CompositionPrincipe actif: brivaracétam.
Excipients:
Comprimés pelliculés:
noyau du comprimé (tous les dosages): croscarmellose sodique, lactose monohydraté, bétadex, lactose anhydre, stéarate de magnésium.
pelliculage du comprimé (tous les dosages): alcool polyvinylique, dioxyde de titane (E171), macrogol 3350, talc et
25 mg: oxyde de fer jaune (E172), oxyde de fer noir (E172).
50 mg: oxyde de fer jaune (E172), oxyde de fer rouge (E172).
75 mg: oxyde de fer jaune (E172), oxyde de fer rouge (E172), oxyde de fer noir (E172).
100 mg: oxyde de fer jaune (E172), oxyde de fer noir (E172).
Solution buvable:
citrate de sodium, acide citrique anhydre, carmellose sodique, sucralose, sorbitol liquide (E420), glycérol (E422), eau purifiée, arôme framboise, conserv.: parahydroxybenzoate de méthyle (E218).
Solution injectable:
acétate de sodium (trihydraté), acide acétique glacial, chlorure de sodium, eau pour préparations injectables.
Forme galénique et quantité de principe actif par unitéChaque comprimé pelliculé contient 10 mg, 25 mg, 50 mg, 75 mg ou 100 mg de brivaracétam.
Solution buvable: 10 mg de brivaracétam par ml.
Solution injectable: 50 mg de brivaracétam par flacon de 5 ml.
Indications/Possibilités d’emploiBriviact est indiqué en association dans le traitement des crises partielles avec ou sans généralisation secondaire chez l'adulte atteint d'épilepsie.
Posologie/Mode d’emploiPosologie
La dose initiale recommandée est de 50 mg/jour. Il est également possible de commencer avec 100 mg/jour, sur la base de l'avis du médecin concernant la nécessité de réduire les crises et en tenant compte des effets indésirables potentiels.
La dose doit être fractionnée en deux doses égales, une le matin et une le soir.
En fonction de la réponse clinique et de la tolérance du patient, la dose peut être ajustée entre 50 mg/jour et 200 mg/jour.
Les patients qui ont oublié une ou plusieurs doses doivent prendre une seule dose dès qu'ils s'en souviennent puis continuer le traitement avec la dose habituelle le matin ou le soir.
Arrêt du traitement
Si le traitement doit être arrêté, il est recommandé de diminuer progressivement la dose de brivaracétam par paliers de 50 mg/jour chaque semaine. Après une semaine de traitement à la dose de 50 mg/jour, une dernière semaine de traitement à la dose de 20 mg/jour est recommandée avant de complètement arrêter le traitement.
Mode d'emploi
Le brivaracétam peut être pris au cours ou en dehors des repas.
La dose journalière est administrée en parties égales, une fois le matin et une fois le soir.
Le brivaracétam peut être initié par voie intraveineuse ou par voie orale. Lors du passage de la voie orale à la voie intraveineuse, ou inversement, la dose journalière totale et la fréquence d'administration doivent être maintenues. Le brivaracétam en solution injectable est une alternative pour les patients chez lesquels l'administration orale est temporairement impossible.
Comprimés pelliculés:
les comprimés pelliculés doivent être avalés avec du liquide sans être croqués.
Solution buvable:
la solution buvable ne doit pas être diluée avant la prise.
Une sonde gastrique ou PEG peut être utilisée pour l'administration de la solution buvable.
Solution injectable:
il n'existe aucune donnée sur l'administration intraveineuse du brivaracétam deux fois par jour sur des périodes de plus de 4 jours.
Bolus intraveineux: le brivaracétam peut être administré en bolus intraveineux sans dilution.
Perfusion intraveineuse: le brivaracétam peut être dilué dans une solution pour perfusion compatible [voir «Remarques concernant la manipulation»] et administré en perfusion intraveineuse de 15 minutes.
Instructions spéciales pour la posologie
Patients âgés:
aucun ajustement posologique n'est nécessaire chez les patients âgés dans la mesure où ils disposent d'une fonction rénale suffisamment bonne (clairance de la créatinine >50 ml/min) [voir «Pharmacocinétique»].
L'expérience clinique avec le brivaracétam chez les patients âgés de ≥65 ans est limitée. Un premier traitement ne devrait pas être administré à des patients âgés de ≥75 ans [voir «Efficacité clinique»].
Patients atteints de maladies rénales:
aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance rénale légère. Les patients atteints d'insuffisance rénale modérée ou sévère ne doivent pas être traités par brivaracétam [voir «Mises en garde et précautions»].
En raison du manque de données, le brivaracétam n'est pas recommandé chez les patients atteints d'insuffisance rénale terminale sous dialyse.
Patients atteints de maladies hépatiques:
l'exposition au brivaracétam est respectivement augmentée de 50%, 57% et 59% en fonction des stades de Child Pugh A, B et C chez les patients présentant une maladie hépatique chronique en comparaison au groupe contrôle sain. Une dose initiale de 50 mg/jour est recommandée. Une dose journalière maximale de 150 mg administrée en deux doses égales est recommandée quel que soit le stade d'insuffisance hépatique.
Pédiatrie:
l'utilisation du brivaracétam chez les enfants et les adolescents n'est pas recommandée car la sécurité et l'efficacité pour ce groupe n'ont pas encore été prouvées.
Contre-indicationsHypersensibilité au brivaracétam, aux substances apparentées ou à l'un des excipients.
Mises en garde et précautionsIdées et comportements suicidaires
Des cas d'idées et de comportements suicidaires ont été rapportés chez des patients traités par des médicaments antiépileptiques, y compris par le brivaracétam, dans plusieurs indications. Une méta-analyse d'études randomisées, contrôlées contre placebo, portant sur des antiépileptiques, a également montré une légère augmentation du risque d'idées et de comportements suicidaires. Le mécanisme de ce risque n'est pas connu et les données disponibles n'excluent pas la possibilité d'une augmentation de ce risque avec le brivaracétam.
Les patients doivent donc être surveillés afin de détecter des signes d'idées et de comportements suicidaires et un traitement approprié devra être envisagé. Il faudra recommander aux patients (et à leurs aidants) de consulter un médecin dans les plus brefs délais en cas de survenue de signes d'idées ou de comportements suicidaires.
