CompositionPrincipes actifs
Liraglutidum
Excipients
Dinatrii phosphas dihydricus, Propylenglycolum, Phenolum, Acidum hydrochloridum/Natrii hydroxidum (q.s. ad pH), Aqua ad iniectabilia.
Ce médicament contient 0.367 mg/ml de sodium.
Indications/Possibilités d’emploiSaxenda est indiqué en complément d'une alimentation hypocalorique et d'une activité physique accrue dans la régulation pondérale chez:
·Les patients adultes présentant un indice de masse corporelle (IMC) de
·≥ 30 kg/m² (obésité) ou
·≥ 27 kg/m² en présence de comorbidités supplémentaires liées au poids (prédiabète ou diabète de type 2, hypertension artérielle ou dyslipidémie).
Il faut arrêter Saxenda si les patients n'ont pas perdu au moins 5% de leur poids corporel après un traitement de 12 semaines avec une dose de 3,0 mg/jour.
·Les adolescents à partir de 12 ans dont le poids est ≥60 kg et en situation d'obésité, conformément aux seuils internationalement reconnus en la matière (correspond à un IMC ≥30 kg/m2 chez l'adulte)*.
*Seuils IMC de l'IOTF pour l'obésité en fonction du sexe pour les adolescents entre 12 et 18 ans.
|
Âge
(ans)
|
Body Mass Index
30 kg/m2
| |
Masculin
|
Féminin
| |
12
|
26.02
|
26.67
| |
12,5
|
26.43
|
27.24
| |
13
|
26.84
|
27.76
| |
13,5
|
27.25
|
28.20
| |
14
|
27.63
|
28.57
| |
14,5
|
27.98
|
28.87
| |
15
|
28.30
|
29.11
| |
15,5
|
28.60
|
29.29
| |
16
|
28.88
|
29.43
| |
16,5
|
29.14
|
29.56
| |
17
|
29.41
|
29.69
| |
17,5
|
29.70
|
29.84
| |
18
|
30.00-
|
30.00
|
Le traitement avec Saxenda doit être interrompu ou réévalué si l'IMC ou la valeur Z de l'IMC des patients ne s'est pas améliorée d'au moins 4% après un traitement de 12 semaines avec une dose de 3.0 mg/jour ou une dose maximale autorisée.
Posologie/Mode d’emploiPosologie usuelle
La dose initiale est de 0.6 mg une fois par jour. La dose doit être augmentée progressivement jusqu'à un maximum de 3.0 mg une fois par jour. Afin d'améliorer la tolérance gastro-intestinale, ceci peut se faire par paliers de 0.6 mg à intervalles d'au moins une semaine (voir Tableau 1). Si le passage à la dose supérieure n'est pas supporté pendant deux semaines consécutives, il faut considérer l'arrêt du traitement. Des doses quotidiennes supérieures à 3.0 mg ne sont pas recommandées.
Ajustement de la posologie / titration
Tableau 1. Schéma d'augmentation de la dose
|
|
Dose
|
Semaines
| |
Augmentation de la dose
sur 4 semaines
|
0.6 mg
|
1
| |
1.2 mg
|
1
| |
1.8 mg
|
1
| |
2.4 mg
|
1
| |
Dose d'entretien
|
3.0 mg
|
Patients présentant un diabète de type 2
Saxenda n'est pas indiqué pour le traitement du diabète de type 2. Saxenda ne doit pas être utilisé en association à un autre agoniste du récepteur du GLP-1.
Lors de l'initiation du traitement par Saxenda, il faut envisager une réduction de la dose de l'insuline utilisée ou des sécrétagogues (tels que les sulfonylurées), afin de réduire le risque d'hypoglycémie. Une autosurveillance glycémique par le patient est nécessaire pour ajuster la dose d'insuline ou des sécrétagogues de l'insuline.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Chez les patients atteints d'une insuffisance hépatique légère ou modérée, aucun ajustement posologique n'est nécessaire. L'utilisation de Saxenda ne peut être recommandée chez les patients atteints d'une insuffisance hépatique sévère et doit se faire avec prudence chez les patients présentant une insuffisance hépatique légère à modérée (voir sous «Mises en garde et précautions» et «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Chez les patients atteints d'une insuffisance rénale légère ou modérée (clairance de la créatinine ≥30 ml/min), aucune adaptation posologique n'est nécessaire. L'expérience clinique avec ce traitement est cependant limitée chez ces patients et Saxenda devrait être utilisé avec prudence dans cette population. L'utilisation de Saxenda ne peut être recommandée chez les patients atteints d'une insuffisance rénale sévère (clairance de la créatinine <30 ml/min), y compris chez les patients présentant une insuffisance rénale terminale (voir sous «Mises en garde et précautions», «Effets indésirables» et «Pharmacocinétique»).
