CompositionPrincipes actifs
Pregabalinum.
Excipients
Gélule à 25 mg: lactosum monohydricum (35 mg), maydis amylum, talcum, gelatina, aqua, titanii dioxidum, natrii laurilsulfas (E487, corresp. 0,006 mg natrium), silica colloidalis anhydrica, lacca, ferrum oxydatum nigrum, propylenglycolum (E1520), hydroxidum kalii.
Gélule à 50 mg: lactosum monohydricum (70 mg), maydis amylum, talcum, gelatina, aqua, titanii dioxidum, natrii laurilsulfas (E487, corresp. 0,008 mg natrium), silica colloidalis anhydrica, lacca, ferrum oxydatum nigrum, propylenglycolum (E1520), hydroxidum kalii.
Gélule à 75 mg: lactosum monohydricum (8,25 mg), maydis amylum, talcum, gelatina, aqua, titanii dioxidum, ferrum oxydatum rubrum, natrii laurilsulfas (E487, corresp. 0,006 mg natrium), silica colloidalis anhydrica, lacca, ferrum oxydatum nigrum, propylenglycolum (E1520), hydroxidum kalii.
Gélule à 100 mg: lactosum monohydricum (11 mg), maydis amylum, talcum, gelatina, aqua, titanii dioxidum, ferrum oxydatum rubrum, natrii laurilsulfas (E487, corresp. 0,008 mg natrium), silica colloidalis anhydrica, lacca, ferrum oxydatum nigrum, propylenglycolum (E1520), hydroxidum kalii.
Gélule à 150 mg: lactosum monohydricum (16,50 mg), maydis amylum, talcum, gelatina, aqua, titanii dioxidum, natrii laurilsulfas (E487, corresp. 0,010 mg natrium), silica colloidalis anhydrica, lacca, ferrum oxydatum nigrum, propylenglycolum (E1520), hydroxidum kalii.
Gélule à 200 mg: lactosum monohydricum (22 mg), maydis amylum, talcum, gelatina, aqua, titanii dioxidum, ferrum oxydatum rubrum, natrii laurilsulfas (E487, corresp. 0,012 mg natrium), silica colloidalis anhydrica, lacca, ferrum oxydatum nigrum, propylenglycolum (E1520), hydroxidum kalii.
Gélule à 300 mg: lactosum monohydricum (33 mg), maydis amylum, talcum, gelatina, aqua, titanii dioxidum, ferrum oxydatum rubrum, natrii laurilsulfas (E487, corresp. 0,015 mg natrium), silica colloidalis anhydrica, lacca, ferrum oxydatum nigrum, propylenglycolum (E1520), hydroxidum kalii.
Indications/Possibilités d’emploiDouleurs neuropathiques
Pregabalin Viatris est indiqué dans le traitement des douleurs neuropathiques périphériques et centrales chez l'adulte.
Des études cliniques ont démontré l'efficacité de la prégabaline contre les douleurs neuropathiques dans la polyneuropathie diabétique, la névralgie postherpétique, ainsi que dans les lésions de la moelle épinière (modèles pour les douleurs neuropathiques d'origine centrale) (voir «Propriétés/Effets», Efficacité clinique).
Épilepsie
Pregabalin Viatris est utilisé dans le traitement adjuvant des crises d'épilepsie partielles comportant ou non une généralisation secondaire chez les patients adultes qui répondent de façon insuffisante à d'autres antiépileptiques.
Troubles anxieux généralisés
Pregabalin Viatris est utilisé pour le traitement des troubles anxieux généralisés de l'adulte.
Posologie/Mode d’emploiLa posologie se situe entre 150 et 600 mg par jour, administrée en deux ou trois prises unitaires.
La prégabaline peut être prise avec ou entre les repas.
Douleurs neuropathiques
|
Traitement initial
|
150 mg par jour (75 mg 2x/jour ou 50 mg 3x/jour)
|
En fonction de la réponse et de la tolérance du patient, la posologie peut être augmentée à 300 mg par jour après un intervalle de 3 à 7 jours, administrés en deux ou trois prises unitaires. Si nécessaire, la dose peut être augmentée à la dose maximale de 600 mg par jour après un intervalle supplémentaire de 7 jours.
Dans les études cliniques réalisées dans les polyneuropathies diabétiques, les doses de 300 mg et 600 mg ont été significativement supérieures au placebo.
Dans les études cliniques réalisées dans les névralgies postherpétiques, les doses de 150 mg, 300 mg et 600 mg ont été significativement supérieures au placebo.
Épilepsie
|
Traitement initial
|
150 mg par jour (75 mg 2x/jour ou 50 mg 3x/jour)
|
En fonction de la réponse et de la tolérance du patient, la posologie peut être augmentée à 300 mg par jour après un intervalle de 1 semaine, administrés en deux ou trois prises unitaires. L'augmentation jusqu'à la dose maximale de 600 mg par jour en deux ou trois prises unitaires est également possible après un délai supplémentaire d'une semaine.
Chez les patients qui présentent des crises partielles, les doses de 300 mg et de 600 mg ont été significativement supérieures au placebo.
Chez les patients qui présentent des crises avec généralisation secondaire, seule la dose maximale de 600 mg a été significativement supérieure au placebo.
Il n'est pas nécessaire de doser la concentration plasmatique de prégabaline pour optimiser le traitement par la prégabaline.
Troubles anxieux généralisés
|
Traitement initial
|
150 mg par jour (75 mg 2x/jour ou 50 mg 3x/jour)
|
En fonction de la réponse clinique et de la tolérance individuelle, la posologie peut être augmentée à 300 mg par jour après un intervalle d'une semaine. La dose peut être augmentée à 450 mg par jour après une semaine supplémentaire. La dose maximale de 600 mg par jour peut être atteinte après un délai supplémentaire d'une semaine.