Insuffisance hépatique
Les données cliniques concernant l'utilisation de brivaracétam chez les patients présentant une insuffisance hépatique préexistante sont limitées.
Des ajustements de la posologie sont recommandés chez les patients atteints d'insuffisance hépatique [voir «Posologie/Mode d'emploi»].
Insuffisance rénale
Les données cliniques concernant l'utilisation de brivaracétam chez les patients présentant une insuffisance rénale modérée ou sévère sont limitées. Le brivaracétam ne devrait pas être utilisé chez les patients présentant une insuffisance rénale modérée ou sévère [voir «Pharmacocinétique/Insuffisance rénale»].
Intolérance au lactose
Les comprimés pelliculés avec 10 mg, 25 mg, 50 mg, 75 mg et 100 mg de brivaracétam contiennent respectivement 88, 94, 189, 283 et 377 mg de lactose. Les patients présentant une intolérance héréditaire rare au galactose, un déficit en lactase de Lapp ou un syndrome de malabsorption du glucose et du galactose ne devraient pas prendre les comprimés pelliculés.
La solution buvable contient du parahydroxybenzoate de méthyle (E218), qui peut provoquer une réaction allergique (éventuellement retardée). La préparation contenant également du sorbitol, la solution orale ne convient pas aux patients présentant une intolérance au fructose. Chaque ml de solution buvable contient 1,16 mg de sodium. Cela est à prendre en compte chez les patients suivant un régime pauvre en sodium.
InteractionsDonnées in vitro
L'hydrolyse du brivaracétam est médiée par des amidases indépendantes des CYP. L'hydroxylation du brivaracétam, principalement médiée par le CYP2C19, semble représenter une voie secondaire d'élimination. L'oxydation médiée par les CYP est responsable d'une proportion limitée de l'élimination du brivaracétam. Pour cette raison, il est peu probable que l'élimination du brivaracétam soit significativement influencée par des inhibiteurs des CYP (p.ex. CYP1A, 2C8, 2C9, 2C19, 2D6 et 3A4).
Inhibition d'enzymes métabolisant des médicaments
Il est peu probable que le brivaracétam inhibe la clairance d'autres médicaments métabolisés par des isoformes du CYP450. Les essais in vitro ont montré que le brivaracétam à des concentrations plasmatiques thérapeutiques n'inhibe que peu ou pas les isoformes du CYP450 (CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 3A4).
Le brivaracétam est un inhibiteur de l'époxyde hydrolase (CI50 = 8,2 µM), ce qui suggère que le brivaracétam peut inhiber l'enzyme in vivo.
Induction d'enzymes métabolisant des médicaments
Le brivaracétam n'a, pour des concentrations allant jusqu'à 10 µM, entraîné que peu ou pas de modifications de l'expression de l'ARNm des CYP1A2, 2B6, 2C9, 2C19, 3A4 et de l'époxyde hydrolase. Une induction de ces enzymes par des concentrations thérapeutiques de brivaracétam (Cmax = 13 µM) ne peut pas être exclue.
Transporteurs
Le brivaracétam n'est pas un substrat de la P-gp, la MRP1 ou la MRP2.
Le brivaracétam n'a pas inhibé les transporteurs P-gp, BCRP, BSEP, MRP2, MATE1, MATE2/K, OATP1B1, OATP1B3, OAT1, OCT1, OCT2 in vitro. Le brivaracétam a inhibé l'OAT3 avec une demi concentration inhibitrice maximale 42 fois supérieure à la Cmax de la dose clinique maximale. Le métabolite principal (métabolite hydroxylé) a entraîné une inhibition statistiquement significative de la BCRP et de l'OCT1 (inhibition de resp. 15 et 39%) à des concentrations suprathérapeutiques in vitro.
Données in vivo
Les études d'interaction n'ont été réalisées que chez des adultes.
Interactions entre le brivaracétam et d'autres médicaments
Le tableau 1 contient une liste des interactions cliniques pertinentes prouvées ou potentielles, mises en évidence par les variations de l'AUC et de la Cmax avec les symboles «↔» (au sein), «↑» (au-dessus) ou «↓» (au-dessous) des limites d'équivalence prédéfinies, le rapport des moyennes géométriques des moindres carrés [moyenne géométrique] avec et sans médicament interagissant ainsi que l'intervalle de confiance à 90% correspondant.
Les interactions médicamenteuses décrites ont été observées soit dans des études de phase 1, soit dans une analyse poolée des concentrations plasmatiques de principes actifs, soit dans des modèles pharmacocinétiques de population issus des données d'études de phase 2/3.
Un ajustement de la posologie n'est pas nécessaire lorsque le brivaracétam est utilisé de façon concomitante avec les médicaments cités dans la liste, à l'exception de la rifampicine, la rifabutine et la rifapentine (voir tableau 1).
Tableau 1: interactions entre le brivaracétam et d'autres médicaments
Rapports des moyennes géométriques de l'AUC et de la Cmax avec/sans médicament interagissant (intervalle de confiance à 90%)
|
Médicament en fonction de l'emploi thérapeutique
|
Effets du médicament sur la concentration plasmatique du brivaracétam
|
Effets du brivaracétam sur la concentration plasmatique du médicament
|
Recommandation en ce qui concerne l'utilisation concomitante avec le brivaracétam
| |
ANTICONVULSIVANTS
| |
Carbamazépine
|
Carbamazépine
600 mg/jour
AUC ↓ 0,74 (0,71; 0,77)
Cmax ↓ 0,87 (0,74; 1,02)
Carbamazépine
200-2400 mg/jour
AUC ↓ 0,74 (0,72; 0,76)
|
Brivaracétam 400 mg/jour
AUC ↔ 0,88 (0,85; 0,90)
Cmax ↔ 0,89 (0,86; 0,93)
Brivaracétam 50-200 mg/jour
Css ↔ 0,94 (0,92; 0,96)
|
Aucun ajustement de la posologie nécessaire
| |
Époxyde de carbamazépine
|
Non pertinent
|
Brivaracétam
400 mg/jour
AUC ↑ 2,57 (2,42; 2,74)
Cmax ↑ 2,64 (2,41; 2,89)
Brivaracétam
50-200 mg/jour
Css ↑ 1,63 (1,58; 1,67)
|
Aucun ajustement de la posologie nécessaire.