Patients âgés (≥65 ans)
Aucun ajustement posologique n'est nécessaire chez les patients âgés. L'expérience de ce traitement chez les patients de ≥75 ans est limitée et l'utilisation n'est pas recommandée chez ces patients (voir sous «Mises en garde et précautions» et «Pharmacocinétique»).
Enfants et adolescents
L'utilisation de Saxenda sur des enfants âgés de moins de 12 ans, ou sur des adolescents dont le poids est inférieur ou égal à 60 kg n'est pas conseillée, en raison du manque de données (voir «Propriétés/Effets»).
Pour les adolescents âgés entre 12 et 18 ans, une augmentation de la dose similaire à celle utilisée chez l'adulte doit être effectuée (voir Tableau 1). La dose doit être augmentée jusqu'à 3.0 mg (dose d'entretien) ou jusqu'à la dose maximale tolérée. Les doses quotidiennes supérieures à 3.0 mg ne sont pas conseillées.
Mode d'administration
Saxenda est uniquement destiné à une administration par voie sous-cutanée. Il ne doit pas être utilisé par voie intraveineuse ou intramusculaire.
Saxenda est utilisé une fois par jour à n'importe quelle heure, indépendamment de l'heure des repas. L'injection peut avoir lieu dans la paroi abdominale, la cuisse ou le bras. Il convient de changer chaque fois de point d'injection dans la partie du corps choisie afin de réduire le risque d'amylose cutanée (voir «Effets indésirables»). Le site et le moment de l'injection peuvent être modifiés sans adaptation de la dose. Toutefois, Saxenda doit être injecté de préférence toujours à la même heure, une fois que le moment le plus propice a été choisi.
Saxenda ne doit pas être mélangé à d'autres médicaments injectables (p.ex. insuline).
En cas d'oubli d'une dose, si l'intervalle avec l'heure habituelle de l'utilisation est inférieur à 12 heures, le patient doit rattraper la dose le plus vite possible. S'il reste moins de 12 heures jusqu'à la prochaine dose prévue, le patient ne doit pas rattraper la dose oubliée, mais poursuivre son schéma posologique d'une administration quotidienne en prenant la prochaine dose prévue. Dans ce cas, aucune dose supplémentaire ne doit être injectée et la dose suivante ne doit pas être augmentée pour compenser la dose oubliée. D'autres remarques concernant la manipulation sont disponibles sous «Remarques particulières». Un mode d'emploi précis est annexé à l'information destinée aux patients.
Contre-indicationsHypersensibilité au liraglutide ou à l'un des excipients selon la «Composition».
Mises en garde et précautionsSaxenda ne doit pas être utilisé à la place de l'insuline chez les patients diabétiques. Des cas d'acidocétose diabétique ont été rapportés chez des patients insulino-dépendants après une interruption rapide de l'administration d'insuline ou une réduction rapide de la dose d'insuline (voir «Posologie/Mode d'emploi»).
On ne dispose d'aucune expérience clinique chez les patients présentant une insuffisance cardiaque de classe IV de la New York Heart Association (NYHA), par conséquent l'utilisation du liraglutide est déconseillée chez ces patients. Les expériences chez les patients atteints d'une insuffisance cardiaque de classes I-III NYHA sont basées sur l'étude LEADER au cours de laquelle ils ont été traités au liraglutide à une dose quotidienne maximale de 1.8 mg.
La sécurité et l'efficacité du liraglutide n'ont pas été prouvées pour la régulation pondérale chez les patients suivants:
·patients âgés de 75 ans ou plus
·patients traités par d'autres produits destinés à la régulation pondérale
·patients dont l'obésité résulte de troubles endocriniens ou de troubles alimentaires tels que la boulimie ou l'hyperphagie boulimique (Binge Eating), ou d'un traitement par des médicaments pouvant provoquer une prise de poids
·patients présentant une insuffisance rénale sévère
·patients présentant une insuffisance hépatique sévère.
