CompositionPrincipe actif
Simoctocog alfa (facteur VIII de coagulation humain recombinant).
Excipients
Saccharose, chlorure de sodium, chlorure de calcium dihydraté, hydrochlorure d'arginine, citrate de sodium dihydraté, poloxamère 188 (soit une teneur totale en sodium de 18,4 mg/flacon).
Solvant: eau pour préparations injectables.
Indications/Possibilités d’emploiTraitement et prophylaxie des hémorragies chez les patients atteints d'hémophilie A (déficit congénital en facteur VIII).
Nuwiq ne contient pas de quantités pharmacologiquement actives de facteur de von Willebrand et il n'est donc pas adapté au traitement du syndrome de von Willebrand-Jürgens.
Posologie/Mode d’emploiLe traitement par Nuwiq sera initié sous la surveillance d'un médecin expérimenté dans la prise en charge des patients atteints d'hémophilie.
Posologie usuelle
La posologie et la durée du traitement de substitution dépendent de la gravité du déficit en facteur VIII, de la localisation et de l'ampleur de l'hémorragie, ainsi que de l'état clinique du patient.
Le nombre d'unités de facteur VIII à administrer est exprimé en Unités Internationales (U.I.), selon le standard actuel de l'OMS pour les concentrés de facteur VIII. L'activité plasmatique du FVIII est exprimée soit en pourcentage (par rapport au plasma humain normal), soit en Unités Internationales (par rapport au standard international pour le FVIII plasmatique). Le test de coagulation en une seule étape tout comme la méthode chromogénique sont appropriés pour mesurer l'activité du FVIII dans le plasma. Les données du test de coagulation en une seule étape sont généralement utilisées dans l'application clinique et pour la comparaison de l'activité avec d'autres préparations de FVIII recombinant et plasmatique.
Une Unité Internationale (U.I.) du FVIII humain est équivalente à la quantité de FVIII présente dans 1 millilitre de plasma humain.
Traitement à la demande
Le calcul de la dose de facteur VIII nécessaire repose sur le résultat empirique qu'une Unité Internationale (UI) de facteur VIII par kg de poids corporel augmente l'activité plasmatique du facteur VIII en moyenne de 2 % par rapport à l'activité normale ou de 2 UI/dl. La dose requise est calculée selon la formule suivante:

La dose et la fréquence d'administration doivent toujours être adaptées de façon individuelle en tenant compte de l'efficacité clinique.
Dans les cas d'événements hémorragiques suivants, l'activité du facteur VIII ne doit pas descendre en dessous du taux d'activité plasmatique indiqué (en % de la normale ou en UI/dl) pendant la période correspondante. Le tableau suivant peut servir de guide pour la détermination des posologies lors des épisodes hémorragiques et des interventions chirurgicales:
|
Intensité de l'hémorragie/Type d'intervention chirurgicale
|
Taux de facteur VIII nécessaire (%) (UI/dl)
|
Fréquences des injections (heures)/Durée de traitement (jours)
| |
Hémorragie
| |
Début d'hémarthroses, saignements musculaires ou buccaux
|
20–40
|
Répéter toutes les 12 à 24 heures pendant au moins 1 jour, jusqu'à la fin de l'épisode hémorragique indiquée par la disparition de la douleur ou l'obtention d'une cicatrisation.
| |
Hémarthroses et hémorragies musculaires plus étendues, ou hématomes
|
30–60
|
Répéter l'injection toutes les 12 à 24 heures pendant 3 à 4 jours ou plus jusqu'à disparition de la douleur et de l'invalidité aiguë.
| |
Hémorragies mettant en jeu le pronostic vital
|
60–100
|
Répéter l'injection toutes les 8 à 24 h jusqu'à disparition du risque vital.
| |
Intervention chirurgicale
| |
Interventions mineures, y compris extractions dentaires
|
30–60
|
Toutes les 24 heures, pendant au moins 1 jour, jusqu'à l'obtention d'une cicatrisation
| |
Interventions chirurgicales majeures
|
80–100
(pré- et postopératoires)
|
Répéter l'injection toutes les 8 à 24 heures jusqu'à obtention d'une cicatrisation adéquate, puis administrer le traitement pendant au moins 7 jours supplémentaires pour maintenir une activité coagulante du facteur VIII entre 30 % et 60 % (UI/dl).
|
Prophylaxie
Les doses habituelles en prophylaxie à long terme des hémorragies chez les patients atteints d'hémophilie A sévère sont de 20 à 40 UI de facteur VIII par kg de poids corporel à des intervalles de 2 à 3 jours. Dans certains cas, en particulier chez les sujets jeunes, il peut s'avérer nécessaire de raccourcir les intervalles entre les doses ou d'augmenter les doses.
