CompositionPrincipes actifs
Talimogene laherparepvec, un virus Herpes simplex de type 1 atténué (HSV-1) produit par la technique de l'ADN recombinant dans des cellules Vero (cellules rénales de singe) et modifié par délétion fonctionnelle de deux gènes (ICP34.5 et ICP47) et insertion de la séquence ADN codant le facteur humain GM-CSF. Le talimogene laherparepvec est un organisme génétiquement modifié.
Excipients
Phosphate disodique dihydraté, phosphate monosodique dihydraté, chlorure de sodium, myo-inositol, sorbitol (E420; 20 mg/ml), eau pour préparations injectables. La teneur en sodium correspond au maximum 7,7 mg par ml.
Indications/Possibilités d’emploiIMLYGIC est indiqué en monothérapie dans le traitement des patients adultes présentant un mélanome non résécable avec métastases régionales ou à distance (stade IIIB, IIIC et IVM1a) sans métastases osseuses, cérébrales, pulmonaires ou autres métastases viscérales.
Posologie/Mode d’emploiLe traitement par IMLYGIC doit être instauré et surveillé par un médecin spécialiste ayant des compétences dans le traitement du cancer.
Afin d'assurer la traçabilité d'IMLYGIC, il convient de documenter pour chaque traitement le nom commercial et le numéro du lot dans le dossier du patient.
IMLYGIC est disponible en flacons à usage unique de 1 ml dans deux concentrations différentes:
·106 (1 million) UFP/ml - uniquement pour la dose initiale
·108 (100 millions) UFP/ml - pour toutes les doses suivantes
Le volume d'injection total par séance de traitement doit être de 4 ml au maximum. La dose initiale recommandée s'élève à 4 ml au maximum d'IMLYGIC à une concentration de 106 (1 million) UFP/ml. Les doses suivantes s'élèvent à 4 ml au maximum d'IMLYGIC à une concentration de 108 (100 millions) UFP/ml.
Le tableau 1 présente le schéma posologique recommandé pour IMLYGIC.
Tableau 1: Schéma posologique recommandé pour IMLYGIC
|
Séance de traitement
|
Intervalle thérapeutique
|
Volume d'injection total maximal
|
Concentration de la dose
|
Ordre de priorité des lésions à injecter
| |
Initiale
|
-
|
Jusqu'à 4 ml
|
106
(1 million) UFP/ml
|
·Traiter tout d'abord la ou les lésions les plus étendues.
·Traiter les autres lésions par ordre de grandeur jusqu'à ce que le volume d'injection maximal soit atteint.
| |
Deuxième
|
3 semaines après le traitement initial
|
Jusqu'à 4 ml
|
108
(100 millions) UFP/ml
|
·Traiter tout d'abord les nouvelles lésions (lésions éventuellement apparues depuis le traitement initial).
·Traiter les autres lésions par ordre de grandeur jusqu'à ce que le volume d'injection maximal soit atteint.
| |
Toutes les séances de traitement suivantes (y compris la reprise du traitement)
|
2 semaines après le traitement précédent
|
Jusqu'à 4 ml
|
108
(100 millions) UFP/ml
|
·Traiter tout d'abord les nouvelles lésions (lésions éventuellement apparues depuis le traitement précédent).
·Traiter les autres lésions par ordre de grandeur jusqu'à ce que le volume d'injection maximal soit atteint.
|
Détermination du volume de la dose d'IMLYGIC (par lésion)
Le volume à injecter dans chaque lésion dépend de la taille de la lésion et doit être déterminé conformément au tableau 2. Le volume d'injection total par séance de traitement ne doit pas dépasser 4 ml.
Tableau 2: Sélection du volume d'injection d'IMLYGIC en fonction de la taille de la lésion
|
Taille de la lésion
(dimension la plus grande)
|
Volume d'IMLYGIC à injecter
| |
> 5 cm
|
Jusqu'à 4 ml
| |
> 2,5 cm à 5 cm
|
Jusqu'à 2 ml
| |
> 1,5 cm à 2,5 cm
|
Jusqu'à 1 ml
| |
> 0,5 cm à 1,5 cm
|
Jusqu'à 0,5 ml
| |
≤ 0,5 cm
|
Jusqu'à 0,1 ml
|
Il est possible que les lésions existantes augmentent de volume ou que de nouvelles lésions apparaissent avant que les patients ne répondent au traitement (voir «Mises en garde et précautions»). Tant qu'il reste une ou des lésions injectables, le traitement par IMLYGIC doit être poursuivi pendant au moins 6 mois, sauf si le médecin considère que le patient ne tire pas de bénéfices du traitement par IMLYGIC ou qu'un autre traitement est nécessaire.
Le traitement par IMLYGIC peut être réinstauré en cas d'apparition de nouvelles lésions après une réponse complète si le médecin considère que le patient pourra tirer un bénéfice du traitement.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique ou rénale
Aucune étude clinique n'a été réalisée pour évaluer les effets d'une insuffisance hépatique ou rénale sur la pharmacocinétique d'IMLYGIC. Cependant, aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance hépatique ou rénale.
Patients âgés
Aucun ajustement posologique n'est nécessaire chez les patients âgés de ≥65 ans (voir «Propriétés/Effets»).
Enfants et adolescents (< 18 ans)
La sécurité et l'efficacité d'IMLYGIC pour les enfants et les adolescents ne sont pas établies. Les données actuelles disponibles pour les patients pédiatriques et les jeunes adultes âgés de 7 à ≤ 21 ans atteints de tumeurs avancées ne touchant pas le système nerveux central pouvant être traitées par injection directe sont décrites à la rubrique «Propriétés/Effets».
Mode d'administration
IMLYGIC doit être administré en injection intralésionnelle dans les lésions cutanées, sous-cutanées et/ou ganglionnaires qui sont visibles, palpables ou détectables par échographie.
Précautions à prendre avant la manipulation ou l'administration du médicament
Ce médicament contient des organismes génétiquement modifiés. Un équipement de protection individuelle doit être porté pendant la préparation et l'administration d'IMLYGIC (voir «Remarques concernant la manipulation et l'utilisation, équipement de protection individuelle, déversements accidentels et élimination des déchets» dans «Remarques particulières»).
Les professionnels de santé immunodéprimés ou les femmes enceintes ne doivent pas administrer IMLYGIC et ne doivent pas avoir de contact direct avec les sites d'injection d'IMLYGIC ou avec les fluides corporels des patients traités (voir «Mises en garde et précautions» et «Contre-indications»).
Il convient de suivre les instructions suivantes lors de la préparation et l'administration d'IMLYGIC:
Avant l'injection
·Décongeler les flacons d'IMLYGIC à une température comprise entre 20°C et 25°C (voir «Remarques particulières concernant le stockage»). Une fois décongelé, IMLYGIC peut être conservé avant son utilisation (voir «Remarques particulières»).
·Prélever dans une seringue la quantité souhaitée d'IMLYGIC à partir du flacon en utilisant une technique aseptique.
·Le site d'injection peut être traité préalablement par un anesthésique local. Un anesthésique injectable peut être injecté à la périphérie de la lésion, mais pas directement dans la lésion.
·Nettoyer la lésion et la zone environnante avec un tampon imbibé d'alcool et laisser sécher.