L'utilisation chez ces patients n'est pas recommandée (voir sous «Posologie/Mode d'emploi»).
Le liraglutide n'a pas été examiné pour la régulation pondérale chez les patients présentant une insuffisance hépatique ou rénale légère ou modérée; il doit donc être utilisé avec prudence chez ces patients (voir sous «Posologie/Mode d'emploi» et «Pharmacocinétique»).
Aspiration liée à une anesthésie générale ou à une sédation profonde
Des cas d'aspiration pulmonaire ont été rapportés chez des patients recevant des agonistes des récepteurs du GLP-1 soumis à une anesthésie générale ou à une sédation profonde, qui avaient pourtant bien respecté les recommandations de jeûne préopératoire. Par conséquent, le risque accru de contenu résiduel gastrique dû au retard de vidange gastrique doit être pris en considération avant d'effectuer des interventions sous anesthésie générale ou sédation profonde.
Maladie inflammatoire de l'intestin et gastroparésie diabétique
L'expérience chez les patients présentant des maladies inflammatoires de l'intestin et une gastroparésie diabétique est limitée. L'utilisation du liraglutide n'est pas recommandée chez ces patients, étant donné qu'elle est associée à des effets indésirables gastro-intestinaux passagers tels que nausées, vomissements et diarrhées.
Pancréatite
Une pancréatite aiguë a été observée avec l'utilisation d'agonistes des récepteurs du GLP-1. Les patients doivent être informés des symptômes caractéristiques de la pancréatite aiguë. En cas de suspicion de pancréatite, le liraglutide doit être arrêté; si la pancréatite aiguë est confirmée, le traitement par le liraglutide ne doit pas être repris. Une augmentation isolée des enzymes pancréatiques sous traitement par Saxenda (sans symptômes caractéristique) n'indique pas nécessairement une pancréatite aiguë (voir «Effets indésirables»).
Lithiase biliaire et cholécystite
Au cours des études cliniques portant sur la régulation pondérale, une survenue accrue de lithiases biliaires et de cholécystites a été observée chez les patients traités par le liraglutide, par rapport aux patients recevant le placebo. Le fait qu'une perte de poids importante puisse augmenter le risque de lithiase biliaire et donc de cholécystite n'expliquait que partiellement la fréquence plus accrue sous le liraglutide. La lithiase biliaire et la cholécystite peuvent conduire à une hospitalisation et à une cholécystectomie. Les patients doivent être informés des symptômes caractéristiques de la lithiase biliaire et de la cholécystite.
Maladies thyroïdiennes
Au cours des études cliniques menées chez des patients atteints de diabète de type 2, des effets indésirables thyroïdiens tels qu'un goitre ont été rapportés, en particulier chez les patients présentant une maladie thyroïdienne préexistante. Le liraglutide doit donc être utilisé avec prudence chez les patients présentant des pathologies thyroïdiennes.
Fréquence cardiaque
Le liraglutide augmente la fréquence cardiaque (voir sous «Propriétés/Effets»). La fréquence cardiaque doit être mesurée à intervalles réguliers conformément à la pratique clinique habituelle. Les patients doivent être informés des symptômes caractéristiques d'une fréquence cardiaque élevée (palpitations ou sensation d'emballement du cœur au repos). Le liraglutide doit être arrêté chez les patients qui présentent une élévation durable cliniquement significative de la fréquence cardiaque au repos.
Déshydratation
Chez les patients traités par des agonistes du récepteur du GLP-1, des signes et des symptômes de déshydratation ont été rapportés, y compris une insuffisance rénale et une insuffisance rénale aiguë. Les patients traités par le liraglutide doivent être avertis du risque potentiel de déshydratation lié aux effets indésirables gastro-intestinaux et doivent prendre des précautions pour éviter une perte hydrique.