Durant le traitement, il est conseillé d'effectuer un contrôle approprié du taux de facteur VIII afin d'ajuster les doses à administrer et la fréquence des injections. Plus particulièrement pour les interventions chirurgicales majeures, un contrôle rigoureux du traitement substitutif par des mesures de la coagulation (activité plasmatique du facteur VIII) est indispensable. Selon les patients, la réponse au traitement par le facteur FVIII peut varier, entraînant des taux de récupération in vivo et des demi-vies différents.
Afin d’assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Patients âgés
Il n'existe que peu d'expérience chez les patients âgés.
Enfants et adolescents
Pour la prévention prolongée des hémorragies chez les patients âgés de moins de 12 ans, une dose de 30 à 50 UI/kg de FVIII tous les 2 jours ou trois fois par semaine est recommandée.
Patients non préalablement traités
La sécurité et l'efficacité de Nuwiq chez des patients non préalablement traités ont été évaluées dans le cadre d'une étude clinique prospective.
Schéma d'administration
Il est recommandé de ne pas administrer plus de 4 ml par minute.
Mode d’administration
Pour administration intraveineuse.
Instructions de reconstitution du médicament avant utilisation, voir le paragraphe «Remarques particulières, remarques concernant la manipulation».
Contre-indicationsHypersensibilité à l'un des principes actifs ou à l'un des excipients mentionnés.
Mises en garde et précautionsHypersensibilité
Comme avec tout médicament contenant des protéines et administré par voie intraveineuse, des réactions allergiques de type hypersensibilité peuvent être constatées. Nuwiq contient des traces d'autres cellules hôtes humaines distinctes du facteur VIII. Si des symptômes d'hypersensibilité apparaissent, il faut indiquer aux patients d'interrompre immédiatement l'administration du produit et de contacter leur médecin. Les patients doivent être informés des signes précoces de réaction d'hypersensibilité incluant urticaire, urticaire généralisée, oppression thoracique, respiration sifflante, hypotension et choc anaphylactique.
En cas de choc, le traitement médical standard de l'état de choc devra être mis en place.
Inhibiteurs
La formation d'anticorps neutralisants (inhibiteurs) du facteur VIII est une complication connue du traitement des patients atteints d'hémophilie A. Ces anticorps sont habituellement des immunoglobulines IgG dirigées contre l'activité pro-coagulante du FVIII et sont quantifiés en unités Bethesda (UB) par ml de plasma à l'aide d'un test modifié. Le risque de développement des anticorps est corrélé à l'exposition au facteur antihémophilique VIII, ce risque étant maximal dans les 20 premiers jours d'exposition (JE). Dans de rares cas, les anticorps peuvent se développer après les 100 premiers JE.
Des cas de réapparition d'anticorps (à faible titre) ont été observés après le remplacement d'un facteur VIII par un autre, chez des patients préalablement traités ayant plus de 100 JE et des antécédents de développement d'anticorps. Il est donc recommandé de surveiller attentivement tous les patients afin de détecter l'apparition d'un anticorps suite à un changement de médicament.
L'immunogénicité de Nuwiq a été évaluée chez des patients atteints d'hémophilie A sévère (<1% FVIII:C), non préalablement traités, dans le cadre d'un essai clinique ouvert prospectif. Sur les 105 patients ayant reçu Nuwiq et ayant été soumis à un dépistage des anticorps neutralisants au moins une fois après le début du traitement, 17 patients (16,2 %) ont développé des anticorps neutralisants à titre élevé et 11 patients (10,5 %) ont développé des anticorps neutralisants à faible titre (dont 5 étaient des anticorps transitoires qui ont été éliminés dans le cadre de la poursuite du traitement prophylactique sans modification de la posologie). Sur les 28 patients ayant développé des anticorps, 25 présentaient moins de 20 JE avant le dépistage. Les délais moyens de développement des anticorps à titre élevé ou à faible titre étaient respectivement de 9,0 JE (fourchette de 4 à 24 JE) et de 12,0 JE (fourchette de 6 à 34 JE). Aucun anticorps neutralisant n'a été détecté chez les patients présentant des mutations non nulles du gène F8.