Interruption de la prise de prégabaline
Dans la pratique clinique habituelle, il est recommandé à l'arrêt du traitement, quelle que soit l'indication, de diminuer progressivement la dose de prégabaline sur une période d'une semaine au moins.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la posologie n'est nécessaire chez les patients présentant une insuffisance hépatique (voir «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
La prégabaline est éliminée de la circulation générale principalement sous forme inchangée, par voie rénale. La clairance de la prégabaline étant directement proportionnelle à la clairance de la créatinine (voir «Pharmacocinétique»), la posologie sera réduite individuellement chez les patients présentant une insuffisance rénale en tenant compte de la clairance de la créatinine (Clcr). Les doses indiquées dans le tableau 1 ont été calculées selon la formule suivante:
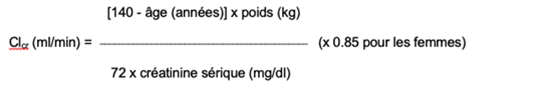
La prégabaline ne doit pas être utilisée chez les patients présentant une insuffisance rénale sévère (Clcr <30 ml/min).
Tableau 1: Adaptation de la posologie de prégabaline selon la fonction rénale
|
Clairance de la créatinine (Clcr) (ml/min)
|
Dose journalière totale de prégabaline*
|
Schéma posologique
| |
|
Dose initiale (mg/jour)
|
Dose maximale (mg/jour)
|
| |
≥60
|
150
|
600
|
en 2 ou 3 prises unitaires
| |
30-60
|
75
|
300
|
en 2 ou 3 prises unitaires
|
* La quantité en mg par prise s'obtient en divisant la dose journalière totale (mg/jour) par le nombre de prises unitaires.
Patients âgés
Une réduction de la posologie n'est nécessaire chez les patients âgés (plus de 65 ans) que s'ils présentent une insuffisance rénale (voir Tableau 1).
Enfants et adolescents
L'innocuité et l'efficacité de la prégabaline n'ont pas été étudiées chez l'enfant et l'adolescent de moins de 18 ans. En conséquence, l'utilisation de Pregabalin Viatris n'est pas recommandée dans ces tranches d'âge.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients (voir «Composition»).
Mises en garde et précautionsLa sécurité d'emploi de la prégabaline n'a pas été étudiée dans les cas d'insuffisance rénale sévère.
La sécurité d'emploi de la prégabaline n'a pas été étudiée chez les patients présentant des troubles de la fonction hépatique (voir «Pharmacocinétique»).
Insuffisance rénale
Des cas d'insuffisance rénale ont été rapportés. Bien que l'effet d'un arrêt du traitement sur la réversibilité d'une insuffisance rénale n'ait pas été systématiquement étudié, des rapports existent sur l'amélioration de la fonction rénale après l'arrêt ou la réduction posologique de la prégabaline.
Insuffisance cardiaque
La prégabaline n'a pas été testée chez des patients présentant une insuffisance cardiaque. En conséquence, l'effet de la prégabaline favorisant les œdèmes pourrait avoir des répercussions défavorables chez ces patients.
Des rapports issus de la surveillance post-marketing font cas d'insuffisance cardiaque chez quelques patients traités par la prégabaline. Ces effets sont observés essentiellement pendant le traitement par la prégabaline pour une indication neuropathique chez les patients âgés dont la fonction cardiovasculaire est altérée. La prégabaline doit être utilisée avec prudence chez ces patients (voir «Effets indésirables»). Lors de traitements à court terme de patients sans maladie cardiaque ou vasculaire périphérique cliniquement significative, aucune association n'a été mise en évidence entre les œdèmes périphériques et les complications cardiovasculaires comme l'hypertension ou l'insuffisance cardiaque.
Diabète
Certains patients diabétiques chez lesquels un traitement par la prégabaline entraîne une prise de poids devront peut-être réajuster leur médication antidiabétique.
Réactions d'hypersensibilité
Dans le cadre de l'expérience post-marketing, des réactions d'hypersensibilité, y compris des cas d'angio-œdèmes, ont été rapportés. La prise de prégabaline doit immédiatement être arrêtée si des symptômes d'angio-œdèmes – tels que gonflement du visage, de la bouche ou des voies respiratoires supérieures – se présentent.
Réactions cutanées médicamenteuses sévères (SCAR, severe cutaneous adverse reactions)
Des réactions cutanées médicamenteuses sévères (SCAR) liées au traitement par la prégabaline ont été rapportées dans de rares cas, notamment un syndrome de Stevens-Johnson (SJS) et une nécrolyse épidermique toxique (NET). Elles peuvent engager le pronostic vital ou être fatales. Au moment de la prescription de la prégabaline, les patients doivent être informés des signes et symptômes et doivent être étroitement suivis pour détecter toute réaction cutanée. En cas d'apparition de signes et de symptômes laissant penser à ces réactions cutanées médicamenteuses, le traitement par la prégabaline doit être immédiatement interrompu et un autre traitement doit être envisagé.
Troubles visuels
Au cours des études contrôlées, les patients sous prégabaline se sont plus souvent plaints d'une vision floue que les patients sous placebo. Dans la majorité des cas, cet effet indésirable a disparu avec la poursuite du traitement. Au cours des études cliniques contrôlées, des examens ophtalmologiques (comprenant des tests de l'acuité visuelle et du champ visuel, de même qu'un examen du fond d'œil en détail) ont été réalisés chez plus de 3600 patients. Une réduction de l'acuité visuelle a été constatée chez 6,5% des patients sous prégabaline contre 4,8% des patients sous placebo. Une modification du champ visuel a été observée chez 12,4% des patients sous prégabaline contre 11,7% des patients sous placebo. Le fond d'œil a permis de constater des modifications chez 1,7% des patients sous prégabaline contre 2,1% des patients sous placebo.