Hypoglycémie chez les patients présentant un diabète de type 2
Les patients présentant un diabète de type 2 qui reçoivent du liraglutide en association à des sécrétagogues tels qu'une sulfonylurée, ou à de l'insuline, présentent un risque accru d'hypoglycémie. Le risque d'hypoglycémie peut être réduit par la diminution de la dose de l'insuline et/ou du sécrétagogue.
Enfants et adolescents
Des épisodes d'hypoglycémie cliniquement pertinente ont été rapportés chez des adolescents (≥12 ans) traités par liraglutide. Les patients doivent être informés des symptômes caractéristiques de l'hypoglycémie et des contre-mesures appropriées.
Composants particuliers
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsInteractions pharmacocinétiques
Évaluation in vitro des interactions médicamenteuses avec le liraglutide
Le liraglutide a montré un très faible potentiel d'interactions pharmacocinétiques avec d'autres principes actifs associés au cytochrome P450 (CYP) et à une liaison aux protéines plasmatiques.
Évaluation in vivo des interactions médicamenteuses avec le liraglutide
Le léger ralentissement de la vidange gastrique observé avec le liraglutide peut influencer l'absorption des médicaments administrés de façon concomitante par voie orale. Les études d'interaction n'ont montré aucun ralentissement cliniquement significatif de l'absorption; par conséquent, aucune adaptation posologique n'est nécessaire.
Les études d'interaction ont été réalisées avec 1.8 mg de liraglutide. L'effet sur la vitesse de vidange gastrique était équivalent pour 1.8 mg de liraglutide et 3 mg de liraglutide (paracétamol AUC0-300 min). Quelques patients traités par le liraglutide ont signalé au moins un épisode sévère de diarrhée. Une diarrhée peut influencer l'absorption concomitante des médicaments pris par voie orale.
Effet de Saxenda sur d'autres médicaments
Warfarine et autres dérivés coumariniques
Aucune étude d'interaction n'a été réalisée. Une interaction cliniquement significative avec des principes actifs peu solubles ou à marge thérapeutique étroite comme la warfarine ne peut être exclue. Lors de l'initiation du traitement par le liraglutide chez les patients sous warfarine ou autres dérivés coumariniques, il est recommandé de surveiller plus fréquemment le Rapport Normalisé International (INR).
Paracétamol (acétaminophène)
Le liraglutide n'a pas entraîné de modification de l'exposition totale au paracétamol après administration d'une dose unique de 1'000 mg. La Cmax du paracétamol a diminué de 31 %; le tmax moyen a augmenté jusqu'à 15 min. Aucune adaptation de la dose n'est nécessaire en cas d'utilisation concomitante de paracétamol.
Atorvastatine
Le liraglutide n'a pas entraîné de modification de l'exposition totale à l'atorvastatine après l'administration d'une dose unique de 40 mg d'atorvastatine. C'est pourquoi aucune adaptation de la dose d'atorvastatine n'est nécessaire lors de l'administration simultanée du liraglutide. Le liraglutide a entraîné une diminution de 38% de la Cmax de l'atorvastatine et un ralentissement du tmax moyen de 1 h à 3 h.
Griséofulvine
Le liraglutide n'a pas entraîné de modification de l'exposition totale à la griséofulvine après l'administration d'une dose unique de 500 mg de griséofulvine. La Cmax de la griséofulvine a augmenté de 37%, alors que le tmax moyen n'a pas changé. Aucun ajustement posologique de la griséofulvine et des autres préparations faiblement solubles et fortement perméables n'est nécessaire.
Digoxine
Après l'administration d'une dose unique de 1 mg de digoxine avec du liraglutide, l'AUC de la digoxine a été réduite de 16 % et la Cmax a diminué de 31 %. Avec le liraglutide, le tmax moyen de la digoxine passait de 1h à 1.5h. Ces résultats indiquent qu'aucun ajustement posologique de la digoxine n'est nécessaire.
Lisinopril
Après l'administration d'une dose unique de 20 mg de lisinopril avec du liraglutide, l'AUC du lisinopril a été réduite de 15 % et la Cmax a diminué de 27 %. Avec le liraglutide, le tmax moyen du lisinopril a été retardé, passant de 6h à 8h. Ces résultats indiquent qu'aucun ajustement posologique du lisinopril n'est nécessaire.