De manière générale, tous les patients traités par un facteur VIII de coagulation doivent faire l'objet d'une surveillance étroite pour détecter l'apparition d'anticorps par un suivi clinique et à l'aide de tests biologiques appropriés. Si le taux de facteur VIII plasmatique attendu n'est pas atteint ou si l'hémorragie n'est pas contrôlée par une dose adéquate, un test de détection d'anticorps du facteur VIII doit alors être effectué. Chez les patients présentant un titre élevé d'anticorps, le traitement de substitution en facteur VIII peut ne pas être efficace et d'autres options thérapeutiques, telles qu'un protocole d'induction de tolérance immune (ITI), doivent être considérées. Le suivi de tels patients doit être effectué par des médecins expérimentés dans la prise en charge de l'hémophilie et des anticorps du facteur VIII.
Événements cardiovasculaires
Chez les patients présentant des facteurs de risque cardiovasculaire, le traitement substitutif par facteur VIII peut augmenter le risque cardiovasculaire.
Complications liées au cathéter
Si un dispositif d'accès veineux central (DAVC) est nécessaire, le risque de complications liées au DAVC, notamment des infections locales, une bactériémie et une thrombose au site du cathéter, doit être pris en compte.
À chaque administration de Nuwiq à un patient, il est fortement recommandé d'enregistrer le nom et le numéro de lot du produit afin de maintenir un lien entre le patient et le numéro de lot du médicament.
Population pédiatrique
Les mises en garde et mesures de précaution indiquées s'appliquent également aux enfants et aux adultes.
Remarques sur les autres composants (teneur en sodium)
Ce médicament contient moins de 1 mmol de sodium (23 mg) par flacon, c'est-à-dire qu'il est presque exempt de sodium.
Toutefois, selon son poids et la posologie, il se peut qu'un patient reçoive plus d'un flacon (voir la rubrique «Forme pharmaceutique et quantité de principe actif par unité » pour le contenu par flacon). Cela doit être pris en considération par les patients qui suivent un régime contrôlé en sodium.
InteractionsAucune étude des interactions de Nuwiq avec d'autres médicaments n'a été réalisée.
Grossesse, allaitementGrossesse
Il n'existe pas de données suffisantes concernant l'emploi chez la femme enceinte.
Il n'existe pas d'expérimentations animales suffisantes concernant les effets sur la grossesse, le développement embryonnaire, le développement fœtal et/ou le développement post-natal. Le risque potentiel pour l'être humain n'est pas connu.
En raison de la rareté des cas d'hémophilie A chez les femmes, il n'existe aucune donnée sur l'utilisation de Nuwiq chez les femmes enceintes. Par conséquent, Nuwiq ne doit être utilisé pendant la grossesse qu'en cas de nécessité absolue.
Allaitement
En raison de la rareté des cas d'hémophilie A chez les femmes, il n'existe aucune donnée sur l'utilisation de Nuwiq chez les femmes allaitantes. Par conséquent, Nuwiq ne doit être utilisé pendant l'allaitement qu'en cas de nécessité absolue.
Fertilité
Il n'existe pas de données concernant l'effet sur la fertilité.
Effet sur l’aptitude à la conduite et l’utilisation de machinesNuwiq n'a aucune influence sur l'aptitude à la conduite ou à l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
Dans de rares cas, des réactions d'hypersensibilité ou allergiques (par exemple, angio-œdème, brûlure et piqûre au site d'injection, frissons, rougeurs au visage et bouffées de chaleur, céphalées, éruption cutanée, hypotension, léthargie, nausées, éruption cutanées, agitation, tachycardie, oppression thoracique, picotement, urticaire, y compris urticaire généralisée, vomissement, respiration sifflante) ont été observées avec les concentrés de facteur VIII. Dans des cas isolés, ces réactions/symptômes peuvent évoluer vers une anaphylaxie sévère (y compris un choc).
Les patients atteints d'hémophilie A peuvent développer des anticorps neutralisants (inhibiteurs) du facteur VIII. L'apparition de tels anticorps se caractérise par une réponse clinique insuffisante. Dans de tels cas, il est recommandé de contacter un centre spécialisé en hémophilie.
Tableau des effets indésirables
Lors d'études cliniques avec Nuwiq chez des enfants (2 à 11 ans, n = 58), des adolescents (12 à 17 ans, n = 3) et des adultes (n = 186) préalablement traités et atteints d'hémophilie A sévère, 15 effets indésirables du médicament (11 chez les adultes et 4 chez les enfants) ont été rapportés au total chez 9 patients (5 adultes et 4 enfants). Dans une étude clinique portant sur 108 patients (âgés de 0 à 146 mois) atteints d'hémophilie A sévère, non préalablement traités, il a été fait état des effets indésirables du médicament suivants: anticorps neutralisants anti-facteur VIII (inhibiteurs) chez 28 patients, pyrexie chez 21 patients, hypersensibilité chez 10 patients, anémie chez 2 patients et anémie hémorragique chez un patient.