Des effets indésirables au niveau de l'œil ont également été rapportés au cours des expériences post-marketing. La plupart du temps, il s'agissait de vision passagèrement floue ou d'autres troubles de l'acuité visuelle. L'arrêt de la prise de prégabaline peut dans ces cas-là entraîner la disparition ou l'amélioration des symptômes visuels.
Encéphalopathie
Des cas d'encéphalopathie ont été rapportés, principalement chez les patients présentant des antécédents qui peuvent favoriser l'apparition d'une encéphalopathie.
Étourdissements, somnolence, perte de conscience, confusion et altération mentale
Le traitement par la prégabaline a été associé à des étourdissements et de la somnolence qui peuvent conduire à la survenue plus fréquente de traumatismes liés à des chutes chez les personnes âgées. Des cas de perte de conscience, de confusion et d'altération de l'état mental ont également été rapportés dans des rapports post-marketing. En conséquence, on conseillera aux patients d'être prudents jusqu'à ce qu'ils soient habitués aux effets potentiels du médicament.
Arrêt du traitement antiépileptique concomitant
Il n'existe pas de données suffisantes permettant un arrêt du traitement antiépileptique concomitant dans le but d'instaurer une monothérapie, lorsqu'un contrôle des crises est atteint avec la prégabaline en association.
Symptômes de sevrage
Après interruption d'un traitement à court ou à long terme par la prégabaline, des symptômes de sevrage ont été observés chez certains patients. Les événements suivants ont été rapportés: troubles du sommeil, céphalées, nausées, diarrhée, symptômes de type grippal, nervosité, dépression, anxiété, douleurs, convulsions, sudation et vertiges (voir «Effets indésirables»). L'apparition de symptômes de sevrage après l'arrêt de la prégabaline peut indiquer une dépendance au médicament (voir «Mésusage, usage nocif et dépendance»). Le patient doit être informé en début de traitement de ce phénomène.
Si la prégabaline doit être arrêtée, il est recommandé de le faire progressivement sur une période d'au moins 1 semaine, indépendamment de l'indication.
Concernant l'arrêt d'un traitement à long terme par la prégabaline, des données suggèrent que l'apparition et la sévérité des symptômes de sevrage peuvent être dose-dépendantes.
Les études actuelles ne permettent pas de fournir des indications précises sur la fréquence et la gravité des symptômes de sevrage observés en fonction de la durée du traitement et de la posologie après l'arrêt d'un traitement à long terme par la prégabaline.
Mésusage, usage nocif et dépendance
Des cas de mésusage, d'usage nocif et de dépendance pouvant également survenir à des doses thérapeutiques ont été rapportés. La prudence est de mise chez les patients ayant des antécédents d'usage nocif de substances, y compris de médicaments (actuels et/ou passés), car ceux-ci présentent un risque accru d'usage nocif de la prégabaline (voir «Propriétés/Effets»).
Ceci est également valable pour les patients avec des antécédents d'affections psychiques.
Avant de prescrire de la prégabaline, le risque de mésusage, d'usage nocif et/ou de dépendance doit être soigneusement évalué pour chaque patient.
Les patients traités par prégabaline doivent être surveillés pour détecter les signes et symptômes d'usage nocif, d'usage incorrect ou de dépendance à la prégabaline (p.ex. développement d'une tolérance, doses croissantes, «drug seeking behaviour») (voir «Effets indésirables»).
Idées et comportement suicidaires
Des idées et un comportement suicidaires ont été rapportés chez des patients traités par des antiépileptiques dans différentes indications. Une méta-analyse d'études randomisées contrôlées par placebo, portant sur des antiépileptiques, a aussi montré un risque légèrement accru de survenue d'idées et de comportement suicidaires. Une étude épidémiologique a relevé un risque accru d'idées suicidaires et de décès par suicide pendant la prise de prégabaline par rapport aux périodes sans prégabaline, surtout dans le groupe d'âge de moins de 55 ans.
Le mécanisme du déclenchement de cet effet indésirable est inconnu et les données disponibles n'excluent pas la possibilité d'une augmentation du risque lors de la prise de prégabaline.
Les patients devront donc être surveillés quant aux signes d'idées et de comportement suicidaires et un traitement approprié devra être envisagé. L'arrêt du traitement par la prégabaline doit être envisagé en cas d'apparition d'idées et de comportement suicidaires. Les patients (et les personnes prenant soin d'eux) devront être avertis de la nécessité de faire appel à un médecin en cas de survenue de signes d'idées ou de comportement suicidaires.
Traitement de douleurs neuropathiques centrales dues à une lésion de la moelle épinière
Les effets indésirables en général et les effets centraux, en particulier la somnolence, ont été plus fréquents lors du traitement de douleurs neuropathiques centrales après lésion de la moelle épinière. Ceci est probablement dû à un effet additif résultant de la comédication (p.ex. spasmolytiques) nécessaire dans ces cas. Il faudra en tenir compte lors de la prescription de prégabaline pour le traitement de cette pathologie.
Dépression respiratoire
Une dépression respiratoire sévère a été rapportée en lien avec l'utilisation de prégabaline. Le risque de survenue de cet effet secondaire sévère peut être accru chez les patients présentant une altération de la fonction respiratoire ou souffrant d'affections respiratoires ou neurologiques, d'insuffisance rénale ainsi que chez les patients qui utilisent concomitamment des dépresseurs du SNC et chez les patients âgés. Chez ces patients, la dose doit être adaptée, le cas échéant (voir «Posologie/Mode d'emploi»).
