Concentration maximale (Cmax)
|
44,1 (±10,8) µM
| |
Demi-vie terminale (t½β)
|
5,1 (±2,4) jours
| |
Clairance totale
|
603 (±129) ml/jour
| |
Volume de distribution à l'état stationnaire
|
3,8 (±1,3) l
|
* n=18 patients
Une analyse pharmacocinétique de population a été effectuée à l'aide de données de 90 patients traités par Respreeza à partir de l'étude RAPID. L'évaluation de la demi-vie moyenne dans la population était de 6,8 jours. Le modèle a montré des concentrations sériques moyennes à l'état d'équilibre (Steady State) de 21,8 µM après une dose de 60 mg/kg/semaine. L'analyse pharmacocinétique de population n'a donné aucune indication sur les effets significatifs de l'âge, du sexe, du poids ou de la concentration de l'inhibiteur de l'alpha1-protéinase endogène dans le sérum à l'inclusion sur la clairance de Respreeza.
Relation pharmacocinétique/pharmacodynamique
Dans une étude clinique contrôlée en double aveugle visant à évaluer la sécurité et l'efficacité biochimique de Respreeza, 44 patients ont été randomisés pour recevoir une dose intraveineuse de 60 mg/kg PC de Respreeza ou un comparateur pendant 10 semaines. Au bout de 10 semaines, les patients dans les deux groupes ont reçu Respreeza pendant encore 14 semaines. Pour compléter l'évaluation de la sécurité on a examiné les patients pendant 24 semaines au total. Les taux sériques résiduels moyens de l'inhibiteur d'alpha1-protéinase à l'état stationnaire (semaine 7-11) des patients traités par Respreeza étaient statistiquement équivalents, dans une plage de ±3 µM, aux taux sériques résiduels des patients traités par la préparation comparative. Des taux sériques résiduels d'inhibiteur de l'alpha1-protéinase supérieurs à 11 µM ont été maintenus dans les deux groupes. La moyenne (± écart-type) du taux sérique résiduel d'inhibiteur de l'alpha1-protéinase à l'état stationnaire était de 17,7 µM (± 2,5 µM) chez les patients traités par Respreeza et de 19,1 µM (±2,2 µM) chez les patients traités par la préparation comparative. La différence entre les groupes Respreeza et préparation comparative n'était pas statistiquement significative.
Un lavage broncho-alvéolaire a été effectué dans un sous-groupe de patients qui avaient été inclus dans cette étude (10 patients traités par Respreeza et 5 traités par la préparation comparative). Les taux d'inhibiteur de l'alpha1-protéinase mesurés dans l'ELF des patients traités par Respreeza n'étaient pas plus bas que chez les patients qui avaient été traités par le produit comparatif, et présentaient une constante augmentation après le traitement.
Le taux ELF de l'inhibiteur de l'alpha1-protéinase endogène et celui d'inhibiteur d'alpha1-protéinase:
Les complexes EN étaient accrus par rapport à la valeur de référence. Dans tous les échantillons des patients, la quantité d'élastase libre était extrêmement faible.
Dans le cadre de la finalisation de l'étude RAPID on a effectué une analyse des taux médians d'inhibiteur de l'alpha1-protéinase atteints et de la diminution de la densité pulmonaire. Cette analyse a montré une relation linéaire inverse entre les taux sériques d'inhibiteur de l'alpha1-protéinase et la diminution annuelle de la densité pulmonaire, mesurée à l'aide d'enregistrements CT ajustés en volume, chez des patients qui avaient reçu une dose intraveineuse de 60 mg/kg PC de Respreeza.
Données précliniquesLa sécurité de Respreeza a été examinée dans plusieurs études précliniques. Les données non cliniques issues des études de pharmacologie de sécurité et de toxicité à court terme ne révèlent aucun risque particulier pour l'Homme. De légères altérations au niveau de la rate, de l'utérus, des poumons ou du cœur sont apparues dans les études sur les rats et/ou les lapins à une dose qui correspondait au double de la dose thérapeutique maximale. Aucune toxicité n'a été constatée à la dose thérapeutique recommandée de 60 mg/kg. La sécurité à long terme ne peut pas être étudiée chez l'animal en raison de la formation d'anticorps contre la protéine humaine. Étant donné que l'inhibiteur de l'alpha1-protéinase humain est un constituant physiologique du sang humain, on considère qu'il n'a pas d'effets carcinogènes, génotoxiques ni tératogènes.