Aucune toxicité n'a été mise en évidence dans les études contrôlées.
| |
Clobazam
|
Interaction non examinée
Attendu: ↔
|
Brivaracétam
100-200 mg/jour
Css ↔ 0,95 (0,85; 1,05)
|
Aucun ajustement de la posologie nécessaire
| |
Clonazépam
|
Interaction non examinée
Attendu: ↔
|
Brivaracétam
100-200 mg/jour
Css ↔ 0,98 (0,87; 1,11)
|
Aucun ajustement de la posologie nécessaire
| |
Lacosamide
|
Interaction non examinée
Attendu: ↔
|
Brivaracétam
50-200 mg/jour
Css ↔ 0,95 (0,90; 1,00)
|
Aucun ajustement de la posologie nécessaire
| |
Lamotrigine
|
Aucune interaction
AUC ↔
|
Brivaracétam
50-200 mg/jour
Css ↔ 1,08 (1,05; 1,11)
Brivaracétam 400 mg
AUC ↔ 1,17 (1,09; 1,25)
Cmax ↔ 1.,10 (1,03; 1,17)
|
Aucun ajustement de la posologie nécessaire
| |
Lévétiracétam
|
Aucune interaction
AUC ↔
|
Brivaracétam
50-200 mg/jour
Css ↔ 1,06 (0,99; 1,13)
|
Aucun ajustement de la posologie nécessaire
| |
Oxcarbazépine
|
Aucune interaction
AUC ↔
|
Brivaracétam
50-200 mg/jour
Css ↔ 1,01 (0,98; 1,05)
|
Aucun ajustement de la posologie nécessaire
| |
Phénobarbital
|
AUC ↓ 0,81 (0,76; 0,86)
|
Brivaracétam
50-200 mg/jour
Css ↔ 1,02 (0,99; 1,05)
|
Aucun ajustement de la posologie nécessaire
| |
Phénytoïne
|
Phénytoïne
250-600 mg/jour
AUC ↓ 0,79 (0,75; 0,83)
|
Brivaracétam
50-200 mg/jour
Css ↔ 1,02 (0,91; 1,14)
Brivaracétam
400 mg/jour
AUC ↑ 1,20 (1,01; 1,42)
Cmax ↑ 1,20 (1,03; 1,40)
|
Aucun ajustement de la posologie nécessaire
| |
Prégabaline
|
Interaction non examinée
Attendu: ↔
|
Brivaracétam
50-200 mg/jour
Css ↔ 1,12 (0,99; 1,27)
|
Aucun ajustement de la posologie nécessaire
| |
Topiramate
|
Aucune interaction
AUC ↔
|
Brivaracétam
50-200 mg/jour
Css ↔ 0,98 (0,94; 1,03)
Brivaracétam 400 mg
AUC ↔ 0,95 (0,89; 1,01)
Cmax ↔ 0,98 (0,93; 1,03)
|
Aucun ajustement de la posologie nécessaire
| |
Acide valproïque
|
Aucune interaction
AUC ↔
|
Brivaracétam
50-200 mg/jour
Css ↔ 1,00 (0,97; 1,03)
|
Aucun ajustement de la posologie nécessaire
| |
Zonisamide
|
Interaction non examinée
Attendu: ↔
|
Brivaracétam
50-200 mg/jour
Css ↔ 1,01 (0,95; 1,07)
|
Aucun ajustement de la posologie nécessaire
| |
AGENTS HYPOLIPÉMIANTS/FIBRATES
| |
Gemfibrozil
|
Gemfibrozil 1200 mg/jour
AUC ↔ 0,95 (0,93; 0,97)
Cmax ↔ 1,01 (0,94; 1,08)
|
Interaction non examinée
Attendu: ↔
|
Aucun ajustement de la posologie nécessaire
| |
ANTIMYCOBACTÉRIENS
| |
Rifampicine
|
Rifampicine 600 mg/jour
AUC ↓ 0,55 (0,53; 0,58)
Cmax ↔ 0.,89 (0,83; 0,95)
|
Interaction non examinée
Attendu: ↔
|
Une élévation de la dose de brivaracétam devrait être prise en compte.
| |
Rifabutine
Rifapentine
|
Interactions non examinées
Attendu: ↓
|
Interactions non examinées
Attendu: ↔
|
Une élévation de la dose de brivaracétam devrait être prise en compte.
| |
PRÉPARATIONS PHYTHOTHÉRAPEUTIQUES
| |
Millepertuis
(Hypericum perforatum)
|
Interaction non examinée
Attendu: ↓
|
Interaction non examinée
Attendu: ↔
|
Un traitement avec du millepertuis doit être initié resp. arrêté avec précaution.
| |
CONTRACEPTIFS ORAUX
| |
Lévonorgestrel (LVN) éthinylestradiol (EES)
|
LVN 150 µg/jour
EES 30 µg/jour
Ctrough ↔ 1,10 (1,01-1,20)
|
Brivaracétam
100 mg/jour
EES AUC ↔ 0,90 (0,86; 0,95)
EES Cmax ↔ 0,96 (0,88; 1,04)
LVN AUC ↔ 0,92 (0,88; 0,97)
LVN Cmax ↔ 0.,95 (0,91; 0,99)
Brivaracétam
400 mg/jour
EES AUC ↓ 0,73 (0,69; 0,78)
EES Cmax ↓ 0,86 (0,79; 0,94)
LVN AUC ↓ 0,78 (0,72; 0,83)
LVN Cmax ↔ 0,90 (0,85; 0,95)
Aucune modification des marqueurs endogènes estradiol, progestérone, hormone lutéinisante, hormone folliculo-stimulante et globuline liant les hormones sexuelles n'a été observée.
|
Aucun ajustement de la posologie nécessaire.