Administration concomitante avec des opiacés
La prudence est de mise en cas de prescription simultanée de prégabaline et d'opiacés en raison du risque de dépression du SNC. Au cours d'une étude cas-témoins menée auprès d'utilisateurs d'opioïdes, les patients qui prenaient de la prégabaline concomitante avec un opioïde présentaient un risque accru de décès lié aux opioïdes par rapport à ceux qui prenaient les opioïdes uniquement (adjusted odds ratio [aOR] 1,68 [IC à 95%, 1,19 à 2,36]). Ce risque accru a été déjà observé à des doses faibles de prégabaline (≤300 mg, aOR 1,52 [IC à 95%, 1,04-2,22]), mais un risque encore plus élevé a été constaté à des doses élevées de prégabaline (>300 mg, aOR 2,51 [IC à 95%, 1,24-5,06]).
Diminution de la fonctionnalité du transit du tractus gastro-intestinal inférieur
Après la commercialisation, des cas de diminution de la fonctionnalité du tractus gastro-intestinal inférieur (p.ex. obstruction intestinale, iléus paralytique, constipation) ont été rapportés lorsque la prégabaline était administrée en association avec des médicaments pouvant entraîner une constipation tels que les analgésiques opioïdes (voir «Interactions»). Lorsque la prégabaline est utilisée en association à des opioïdes, des mesures de prévention de la constipation doivent être envisagées (en particulier chez les femmes et les patients âgés).
Femmes en âge de procréer/contraception
L'utilisation de prégabaline au cours du premier trimestre de grossesse peut provoquer des malformations congénitales majeures (définies par le réseau European Surveillance of Congenital Anomalies [EUROCAT], version 2014) chez l'enfant à naître et avoir un effet négatif sur le développement neurologique post-natal ainsi que sur le poids de naissance. La prégabaline ne doit pas être utilisée pendant la grossesse, sauf si le bénéfice pour la mère l'emporte clairement sur les risques potentiels pour le fœtus. Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant le traitement (voir «Grossesse, Allaitement»).
Excipients revêtant un intérêt particulier
Pregabalin Viatris contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne devraient pas prendre Pregabalin Viatris.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par gélule, c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsLa prégabaline est éliminée par les reins, principalement sous forme inchangée, et n'est pratiquement pas métabolisée par l'organisme humain (<2% d'une dose sont retrouvés sous forme de métabolites dans les urines). In vitro, la prégabaline n'inhibe pas le métabolisme des médicaments et ne se lie pas aux protéines plasmatiques. Il est donc peu probable que la prégabaline provoque des interactions médicamenteuses ou en soit l'objet.
Par conséquent, aucune interaction pharmacocinétique cliniquement significative n'a été observée dans les études in vivo entre la prégabaline et la phénytoïne, la carbamazépine, l'acide valproïque, la lamotrigine, la gabapentine, le lorazépam, l'oxycodone ou l'éthanol. De plus, des analyses ont montré que les trois classes de médicaments les plus souvent utilisés que sont les antidiabétiques oraux, les diurétiques et l'insuline, de même que les antiépileptiques souvent administrés que sont la phénytoïne, la carbamazépine, l'acide valproïque, la lamotrigine, le phénobarbital, la tiagabine et le topiramate, n'avaient pas d'effet cliniquement significatif sur la clairance de la prégabaline. De même, ces analyses ont montré que la prégabaline n'a pas d'influence cliniquement significative sur la clairance des substances suivantes: phénytoïne, carbamazépine, acide valproïque, lamotrigine, topiramate et phénobarbital.
L'administration concomitante de prégabaline avec les contraceptifs oraux à base de noréthistérone et/ou d'éthinylestradiol n'influence pas les paramètres pharmacocinétiques à l'état d'équilibre de l'une ou l'autre de ces substances.
La prégabaline peut potentialiser les effets de l'éthanol et du lorazépam. Dans des études cliniques contrôlées, l'administration de doses orales multiples de prégabaline en même temps que l'oxycodone, le lorazépam ou l'éthanol n'a pas provoqué d'effets cliniquement pertinents sur la fonction respiratoire.
Une insuffisance respiratoire, un coma et des décès ont été rapportés dans le cadre de l'expérience post-marketing chez les patients prenant simultanément de la prégabaline et d'autres médicaments à effet dépresseur sur le SNC (en particulier des opioïdes), y compris chez les patients toxicomanes.
L'altération de la fonction cognitive et motrice globale provoquée par l'oxycodone semble être accentuée par la prégabaline.
En cas d'administration simultanée de prégabaline et de médicaments pouvant provoquer une constipation (p.ex. opioïdes), une insuffisance fonctionnelle du tractus gastro-intestinal inférieur a été rapportée (p.ex. iléus, iléus paralytique, constipation) dans le cadre des expériences post-marketing.
Aucune étude pharmacodynamique spécifique n'a été conduite chez des volontaires âgés.
Grossesse, allaitementGrossesse
On ne dispose que de données limitées sur l'administration de la prégabaline chez la femme enceinte.
Les données issues d'une étude observationnelle portant sur plus de 2700 expositions à la prégabaline pendant la grossesse, obtenues grâce à des données collectées régulièrement dans les registres administratifs et de santé danois, finlandais, norvégiens et suédois, n'ont révélé aucun risque significativement plus élevé de malformations congénitales majeures (définies dans l'European Surveillance of Congenital Anomalies [EUROCAT], version 2014), d'effets indésirables sur la naissance (p.ex. mortinaissances) ou de développement neurologique post-natal anormal après une exposition à la prégabaline pendant la grossesse (voir ci-après).
Malformations congénitales majeures (définies dans l'European Surveillance of Congenital Anomalies [EUROCAT], version 2014)
Dans les méta-analyses standard, le rapport de prévalence ajusté (aPR) et l'intervalle de confiance (IC) à 95% s'élevaient à 1,14 (0,96-1,35) pour les grossesses exposées à la prégabaline en monothérapie au cours du premier trimestre par rapport aux grossesses non exposées à des antiépileptiques.