Injection
·Administrer IMLYGIC en injection intralésionnelle dans les lésions cutanées, sous-cutanées et/ou ganglionnaires qui sont visibles, palpables ou détectables par échographie.
·Déterminer le volume d'injection pour chaque lésion en utilisant le tableau 2.
·Choisir un seul point d'insertion et injecter IMLYGIC en éventail, aussi loin que la portée radiale de l'aiguille dans la lésion le permet afin d'obtenir une dispersion régulière et complète. Plusieurs points d'insertion peuvent être utilisés si la taille d'une lésion est supérieure à la portée de l'aiguille.
|
Lésions cutanées
|
Lésions sous-cutanées
|
Lésions ganglionnaires
| |
|
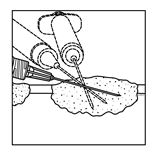
|
|

|
|
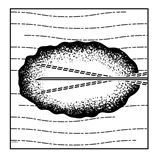
| |
Figure 1: Injection dans des lésions cutanées
|
Figure 2: Injection dans des lésions sous-cutanées
|
Figure 3: Injection dans des lésions ganglionnaires
|
·Disperser IMLYGIC uniformément et en totalité dans la lésion en tirant l'aiguille vers l'arrière sans la sortir complètement de la lésion. Rediriger l'aiguille aussi souvent que nécessaire tout en injectant le reste de la dose. Continuer jusqu'à ce que la dose complète soit dispersée uniformément et en totalité.
·Lors du retrait de l'aiguille l'extraire lentement de la lésion pour éviter un écoulement ou une éclaboussure d'IMLYGIC au point d'insertion.
·Répéter ces étapes pour les autres lésions à injecter. Utiliser une aiguille neuve à chaque fois que l'aiguille est retirée complètement d'une lésion et à chaque nouvelle lésion injectée.
Après l'injection
·Exercer une pression sur le site d'injection avec une compresse de gaze stérile pendant au moins 30 secondes.
·Tamponner le site d'injection et la zone environnante avec un tampon imbibé d'alcool et couvrir la lésion injectée d'une compresse absorbante et d'un pansement occlusif sec.
D'autres instructions concernant l'utilisation, la manipulation et l'élimination figurent sous voir «Remarques particulières».
Contre-indicationsIMLYGIC est contre-indiqué chez:
·Les patients ayant des antécédents d'hypersensibilité au talimogene laherparepvec ou à l'un de ses excipients (voir «Composition»).
·Les patients présentant une immunosuppression sévère (p.ex. les patients présentant une immunodéficience sévère, congénitale ou acquise, cellulaire et/ou humorale) (voir «Mises en garde et précautions» et «Données précliniques»).
Mises en garde et précautionsIMLYGIC est soumis à des mesures spécifiques de minimisation du risque. Le médecin traitant reçoit une brochure spécifiquement destinée aux médecins («Physician Education Booklet») contenant des informations sur les risques d'une transmission de l'herpès et les complications de l'herpès ainsi que sur l'utilisation et la manipulation sûres d'IMLYGIC. De plus, il est demandé au médecin de remettre au patient concerné une brochure patient, une carte patient et une information destinée au patient.
Patients précédemment traités
Les données d'efficacité d'IMLYGIC en traitement de seconde ligne ou en traitement de ligne ultérieure sont limitées.
Infections herpétiques disséminées
Des infections herpétiques disséminées, y compris des cas graves, ont été rapportées chez des patients traités par IMLYGIC (voir «Effets indésirables»).
IMLYGIC n’a pas été étudié chez les patients immunodéprimés. Sur la base des données épidémiologiques, il est possible que les patients immunodéprimés (p. ex. les patients présentant une infection par le VIH/SIDA, une leucémie, un lymphome, une immunodéficience variable commune ou les patients ayant un besoin chronique de stéroïdes à dose élevée ou d’autres immunosuppresseurs) présentent un risque accru d’infection herpétique disséminée. Les bénéfices et les risques du traitement doivent être évalués avant d’utiliser IMLYGIC chez les patients immunodéprimés.
Sur la base des données chez l’animal, il est possible que les patients sévèrement immunodéprimés présentent un risque accru d’infection herpétique disséminée. Ces patients ne doivent donc pas être traités par IMLYGIC (voir «Contre-indications» et «Données précliniques»).
Exposition accidentelle à IMLYGIC
Une exposition accidentelle peut entraîner la transmission d'IMLYGIC et une infection herpétique. Les professionnels de santé et l'entourage proche du patient (p.ex. les membres de la famille, les soignants, les partenaires sexuels ou les personnes partageant le même lit) doivent éviter tout contact direct avec les lésions injectées ou avec les fluides corporels des patients traités pendant toute la période de traitement et jusqu'à 30 jours après la dernière administration du traitement (voir «Exposition accidentelle» dans «Remarques particulières»). Des cas de piqûre d'aiguille accidentelle et d'éclaboussures de solution injectable ont été rapportés chez des professionnels de santé pendant la préparation et l'administration d'IMLYGIC.
Les personnes de l'entourage proche du patient qui sont enceintes ou immunodéprimées ne doivent pas changer les pansements du patient ou nettoyer le site d'injection. Les femmes enceintes, les nouveau-nés et les sujets immunodéprimés ne doivent pas entrer en contact avec les matériels potentiellement contaminés.
Il convient de s'assurer que les patients sont capables de couvrir les sites d'injection avec des pansements occlusifs (voir «Remarques concernant la manipulation et l'utilisation, équipement de protection individuelle, déversements accidentels et élimination des déchets» dans «Remarques particulières»). Il faut également indiquer aux patients de ne pas toucher ou gratter les sites d'injection, car cela pourrait entraîner le transfert accidentel d'IMLYGIC vers d'autres régions du corps ou vers des personnes de leur entourage proche.
Bien qu'on ne sache pas si IMLYGIC peut être transmis par voie sexuelle, il est connu que ce mode de transmission est possible pour le HSV-1 type sauvage. Il faut indiquer aux patients d'utiliser un préservatif en latex lors des contacts sexuels afin de prévenir une transmission possible d'IMLYGIC. Il faut indiquer aux femmes en âge de procréer d'utiliser une méthode contraceptive fiable pour éviter une grossesse pendant le traitement (voir «Grossesse, allaitement»).
Il faut indiquer aux personnels soignants de porter des gants de protection lorsqu'ils aident les patients à appliquer ou changer les pansements occlusifs et de respecter les précautions pour l'élimination des pansements et des matériels de nettoyage usagés (voir «Remarques concernant la manipulation et l'utilisation, équipement de protection individuelle, déversements accidentels et élimination des déchets» dans «Remarques particulières»).
En cas d'exposition accidentelle à IMLYGIC, il convient d'observer les instructions indiquées dans «Remarques particulières». En cas d'apparition de signes ou symptômes d'infection herpétique, les personnes concernées doivent consulter un professionnel de santé. Si des lésions herpétiques sont suspectées, les patients, les personnes de l'entourage et les professionnels de santé ont la possibilité de demander au titulaire de l'autorisation de mise sur le marché d'effectuer des tests complémentaires afin de permettre une meilleure caractérisation de l'infection.