Une perte de l'effet des contraceptifs oraux après administration de 200 mg de brivaracétam par jour ne peut pas être exclue.
| |
SOMNIFÈRES
| |
Midazolam
|
Interaction non examinée
Attendu: ↔
|
Brivaracétam
150 mg/jour
AUC ↔ 1,08 (0,97; 1,21)
Cmax ↑ 1,49 (1,18; 1,87)
|
Aucun ajustement de la posologie nécessaire
|
Notes:
·Les effets de la carbamazépine, du phénobarbital et de la phénytoïne sur le brivaracétam ont été examinés dans une analyse rétrospective de pharmacocinétique de population et étaient statistiquement significatifs.
·Les effets de la lamotrigine, du lévétiracétam, de l'oxcarbazépine, du topiramate et de l'acide valproïque ont été examinés dans une analyse rétrospective de pharmacocinétique de population et n'étaient pas statistiquement significatifs.
·Css = concentration plasmatique à l'état d'équilibre obtenue lors du suivi thérapeutique pharmacologique des patients; les données sont basées sur des doses thérapeutiques de brivaracétam de 50 à 200 mg/jour
·Ctrough = concentration avant l'administration de la dose à l'état d'équilibre
Autres
L'ampleur de la résorption du brivaracétam n'est pas influencée par les aliments.
Une étude d'interaction pharmacocinétique et pharmacodynamique entre le brivaracétam 200 mg en dose unique et l'éthanol 0,6 g/l en perfusion continue chez des sujets sains a montré que le brivaracétam a presque doublé l'effet de l'alcool sur les fonctions psychomotrices, l'attention et la mémoire. La prise de brivaracétam avec de l'alcool n'est pas recommandée.
Grossesse/AllaitementFemmes en âge de procréer
Les médecins doivent parler de planification familiale et de contraception avec les femmes en âge de procréer traitées par brivaracétam.
Une perte de l'effet des contraceptifs oraux ne pouvant pas être exclue, des méthodes contraceptives non hormonales devraient être utilisées.
Si une femme prévoit une grossesse, l'utilisation de brivaracétam devra être soigneusement réévaluée.
Grossesse
Il n'existe pas de données suffisantes concernant l'utilisation du brivaracétam chez la femme enceinte.
Il n'existe pas de données concernant le passage transplacentaire chez l'humain, mais les études chez le rat ont montré que le brivaracétam traverse le placenta [voir «Données précliniques»]. Le risque potentiel pour l'être humain n'est pas connu.
Les études chez l'animal, que cela soit chez le rat ou le lapin, n'ont détecté aucun potentiel tératogène du brivaracétam [voir «Données précliniques»].
Le brivaracétam a été utilisé en association dans des études cliniques et a entraîné une augmentation dose-dépendante de la concentration du métabolite actif époxycarbamazépine lors d'une administration concomitante avec la carbamazépine [voir «Interactions»]. Il n'existe pas de données suffisantes pour déterminer la pertinence clinique de cet effet pendant la grossesse.
Le brivaracétam ne doit pas être administré pendant la grossesse, sauf lorsque cela est clairement nécessaire.
Allaitement
On ignore si le brivaracétam passe dans le lait maternel humain. Les études chez le rat ont mis en évidence l'excrétion du brivaracétam dans le lait [voir «Données précliniques»].
Comme de nombreux médicaments passent dans le lait maternel humain, la décision concernant l'interruption de l'allaitement ou l'interruption de l'administration de brivaracétam devra être prise en tenant compte des bénéfices du médicament pour la mère.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude relative aux effets sur l'aptitude à la conduite et l'utilisation de machines n'a été effectuée.
Le brivaracétam peut avoir un effet sur l'aptitude à la conduite et l'utilisation de machines.
En raison des différences possibles de sensibilité individuelle, certains patients pourraient présenter une somnolence, des sensations vertigineuses et d'autres troubles du système nerveux central au début du traitement ou après une augmentation de la dose. Il faut suggérer aux patients de ne pas conduire de véhicules ni d'utiliser de machines potentiellement dangereuses avant d'être familiarisés avec les effets du brivaracétam sur leur capacité à effectuer ce type d'activités.
Effets indésirablesDans les études contrôlées et non contrôlées menées chez des patients épileptiques, 2388 patients ont au total reçu du brivaracétam. Parmi ces patients, 1740 ont été traités pendant ≥ 6 mois, 1363 pendant ≥ 12 mois, 923 pendant ≥ 24 mois, 733 pendant ≥ 36 mois et 569 pendant ≥ 60 mois (5 ans).
Dans les études d'association poolées contrôlées contre placebo menées chez 1558 patients adultes avec des crises partielles (1099 patients ont reçu du brivaracétam, 459 un placebo), des effets indésirables sont survenus chez 68,3% des patients traités par brivaracétam et 62,1% des patients traités par placebo.
Les effets indésirables les plus fréquemment rapportés (>10%) avec le brivaracétam étaient une somnolence (14,3%) et des sensations vertigineuses (11,0%). L'intensité était en règle générale légère à modérée. Somnolence et fatigue (8,2%) étaient plus fréquemment rapportées à une dose plus élevée. Les types d'effets indésirables rapportés au cours des sept premiers jours de traitement ressemblaient à ceux de l'ensemble de la période de traitement.
Le taux d'interruption suite à des effets indésirables était de 6,0%, 7,4% et 6,8% chez les patients qui avaient reçu respectivement 50 mg/jour, 100 mg/jour et 200 mg/jour de brivaracétam et de 3,5% chez les patients qui avaient reçu le placebo. Les effets indésirables qui entraînaient le plus fréquemment l'interruption du traitement par brivaracétam étaient les sensations vertigineuses (0,8%) et les convulsions (0,8%).
La liste suivante présente les effets indésirables des études cliniques poolées contrôlées contre placebo avec le brivaracétam par classe de système d'organes et fréquence.
La fréquence est définie comme suit: «très fréquent» (≥1/10), «fréquent» (≥1/100, <1/10,), «peu fréquent» ( ≥1/1000, <1/100). Dans les groupes individuels de fréquence, les effets indésirables sont indiqués par degré de sévérité décroissant.
Infections et infestations
Fréquent: grippe.
Affections hématologiques et du système lymphatique
Peu fréquent: neutropénie.