Contraceptifs oraux
Après l'administration d'une dose unique d'un contraceptif oral, le liraglutide a diminué la Cmax de l'éthinylestradiol et du lévonorgestrel de respectivement 12 % et 13 %. Le tmax des deux substances s'est prolongé de 1.5 h sous le liraglutide. Aucun effet cliniquement significatif concernant l'AUC de l'éthinylestradiol ou du lévonorgestrel n'a été observé. L'effet contraceptif n'est donc apparemment pas altéré lors de l'administration simultanée du liraglutide.
Grossesse, allaitementGrossesse
Il n'existe pas suffisamment de données concernant l'utilisation du liraglutide durant la grossesse. Des études d'expérimentation animale ont montré une toxicité sur la reproduction (voir sous «Données précliniques»). Le risque potentiel pour l'être humain n'est pas connu.
Saxenda ne doit pas être utilisé durant la grossesse, à moins que cela ne soit clairement nécessaire. En cas de projet de grossesse ou en cas de grossesse, le traitement par Saxenda devra être interrompu.
Allaitement
On ignore si le liraglutide passe dans le lait maternel. Des études d'expérimentation animale ont montré que de petites quantités de liraglutide et de métabolites de structure similaire passent dans le lait maternel. Des études précliniques réalisées chez de jeunes rats allaités ont mis en évidence un ralentissement de la croissance néonatale lié au traitement (voir sous «Données précliniques»). En raison du manque d'expérience, Saxenda ne devra pas être utilisé pendant l'allaitement.
Fertilité
Mise à part une légère réduction du taux d'implantation, les études animales sur la fertilité n'ont montré aucun effet néfaste direct (voir sous «Données précliniques»).
Il n'existe pas suffisamment de données cliniques relatives à l'influence de la fertilité chez l'être humain.
Effet sur l’aptitude à la conduite et l’utilisation de machinesSaxenda n'a aucune influence ou a une influence négligeable sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
La sécurité de Saxenda a été évaluée dans le cadre de 5 études cliniques en double aveugle et contrôlées par placebo dans lesquelles ont été inclus 5'813 patients adultes, en surpoids ou obèses présentant au moins un facteur de comorbidité lié au poids. Dans l'ensemble, les effets indésirables signalés le plus fréquemment lors du traitement par Saxenda concernaient des effets indésirables gastro-intestinaux (voir sous «Description de certaines réactions indésirables»).
Effets indésirables présentés sous forme de tableau
Le Tableau 2 répertorie les effets indésirables rapportés au cours des études contrôlées de phase 2 et de phase 3 sur des adultes et de rapports post-marketing Les effets indésirables sont énumérés par classe de systèmes d'organes et par fréquence. La fréquence des effets indésirables se base sur les données consolidées des études de phase 2 et de phase 3.
Les catégories de fréquences sont définies de la manière suivante: «très fréquents» (≥1/10), «fréquents» (≥1/100 à <1/10), «occasionnels» (≥1/1000 à <1/100), «rares» (≥1/10'000 à <1/1000), «très rares» (<1/10'000), «Fréquence inconnue» (ne peut être estimée sur la base des données disponibles).
Au sein de chaque groupe de fréquence, les réactions indésirables sont présentées suivant un ordre décroissant de gravité.