Infection herpétique chez les patients traités par IMLYGIC
Des infections herpétiques (entre autres, herpès labial et kératite herpétique) et des cas graves d’infection herpétique disséminée ont été rapportés chez des patients traités par IMLYGIC (voir «Effets indésirables»). Les symptômes d’infection locale ou systémique pouvant être liés à IMLYGIC devraient être comparables à ceux causés par les infections par le HSV-1 type sauvage.
On sait que les sujets porteurs d'une infection par le HSV-1 type sauvage ont un risque à vie d'infection herpétique symptomatique due à la réactivation du HSV-1 type sauvage latent. La possibilité d'infection herpétique symptomatique due à une réactivation potentielle d'IMLYGIC doit être prise en compte.
Il faut indiquer aux patients qui développent des infections herpétiques de suivre les pratiques d'hygiène habituelles pour prévenir la transmission du virus.
IMLYGIC est sensible à l'aciclovir. Les bénéfices et les risques du traitement par IMLYGIC doivent être évalués avant l'administration d'aciclovir ou d'autres antiviraux indiqués dans le traitement des infections herpétiques. Ces principes actifs pourraient nuire à l'efficacité du traitement s'ils sont administrés par voie systémique ou par voie topique directement sur le site d'injection.
Cellulite au site d'injection
Une nécrose ou une ulcération du tissu tumoral peut survenir après le traitement par IMLYGIC. Des cas de cellulite et d'infections bactériennes systémiques ont été rapportés. Des soins attentifs de la plaie et des mesures de prévention des infections sont recommandés, en particulier si la nécrose entraîne des plaies ouvertes.
Troubles de la cicatrisation au site d'injection
Des troubles de la cicatrisation au site d'injection ont été rapportés dans les études cliniques. IMLYGIC peut augmenter le risque de troubles de la cicatrisation chez les patients présentant des facteurs de risque sous-jacents (p.ex. des antécédents de radiothérapie au site d'injection ou des lésions situées dans des régions mal vascularisées).
En cas d'apparition d'une infection persistante ou d'un retard de cicatrisation, les bénéfices et les risques d'IMLYGIC doivent être évalués avant de poursuivre le traitement.
Événements d'origine immunologique
Dans les études cliniques, des événements d'origine immunologique incluant glomérulonéphrite, vascularite, pneumopathie inflammatoire, aggravation d'un psoriasis et vitiligo ont été rapportés chez des patients traités par IMLYGIC.
Les bénéfices et les risques d'IMLYGIC doivent être évalués avant l'instauration du traitement chez les patients présentant une maladie auto-immune sous-jacente ou avant la poursuite du traitement chez les patients qui développent des événements d'origine immunologique.
Plasmocytome au site d'injection
Des cas de plasmocytome à proximité du site d'injection ont été rapportés après l'administration d'IMLYGIC. Les bénéfices et les risques d'IMLYGIC doivent être évalués chez les patients atteints d'un myélome multiple ou les patients développant un plasmocytome pendant le traitement.
Affections respiratoires obstructives
Des affections respiratoires obstructives ont été rapportées après le traitement par IMLYGIC. La prudence est de rigueur en cas d'injection de lésions à proximité des voies aériennes supérieures.
Hémorragies hépatiques lors de l’administration intrahépatique par voie transcutanée
IMLYGIC n’est pas indiqué pour une administration intrahépatique par voie transcutanée.
Lors d’études cliniques, des cas d’hémorragies hépatiques ayant entraîné une hospitalisation et le décès du patient ont été rapportés chez des patients ayant reçu des injections intrahépatiques d’IMLYGIC par voie transcutanée.
Pseudoprogression
Chez les patients traités par IMLYGIC, il est possible que les lésions – à la fois celles ayant fait l'objet d'une injection et les lésions existantes n'ayant pas fait l'objet d'une injection – augmentent de volume initialement ou que de nouvelles lésions apparaissent avant que les patients ne répondent au traitement.
Patients séronégatifs pour le HSV-1
L'incidence de la pyrexie, des frissons et du syndrome pseudogrippal a été plus élevée chez les patients qui étaient séronégatifs pour le HSV-1 avant le traitement que chez ceux qui étaient séropositifs pour le HSV-1 à l'inclusion, en particulier pendant les 3 premiers mois du traitement (voir «Effets indésirables»).
Mises en garde concernant d'autres composants
Ce médicament contient 20 mg de sorbitol (E420) par flacon de 1 ml. L'effet additif des produits administrés concomitamment contenant du sorbitol (ou du fructose) et l'apport alimentaire de sorbitol (ou de fructose) doivent être pris en compte.
Ce médicament ne doit pas être utilisé chez les patients présentant une intolérance au fructose, une maladie héréditaire rare.
Ce médicament contient 7,7 mg de sodium par flacon de 1 ml, ce qui équivaut à 0,4% de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte.
Traitement combiné
Aucune donnée démontrant la sécurité et l'efficacité d'IMLYGIC en traitement combiné n'est disponible à ce jour. IMLYGIC doit donc être administré uniquement en monothérapie.
InteractionsAucune étude d'interactions n'a été réalisée avec IMLYGIC. L'aciclovir et les autres antiviraux peuvent nuire à l'efficacité du traitement, qu'ils soient administrés par voie systémique ou par voie topique directement sur le site d'injection. Les bénéfices et les risques d'un traitement par IMLYGIC doivent être évalués avant l'administration d'aciclovir ou d'autres antiviraux indiqués dans le traitement des infections herpétiques.
Grossesse, allaitementTransmission d'IMLYGIC par contact sexuel
Il faut indiquer à tous les patients d'utiliser un préservatif en latex lors des rapports sexuels afin de prévenir une transmission éventuelle d'IMLYGIC (voir «Mises en garde et précautions»).
Contraception
Il faut indiquer aux femmes en âge de procréer d'utiliser une méthode contraceptive fiable pour éviter une grossesse pendant le traitement par IMLYGIC.
Grossesse
Aucune étude avec IMLYGIC n'a été réalisée chez la femme enceinte. Les expérimentations animales n'ont révélé aucun effet sur le développement embryo-fœtal (voir «Données précliniques»). Par mesure de précaution, il est préférable d'éviter l'utilisation du talimogene laherparepvec pendant la grossesse.
En cas d'infection par le HSV-1 type sauvage chez une femme enceinte (infection primaire ou réactivation), le virus peut éventuellement franchir la barrière placentaire. Il existe également un risque de transmission pendant l'accouchement en raison de l'excrétion virale (viral shedding). Les infections par le HSV-1 type sauvage ont été associées à des effets indésirables graves, incluant défaillance multiviscérale et décès, si le fœtus ou le nouveau-né contracte une infection par le virus herpétique de type sauvage. Bien qu'il n'existe pas de données cliniques à ce jour concernant des infections par IMLYGIC chez des femmes enceintes, il pourrait exister un risque pour le fœtus ou le nouveau-né si IMLYGIC agit de la même façon.
Des métastases transplacentaires du mélanome malin peuvent survenir. IMLYGIC étant conçu pour pénétrer dans le tissu tumoral et s'y répliquer, il pourrait exister un risque d'exposition du fœtus à IMLYGIC à partir du tissu tumoral qui a franchi la barrière placentaire.