Troubles du métabolisme et de la nutrition
Fréquent: diminution de l'appétit.
Affections psychiatriques
Fréquent: dépression, anxiété, insomnie, irritabilité.
Peu fréquent: idées suicidaires, agressivité, agitation.
Affections du système nerveux
Très fréquent: sensations vertigineuses, somnolence
Fréquent: convulsion, vertiges
Affections respiratoires, thoraciques et médiastinales
Fréquent: infections des voies respiratoires supérieures, toux.
Affections gastro-intestinales
Fréquent: nausées, vomissements, constipation.
Troubles généraux et anomalies au site d'administration
Fréquent: fatigue.
Description d'effets indésirables sélectionnés
Une neutropénie a été observée chez 0,5% (6/1099) des patients traités par brivaracétam et 0% (0/459) des patients sous placebo. Quatre de ces sujets présentaient une diminution du nombre de neutrophiles à l'inclusion, et ont présenté une diminution supplémentaire du nombre de neutrophiles après l'introduction du brivaracétam. Aucun des 6 cas de neutropénie n'était sévère, n'a requis de traitement spécifique ou n'a conduit à l'arrêt du traitement par brivaracétam et aucun n'a présenté d'infections associées.
Des cas d'idées suicidaires ont été rapportés chez 0,3% (3/1099) des patients traités par brivaracétam et 0,7% (3/459) des patients sous placebo. Dans les études cliniques à court terme du brivaracétam menées chez des patients atteints d'épilepsie, aucun cas de suicide ou de tentative de suicide n'a été observé. Ces événements ont toutefois tous deux été rapportés dans les études d'extension en ouvert.
Administration intraveineuse
Les effets indésirables lors de l'administration intraveineuse ressemblaient en général à ceux d'une administration orale. Lors de l'administration intraveineuse, 2,8% des patients ont présenté des douleurs au site de perfusion.
Études d'extension en ouvert
Le profil de sécurité dans les études d'extension en ouvert (jusqu'à 8 ans) a été comparable à celui observé dans les études à court terme contrôlées contre placebo.
SurdosageSymptômes
L'expérience clinique sur le surdosage avec du brivaracétam chez l'homme est limitée. Somnolence et sensations vertigineuses ont été rapportées chez un volontaire sain ayant reçu une dose unique de 1400 mg de brivaracétam.
Mesures indiquées en cas de surdosage
Il n'existe pas d'antidote spécifique en cas de surdosage de brivaracétam. Le traitement d'un surdosage doit comprendre des mesures générales de soutien. Moins de 10% du brivaracétam étant éliminés dans les urines, l'hémodialyse ne devrait pas améliorer significativement l'élimination du brivaracétam [voir «Pharmacocinétique»].
Propriétés/EffetsCode ATC: N03AX23
Mécanisme d'action
Le brivaracétam présente une affinité élevée et sélective pour la protéine 2A des vésicules synaptiques (SV2A), une glycoprotéine transmembranaire présente dans les neurones et cellules endocrines au niveau présynaptique. La liaison à SV2A est considérée comme étant le principal mécanisme de l'activité anticonvulsivante du brivaracétam.
Pharmacodynamique
Impact sur l'intervalle QT
Chez les sujets sains, il n'y a eu aucun indice que le brivaracétam prolonge l'intervalle QTc aux doses de 150 mg/jour et 800 mg/jour (4× la dose journalière maximale recommandée).
Efficacité clinique
L'efficacité du brivaracétam en association dans le traitement des crises d'origine focale a été établie dans trois études multicentriques, randomisées en double aveugle (N01252, N01253 et N01358), contrôlées contre placebo, à dose fixe. Dans ces études, la dose quotidienne de brivaracétam était de 5 à 200 mg/jour. Toutes les études comportaient une période initiale de 8 semaines suivie d'une période de traitement de 12 semaines sans augmentation de dose. Parmi les 1558 patients participant à l'étude, 1099 ont reçu du brivaracétam. Selon les critères d'inclusion de l'étude, les patients devaient présenter des crises non contrôlées d'origine focale malgré un traitement par 1 ou 2 antiépileptiques concomitants. Les patients devaient présenter au moins 8 crises d'origine focale pendant la période initiale.
Les antiépileptiques les plus fréquemment utilisés au moment de l'inclusion dans les études ont été: carbamazépine (40,6%), lamotrigine (25,2%), valproate (20,5%), oxcarbazépine (16,0%), topiramate (13,5%), phénytoïne (10,2%) et lévétiracétam (9,8%) [voir «Efficacité clinique/Traitement par lévétiracétam»]. La fréquence médiane des crises durant la période initiale sur l'ensemble des trois études était de 9 crises en 28 jours. La durée moyenne de l'épilepsie des patients était d'environ 23 ans.
Le tableau 2 présente une synthèse des résultats d'efficacité.
Les résultats du pourcentage de réduction, en comparaison au placebo, de N01252 et N01253 se basent sur la fréquence des crises d'origine focale en l'espace de 28 jours, pour permettre la comparaison avec les résultats de N01358, bien que les analyses primaires de l'efficacité de N01252 et N01253 reposent sur la fréquence des crises d'origine focale en l'espace de 7 jours. Les résultats concernant la significativité statistique n'ont pas été influencés par la modification de la durée en fonction de laquelle la fréquence des crises d'origine focale a été standardisée pour N01252 et N01253.