Le Tableau 1 ci-dessous est présenté conformément à la classification des systèmes d'organes MedDRA (CSO et termes préconisés).
La fréquence a été définie selon les critères suivants: très fréquent (≥1/10); fréquent (≥1/100 à <1/10); occasionnel (≥1/1'000 à <1/100); rare (≥1/10'000 à <1/1'000); très rare (<1/10'000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Au sein de chaque groupe de fréquence, les effets indésirables sont indiqués par degré de gravité décroissant.
Tableau 1. Fréquence de survenue des effets indésirables, par patient, lors des études cliniques
|
Classe de systèmes d'organes MedDRA
|
Effets indésirables
|
Fréquence
| |
Troubles du système sanguin et lymphatique
|
Anémie
|
occasionnel*
| |
Anticorps anti-facteur VIII
|
occasionnel (chez les patients préalablement traités)#
très fréquent (chez les patients non préalablement traités)#
| |
Anémie hémorragique
|
occasionnel*
| |
Affections du système immunitaire
|
Hypersensibilité
|
fréquent*
| |
Affections du système nerveux
|
Sensation de vertige;
Céphalées;
Paresthésies
|
occasionnel*
| |
Affections de l'oreille et du labyrinthe
|
Vertiges
|
occasionnel*
| |
Affections respiratoires, thoraciques et médiastinales
|
Dyspnée
|
occasionnel*
| |
Affections gastro-intestinales
|
Sécheresse buccale
|
occasionnel*
| |
Affections musculo-squelettiques et systémiques
|
Lombalgie
|
occasionnel*
| |
Troubles généraux et anomalies au site d'administration
|
Fièvre
|
fréquent*
| |
Douleur thoracique;
Inflammation au site d'administration;
Douleurs au site d'administration;
Malaise
|
occasionnel*
| |
Investigations
|
Présence d'anticorps non neutralisants dirigés contre le facteur VIII (chez les patients non préalablement traités)
|
occasionnel*
|
* Calculé comme patients ayant des effets indésirables du médicament sur le nombre total de 355 patients étudiés, dont 247 patients préalablement traités et 108 patients non préalablement traités.
# La fréquence est déterminée d'après des études sur tous les produits FVIII, menées auprès de patients atteints d'hémophilie A sévère.
Description de certains effets indésirables
Un anticorps non neutralisant dirigé contre le facteur VIII a été observé chez un patient adulte (voir le Tableau 1). L'échantillon a été testé par le laboratoire central à 8 dilutions. Le résultat n'était positif qu'au facteur de dilution 1 et le titre d'anticorps était très faible. Aucune activité inhibitrice, telle que mesurée par le test de Bethesda modifié, n'a été détectée chez ce patient. L'efficacité clinique et la récupération in vivo de Nuwiq n'ont pas été affectées chez ce patient.
Population pédiatrique
On peut supposer que la fréquence, la nature et la gravité des effets indésirables chez les enfants soient les mêmes que chez les adultes.
La déclaration de suspicion d'effets indésirables après l'autorisation de mise sur le marché est d'une grande importance. Elle permet de réévaluer en permanence le rapport bénéfice/risque du médicament. Les professionnels de la santé sont invités à signaler tout soupçon d'effet indésirable nouveau ou grave via le portail en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur le site www.swissmedic.ch.
SurdosageAucun cas de surdosage n'a été rapporté.
Propriétés/EffetsCode ATC
B02BD02
Mécanisme d'action
Le simoctocog alfa (facteur VIII de coagulation humain [ADNr]) est une protéine purifiée constituée de 1440 acides aminés. La séquence d'acides aminés est comparable à la forme 90 kDa et 80 kDa du facteur VIII plasmatique humain (c'est-à-dire sans le domaine B). Nuwiq est produit par la technique de l'ADN recombinant sur cellules rénales embryonnaires humaines (HEK) 293F génétiquement modifiées. Aucun produit d'origine animale ou humaine n'est ajouté au cours du procédé de fabrication ou au médicament final.