Si IMLYGIC est utilisé pendant la grossesse, ou en cas de survenue d'une grossesse pendant le traitement par IMLYGIC, la patiente doit être informée des risques possibles pour le fœtus et/ou le nouveau-né.
Allaitement
On ne sait pas si IMLYGIC est excrété dans le lait maternel. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre/de s'abstenir du traitement avec IMLYGIC en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Aucune étude clinique n'a été réalisée pour examiner les effets du talimogene laherparepvec sur la fertilité.
Effet sur l’aptitude à la conduite et l’utilisation de machinesIMLYGIC pourrait avoir une légère influence sur l'aptitude à la conduite et l'utilisation de machines. Compte tenu de la possibilité d'effets indésirables tels que des vertiges ou un état confusionnel (voir «Effets indésirables»), il doit être conseillé aux patients de faire preuve de prudence lors de la conduite de véhicules ou de l'utilisation de machines jusqu'à ce qu'ils soient certains qu'IMLYGIC n'entraîne chez eux aucun effet défavorable.
Effets indésirablesLa sécurité d'IMLYGIC a été évaluée dans l'étude pivot chez 419 patients (IMLYGIC: N = 292, GM-CSF: N = 127) qui ont reçu au moins une dose du médicament à l'étude (voir «Propriétés/Effets»). La durée médiane d'exposition à IMLYGIC était de 23 semaines (5,3 mois). Vingt-six (26) patients ont été exposés à IMLYGIC pendant au moins un an.
Les effets indésirables les plus fréquemment rapportés (≥25%) chez les patients traités par IMLYGIC étaient: fatigue (50,3%), frissons (48,6%), fièvre (42,8%), nausées (35,6%), symptômes pseudogrippaux (30,5%) et douleur au site d'injection (27,7%). Au total, quatre-vingt-dix-huit pour cent (98%) de ces effets indésirables ont été rapportés comme étant de sévérité légère à modérée. L'effet indésirable de grade 3 ou supérieur le plus fréquent était la cellulite (2,1%) (voir «Mises en garde et précautions»).
Les effets indésirables ont été identifiés au cours des études cliniques menées chez des patients présentant un mélanome, traités par IMLYGIC versus GM-CSF et durant l'expérience post-commercialisation. L'incidence des effets indésirables est présentée par classe de systèmes d'organes et par fréquence. Les fréquences sont définies comme suit: très fréquents (≥1/10), fréquents (≥1/100 et < 1/10) et occasionnels (≥1/1'000 et < 1/100). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
Infections et infestations
Fréquents: cellulite**, infections herpétiques***.
Occasionnels: infections au site d'incision.
Tumeurs bénignes, malignes et non précisées
Fréquents: douleur tumorale, néoplasme infecté.
Occasionnels: plasmocytome au site d'injection**.
Affections hématologiques et du système lymphatique
Très fréquents: œdème périphérique (12,0%).
Fréquents: anémie.
Affections du système immunitaire
Fréquents: événements d'origine immunologique†**.
Occasionnels: hypersensibilité.
Troubles du métabolisme et de la nutrition
Fréquents: déshydratation.
Affections du système nerveux
Très fréquents: céphalées (18,8%).
Fréquents: état confusionnel, anxiété, dépression, vertiges, insomnie.
Affections oculaires
Occasionnels: kératite herpétique.
Affections de l'oreille et du labyrinthe
Fréquents: douleurs auriculaires.
Affections cardiaques
Fréquents: tachycardie.
Affections vasculaires
Fréquents: thrombose veineuse profonde, hypertension, rougeur cutanée.
Affections respiratoires, thoraciques et médiastinales
Très fréquents: toux (10,6%).
Fréquents: dyspnée, douleur oro-pharyngée, infection des voies respiratoires supérieures.
Occasionnels: affection obstructive des voies aériennes, pneumopathie inflammatoire.
Affections gastro-intestinales
Très fréquents: vomissements (21,2%), diarrhée (18,8%), constipation (11,6%), nausées (35,6%).
Fréquents: douleurs abdominales, gêne abdominale.
Affections de la peau et du tissu sous-cutané
Fréquents: vitiligo, rash cutané, dermatite.
Occasionnels: dermatite granulomateuse.
Affections musculo-squelettiques et du tissu conjonctif
Très fréquents: myalgie (17,5%), arthralgies (17,1%), extrémités douloureuses (16,4%).
Fréquents: dorsalgie, douleur inguinale.
Troubles généraux et anomalies au site d'administration
Très fréquents: syndrome grippal (30,5%)**, fièvre (42,8%), frissons (48,6%), fatigue (50,3%), douleurs (16,1%), réactions au site d'injection*.
Fréquents: malaise, douleur axillaire.
Investigations
Fréquents: perte de poids.
Lésions, intoxications et complications d'interventions
Fréquents: complications au niveau de la plaie, plaie suintante, contusion, douleurs liées à l'intervention.
* Les réactions au site d'injection comprennent: très fréquents douleurs (27,7%), fréquents rougeurs, hémorragies, gonflements, réactions, inflammations, sécrétions, suintement, occasionnels sensation de chaleur.
† Les évènements indésirables d'origine immunologique incluent: occasionnels vascularite, pneumopathie inflammatoire, aggravation d'un psoriasis et glomérulonéphrite.
** voir Description de certains effets indésirables.
*** Infections herpétiques (entre autres, herpès buccal).
Description de certains effets indésirables
Événements d'origine immunologique
Les événements d'origine immunologique rapportés dans l'étude clinique pivot consistaient en un cas d'aggravation du psoriasis chez un patient ayant des antécédents de psoriasis, un cas de pneumopathie inflammatoire chez un patient ayant des antécédents de maladie auto-immune, un cas de vascularite et deux cas de glomérulonéphrite dont un cas présentant une insuffisance rénale aiguë.
Plasmocytome
Dans les études cliniques, un cas de plasmocytome au site d'injection a été observé chez un patient chez lequel un myélome multiple a été diagnostiqué.
Cellulite
Dans l'étude pivot, les événements de cellulite rapportés n'ont pas conduit à un arrêt durable du traitement par IMLYGIC. Des soins attentifs de la plaie et des mesures de prévention des infections sont recommandés, en particulier si la nécrose entraîne des plaies ouvertes.
Symptômes pseudogrippaux
Des symptômes pseudogrippaux sont survenus chez 90% des patients traités par IMLYGIC. La fièvre, les frissons et le syndrome grippal, qui peuvent survenir à tout moment pendant le traitement, se sont généralement résolus en 72 heures. Ces événements ont été rapportés plus fréquemment pendant la période des six premiers traitements, en particulier chez les patients qui étaient séronégatifs pour le HSV-1 à l'inclusion.
Population pédiatrique
Une étude clinique de phase I (étude 20110261) a été menée chez 15 patients pédiatriques et jeunes adultes âgés de 7 à ≤ 21 ans atteints de tumeurs avancées ne touchant pas le système nerveux central pouvant être traitées par injection directe (voir «Propriétés/Effets»). Les données de sécurité étaient cohérentes avec la maladie sousjacente des patients et le profil de sécurité connu du talimogene laherparepvec chez les adultes.