Tableau 2. Effets indésirables rapportés au cours d'études contrôlées de phase 2 et de phase 3 sur des adultes et de rapports post-marketing
|
Classes de systèmes d'organes selon MedDRA
|
Fréquence
|
Effet indésirable
| |
Affections du système immunitaire
|
Rares
|
Réaction anaphylactique
| |
Troubles du métabolisme et de la nutrition
|
Fréquents
|
Hypoglycémie*
| |
Occasionnels
|
Déshydratation
| |
Affections psychiatriques
|
Fréquents
|
Insomnie**
| |
Affections du système nerveux
|
Très fréquents
|
Céphalées
| |
Fréquents
|
Vertiges
Dysgueusie
| |
Affections cardiaques
|
Occasionnels
|
Tachycardie
| |
Affections gastro-intestinales
|
Très fréquents
|
Nausées
Vomissements
Diarrhées
Constipation
| |
Fréquents
|
Sécheresse buccale
Dyspepsie
Gastrite
Reflux gastro-œsophagien
Douleurs abdominales hautes
Flatulences
Éructations
Distension abdominale
| |
Occasionnels
|
Vidange gastrique retardée
Pancréatite***
| |
Fréquence inconnue
|
Obstruction intestinale†a
| |
Affections hépatobiliaires
|
Fréquents
|
Lithiase biliaire***
| |
Occasionnels
|
Cholécystite***
| |
Investigations
|
Fréquents
|
Lipase augmentée
Amylase augmentée
| |
Affections de la peau et du tissu sous-cutané
|
Fréquents
|
Éruption cutanée
| |
Occasionnels
|
Urticaire
| |
Affections du rein et des voies urinaires
|
Rares
|
Insuffisance rénale aiguë
Insuffisance rénale
| |
Fréquence inconnue
|
Amyloïdose cutanée†
| |
Troubles généraux et anomalies au site d'administration
|
Fréquents
|
Réactions au site d'injection
Asthénie
Épuisement
| |
Occasionnels
|
Malaise
| |
* Hypoglycémie (basée sur les symptômes rapportés par les patients eux-mêmes et non confirmée par une mesure de la glycémie) rapportée chez les patients ne présentant pas de diabète de type 2 traités par Saxenda en association à un régime alimentaire et à de l'activité physique. Voir sous «Description de certains effets indésirables» pour plus d'informations.
** L'insomnie a été principalement observée pendant les 3 premiers mois de traitement.
*** voir sous «Mises en garde et précautions».
† Effet indésirable rapporté après la mise sur le marché.
a) Terme groupé couvrant les événements indésirables obstruction intestinale, iléus et obstruction de l'intestine grêle.
|
Description de certains effets indésirables
Tumeurs bénignes, malignes et non précisées (y compris kystes et polypes)
Occasionnels – Cancer du sein
Dans le cadre des études cliniques portant sur Saxenda, un cancer du sein a été rapporté chez 17 (0.7 %) des 2379 femmes traitées par Saxenda par rapport à 3 (0.2 %) des 1300 femmes recevant le placebo, y compris des cancers invasifs (13 femmes traitées par Saxenda et 2 femmes recevant le placebo) et des carcinomes canalaires in situ (4 femmes traitées par Saxenda et 1 femme recevant le placebo). La plupart des cancers étaient positifs pour les récepteurs à estrogènes et progestérone. Les cas n'étaient pas assez nombreux pour qu'on puisse déterminer s'ils étaient liés à Saxenda. En outre, les données disponibles sont insuffisantes pour déterminer si Saxenda a des effets sur une néoplasie du sein préexistante.
Occasionnels – carcinome papillaire de la thyroïde
Dans le cadre des études cliniques portant sur Saxenda, un cancer papillaire de la thyroïde a été rapporté chez 8 (0.2 %) des 3'291 patients traités par Saxenda par rapport à l'absence de cas chez les 1'843 patients recevant le placebo. Parmi ces carcinomes papillaires de la thyroïde, quatre avaient un diamètre maximal inférieur à 1 cm, et 4 ont été diagnostiqués sur des échantillons tissulaires après thyroïdectomie effectuée en raison de résultats obtenus avant le traitement par Saxenda.
Occasionnels – tumeurs colorectales
Dans les études cliniques portant sur Saxenda, des tumeurs colorectales bénignes (principalement des adénomes coliques) ont été rapportées chez 20 (0.6 %) des 3'291 patients traités par Saxenda par rapport à 7 (0.4 %) des 1'843 patients recevant le placebo. Six cas de carcinome colorectal ont été rapportés, 5 (0.2 %) chez les patients traités par Saxenda et un (0.1 %) chez les patients recevant le placebo.
Affections du système immunitaire
Rares – Réactions anaphylactiques
Quelques cas de réactions anaphylactiques comportant des symptômes tels qu'hypotension artérielle, palpitations, dyspnée et œdèmes ont été rapportés lors de l'utilisation du liraglutide après la mise sur le marché. Les réactions anaphylactiques peuvent mettre la vie en danger. En cas de suspicion de réaction anaphylactique, il faut arrêter le liraglutide et ne plus reprendre le traitement (voir sous «Contre-indications»).