L'hémophilie A est un trouble héréditaire de la coagulation sanguine lié au sexe, dû à une diminution du taux de facteur VIII:C et qui se caractérise par des saignements abondants au niveau des articulations, des muscles ou des organes internes, soit spontanément soit suite à un traumatisme accidentel ou chirurgical. Grâce à un traitement de substitution, le taux plasmatique en facteur VIII est augmenté, ce qui permet ainsi une correction temporaire du déficit en facteur VIII et de la tendance au saignement.
Pharmacodynamique
Le complexe facteur VIII / facteur de von Willebrand se compose de deux molécules (facteur VIII et facteur de von Willebrand) aux fonctions physiologiques distinctes. Lorsqu'il est injecté à un patient atteint d'hémophilie A, le facteur VIII se fixe sur le facteur de von Willebrand présent dans la circulation sanguine du patient. Le facteur VIII activé agit comme cofacteur du facteur IX activé, accélérant la conversion du facteur X en facteur X activé. Le facteur X activé convertit la prothrombine en thrombine. La thrombine convertit ensuite le fibrinogène en fibrine, ce qui aboutit à la formation d'un caillot.
Efficacité clinique
L'immunogénicité de Nuwiq a été évaluée dans des études cliniques réalisées chez 135 patients atteints d'hémophilie A sévère préalablement traités (74 adultes et 61 patients pédiatriques). Des anticorps non neutralisants relatifs au facteur VIII ont pu être détectés chez 4 patients. 3 des 4 patients présentaient déjà des anticorps non neutralisants avant l'administration de Nuwiq. Des anticorps non neutralisants dirigés contre le facteur VIII ont été détectés chez un patient adulte. L'échantillon a été testé au travers de 8 dilutions par un laboratoire centralisé. Uniquement avec un facteur de dilution de 1, le résultat du test était positif et le titre d'anticorps était très faible. Une activité inhibitrice, selon le test de Bethesda modifié, n'a pas pu être décelée chez ce patient. L'efficacité clinique et la récupération in vivo de Nuwiq n'ont pas été altérées chez le patient.
Dans une étude clinique réalisée chez 32 patients adultes atteints d'hémophilie A sévère, la dose mensuelle moyenne de Nuwiq lors du traitement prophylactique était de 468,7 UI/kg/mois. La dose moyenne de traitement d'épisodes hémorragiques était de 33,0 UI/kg pour les saignements intermittents chez les patients recevant un traitement prophylactique. Dans une autre étude clinique, 22 patients adultes ont été traités à la demande. Au total, 986 épisodes hémorragiques ont été traités avec une dose moyenne de 30,9 UI/kg. En règle générale, les saignements mineurs nécessitaient des doses légèrement inférieures, et les saignements plus abondants nécessitaient des doses moyennes jusqu'à trois fois plus élevées.
Prophylaxie individualisée: la prophylaxie individualisée sur base de la PK a été évaluée chez 66 patients adultes déjà traités (PTP) et présentant une hémophilie A sévère. Après une phase de prophylaxie standard de 1 à 3 mois (administration un jour sur deux ou 3 fois par semaine), 44 patients (67 %) sont passés à un schéma d'administration basé sur leur évaluation PK, et 40 patients ont terminé les 6 mois de prophylaxie selon le schéma d'administration et de traitement qui leur avait été attribué. Parmi ces patients, 34 (85 %) ont été traités deux fois par semaine ou moins. 33 patients (82,5 %) n'ont présenté aucune hémorragie et 36 patients (90,0 %) n'ont eu aucun saignement spontané. Le taux de saignement annualisé (TSA; moyen ± ET) était de 1,2 ± 3,9 et la dose moyenne ± ET était de 52,2 ± 12,2 UI/kg par injection et 99,7 ± 25,6 UI/kg par semaine.
Il est à noter que le taux de saignement annualisé (TSA) n'est pas comparable entre les différents concentrés de facteur et entre les différentes études cliniques.