Remarques particulièresIncompatibilités
Ce médicament ne peut être mélangé qu'aux médicaments mentionnés dans la rubrique «Remarques concernant la manipulation».
Stabilité
Respreeza est stable jusqu'à la date de péremption indiquée après «EXP» sur l'étiquette et sur l'emballage. Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Stabilité après ouverture
Pour des raisons microbiologiques, la préparation prête à l'emploi doit être utilisée immédiatement après reconstitution. La stabilité chimique et physique pendant 3 heures à la température ambiante (jusqu'à 25 °C) a été néanmoins démontrée. La préparation prête à l'emploi ne doit pas être congelée.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 25 °C. Ne pas congeler. Tenir hors de la portée des enfants.
Remarques concernant la manipulation
Remarques générales
·La reconstitution doit être réalisée conformément aux instructions sous-mentionnées.
·La reconstitution, l'administration et la manipulation doivent s'effectuer avec soin et dans des conditions aseptiques.
·N'utiliser aucun des dispositifs médicaux stériles joints pour la reconstitution si l'emballage est ouvert ou défectueux.
·Avant l'administration, contrôlez la présence de particules et de décolorations dans la solution reconstituée.
·La solution doit être incolore, limpide ou légèrement opalescente et exempte de particules visibles.
·La poudre lyophilisée doit être reconstituée avec un solvant stérile (eau pour préparations injectables). La reconstitution d'un flacon de produit de 1000 mg doit être effectuée dans les 5 minutes. La reconstitution d'un flacon de produit de 4000 mg doit être effectuée dans les 10 minutes.
Veuillez suivre les instructions ci-après pour la reconstitution:
|
1. Assurez-vous que le flacon perforable de Respreeza contenant la poudre et le flacon perforable contenant le solvant sont à la température ambiante (jusqu'à 25 °C).
|
| |
2. Retirez la capsule en matière plastique du flacon contenant le solvant.
|
| |
3. Nettoyez le bouchon en caoutchouc du flacon perforable contenant le solvant avec une solution antiseptique et laissez-le sécher.
|
| |
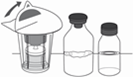
4. Ouvrez le dispositif de transfert 20/20 (Mix2Vial™) avec filtre intégré en retirant l'opercule (Figure 1).
Ne pas retirer le dispositif de transfert de l'emballage sous blister.
|
Figure 1
|
| |
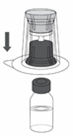
5. Poser le flacon contenant le solvant sur une surface plane et propre et le tenir fermement. Saisir le dispositif de transfert avec l'emballage sous blister et percer verticalement le bouchon du flacon contenant le solvant avec l'extrémité bleue de l'adaptateur (Figure 2).
|
Figure 2
|
| |
6. Retirer avec précaution l'emballage sous blister du dispositif de transfert, en saisissant le sceau du blister et en le retirant verticalement vers le haut. Assurez-vous que seul l'emballage sous blister est retiré et non le dispositif de transfert (Figure 3).
|
Figure 3
|

| |
7. Retirez le capuchon en plastique du flacon perforable de Respreeza.
|
| |
8. Nettoyez le bouchon en caoutchouc du flacon de Respreeza avec une solution antiseptique et laissez-le sécher.
|
| |
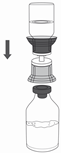
9. Placer le flacon de Respreeza sur une surface plane et rigide. Retourner le flacon contenant le solvant avec le dispositif de transfert attaché et percer verticalement le bouchon du flacon de Respreeza avec l'extrémité transparente de l'adaptateur (Figure 4). Le solvant s'écoule automatiquement dans le flacon de Respreeza.