Tableau 2: principaux résultats d'efficacité sur la fréquence des crises d'origine focale, en l'espace de 28 jours
|
Étude
|
Placebo
|
Brivaracétam
* Statistiquement significatif (valeur p)
| |
50 mg/jour
|
100 mg/jour
|
200 mg/jour
| |
Étude N01253(1)
| |
|
n = 96
|
n = 101
|
|
| |
Pourcentage de réduction par rapport au placebo (%)
|
NA
|
22,0*
(p=0,004)
|
~
|
~
| |
Taux de répondeurs à 50%** (%)
|
16,7
|
32,7*
(p=0,008)
|
~
|
~
| |
Pourcentage de réduction médian à partir de la valeur initiale (%)
|
17,8
|
30,5*
(p=0.003)
|
~
|
~
| |
Étude N01252(1)
| |
|
n = 100
|
n = 99
|
n = 100
|
| |
Pourcentage de réduction par rapport au placebo (%)
|
NA
|
9,2
(p=0,274)
|
20,5(2)
(p=0,01)
|
~
| |
Taux de répondeurs à 50%** (%)
|
20,0
|
27,3
(p=0,372)
|
36,0(2)
(p=0,023)
|
~
| |
Pourcentage médian de réduction à partir de la valeur initiale (%)
|
17,0
|
26,8
(p=0,092)
|
32,5(2)
(p=0,004)
|
~
| |
Étude N01358
| |
|
n = 259
|
|
n = 252
|
n = 249
| |
Pourcentage de réduction par rapport au placebo (%)
|
NA
|
~
|
22,8*
(p<0,001)
|
23,2*
(p<0,001)
| |
Taux de répondeurs à 50%** (%)
|
21,6
|
~
|
38,9
(p<0,001)
|
37,8
(p<0,001)
| |
Pourcentage médian de réduction à partir de la valeur initiale (%)
|
17,6
|
~
|
37,2*
(p<0,001)
|
35,6*
(p<0,001)
|
n = patients randomisés ayant reçu au moins une dose du médicament expérimental
~ Dose non étudiée
* Statistiquement significatif
** Taux de répondeurs à 50%: pourcentage de patients chez lesquels il y a eu une réduction de la fréquence des crises d'origine focale d'au moins 50% en l'espace de 28 jours, de la valeur initiale à la période de traitement.
(1) Environ 20% des patients ont reçu un traitement concomitant par lévétiracétam.
(2) Le critère primaire de N01252 n'a pas atteint la significativité statistique selon la procédure de test séquentiel, lequel exigeait la significativité statistique à 0,050 pour la dose de 50 mg/jour de brivaracétam par rapport au placebo avant les tests avec 100 mg/jour de brivaracétam. La dose de 100 mg/jour a été nominalement significative.
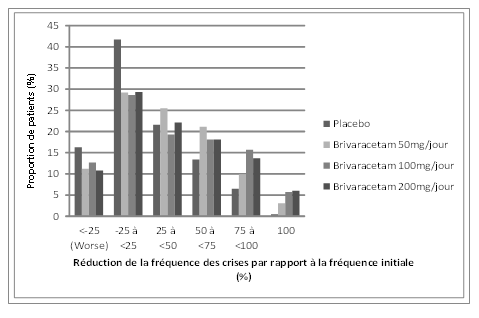
La figure 1 présente la proportion de patients (excluant les patients traités de façon concomitante par lévétiracétam) classée par pourcentage de réduction de la fréquence des crises d'origine focale par période de 28 jours par rapport à la fréquence initiale dans les trois études. Les patients avec une augmentation de plus de 25% des crises d'origine focale sont représentés à gauche du graphique sous «aggravation» (worse). Les patients avec une amélioration en termes de réduction de la fréquence des crises d'origine focale par rapport à la fréquence initiale apparaissent dans les quatre catégories de droite. Les pourcentages de patients ayant obtenu une réduction d'au moins 50% de la fréquence des crises ont été respectivement de 20,3%, 34,2%, 39,5% et 37,8% dans le groupe placebo et les groupes brivaracétam 50 mg/jour, 100 mg/jour et 200 mg/jour.
Figure 1: proportion de patients classée par pourcentage de réduction de la fréquence des crises par rapport à la fréquence initiale dans les groupes brivaracétam et placebo pendant 12 semaines issue des trois études pivots en double aveugle
Dans une analyse poolée des trois études pivots, aucune différence d'efficacité (mesurée par le taux de répondeurs à 50%) n'a été observée lorsque le brivaracétam était associé à des antiépileptiques inducteurs ou non inducteurs.
Dans les études cliniques, 2,5% (4/161), 5,1% (17/332) et 4,0% (10/249) des patients recevant respectivement du brivaracétam 50 mg/jour, 100 mg/jour et 200 mg/jour sont devenus libres de crises pendant la période de traitement de 12 semaines versus 0,5% (2/418) des patients recevant le placebo.
L'efficacité du brivaracétam en monothérapie n'a pas été examinée. Le brivaracétam ne doit pas être administré en monothérapie.
Traitement par lévétiracétam
Dans les études N01252 et N01253, le lévétiracétam a été l'antiépileptique concomitant administré chez environ 20% des patients. Chez les patients recevant un traitement concomitant par lévétiracétam, aucun bénéfice du brivaracétam par rapport au placebo n'a été observé. Aucun problème de sécurité ou de tolérance supplémentaire n'a été observé.
Dans la troisième étude (N01358), le lévétiracétam n'était pas autorisé en tant qu'antiépileptique concomitant. Une analyse prédéfinie a démontré l'efficacité cliniquement significative du brivaracétam en comparaison au placebo. L'efficacité était cependant plus faible chez les patients ayant au préalable reçu du lévétiracétam que chez les patients sans traitement préalable par lévétiracétam. L'efficacité plus faible observée chez ces patients en comparaison aux patients naïfs de traitement par lévétiracétam a probablement été due au nombre plus élevé d'antiépileptiques pris précédemment et à la fréquence des crises plus élevée durant la période initiale.
Dans les données poolées sur l'efficacité, les patients recevant un traitement concomitant par lévétiracétam ont été exclus.
Groupes de patients particuliers
Personnes âgées
Les trois études en double aveugle contrôlées contre placebo ont inclus 38 (2,4%) personnes âgées de ≥65 ans. Parmi ces patients de ≥65 ans, 30 ont été traités par brivaracétam. Au total, 2 patients de ≥75 ans ont été traités par brivaracétam [voir «Instructions spéciales pour la posologie/Patients âgés»].
Enfants et adolescents
L'efficacité et la sécurité du brivaracétam pour les enfants et les adolescents n'ont pas encore été établies.
Études d'extension en ouvert
Toutes études confondues, 81,7% des patients ayant terminé les études randomisées ont été inclus dans les études d'extension en ouvert à long terme. À partir de l'inclusion dans les études randomisées, 5,3% des sujets traités par brivaracétam pendant 6 mois (n=1500) ont été libres de crises versus 4,6% et 3,7% des patients traités pendant respectivement 12 mois (n=1188) et 24 mois (n=847).