Sécurité et efficacité chez les patients pédiatriques
Les données ont été obtenues chez 29 enfants préalablement traités âgés de 2 à 5 ans, 31 enfants âgés de 6 à 12 ans et un adolescent de 14 ans. La dose moyenne par perfusion prophylactique était de 37,8 UI/kg. Vingt patients ont utilisé des doses moyennes de plus de 45 UI/kg. La dose moyenne mensuelle de Nuwiq pour la prophylaxie était de 521,9 UI/kg. La dose moyenne de Nuwiq nécessaire pour traiter les hémorragies chez les enfants (43,9 UI/kg) était supérieure à la dose moyenne nécessaire chez les adultes (33,0 UI/kg). Une dose moyenne plus élevée a été nécessaire pour traiter les hémorragies modérées à majeures par rapport aux hémorragies mineures (78,2 UI/kg contre 41,7 UI/kg). Les jeunes enfants ont en général eu besoin de doses moyennes plus élevées (6-12 ans: 43,9 UI/kg; 2-5 ans: 52,6 UI/kg). Ces données ont été corroborées par le suivi à long terme de 49 de ces enfants qui ont bénéficié d'une période de traitement supplémentaire médiane d'environ 30 mois (fourchette de 9,5 à 52 mois). Au cours de cette période, 45 % des enfants n'ont présenté aucun saignement spontané.
PharmacocinétiqueAbsorption
Non applicable.
Distribution
Le FVIII se distribue dans le plasma.
Métabolisme
Non applicable.
Élimination
Voir les tableaux 2 à 4.
Tableau 2. Paramètres pharmacocinétiques de Nuwiq (dose: 50 UI/kg) chez des patients adultes préalablement traités (âgés de 18 à 65 ans) atteints d'hémophilie A sévère (n = 20)
|
Paramètre pharmacocinétique
|
Méthode chromogénique
|
Test de coagulation en une étape
| |
Moyenne ± ET
|
Moyenne ± ET
| |
ASC (h*UI/ml/(UI/kg))
|
0,39 ± 0,14
|
0,37 ± 0,11
| |
T½ (h)
|
14,7 ± 10,4
|
17,0 ± 11,8
| |
IVR (%/UI/kg)
|
2,5 ± 0,4
|
2,2 ± 0,3
| |
CL (ml/h/kg)
|
3,0 ± 1,2
|
2,9 ± 1,0
|
ASC = Aire sous la courbe (FVIII:C), T½ = demi-vie terminale,
IVR = récupération in vivo incrémentale, Cl = Clairance, ET = écart-type
Cinétique des groupes de patients particuliers
Sous-groupes ajustés sur le poids
Chez les patients adultes pré-obèses (IMC 25-30 kg/m2) et chez les patients obèses (IMC >30 kg/m2) de l'étude de pharmacocinétique, la clairance était supérieure à celle des patients ayant un poids normal.
Population pédiatrique
Tableau 3. Paramètres pharmacocinétiques de Nuwiq (dose: 50 UI/kg) chez des enfants préalablement traités âgés de 6 à 12 ans atteints d'hémophilie A sévère (n = 12)
|
Paramètre pharmacocinétique
|
Méthode chromogénique
|
Test de coagulation en une étape
| |
Moyenne ± ET
|
Moyenne ± ET
| |
ASC (h*UI/ml/(UI/kg))
|
0,25 ± 0,1
|
0,26 ± 0,1
| |
T½(h)
|
10,0 ± 1,9
|
13,1 ± 2,6
| |
IVR (%/UI/kg)
|
1,9 ± 0,4
|
1,6 ± 0,4
| |
CL (ml/h/kg)
|
4,3 ± 1,2
|
4,1 ± 0,9
|
ASC = Aire sous la courbe (FVIII:C), T½ = demi-vie terminale,
IVR = récupération in vivo incrémentale, Cl = Clairance, ET = écart-type
Tableau 4. Paramètres pharmacocinétiques de Nuwiq (dose: 50 UI/kg) chez des enfants préalablement traités âgés de 2 à 5 ans atteints d'hémophilie A sévère (n = 13)
|
Paramètre pharmacocinétique
|
Méthode chromogénique
|
Test de coagulation en une étape
| |
Moyenne ± ET
|
Moyenne ± ET
| |
ASC (h*UI/ml/(UI/kg))
|
0,22 ± 0,1
|
0,22 ± 0,1
| |
T½(h)
|
9,5 ± 3,3
|
11,9 ± 5,4
| |
IVR (%/UI/kg)
|
1,9 ± 0,3
|
1,6 ± 0,2
| |
CL (ml/h/kg)
|
5,4 ± 2,4
|
5,4 ±2,3
|
ASC = Aire sous la courbe (FVIII:C), T½ = demi-vie terminale,
IVR = récupération in vivo incrémentale, Cl = Clairance, ET = écart-type
Comme la littérature le mentionne, la récupération et la demi-vie étaient plus faibles chez les jeunes enfants que chez les adultes, et la clairance était plus élevée, ce qui peut être dû en partie au fait que le volume plasmatique par kilogramme de poids corporel est supérieur chez les patients plus jeunes.