Annonce d'effets indésirables
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucune expérience clinique de surdosage avec IMLYGIC n'a été rapportée. Des doses allant jusqu'à 4 ml à une concentration de 108 UFP/ml toutes les deux semaines ont été administrées dans les études cliniques sans signes de toxicité dose-limitante. La dose maximale pouvant être administrée sans risque n'a pas été déterminée. En cas de surdosage suspecté ou d'administration intraveineuse accidentelle, un traitement symptomatique, p.ex. par aciclovir ou d'autres antiviraux, doit être administré au patient (voir «Mises en garde et précautions») et des mesures de soutien doivent être instaurées si nécessaire.
Propriétés/EffetsCode ATC
L01XL02
Mécanisme d'action
Le talimogene laherparepvec est un virus oncolytique dérivé du HSV-1. Le talimogene laherparepvec a été modifié de façon à se répliquer dans les tumeurs et à produire la protéine humaine GM-CSF stimulant les réponses immunitaires. Le talimogene laherparepvec provoque la mort des cellules tumorales et la libération d'antigènes provenant de cellules tumorales. On pense qu'avec le GM-CSF, il stimule une réponse immunitaire antitumorale systémique et une réponse des lymphocytes T effecteurs. Des souris qui présentaient une régression complète de leurs tumeurs primaires après le traitement ont été résistantes à une nouvelle exposition tumorale ultérieure.
Les modifications du talimogene laherparepvec par rapport au HSV-1 incluent la délétion des gènes ICP34.5 et ICP47. Tandis que les réponses immunitaires antivirales protègent les cellules saines après l'infection par le talimogene laherparepvec, il a été observé que les tumeurs sont sensibles aux lésions et à la mort cellulaire causées par les virus HSV-1 déficients en gène ICP34.5, dont le talimogene laherparepvec. La délétion d'ICP47 prévient la régulation négative des molécules de présentation de l'antigène et augmente l'expression du gène US11 du HSV, ce qui stimule la réplication virale dans les cellules tumorales.
Efficacité et sécurité cliniques
Étude pivot
La sécurité et l'efficacité d'IMLYGIC en monothérapie par rapport au GM-CSF administré par voie sous-cutanée ont été évaluées dans une étude clinique de phase III internationale, randomisée, en ouvert, menée chez des patients présentant un mélanome de stade IIIB, IIIC ou IV considéré comme non résécable chirurgicalement. Un traitement systémique antérieur du mélanome était autorisé, mais n'était pas obligatoire. Les patients présentant des métastases cérébrales évolutives, des métastases osseuses, une atteinte viscérale étendue, un mélanome oculaire ou muqueux primaire, des signes d'immunosuppression ou qui recevaient un traitement anti-herpétique systémique étaient exclus de l'étude.
Les patients étaient randomisés selon un rapport 2:1 et ont reçu soit IMLYGIC soit GM-CSF (N = 436; IMLYGIC: N = 295, GM-CSF: N = 141). IMLYGIC était administré en injection intralésionnelle à la concentration initiale de 106 (1 million) UFP/ml le jour 1, puis à la concentration de 108 (100 millions) UFP/ml le jour 21 et toutes les deux semaines ensuite, à une dose allant jusqu'à 4 ml. Le GM-CSF était administré par voie sous-cutanée en cycles répétés à la dose 125 µg/m2 par jour pendant 14 jours suivis d'une phase sans traitement de 14 jours.
Pour permettre l'apparition des effets antitumoraux dus à la réponse immunitaire retardée, les patients étaient traités pendant une durée minimale de 6 mois ou jusqu'à ce qu'il n'y ait plus de lésions injectables. Pendant cette période, le traitement a été poursuivi même en cas d'augmentation de la taille des lésions existantes et/ou d'apparition de nouvelles lésions, à moins que le patient ait développé une toxicité intolérable ou que l'état clinique du patient ait nécessité l'instauration d'un nouveau traitement. Après 6 mois de traitement, les patients devaient continuer le traitement jusqu'à constater une progression de la maladie cliniquement pertinente (c.-à-d. progression de la maladie associée à une diminution du score de performance et/ou nécessité d'instaurer un autre traitement selon l'avis de l'investigateur).
Les patients présentant une réponse après 12 mois de traitement pouvaient poursuivre le traitement pendant une durée supplémentaire allant jusqu'à 6 mois. La durée moyenne (ET) de traitement dans la population en intention de traiter (ITT) était de 15,76 semaines (15,79) dans le bras GM-CSF et de 26,83 semaines (18,39) dans le bras IMLYGIC. Le critère principal d'évaluation était le taux de réponse durable (TRD) [défini comme le pourcentage de patients présentant une réponse complète (RC) ou une réponse partielle (RP) maintenue en permanence pendant au moins 6 mois] selon une évaluation centralisée en aveugle des patients répondeurs. Les critères d'évaluation secondaires comprenaient la survie globale (SG), le taux de réponse globale (TRG) [RP+RC], le délai de réponse, la durée de la réponse et le temps jusqu'à l'échec thérapeutique (temps depuis la randomisation jusqu'au premier épisode de progression de la maladie cliniquement significative sans réponse après l'événement de progression, ou jusqu'au décès).
L'âge moyen était de 63 ans (intervalle: de 22 à 94 ans), 26,5% des patients étant âgés de plus de 65 ans et 23,3% de plus de 74 ans. Les patients étaient en majorité caucasiens (98%). Les hommes constituaient 57% de la population de l'étude et 70% des patients avaient un score ECOG de 0 à l'inclusion. Parmi les patients inclus, 22% des patients présentaient une maladie de stade IVM1c et 53% des patients avaient reçu un traitement antérieur du mélanome tel qu'une chimiothérapie ou une immunothérapie à base de cytokines, en plus de la chirurgie, du traitement adjuvant ou de la radiothérapie. Au total, 58% des patients inclus dans l'étude étaient séropositifs pour le HSV-1 type sauvage à l'inclusion et 32,6% étaient séronégatifs. Le statut sérologique HSV-1 des 9,4% restants n'était pas connu.
Le traitement par IMLYGIC a entraîné une augmentation statistiquement significative du TRD dans la population ITT (voir tableau 3).
Tableau 3: Synthèse des résultats dans la population en ITT de l'étude pivot avec IMLYGIC
|
|
Critère d'évaluation de l'étude
|
IMLYGIC N = 295
|
GM-CSF N = 141
| |
Taux de réponse durable
|
principal
|
16,3% (n = 48)
(IC à 95%: 12,1, 20,5)
|
2,1% (n = 3)
(IC à 95%: 0,0, 4,5)
| |
Odds ratio 8,9; (IC à 95%: 2,7, 29,2)
p < 0,0001
| |
Taux de réponse global
(% de RC, % de RP)
|
secondaire
|
26,4% (n = 78)
(IC à 95%: 21,4%, 31,5%)
(RC 10,8%, RP 15,6%)
|
5,7% (n = 8)
(IC à 95%: 1,9%, 9,5%)
(RC 0,7%, RP 5%)
| |
Survie globale
|
secondaire
|
Médiane 23,3
(IC à 95%: 19,5, 29,6) mois
|
Médiane 18,9
(IC à 95%: 16,0, 23,7) mois
| |
HR: 0,79; (IC à 95%: 0,62, 1,00) p = 0,051
| |
Durée de la réponse (réponse durable au moment du dernier bilan tumoral)
|
secondaire
|
Non atteinte
(intervalle: > 0,0 à > 16,8 mois)
|
Médiane 2,8 mois
(intervalle: 1,2 à > 14,9 mois)
| |
HR: 0,46; (IC à 95%: 0,35, 0,60)
| |
Délai de réponse (médiane)
|
secondaire
|
4,1 mois
|
3,7 mois
| |
Temps jusqu'à l'échec du traitement (médiane)
|
secondaire
|
8,2 mois
(IC à 95%: 6,5, 9,9)
|
2,9 mois
(IC à 95%: 2,8, 4,0)
| |
HR: 0,42; (IC à 95%: 0,32, 0,54)
|
Chez 56 des répondeurs traités par IMLYGIC (72%), la réponse était toujours en cours au moment de l'analyse principale. Parmi les répondeurs, 42 (54%) ont présenté une augmentation de ≥25% de la taille totale des lésions existantes et/ou ont développé une ou plusieurs nouvelles lésions avant d'obtenir finalement une réponse (pseudoprogression).