Potentiel d'abus
Bien que les études réalisées avec le brivaracétam n'ont pas montré de potentiel d'abus, cette possibilité ne peut pas être complètement exclue.
Dépendance
Dans une évaluation poolée des études d'association contrôlées contre placebo, il n'y avait aucune indication d'un potentiel de dépendance physique ou de symptômes de sevrage provoqués par le brivaracétam.
PharmacocinétiqueAbsorption
Après administration orale, le brivaracétam est rapidement et complètement absorbé. La biodisponibilité absolue du comprimé pelliculé est de 103%. Le tmax moyen à jeun est de 1 heure pour les comprimés pelliculés (intervalle du tmax 0,25 à 3 h).
L'administration pendant un repas riche en graisses a ralenti la vitesse d'absorption du brivaracétam tandis que l'ampleur de l'absorption n'était pas modifiée.
Les comprimés pelliculés, la solution buvable et la solution injectable sont bioéquivalents.
Après administration de doses uniques de 10 mg-600 mg, l'AUC du brivaracétam augmente proportionnellement à la dose administrée. L'administration de doses uniques plus élevées entraîne une augmentation légèrement disproportionnée de l'AUC.
Après administration de 2,5 mg à 200 mg deux fois par jour (dose journalière 5 mg-400 mg) pendant 10 à 14 jours, l'AUC du brivaracétam augmente également proportionnellement à la dose administrée. L'administration de 400 mg deux fois par jour entraîne après plusieurs doses une augmentation de l'exposition au brivaracétam légèrement inférieure à une augmentation proportionnelle à la dose.
Lors d'une administration deux fois par jour, l'état d'équilibre est atteint en l'espace de 2 jours.
Distribution
La liaison du brivaracétam aux protéines plasmatiques est faible (≤20%). Le volume de distribution est de 0,5 l/kg, une valeur proche de celle de l'eau corporelle totale.
Métabolisme
Le brivaracétam est métabolisé principalement par hydrolyse de la fraction amide pour former l'acide carboxylique correspondant et secondairement par hydroxylation sur la chaîne latérale propyle. L'hydrolyse de la fraction amide entraînant la formation du métabolite acide carboxylique fait intervenir des amidases hépatiques et extra-hépatiques (EC3.5.1.4). In vitro, l'hydroxylation du brivaracétam est principalement due au CYP2C19. In vivo chez l'homme, la formation du métabolite hydroxylé est diminuée de 2 ou 10 fois chez les sujets présentant des mutations inefficaces du CYP2C19 tandis que l'AUC du brivaracétam est augmentée respectivement de 22% ou 42% chez les sujets porteurs de mutations impliquant une perte de fonction sur l'un ou les deux allèles, en comparaison aux sujets avec l'enzyme de type sauvage. Les inhibiteurs du CYP2C19 n'ont par conséquent probablement pas d'impact significatif sur la pharmacocinétique du brivaracétam. Un autre métabolite (le métabolite hydroxyacide) est en majeure partie créé par l'hydroxylation de la chaîne latérale propyle du métabolite acide carboxylique (principalement par le CYP2C9). L'AUC des métabolites hydroxy, acide et hydroxyacide correspond respectivement jusqu'à 10%, 8% et 2% de la substance mère. Les 3 métabolites ne sont pas pharmacologiquement actifs.
Élimination
Le brivaracétam est éliminé essentiellement par métabolisme et excrétion dans les urines. Après administration d'une dose unique marquée au 14C, 96,8% de la dose radioactive administrée est éliminée dans les urines et 0,7% dans les selles. 34% de la dose radioactive est éliminée dans les urines en l'espace de 48 heures sous forme de métabolite acide carboxylique. La proportion de brivaracétam inchangé éliminée dans les urines a été de 9%.
La demi-vie plasmatique (t½) est d'environ 9 heures.
Cinétique chez certains groupes de patients
Personnes âgées
Dans une étude chez des patients âgés (de 65 à 79 ans avec une clairance de la créatinine comprise entre 53 et 98 ml/min/1,73 m2) recevant 400 mg de brivaracétam par jour en deux prises, la demi-vie plasmatique du brivaracétam a respectivement été de 7,9 heures et 9,3 heures dans les groupes âgés de 65 à 75 ans et de >75 ans. La clairance plasmatique du brivaracétam à l'état d'équilibre a été légèrement inférieure (0,76 ml/min/kg) à celle observée chez des sujets sains, jeunes et de sexe masculin (0,83 ml/min/kg). Des adaptations de la dose ne sont pas nécessaires [voir «Posologie/Mode d'emploi»].
Insuffisance rénale
Une étude chez des patients présentant une insuffisance rénale sévère (clairance de la créatinine <30 ml/min/1,73 m2 ne nécessitant pas de dialyse) a montré que l'AUC de la concentration plasmatique du brivaracétam en fonction du temps était légèrement augmentée (+21%) par rapport aux sujets témoins sains, tandis que les AUC des concentrations plasmatiques des métabolites acide, hydroxy et hydroxyacide en fonction du temps étaient augmentées de respectivement 3, 4 et 21 fois. La clairance rénale de ces métabolites inactifs était diminuée de 10 fois. Des adaptations de la dose ne sont pas nécessaires chez les patients présentant une insuffisance rénale légère [voir «Posologie/Mode d'emploi»]. Le brivaracétam ne doit pas être administré en cas d'insuffisance rénale modérée ou sévère.
Le brivaracétam n'a pas été étudié chez les patients sous hémodialyse.
Insuffisance hépatique
Une étude de pharmacocinétique chez des patients présentant une cirrhose (classes A, B et C de Child-Pugh) a montré des augmentations de l'exposition au brivaracétam comparables, quelle que soit la sévérité de la maladie (50%, 57% et 59%) en comparaison aux sujets sains. Des ajustements de la posologie sont recommandés chez les patients atteints d'insuffisance hépatique [voir «Posologie/Mode d'emploi»].
Sexe
Il n'y a pas de différences spécifiques de la pharmacocinétique du brivaracétam en fonction du sexe.