Effets sur la naissance et troubles du développement neurologique post-natal
Aucun effet statistiquement significatif n'a été constaté en ce qui concerne une mortinaissance, un faible poids de naissance, une prématurité, une taille ou un poids insuffisant pour l'âge gestationnel (c.-à-d. un nouveau-né dont le poids de naissance ou la taille se situe dans la fourchette inférieure de la distribution normale pour l'âge gestationnel correspondant), un faible score d'Apgar et une microcéphalie.
Dans une population pédiatrique ayant été exposée in utero, aucun risque accru de trouble du déficit de l'attention avec ou sans hyperactivité (TDAH), de trouble du spectre de l'autisme (TSA) et de déficience intellectuelle n'a été mis en évidence au cours de l'étude.
Des études effectuées chez l'animal ont montré une toxicité sur la reproduction (voir «Données précliniques»). En conséquence, la prégabaline ne doit pas être administrée pendant la grossesse, sauf si le bénéfice pour la mère l'emporte clairement sur les risques potentiels pour le fœtus. Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant le traitement (voir «Mises en garde et précautions»).
Allaitement
La prégabaline passe dans le lait maternel (voir données sous «Pharmacocinétique»). La sécurité d'emploi de la prégabaline chez les nourrissons est inconnue, il est donc recommandé de ne pas allaiter pendant le traitement. Tenant compte de l'utilité de l'allaitement pour l'enfant d'une part et la nécessité et des bénéfices du traitement pour la mère d'autre part, on décidera soit de l'arrêt de l'allaitement soit de l'interruption du traitement pendant l'allaitement.
Effet sur l’aptitude à la conduite et l’utilisation de machinesLa prégabaline peut induire des étourdissements et une somnolence. Il est donc conseillé aux patients de ne pas conduire, de ne pas utiliser de machines complexes ni d'entreprendre d'autres activités potentiellement dangereuses tant qu'ils ignorent si ce médicament altère leur capacité à effectuer ces activités.
Effets indésirablesLes effets indésirables mentionnés proviennent d'études cliniques sur les douleurs neuropathiques, l'épilepsie, le trouble anxieux généralisé et la fibromyalgie portant sur plus de 12'000 patients, ainsi que de données issues de l'expérience post-marketing.
Les effets indésirables les plus fréquemment rapportés dans les études cliniques ont été les étourdissements (30,7%), la somnolence (16,4%), la vision floue (6,7%), la sécheresse buccale (8,6%), la constipation (5,3%), la fatigue (6,7%), l'œdème périphérique (5,7%) et la prise de poids (7,2%). La sévérité des effets indésirables était en règle générale légère à modérée.
Le taux d'arrêt du traitement en raison d'effets indésirables dans toutes les études contrôlées se situe à 14% chez les patients sous prégabaline et à 5% chez les patients sous placebo. Les effets indésirables les plus fréquemment rapportés ayant entraîné l'arrêt du traitement par la prégabaline étaient les étourdissements et la somnolence.
La liste ci-dessous énumère les effets indésirables apparus dans le cadre du programme d'études cliniques et de l'expérience post-marketing. Les données issues de l'expérience post-marketing décrivent les effets secondaires notifiés spontanément, décrits dans la littérature et signalés par les autorités à travers le monde.
L'attribution à une catégorie de fréquence se fait sur la base d'études contrôlées.
Les effets indésirables sont rangés par classe de système d'organes et par fréquence [très fréquents (≥1/10), fréquents (≥1/100, <1/10), occasionnels (≥1/1000, <1/100), rares (≥1/10'000, <1/1000), très rares (<1/10'000), fréquence inconnue (ne peut pas être estimée sur la base des données disponibles)].
Infections et infestations
Fréquents: rhinopharyngite.
Affections hématologiques et du système lymphatique
Occasionnels: neutropénie, diminution du nombre de plaquettes.
Rares: diminution du nombre de leucocytes.
Affections du système immunitaire
Fréquence inconnue: hypersensibilité*, angioœdème*, réaction allergique*.
Troubles du métabolisme et de la nutrition
Fréquents: augmentation de l'appétit, prise de poids.
Occasionnels: anorexie, hypoglycémie, hyperglycémie, perte de poids, aggravation d'un état métabolique diabétique.
Affections psychiatriques
Fréquents: euphorie, confusion, irritabilité, dépression, désorientation, insomnie, baisse de la libido.
Occasionnels: hallucinations, excitation, agitation, abattement, humeur élevée, humeur changeante, dépersonnalisation, rêves anormaux, difficulté à trouver ses mots, hausse de la libido, anorgasmie.
Rares: attaques de panique, désinhibition, apathie.
Fréquence inconnue: usage nocif et dépendance*, idées suicidaires*, comportement suicidaire*, suicide*.
Affections du système nerveux
Très fréquents: étourdissements (28%), somnolence (15,7%).
Fréquents: ataxie, troubles de la coordination, tremblements, dysarthrie, amnésie, troubles de la mémoire, troubles de l'attention, paresthésies, hypoesthésies, sédation, troubles de l'équilibre, léthargie.
Occasionnels: syncopes, myoclonies, hyperactivité psychomotrice, dyskinésie, vertige orthostatique, tremblement intentionnel, nystagmus, troubles cognitifs, troubles de l'élocution, diminution des réflexes, hyperesthésies, sensation de brûlure.
Rares: stupeur, parosmie, hypokinésie, agueusie, dysgraphie.
Fréquence inconnue: céphalées*, perte de conscience*, altération de l'état psychique*, encéphalopathie*.
Affections oculaires
Fréquents: vision trouble, diplopie.
Occasionnels: perte de la vision périphérique (vision «en tunnel»), troubles de la vision, gonflement oculaire, pertes de champ visuel, diminution de l'acuité visuelle, douleurs oculaires, amblyopie, photopsie, xérophtalmie, larmoiement, irritations oculaires.