ATTENTION: Assurez-vous que tout le solvant ait été transféré dans le flacon de Respreeza.
|
Figure 4
|
| |
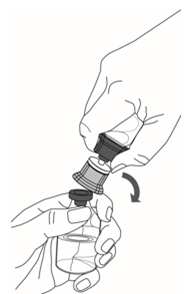
10. Veuillez suivre les étapes suivantes pour retirer entièrement le dispositif de transfert du flacon de Respreeza:
·D'une main, bien saisir le flacon de Respreeza et de l'autre main le flacon de solvant avec l'adaptateur bleu (Figure 5).
·Plier sur le côté le dispositif de transfert complet avec le flacon de solvant jusqu'à ce qu'il se détache du flacon de Respreeza (Figure 5).
Éliminer le flacon de solvant avec l'ensemble du dispositif de transfert.
|
Figure 5
|
| |
11. Faites tourner avec précaution le flacon de Respreeza jusqu'à dissolution complète de la poudre (Figure 6). NE PAS SECOUER. Faites attention de ne pas toucher le bouchon de caoutchouc.
|
Figure 6
|
| |
12. Vérifiez visuellement la solution reconstituée. La solution doit être claire, incolore à jaune pâle et exempte de particules visibles. Ne pas utiliser de solutions décolorées ou troubles ou contenant des particules.
|
|
Si plus d'un flacon de Respreeza est nécessaire pour obtenir la dose requise, vous devez répéter les instructions 1 à 11 indiquées plus haut avec un nouveau dispositif de transfert Mix2Vial non utilisé et une nouvelle boîte.
Utilisez pour chaque flacon de Respreeza un dispositif de transfert Mix2Vial non utilisé et un flacon de solvant stérile.
Le transfert des solutions reconstituées provenant des flacons perforables dans le récipient d'administration (p.ex. poche de perfusion vide ou bouteille en verre (non jointes) au moyen d'un dispositif de transfert pour perfusions intraveineuses disponible dans le commerce (non joint) doit se faire dans des conditions aseptiques.
Administration:
La solution reconstituée doit être administrée au moyen d'un dispositif de perfusion i.v. (non inclus dans l'emballage)
|
1. Assurez-vous que le capuchon évacuateur d'air et la molette de réglage sur le dispositif de perfusion sont fermés. Insérez la pointe du dispositif de perfusion PERPENDICULAIREMENT dans le flacon de produit Respreeza en le tournant légèrement, ou fixez le dispositif de perfusion au récipient d'administration
| |
2. Soulevez le flacon de produit Respreeza ou le récipient d'administration (si vous utilisez une poche à perfusion i.v., suspendez-la à un pied à perfusion).
| |
3. Compressez la chambre compte-gouttes jusqu'à ce que Respreeza la remplisse jusqu'à moitié.
| |
4. Ouvrez gentiment la molette de réglage du kit de perfusion et laissez Respreeza s'écouler jusqu'à ce qu'il n'y ait plus de bulles d'air au bout du tube.
| |
5. Fermez la molette de réglage.
| |
6. Connectez le dispositif de perfusion au dispositif d'injection (par ex. canule papillons/à ailettes ou cathéter à perfusion) et ouvrez à nouveau la molette de réglage.
| |
7. Administrez la solution reconstituée dans une veine. La solution doit être administrée à un débit d'environ 0,08 ml/kg/min, en fonction de votre état clinique et de votre bien-être. L'administration de la dose recommandée de 60 mg/kg prend à peu près 15 minutes.
|
Tout médicament non utilisé et tout déchet doivent être éliminés conformément aux réglementations locales.
Numéro d’autorisation65337 (Swissmedic).
Présentation1000 mg de lyophilisat et solvant (20 ml) (B)
4000 mg de lyophilisat et solvant (76 ml) (B)
Une boîte contient:
·Un flacon perforable avec poudre
·Un flacon perforable avec solvant
·Un dispositif de transfert avec filtre intégré 20/20
Titulaire de l’autorisationCSL Behring AG, Berne.
Mise à jour de l’informationMars 2020.
|