Origine ethnique
L'origine ethnique n'a pas eu d'influence significative sur la pharmacocinétique du brivaracétam dans un modèle de pharmacocinétique de population issu des données de patients atteints d'épilepsie (blanc, noir/afro-américain, asiatique, amérindien [USA et Alaska], espagnol/latino-américain).
Données précliniquesLa dose orale non mortelle maximale chez le rat après une administration unique était >1000 mg/kg. Aucun signe de toxicité n'a été identifié à une dose de 500 mg/kg.
Dans des études conventionnelles de sécurité pharmacologique, les effets prédominants étaient liés au SNC (principalement dépression transitoire du SNC et diminution de l'activité locomotrice spontanée). Ces effets ont été observés à partir de 100 mg/kg, ce qui représente plusieurs fois (plus de 50 fois) la dose pharmacologiquement efficace de 2 mg/kg. De plus, les fonctions d'apprentissage et de mémoire n'ont pas été affectées par le brivaracétam.
Aucun effet indésirable hépatique n'a été observé chez le rat et le singe après administration chronique de brivaracétam à des expositions nettement supérieures (5 à 42 fois) à l'exposition humaine avec la dose clinique de 200 mg/jour. L'administration de brivaracétam chez le chien a entraîné des effets hépatotoxiques, principalement une porphyrie, à une exposition similaire à celle d'une dose clinique de 200 mg/jour chez l'homme. Les données collectées sur le brivaracétam et un composé structurellement apparenté indiquent toutefois que les modifications hépatiques (principalement porphyrie) chez le chien se sont développées par des mécanismes non pertinents chez l'homme.
Les expériences in vitro sur des bactéries et des cellules de mammifères et in vivo chez le rat et la souris n'ont pas mis en évidence d'effets mutagènes ou clastogènes.
Une étude de carcinogenèse chez le rat n'a pas montré de potentiel cancérogène. Une étude chez la souris a montré une légère augmentation de la fréquence de tumeurs hépatiques chez la souris mâle, considérées comme étant dues à un mode d'action non génotoxique lié à l'induction d'enzymes hépatiques par des agents de type phénobarbital, un phénomène connu chez le rongeur et lui étant spécifique.
Le brivaracétam n'a pas eu d'effet sur la fertilité mâle ou femelle et n'a pas montré de potentiel tératogène chez le rat ou le lapin. Une embryotoxicité a été observée chez le lapin à une dose maternotoxique correspondant à un niveau d'exposition 8 fois supérieur à l'exposition clinique. Chez le rat, il a été montré que le brivaracétam traverse le placenta et est excrété dans le lait des femelles.
Chez les rats juvéniles, des effets indésirables sur le développement (mortalité, symptômes cliniques, diminution du poids corporel et du poids du cerveau) ont été observés à la dose maximale testée de 600 mg/kg/jour.
Aucun effet négatif fonctionnel ou histopathologique sur le système nerveux n'a été constaté. Le NOAEL a été estimé à 300 mg/kg/jour (ce qui correspond à 3 à 20 fois l'exposition humaine pour une dose clinique de 200 mg/jour). Chez les chiens juvéniles, la dose de 100 mg/kg/jour a induit des anomalies hépatiques similaires à celles observées chez les animaux adultes.
Aucun effet négatif n'a été observé au niveau des paramètres standard de croissance ou de développement. Le NOAEL a été estimé à 30 mg/kg/jour (ce qui correspond à 1 à 3 fois l'exposition humaine pour une dose clinique de 200 mg/jour). Une exposition similaire au brivaracétam a été atteinte chez les animaux juvéniles et adultes lors du NOAEL, hormis au jour 4 après la naissance (PND4), où l'exposition était plus élevée que chez les animaux adultes.
Les études chez le rat n'ont pas montré de potentiel d'abus ou de dépendance.
Remarques particulièresIncompatibilités
La solution injectable Briviact ne doit pas être mélangée avec d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques concernant le stockage
Tenir hors de la portée des enfants.
Comprimés pelliculés: ne pas conserver au-dessus de 30 °C.
Solution buvable et solution injectable: conserver à 15-30 °C.
Solution buvable: conserver dans le récipient d'origine. Après la première ouverture: à utiliser dans les 5 mois.
Remarques concernant la manipulation
Solution injectable
La solution injectable Briviact étant destinée à un usage unique et ne contenant pas d'agents conservateurs, utiliser la solution immédiatement après avoir brisé le sceau du flacon et éliminer la solution non utilisée. La solution injectable ne doit pas être utilisée si des particules sont visibles ou si elle est colorée.
Dilution avec des solutions pour perfusion
La solution injectable Briviact peut être diluée avec une solution pour perfusion de chlorure de sodium 9 mg/ml (0,9%), une solution pour perfusion de glucose 50 mg/ml (5%) ou une solution pour perfusion de lactate de Ringer. La stabilité physique et chimique de la solution diluée, conservée dans des poches en PVC ou en polyoléfine à température ambiante pendant 24 heures, a été démontrée à des concentrations de 0,1 à 1,5 mg de brivaracétam par millilitre.
Les solutions diluées ne contiennent pas de conservateurs et doivent, pour des raisons microbiologiques, immédiatement être utilisées après dilution, sauf si la méthode de dilution exclut le risque d'une contamination microbiologique. Si la solution n'est pas utilisée immédiatement après dilution, le délai d'utilisation et les conditions de conservation relèvent de la responsabilité de l'utilisateur.
Numéro d’autorisation65830, 65831, 65832 (Swissmedic).
PrésentationComprimés pelliculés:
10 mg: emballages de 14 comprimés pelliculés [B]
25 mg, 50 mg, 75 mg et 100 mg: emballages de 56 comprimés pelliculés [B]
Emballages hospitaliers de 100 comprimés pelliculés individuels à 100 mg [B]
Solution buvable (10 mg/ml):
Emballage de 300 ml de solution (avec seringue pour administration de 10 ml et adaptateur) [B]
Solution injectable (50 mg/5 ml):
Emballage de 10 flacons de 5 ml [B]
Titulaire de l’autorisationUCB-Pharma SA, Bulle.
Mise à jour de l’informationMai 2016.
|