Rares: oscillopsie, altération de la vision spatiale, mydriase, strabisme, photosensibilité.
Fréquence inconnue: kératite*.
Affections de l'oreille et du labyrinthe
Fréquents: vertiges.
Rares: hyperacousie.
Affections cardiaques
Occasionnels: tachycardie, bloc AV du 1er degré, bradycardie sinusale.
Rares: tachycardie sinusale, arythmie sinusale.
Fréquence inconnue: insuffisance cardiaque*.
Affections vasculaires
Occasionnels: hypotension, hypertension, rougissement, bouffées de chaleur au visage et au cou, froideur des extrémités.
Affections respiratoires, thoraciques et médiastinales
Occasionnels: dyspnée, épistaxis, toux, obstruction nasale, rhinite, ronflement.
Rares: sensation de gorge serrée, nez sec.
Fréquence inconnue: œdème pulmonaire*, dépression respiratoire*.
Affections hépatobiliaires
Occasionnels: augmentation de l'alanine aminotransférase et de l'aspartate aminotransférase.
Fréquence inconnue: ictère, insuffisance hépatique, hépatite.
Affections gastro-intestinales
Fréquents: vomissements, constipation, flatulences, météorisme, sécheresse buccale.
Occasionnels: reflux gastro-œsophagien, salivation, hypoesthésie buccale.
Rares: ascite, pancréatite, dysphagie.
Fréquence inconnue: nausée*, diarrhée*, tuméfaction de la langue*, diminution de la fonctionnalité du tractus gastro-intestinal inférieur*.
Affections de la peau et du tissu sous-cutané
Occasionnels: éruption papulaire, urticaire, sudation.
Rares: sueurs froides.
Fréquence inconnue: tuméfaction du visage*, prurit*, réactions cutanées médicamenteuses sévères (SCARs, severe cutaneous adverse reactions), y compris syndrome de Stevens-Johnson (SJS) et nécrolyse épidermique toxique (NET)*.
Affections musculosquelettiques et du tissu conjonctif
Fréquents: crampes musculaires, arthralgies, dorsalgies, douleurs des extrémités, spasmes cervicaux.
Occasionnels: tuméfactions articulaires, myalgies, spasmes musculaires, douleurs nucales, rigidité musculaire, élévations de la créatine phosphokinase.
Rares: rhabdomyolyse.
Affections du rein et des voies urinaires
Occasionnels: incontinence urinaire, dysurie.
Rares: défaillance rénale, oligurie, élévation du taux de créatinine.
Fréquence inconnue: rétention urinaire*.
Affections des organes de reproduction et du sein
Occasionnels: dysfonction érectile, troubles de la fonction sexuelle, éjaculation retardée, dysménorrhée.
Rares: douleur mammaire, aménorrhée, écoulement mammaire, augmentation du volume des seins.
Fréquence inconnue: gynécomastie*.
Troubles généraux et anomalies au site d'administration
Fréquents: œdèmes périphériques, œdèmes, chutes, troubles de la marche, sensation d'ébriété, trouble sensoriel, épuisement.
Occasionnels: œdèmes généralisés, sensation d'oppression thoracique, douleurs, fièvre, soif, frissons, asthénie.
Fréquence inconnue: malaise*.
Investigations
Occasionnels: hypokaliémie.
* Effets indésirables issus de l'expérience post-marketing.
Description d'effets indésirables spécifiques et informations complémentaires
Populations particulières
Patients âgés (plus de 65 ans)
Le traitement par la prégabaline a été associé à des étourdissements et de la somnolence qui peuvent conduire à la survenue plus fréquente de traumatismes liés à des chutes chez les personnes âgées.
Patients atteints de diabète sucré
Dans six études contrôlées sur le traitement de la neuropathie diabétique par la prégabaline pendant 5 à 12 semaines, une prise de poids d'au moins 7% du poids corporel a été observée chez 5,2% des patients diabétiques sous traitement par la prégabaline. Cet effet était dose-dépendant (3,4% sous 150 mg contre 7,5% sous 600 mg de dose quotidienne). L'incidence augmente en outre avec la durée du traitement et a concerné jusqu'à 31,3% des patients dans des études contrôlées et non contrôlées à long terme.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLors de surdosages (1,7 g jusqu'à 15 g), aucun événement indésirable inattendu n'a été rapporté.
Signes et symptômes
Les effets indésirables les plus fréquemment rapportés dans l'expérience post-marketing lorsque la prégabaline a été prise à doses trop élevées regroupent troubles affectifs, somnolence, états confusionnels, dépression, besoin de bouger et agitation. De rares cas de coma ont été rapportés. Des crises convulsives ont également été rapportées.
Traitement
Le traitement d'un surdosage de prégabaline comprendra des mesures générales de soutien et une hémodialyse si nécessaire.
Propriétés/EffetsCode ATC
N02BF02
Mécanisme d'action
La substance active prégabaline est un analogue de l'acide gamma-aminobutyrique (GABA) répondant à la dénomination chimique (S)-3-(aminométhyl)-5-acide méthylhexanoïque.
Pharmacodynamique
Les études réalisées in vitro ont montré que la prégabaline se lie à une sous unité (protéine α2-δ) du canal calcique voltage-dépendant dans le système nerveux central et déloge ainsi effectivement la [3H]-gabapentine. Bien que le mécanisme d'action précis ne soit pas encore élucidé, on a pu montrer que la prégabaline diminue la libération de différents neurotransmetteurs y compris le glutamate, la noradrénaline et la substance P, réduisant ainsi l'irritabilité neuronale dans le système nerveux central.