Données précliniquesLes études toxicologiques ont montré que l'administration intraveineuse locale et l'exposition systémique étaient bien tolérées chez les animaux de laboratoire (rats et singes cynomolgus). L'implication de l'administration répétée chez le singe du Simoctocog alfa dans la diminution de poids du thymus reste incertaine.
En raison de la réponse immunitaire aux protéines hétérologues attendues chez toutes les espèces de mammifères non humaines, il n'a été mené aucune étude spécifique à doses répétées avec Nuwiq sur une période prolongée (telles que des études sur la toxicité de reproduction, la toxicité chronique et la cancérogénicité).
Aucune étude sur le potentiel mutagène de Nuwiq n'a été réalisée.
Des études ex vivo effectuées à l'aide d'un kit de test commercial pour quantifier la réponse des lymphocytes T aux protéines thérapeutiques indiquent une immunogénicité au moins aussi marquée que celle des produits comparables.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments.
Utiliser exclusivement le dispositif de perfusion fourni, car un échec du traitement peut survenir suite à l'adsorption du facteur VIII de coagulation humain sur les surfaces internes de certains dispositifs de perfusion.
Stabilité
Le médicament ne doit pas être utilisé après la date de péremption indiquée sur l'emballage après la mention «Verw. bis/EXP».
Pendant la durée de conservation, la préparation peut être entreposée à température ambiante (jusqu'à 25 °C) pendant une durée ne dépassant pas 1 mois. Le produit ne doit pas être replacé au réfrigérateur après en avoir été sorti. Veuillez noter la date de début de conservation à température ambiante sur l'emballage. Le flacon doit être conservé dans son emballage d'origine afin de le protéger de la lumière.
Après la reconstitution
Du point de vue microbiologique, le médicament doit être utilisé immédiatement après la reconstitution. À défaut, la solution reconstituée conservée à une température ambiante ≤25 °C doit être utilisée dans un délai de 3 heures. La solution reconstituée doit être conservée dans le flacon.
Les préparations inutilisées, qui sont restées à température ambiante plus de 3 heures, doivent être éliminées.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Ne pas congeler.
Conserver le récipient dans son carton pour le protéger de la lumière.
Tenir hors de portée des enfants.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique «Remarques particulières: conservation — après la reconstitution».
Traitement des déchets
Tout produit non utilisé ou déchet doit être éliminé conformément aux exigences locales.
Remarques concernant la manipulation
La poudre ne doit être reconstituée qu'avec le solvant fourni (2,5 ml d'eau pour préparations injectables) en utilisant le dispositif de perfusion fourni. Le flacon doit être agité doucement par rotation jusqu'à dissolution complète de la poudre. Après reconstitution, la solution est aspirée dans la seringue.
Avant toute administration, inspecter minutieusement le médicament reconstitué pour déceler des particules en suspension ou une décoloration. Le médicament reconstitué est une solution limpide et incolore, exempte de particules étrangères, dont le pH est compris entre 6,5 et 7,5. Ne pas utiliser de solutions troubles ou présentant des dépôts.
Reconstitution
1.Amenez à température ambiante la seringue de solvant (eau pour préparations injectables) et la poudre du flacon fermé avant toute utilisation. Pour cela, tenez-les dans vos mains jusqu'à ce qu'elles soient aussi chaudes que vos mains. Ne pas utiliser d'autres moyens pour chauffer le flacon et la seringue. Cette température doit être maintenue au cours de la reconstitution.
2.Enlevez l'opercule amovible en plastique du flacon de poudre afin de découvrir la partie centrale du bouchon en caoutchouc. Ne pas retirer le bouchon gris ni la bague métallique (capsule à sertir) entourant la partie supérieure du flacon.

3.Nettoyez le dessus du flacon à l'aide d'un tampon imbibé d'alcool. Laissez sécher.
4.Retirez le papier protecteur de l'emballage du dispositif de transfert pour flacon. Ne pas retirer le dispositif de son emballage.

5.Placez et maintenez le flacon de poudre sur une surface plane. Tout en tenant l'emballage du dispositif de transfert, placez le dispositif au-dessus du centre du bouchon en caoutchouc du flacon de poudre. Appuyez fermement sur l'emballage du dispositif de transfert jusqu'à ce que la pointe du dispositif pénètre dans le bouchon en caoutchouc. Une fois l'opération terminée, le dispositif de transfert s'enclenche sur le flacon.