Dans une analyse effectuée pour évaluer l'activité systémique d'IMLYGIC, 27 patients sur 79 (34,2%) ont présenté une diminution totale de ≥50% des lésions non viscérales non traitées par injection d'IMLYGIC et 8 patients sur 71 (11,3%) ont présenté une diminution totale de ≥50% des lésions viscérales non traitées par IMLYGIC.
Au moment du suivi médian de 44,4 mois, la survie globale médiane était de 23,3 mois (intervalle: 19,5, 29,6 mois) sous IMLYGIC et de 18,9 mois (intervalle: 16,0, 23,7 mois) sous GM-CSF (Hazard Ratio [HR] 0,79 (IC à 95%: 0,62, 1,00), p = 0,051, figure 4). Le taux de survie globale à 1, 2, 3 et 4 ans était plus élevé chez les patients affectés au bras IMLYGIC (73,7%, 49,8%, 38,6% et 32,6%) que chez les patients ayant reçu le GM-CSF (69,1%, 40,3%, 30,1% et 21,3%) (voir tableau 4).
Dans l'ensemble, aucune différence de sécurité ou d'efficacité n'a été observée entre les patients âgés (≥65 ans) et les patients adultes plus jeunes.
Figure 4: Courbe de Kaplan-Meier − survie globale (population en ITT)

Les patients qui n'avaient pas été enregistrés comme décédés ont été inclus comme censurés.
Des analyses exploratoires en sous-groupes du TRD et de la survie globale en fonction du stade de la maladie ont également été effectuées (voir figure 5 et tableau 4). Bien que l'étude pivot n'ait pas eu la puissance nécessaire pour évaluer l'efficacité dans ces sous-groupes d'individus, le bénéfice du traitement par IMLYGIC a été plus important chez les patients ne présentant pas d'atteinte viscérale que chez ceux présentant une maladie plus avancée.
Tableau 4: Synthèse des résultats d'une analyse exploratoire de l'étude pivot avec IMLYGIC
|
|
TRD (%)
|
TRG (%)
|
SG (hazard ratio)
| |
IMLYGIC
|
GM-CSF
|
IMLYGIC
|
GM-CSF
|
IMLYGIC versus GM-CSF
| |
Stade IIIB/IIIC/ Stade IVM1a
(IMLYGIC: n = 163; GM-CSF: n = 86)
|
25,2
|
1,2
|
40,5
|
2,3
|
0,57 (IC à 95%: 0,40; 0,80)
| |
Stade IVM1b/IVM1c
(IMLYGIC: n = 131; GM-CSF: n = 55)
|
5,3
|
3,6
|
9,2
|
10,9
|
1,07 (IC à 95%: 0,75; 1,52)
|
Figure 5: Courbe de Kaplan-Meier de la survie globale par bras de traitement randomisé pour les stades IIIB, IIIC et IVM1a (analyse exploratoire)

Les patients censurés sont indiqués par des barres verticales I.
Les patients qui n'avaient pas été enregistrés comme décédés ont été inclus comme censurés.
La population en ITT comprend tous les patients qui ont été randomisés pour recevoir un traitement à l'étude. Les patients sont analysés au moyen du traitement randomisé.
Un mois = 365,25/12 jours. NE = non évaluable.
En raison de la nature exploratoire de l'analyse et sur la base des données actuelles, il n'est pas établi qu'IMLYGIC soit associé à un effet sur la survie globale.
Population pédiatrique
Une étude de phase I multicentrique, en ouvert, à doses décroissantes (étude 20110261) a évalué la sécurité et l’efficacité du talimogene laherparepvec chez des patients pédiatriques atteints de tumeurs avancées ne touchant pas le système nerveux central pouvant être traitées par injection directe. Un total de 15 patients pédiatriques et de jeunes adultes âgés de 7 à ≤ 21 ans divisés en deux cohortes, la cohorte A1 (13 patients âgés de 12 à ≤ 21 ans) et la cohorte B1 (2 patients âgés de 7 à < 12 ans), ont reçu du talimogene laherparepvec au cours de l’étude. Le schéma posologique était cohérent avec la dose de talimogene laherparepvec recommandée chez l’adulte.
Treize patients ont été inclus dans l’ensemble d’analyse de la toxicité doselimitante (TDL). Aucun patient n’a présenté de TDL pendant la période d’évaluation de la TDL. L’ensemble des patients (15 patients, 100, 0%) ont présenté au moins 1 événement indésirable apparu sous traitement, et 8 patients (53,3%) ont présenté des événements indésirables de grade ≥ 3.
Aucune réponse n’a été observée; le taux de réponse globale (TRG) d’après le critère irRC-RECIST (critères de réponse immunologique/critères d’évaluation de la réponse au traitement des tumeurs solides) modifié était de 0% (IC 95%: 0,0;21,8).
PharmacocinétiqueLe talimogene laherparepvec est un virus HSV-1 génétiquement modifié et capable de se répliquer. Par conséquent, sa pharmacocinétique et sa biodistribution dépendent du site d'injection intralésionnelle, de la réplication sélective au sein de la tumeur et de la libération à partir du tissu tumoral.
Absorption
Après injection locale dans les tumeurs, la captation cellulaire du talimogene laherparepvec s'effectue par l'intermédiaire des récepteurs du HSV-1 présents sur les cellules tumorales et sur les cellules saines. Puisque le talimogene laherparepvec est injecté par voie intratumorale et se réplique dans la tumeur, la biodisponibilité et la concentration systémique de talimogene laherparepvec ne sont pas prédictives de l'activité du principe actif et n'ont donc pas été évaluées.
Métabolisme et Élimination
Le talimogene laherparepvec est éliminé par les mécanismes de défense généraux de l'hôte (p.ex. autophagie, réponses immunitaires adaptatives). Le talimogene laherparepvec est dégradé par les voies endogènes typiques du catabolisme des protéines et de l'ADN. Comme lors des autres infections par le HSV-1 type sauvage, un pool latent d'ADN du talimogene laherparepvec peut persister dans le corps cellulaire des neurones innervant les sites d'injection. Par conséquent, la survenue d'une infection latente par le talimogene laherparepvec ne peut être exclue.