Les résultats concernant les modèles animaux atteints de neuropathies suggèrent que la prégabaline réduit la libération de neurotransmetteurs pro-nociceptifs calcium-dépendante dans la moelle épinière, éventuellement par une interruption du transport du calcium et/ou des flux de calcium. L'expérience sur d'autres modèles animaux indique que l'effet anti-nociceptif de la prégabaline est médié via une interaction avec des voies noradrénergique et sérotoninergiques descendantes.
Efficacité clinique
Douleurs neuropathiques
L'efficacité de la prégabaline a été évaluée dans le traitement des douleurs neuropathiques dans 12 études multicentriques réalisées en double aveugle et contrôlées contre placebo d'une durée allant jusqu'à 13 semaines avec une administration 2 fois par jour, ou jusqu'à 8 semaines avec une administration 3 fois par jour. Au total, 2912 patients ont été inclus dans les 12 études. Les études incluaient des patients qui présentaient des douleurs d'intensité modérée à forte.
Dans les études cliniques sur les polyneuropathies diabétiques allant jusqu'à 13 semaines, le critère de jugement moyen sur l'échelle de la douleur s'est amélioré significativement par rapport au placebo de -1,3 à -1,5 avec 300 mg/j et de -1,0 à -1,5 avec 600 mg/j. Une baisse significative des douleurs par rapport au placebo a été établie au cours de la première semaine et s'est maintenue pendant toute la durée du traitement. Le taux de sujets répondeurs (baisse de 50% sur l'échelle de la douleur) atteignait 33-46% pour une dose de 300 mg/j et 39-48% pour une dose de 600 mg/j comparé à 15-30% sous placebo.
Dans les études cliniques sur la névralgie post-herpétique pendant 13 semaines, le critère de jugement moyen sur l'échelle de la douleur s'est amélioré significativement par rapport au placebo de -0,9 à -1,2 avec 150 mg/j, de -1,1 à -1,6 avec 300 mg/j et de -1,7 à -1,8 avec 600 mg/j. Une baisse significative des douleurs par rapport au placebo a été mise en évidence au cours de la première semaine et s'est maintenue pendant toute la durée du traitement. Le taux de sujets répondeurs (baisse de 50% sur l'échelle de la douleur) atteignait 22-26% pour une dose de 150 mg/j, 26-28% pour une dose de 300 mg/j et 38-50% pour une dose de 600 mg/j comparé à 9-20% sous placebo.
Dans une étude clinique sur des lésions de la moelle épinière menée sur 12 semaines, le critère de jugement moyen sur l'échelle de la douleur s'est amélioré significativement par rapport au placebo de -1,53 sur l'échelle numérique d'évaluation de la douleur, qui compte 11 points. Le taux de sujets répondeurs (baisse de 50% sur l'échelle de la douleur) atteignait 22% chez les patients traités par la prégabaline, comparé à 7% chez les patients sous placebo.
Épilepsie
L'efficacité de la prégabaline comme traitement adjuvant a été évaluée dans trois études multicentriques, randomisées, menées en double aveugle et contrôlées contre placebo d'une durée de 12 semaines chez 1052 patients traités par une posologie répartie en deux et/ou en trois prises quotidiennes. Les patients présentaient des crises réfractaires partielles avec ou sans généralisation secondaire ainsi qu'initialement une fréquence moyenne de crises entre 19 et 27 crises et une fréquence médiane de crises entre 9 et 12 crises en 28 jours.
L'efficacité de la prégabaline dans l'épilepsie a été démontrée dans toutes les études par la réduction du nombre de crises par rapport au placebo. Les patients répondeurs étaient ceux qui ont vu la fréquence de leurs crises partielles réduite de ≥50% pendant le traitement par rapport à la fréquence initiale. Les taux des répondeurs étaient de 14 à 31% avec 150 mg/j, de 40% avec 300 mg/j et de 43 à 51% avec 600 mg/j comparé à 6-14% sous placebo, ce qui indique un effet dose-dépendant.
Chez les patients présentant des crises généralisées secondaires, seule la dose maximale de prégabaline de 600 mg/j était significativement supérieure au placebo.
Troubles anxieux généralisés
La prégabaline a été évaluée au cours de six études contrôlées portant sur une durée de 4-6 semaines, une étude de 8 semaines chez des patients âgés ainsi qu'une étude à long terme sur la prévention des rechutes avec une phase de prévention des rechutes en double aveugle de 6 mois.
Une amélioration des symptômes des troubles anxieux généralisés sur la base de l'échelle de Hamilton (Hamilton Anxiety Rating Scale, HAM-A) a été observée pendant la première semaine.
Dans les études cliniques contrôlées portant sur une période de 4-8 semaines, 52% des patients traités par la prégabaline et 38% des patients sous placebo ont présenté une amélioration d'au moins 50% du score global HAM-A par rapport aux valeurs initiales.
PharmacocinétiqueLes caractéristiques pharmacocinétiques à l'état d'équilibre de la prégabaline sont similaires chez les volontaires sains, chez les patients épileptiques recevant des médicaments antiépileptiques ainsi que chez les patients souffrant de douleurs chroniques.
Absorption
La prégabaline est rapidement absorbée lorsqu'elle est administrée à jeun. Les pics plasmatiques sont atteints dans l'heure suivant l'administration d'une dose unique ou de doses multiples. La biodisponibilité orale de la prégabaline est estimée à ≥90% et est indépendante de la dose. Après administration répétée du produit, l'état d'équilibre est atteint dans un délai de 24 à 48 heures. Le taux d'absorption de la prégabaline diminue lorsque le médicament est administré pendant un repas, entraînant une diminution de la Cmax de 25-30% environ et un retard du tmax de 2,5 heures environ. Toutefois, l'administration de la prégabaline au cours du repas n'a aucun effet cliniquement significatif sur son taux global d'absorption.