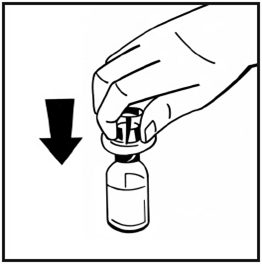
6.Retirez le papier protecteur de l'emballage de la seringue préremplie. Tenez la tige du piston par l'extrémité et ne touchez pas l'axe. Fixez l'extrémité filetée de la tige du piston à la seringue de solvant. Faites tourner la tige du piston dans le sens des aiguilles d'une montre jusqu'à ce qu'une légère résistance soit ressentie.
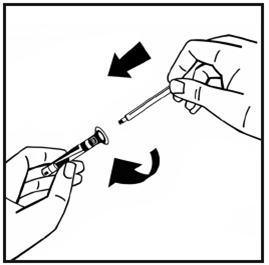
7.Retirez l'embout protecteur d'inviolabilité en plastique de la seringue de solvant après avoir rompu la zone perforée du capuchon. Ne pas toucher l'intérieur de l'opercule ni l'embout de la seringue. Si la solution n'est pas utilisée immédiatement, fermez la seringue remplie, au moyen de l'embout, pour la conserver.
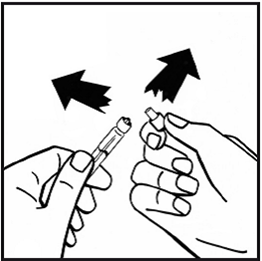
8.Retirez l'emballage du dispositif de transfert et jetez-le.
9.Fixez la seringue de solvant au dispositif de transfert en poussant fermement et en tournant dans le sens des aiguilles d'une montre jusqu'à ce qu'une légère résistance soit ressentie.

10.Injectez lentement tout le solvant dans le flacon de poudre en appuyant sur la tige du piston.
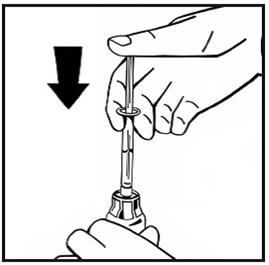
11.Pour dissoudre la poudre, secouez ou remuez délicatement le flacon circulairement pendant quelques secondes sans extraire la seringue. Ne pas agiter. Attendez que la poudre soit totalement dissoute.
12.Avant toute administration, vérifiez visuellement l'absence de particules dans la solution obtenue. La solution doit être limpide, incolore et pratiquement exempte de particules visibles. Ne pas utiliser de solutions troubles ou présentant des dépôts.
13.Retournez le flacon fixé à la seringue, et aspirez lentement la solution dans la seringue. Veillez à ce que la totalité du flacon soit transférée dans la seringue.

14.Détachez la seringue remplie du dispositif de transfert en tournant dans le sens contraire des aiguilles d'une montre, et jetez le flacon vide.
15.La solution est à présent prête pour une utilisation immédiate. Ne pas la mettre au réfrigérateur.
16.Nettoyez le site d'injection choisi avec l'un des tampons imbibés d'alcool fournis.
17.Fixez la tubulure du nécessaire à perfusion fourni à la seringue. Introduisez l'aiguille du nécessaire à perfusion dans la veine choisie. Si vous avez utilisé un garrot pour rendre la veine plus visible, enlevez le garrot avant de commencer l'injection de la solution. Du sang ne doit pas pénétrer dans la seringue, en raison du risque de formation de caillots de fibrine.
18.Injectez lentement la solution dans la veine, le débit ne doit pas excéder 4 ml par minute.
Si vous utilisez plus d'un flacon de poudre au cours d'un même traitement, vous pouvez réutiliser la même aiguille d'injection. Le dispositif de transfert et la seringue sont à usage unique.
Numéro d’autorisation65551 (Swissmedic).
PrésentationNuwiq est disponible en emballages contenant respectivement 1 flacon de 250 UI (100 UI/ml), 500 UI (200 UI/ml), 1000 UI (400 UI/ml) et 2000 UI (800 UI/ml).
Chaque emballage contient:
1 flacon contenant 250/500/1000/2000 Unités Internationales de Simoctocog alfa, 1 seringue préremplie de 2,5 ml d'eau pour préparations injectables, 1 dispositif de transfert pour le flacon, une aiguille (à ailettes) et deux tampons imbibés d'alcool.
Catégorie de remise B
Titulaire de l’autorisationOctapharma AG
8853 Lachen
Mise à jour de l’informationNovembre 2023
|