Biodistribution (dans l'organisme) et excrétion virale (excrétion/sécrétion)
L'ADN du talimogene laherparepvec a été quantifié à l'aide d'une méthode par la réaction en chaîne par polymérase quantitative (qPCR) très sensible et spécifique qui peut ne pas être corrélée au risque d'infection virale. Le talimogene laherparepvec a également été quantifié chez des patients sélectionnés au cours des études cliniques en utilisant des dosages de la charge virale aux sites d'injection et dans certains cas de lésions herpétiques éventuelles.
Biodistribution clinique, élimination et excrétion
La biodistribution et l'excrétion du talimogene laherparepvec administré par voie intralésionnelle ont été évaluées dans une étude clinique qui a mesuré le taux d'ADN du talimogene laherparepvec dans le sang, les urines, le site d'injection, l'extérieur des pansements occlusifs, la muqueuse buccale, la région anogénitale et les lésions herpétiques suspectées. Soixante patients présentant un mélanome ont reçu une injection intralésionnelle d'IMLYGIC à une dose et un schéma identiques à ceux de l'étude pivot (voir «Efficacité et sécurité cliniques»). Des échantillons des pansements occlusifs ont été recueillis pendant le traitement. Des échantillons de sang et d'urine ont été recueillis pendant le traitement et pendant une période allant jusqu'à 30 jours après la fin du traitement. Des échantillons du site d'injection, de la muqueuse buccale et de la région anogénitale ont été recueillis pendant le traitement et pendant une période allant jusqu'à 60 jours après la fin du traitement. Des échantillons de lésions herpétiques suspectées ont été recueillis à chaque fois qu'un patient a présenté des lésions d'origine herpétique suspectée. Si les analyses qPCR pour l'ADN du talimogene laherparepvec étaient positives, une détermination de la DICT50 était effectuée pour mesurer la charge virale. Chez les 60 patients traités, les données indiquent que l'ADN du talimogene laherparepvec était présent dans tous les sites pendant l'étude (voir tableau 5).
Tableau 5: Patients ayant un taux d'ADN détectable pendant le traitement
|
Fluide corporel/site
|
Patients ayant un taux d'ADN détectable pendant le traitement
(n = 60)
| |
Sang
|
59 (98%)
| |
Urine
|
19 (32%)
| |
Site d'injection
|
60 (100%)
| |
Extérieur du pansement occlusif
|
48 (80%)
| |
Muqueuse buccale
|
8 (13%)
| |
Région anogénitale
|
5 (19%)a
|
a Pour la région anogénitale, 26 patients ont fait l'objet de tests pour détecter la présence de l'ADN d'IMLYGIC.
La proportion d'échantillons et de patients ayant un taux d'ADN du talimogene laherparepvec était la plus élevée pendant le cycle 2 du traitement pour le sang, l'urine, le site d'injection et les pansements occlusifs, pendant le cycle 1 du traitement pour la muqueuse buccale et pendant les cycles 1 et 2 pour la région anogénitale. Chez les patients ayant un taux d'ADN du talimogene laherparepvec détectable dans le sang, l'urine, la muqueuse buccale et la région anogénitale, aucun échantillon ne contenait d'ADN du talimogene laherparepvec détectable 30 jours après la fin du traitement. Chez les patients ayant un taux d'ADN détectable dans les lésions traitées, l'ADN du talimogene laherparepvec n'était détectable dans aucun échantillon 60 jours après la fin du traitement.
Au total, 3 des 19 patients présentant des lésions d'origine herpétique suspectée avaient un taux détectable d'ADN du talimogene laherparepvec à tout moment de l'étude.
L'activité virale a été mesurée dans les échantillons qui étaient positifs pour l'ADN du talimogene laherparepvec provenant du site d'injection, des pansements occlusifs, de la muqueuse buccale, de la région anogénitale et des lésions herpétiques suspectées. Une activité virale n'a été détectée dans aucun des échantillons des pansements occlusifs, de la muqueuse buccale, de la région anogénitale et des lésions herpétiques suspectées. Le virus talimogene laherparepvec infectieux a été détecté au niveau du site d'injection chez 7 (11%) patients à plusieurs moments de l'étude; aucun échantillon n'a été positif pour la charge virale après le cycle 2 ou après la fin du traitement.
Cinétique pour certains groupes de patients
Aucune étude pharmacocinétique n'a été réalisée avec le talimogene laherparepvec chez des groupes particuliers de patients.
Données précliniquesLe talimogene laherparepvec, administré à des doses uniques ou répétées allant jusqu'à 4 × 108 UFP/kg ou 107 UFP/dose (60 fois la dose clinique maximale recommandée) en injection sous-cutanée, intraveineuse ou intratumorale, a été bien toléré chez des souris, des rats et des chiens immunocompétents. Les effets observés chez les souris après l'injection répétée de talimogene laherparepvec étaient de manière générale modérés, limités à des réactions tissulaires locales au site d'injection, et correspondaient aux réactions inflammatoires attendues lors d'infections virales (p.ex. modifications passagères des populations de leucocytes, hyperplasie lymphoïde dans la rate et augmentation de l'hématopoïèse). Ils se sont améliorés avec la poursuite du traitement ou après la fin du traitement. Il n'a pas été observé de neuropathie ni d'effets indésirables neurologiques. Dans une étude in vivo en injection intracérébrale, le talimogene laherparepvec a été 10 000 fois moins neurovirulent qu'une dose de HSV-1 type sauvage qui était létale dans 50% des cas chez la souris.
Le talimogene laherparepvec a été injecté dans différentes xénogreffes tumorales à des doses allant jusqu'à 2 × 108 UFP/kg (30 fois la dose clinique maximale recommandée) chez des souris immunodéficientes (souris nudes et souris SCID). Une infection virale systémique létale a été observée chez jusqu'à 20% des souris nudes (principalement déficientes en fonction lymphocytaire T) et chez 100% des souris SCID (déficientes en lymphocytes T et B). Dans toutes les études, une infection virale disséminée létale a été observée chez 14% des souris nudes après le traitement par le talimogene laherparepvec à des doses 10 à 100 fois supérieures à celles entraînant 100% de létalité avec le HSV-1 type sauvage.
Mutagénicité
Le potentiel génotoxique du talimogene laherparepvec n'a pas été évalué dans des études à long terme chez l'animal ou chez l'homme. Le HSV-1 type sauvage ne s'intégrant pas dans le génome de l'hôte, le risque de mutagenèse insertionnelle avec le talimogene laherparepvec est négligeable.
Carcinogénicité
Le potentiel cancérogène du talimogene laherparepvec n'a pas été évalué dans des études à long terme chez l'animal ou chez l'homme. Cependant, les données disponibles pour le talimogene laherparepvec et le HSV-1 type sauvage n'indiquent pas de risque cancérogène chez l'homme.
Toxicité sur la reproduction et le développement
Aucun effet n'a été mis en évidence sur les tissus des organes reproducteurs mâles ou femelles après un traitement chez des souris adultes à des doses allant jusqu'à 4 × 108 UFP/kg (60 fois la dose clinique maximale sur une base UFP/kg). Aucun effet sur le développement embryonnaire et fœtal n'a été observé chez des souris en gestation ayant reçu du talimogene laherparepvec pendant la période d'organogenèse à des doses allant jusqu'à 4 × 108 (400 millions) UFP/kg (60 fois la dose clinique maximale sur une base UFP/kg). Des quantités négligeables (< 0,001% des concentrations plasmatiques maternelles) d'ADN du talimogene laherparepvec ont été détectées dans le sang fœtal.