Distribution
Les études précliniques ont montré que la prégabaline traverse rapidement la barrière hémato-encéphalique chez la souris, le rat et le singe. La prégabaline traverse le placenta chez la rate et est présente dans le lait des rates allaitantes. Chez l'être humain, le volume de distribution après administration orale est de 0,56 l/kg environ. La prégabaline ne se lie pas aux protéines plasmatiques.
Métabolisme
La prégabaline est métabolisée de manière insignifiante chez l'être humain. Après administration d'une dose de prégabaline radiomarquée, environ 98% se trouvent dans l'urine sous forme inchangée. Le dérivé N-méthylé, le principal métabolite de la prégabaline retrouvé dans l'urine, représente 0,9% de la dose. Aucun signe de racémisation de l'énantiomère S de la prégabaline en énantiomère R n'a été mis en évidence dans les études précliniques.
Élimination
La prégabaline est éliminée de la circulation générale principalement par voie rénale sous forme inchangée.
La demi-vie d'élimination de la prégabaline est de 6,3 heures en moyenne. La clairance plasmatique et la clairance rénale de la prégabaline sont directement proportionnelles à la clairance de la créatinine (voir «Cinétique pour certains groupes de patients», Troubles de la fonction rénale).
L'adaptation de la dose de prégabaline est nécessaire chez les patients présentant une insuffisance rénale (voir «Posologie/Mode d'emploi», Tableau 1).
Linéarité/non-linéarité
La prégabaline présente une pharmacocinétique linéaire aux doses journalières recommandées. La variabilité pharmacocinétique interindividuelle observée avec la prégabaline est faible (<20%). Les paramètres de pharmacocinétique après dose multiple sont extrapolables à partir de ceux obtenus lors de l'administration d'une dose unique.
Cinétique pour certains groupes de patients
Sexe
Les études cliniques ont montré que le sexe n'a aucune influence cliniquement significative sur les concentrations plasmatiques de prégabaline.
Troubles de la fonction hépatique
Aucune étude pharmacocinétique spécifique n'a été menée chez les patients présentant une insuffisance hépatique. Parce que la prégabaline n'est quasiment pas métabolisée et qu'elle est essentiellement excrétée sous forme inchangée dans l'urine, une insuffisance hépatique ne devrait pas modifier significativement les concentrations plasmatiques de prégabaline. Il faut cependant noter que la sécurité d'emploi de la prégabaline n'a pas été étudiée chez les patients qui présentent des troubles de la fonction hépatique.
Troubles de la fonction rénale
La clairance de la prégabaline est directement proportionnelle à la clairance de la créatinine. L'administration de la moitié de la dose est indiquée chez les patients qui présentent une insuffisance rénale modérée (voir «Posologie/Mode d'emploi», Tableau 1).
Patients âgés
La clairance de la prégabaline tend à diminuer avec l'âge. Cette diminution de la clairance orale de la prégabaline correspond à la diminution de la clairance de la créatinine liée à l'âge. Une réduction de la dose de prégabaline peut s'avérer nécessaire chez les patients qui présentent une diminution de la fonction rénale due à l'âge (voir «Posologie/Mode d'emploi», Tableau 1).
Allaitement
La pharmacocinétique de 300 mg de prégabaline par jour (150 mg toutes les 12 heures) a été étudiée chez 10 femmes qui allaitent (≥12 semaines post-partum). La lactation n'a eu qu'un effet minime ou nul sur la pharmacocinétique de la prégabaline. Dans le lait, la concentration moyenne à l'état d'équilibre représentait environ 76% de la concentration plasmatique maternelle. La dose de prégabaline quotidienne moyenne estimée chez le nourrisson (en partant d'une consommation de lait moyenne de 150 ml/kg/j) était de 0,31 mg/kg/j, ce qui représente, en se basant sur les mg/kg, environ 7% de la dose maternelle.
Données précliniquesDans les études de toxicité à doses répétées menées chez le rat et le singe, des effets sur le SNC ont été observés, comprenant une hypoactivité, une hyperactivité et une ataxie.
Lors d'une exposition supérieure à plus de 5 fois l'exposition humaine à la dose maximale recommandée au cours des expériences à long terme sur des rats albinos, l'incidence d'atrophie rétinienne, communément observée chez les animaux les plus âgés, a été plus élevée.
La prégabaline ne s'est pas révélée tératogène chez la souris, le rat et le lapin. Une toxicité fœtale chez le rat et le lapin est uniquement apparue lors d'exposition à des doses largement supérieures à celles administrées chez l'être humain. Dans les études de toxicité pré- et postnatales effectuées chez le rat, la prégabaline a induit une toxicité sur le développement de la descendance à des expositions 5 fois supérieures à la dose maximale recommandée chez l'être humain.
La prégabaline n'est pas génotoxique comme le montrent les résultats d'une batterie de tests in vitro et in vivo.
Les résultats obtenus lors d'une étude réalisée chez le rat permettent de déduire que la prégabaline ne présente aucun risque d'effets cancérigènes pour l'espèce humaine. La pertinence des hémangiosarcomes observés chez la souris ne peut actuellement être déterminée de façon concluante pour l'être humain.
Remarques particulièresConservation
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver à température ambiante (15-25 °C) et hors de portée des enfants.
Numéro d’autorisation65678 (Swissmedic).
PrésentationPregabalin Viatris 25 mg: 14, 56 gélules. [B]
Pregabalin Viatris 50 mg: 14, 84 gélules. [B]
Pregabalin Viatris 75 mg: 14, 56 gélules. [B]
Pregabalin Viatris 100 mg: 84 gélules. [B]
Pregabalin Viatris 150 mg: 56, 168 gélules. [B]
Pregabalin Viatris 200 mg: 84 gélules. [B]
Pregabalin Viatris 300 mg: 56 et 168 gélules. [B]
Titulaire de l’autorisationViatris Pharma GmbH, 6312 Steinhausen.
Mise à jour de l’informationMars 2025.
[Version 109 F]
|