Biodistribution/excrétion
Chez la souris, l'ADN du talimogene laherparepvec a été détecté après administration intralésionnelle dans environ 40% des échantillons tumoraux et dans ≤20% des échantillons de sang et de tissu organique (p.ex. rate, ganglions lymphatiques, foie, cœur et reins). L'ADN du talimogene laherparepvec a été détecté dans ≤2% des échantillons de cerveau, d'ovaire et de glandes salivaires. Il n'a pas été détecté dans la moelle osseuse, dans les yeux, les tissus sécréteurs (glandes lacrymales, muqueuse nasale) ou dans les excréments. La concentration d'ADN du talimogene laherparepvec la plus élevée a été constatée dans les lésions. Tous les autres tissus présentaient une concentration d'ADN du talimogene laherparepvec nettement plus faible (< 0,5% de la concentration maximale observée dans les tumeurs). Dans des tumeurs traitées, l'ADN du talimogene laherparepvec a pu être détecté jusqu'à 84 jours après la dernière dose. Il était cependant entièrement décomposé dans la majorité des échantillons de sang (94%) sept jours après la dernière dose.
Chez la souris, l'ADN du talimogene laherparepvec a été détecté dans environ 8% des échantillons de nerfs périphériques après administration intraveineuse.
Remarques particulièresIncompatibilités
Aucune étude de tolérance n'ayant été effectuée, ce médicament ne doit pas être mélangé à d'autres médicaments.
Conservation
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Préparation et conservation avant utilisation
IMLYGIC doit être utilisé aussi vite que possible après la décongélation.
Après décongélation, IMLYGIC est stable s'il est conservé à une température comprise entre 2°C et 25°C, à l'abri de la lumière, dans le flacon d'origine, dans une seringue, ou dans le flacon d'origine puis dans une seringue. Les durées de conservation indiquées dans le tableau 6 et le tableau 7 ne doivent pas être dépassées.
·Si IMLYGIC décongelé est conservé dans le flacon d'origine puis dans une seringue:
·La même plage de température doit être maintenue pendant toute la durée de stockage jusqu'à l'utilisation.
·La durée de conservation dans une seringue à une température allant jusqu'à 25°C ne doit pas excéder 2 heures pour 106 (1 million) UFP/ml et 4 heures pour 108 (100 millions) UFP/ml (voir tableau 6).
·La durée maximale cumulée de conservation (durée de conservation dans le flacon plus durée de conservation dans une seringue) ne doit pas dépasser la durée indiquée dans le tableau 7.
Après décongélation, IMLYGIC ne doit pas être recongelé. Jeter tout flacon ou seringue d'IMLYGIC décongelé, conservé plus longtemps que les durées indiquées ci-dessous.
Tableau 6: Durée maximale de conservation d'IMLYGIC décongelé dans une seringue
|
|
106 (1 million) UFP/ml
|
108 (100 millions) UFP/ml
| |
2°C à 8°C
|
8 heures
|
8 heures
| |
Jusqu'à 25°C
|
2 heures
|
4 heures
|
Tableau 7: Durée maximale cumulée de conservation (durée de conservation dans le flacon plus durée de conservation dans une seringue) d'IMLYGIC décongelé
|
|
106 (1 million) UFP/ml
|
108 (100 millions) UFP/ml
| |
2°C à 8°C
|
24 heures
|
1 semaine [7 jours]
| |
Jusqu'à 25°C
|
12 heures
|
24 heures
|
Remarques particulières concernant le stockage
Conserver et transporter congelé entre -90°C et -70°C.
Conserver dans son carton d'origine à l'abri de la lumière.
Conserver hors de portée des enfants.
Décongélation des flacons d'IMLYGIC
·Avant utilisation, décongeler les flacons d'IMLYGIC congelés à une température comprise entre 20°C et 25°C jusqu'à ce qu'IMLYGIC soit liquide. Selon les prévisions, la durée pour atteindre une décongélation complète des flacons est de 30 à 70 minutes, selon la température ambiante. Remuer doucement. Ne PAS agiter.
·Les flacons d'IMLYGIC doivent être décongelés et conservés dans leur carton d'origine jusqu'à l'utilisation afin de protéger le contenu de la lumière.
Remarques concernant la manipulation et l'utilisation, équipement de protection individuelle, déversements accidentels et élimination des déchets
Suivre les directives locales concernant la manipulation et l'utilisation, l'équipement de protection individuelle, les déversements accidentels et l'élimination des déchets.
·Porter des vêtements de protection ou une blouse de laboratoire, des lunettes de protection ou un masque et des gants pendant la préparation et l'administration d'IMLYGIC. Couvrir toutes les plaies exposées avant l'administration. Éviter tout contact avec la peau, les yeux ou les muqueuses.
·Après administration, changer de gants avant d'appliquer les pansements occlusifs sur les lésions traitées. Nettoyer l'extérieur des pansements occlusifs avec un tampon imbibé d'alcool. Il est recommandé que les sites d'injection restent couverts en permanence, dans la mesure du possible, par un pansement étanche à l'air et à l'eau. Pour réduire le risque de transmission virale, les patients doivent couvrir le site d'injection pendant au moins 8 jours après le dernier traitement, ou plus longtemps en cas de suintement ou d'exsudation du site d'injection. Indiquer aux patients d'appliquer le pansement conformément aux instructions du professionnel de santé et de remplacer le pansement s'il se détache.
·Éliminer tous les matériels ayant été en contact avec IMLYGIC (p.ex. flacon, seringue, aiguille, pansements) conformément aux directives locales.
Exposition accidentelle
·En cas de contact professionnel accidentel avec IMLYGIC (par projection dans les yeux ou sur les muqueuses) pendant la préparation ou l'administration, rincer à l'eau claire pendant au moins 15 minutes. En cas d'exposition d'une peau lésée ou par piqûre d'aiguille, nettoyer abondamment le site touché à l'eau et au savon et/ou avec un désinfectant.
·Éliminer tout déversement d'IMLYGIC avec un agent virucide et du matériel absorbant.
·Recommander aux patients de placer les pansements et les matériels de nettoyage usagés dans un sac en plastique refermable, car ceux-ci peuvent être contaminés, et d'éliminer le sac avec les ordures ménagères.
Éliminer tout médicament ou déchet conformément aux exigences nationales.
Numéro d’autorisation65812 (Swissmedic)
PrésentationIMLYGIC est proposé dans deux présentations différentes:
Flacon à usage unique inséré définitivement dans un manchon en plastique transparent
OU
Flacon à usage unique sans manchon en plastique transparent
IMLYGIC 106 unités formant plage (UFP)/ml: 1 flacon (avec bouchon vert clair). [A]
IMLYGIC 108 unités formant plage (UFP)/ml: 1 flacon (avec bouchon bleu roi). [A]
Titulaire de l’autorisationAmgen Switzerland AG, Risch
Domicile: 6343 Rotkreuz
Mise à jour de l’informationAvril 2025
Version #270924
|