Troubles du métabolisme et de la nutrition
Très fréquents – Hypoglycémie chez les patients présentant un diabète de type 2 (23 %)
Au cours d'une étude clinique menée chez des patients obèses ou en surpoids présentant un diabète de type 2 et traités par Saxenda en association à un régime alimentaire et à de l'activité physique, des évènements hypoglycémiques sévères (nécessitant l'intervention d'un tiers) ont été rapportés par 0.7 % des patients traités par Saxenda, mais uniquement chez les patients traités de façon concomitante par une sulfonylurée. De plus, chez ces patients, des hypoglycémies symptomatiques documentées ont été rapportées par 43.6 % des patients traités par Saxenda et par 27.3 % des patients recevant un placebo. Parmi les patients non traités de manière concomitante par une sulfonylurée, 15.7 % des patients traités par Saxenda et 7.6 % des patients recevant un placebo ont rapporté des événements hypoglycémiques symptomatiques documentés (définis par une glycémie ≤3.9 mmol/l accompagnée de symptômes).
Très fréquents - hypoglycémie chez les patients atteints de diabète sucré de type 2 traités à l'insuline
Dans le cadre d'une étude clinique menée auprès de patients en surpoids ou obèses atteints de diabète sucré de type 2, traités à l'insuline et à Saxenda en association avec un régime alimentaire et une activité physique et traités par jusqu'à deux ADO, 1.5 % d'hypoglycémies sévères (nécessitant une aide extérieure) ont été signalées sous Saxenda. Dans cette étude, 47.2 % d'hypoglycémies symptomatiques (définie par un taux de glucose plasmatique de ≤3.9 mmol/l accompagné de symptômes) ont été signalées avec Saxenda et 51.8 % avec le placebo. Dans le sous-groupe de patients traités en plus par une sulfonylurée, 60.9 % des patients sous Saxenda ont signalé des événements hypoglycémiques symptomatiques documentés et 60% sous placebo.
Fréquents – Hypoglycémie chez les patients ne présentant pas de diabète de type 2
Au cours des études cliniques menées chez des patients obèses ou en surpoids ne présentant pas de diabète de type 2 et traités par Saxenda en association à un régime alimentaire et à de l'activité physique, aucun événement hypoglycémique sévère (nécessitant l'intervention d'un tiers) n'a été rapporté. Des symptômes compatibles avec une hypoglycémie ont été rapportés par 1.6 % des patients traités par Saxenda et par 1.1 % des patients recevant le placebo; ces événements n'ont toutefois pas été confirmés par une mesure de la glycémie. La majorité de ces événements était d'intensité légère.
Affections cardiaques
Occasionnels – Tachycardie
Au cours des études cliniques, une tachycardie a été rapportée chez 0.6 % des patients traités par Saxenda et chez 0.1 % des patients recevant le placebo. La plupart de ces évènements étaient d'intensité légère à modérée. Les évènements apparaissaient uniquement de manière isolée et la majorité d'entre eux s'est résolue lors de la poursuite du traitement par Saxenda.
Fréquents - Hypotension
Les effets indésirables liés à l'hypotension artérielle (c'est-à-dire les rapports d'hypotension, hypotension orthostatique, de collapsus circulatoire et de chute de tension artérielle) ont été rapportés plus fréquemment (1.1 %) au cours des études cliniques menées sur Saxenda que lors de l'utilisation du placebo (0.5 %). Les baisses de la tension artérielle systolique inférieures à 80 mmHg ont été rapportées chez 4 (0.1 %) patients traités par Saxenda alors qu'elles n'étaient rapportées chez aucun patient recevant le placebo. Chez l'un des patients traités par Saxenda, l'hypotension artérielle est apparue dans un contexte d'effets indésirables gastro-intestinaux et d'insuffisance rénale.
Affections gastro-intestinales
Très fréquents – Nausées (39.3 %), vomissements (15.7 %), diarrhée (20.9 %), constipation (19.4 %)
Fréquents – Sécheresse buccale, dyspepsie, gastrite, reflux gastroœsophagien, douleurs épigastriques, flatulence, éructations, distension abdominale
Occasionnels – Vidange gastrique retardée
La plupart des évènements gastro-intestinaux étaient légers à modérés, passagers, et ne nécessitaient pas, pour la majorité, d'arrêt du traitement. Les réactions apparaissaient généralement au cours de la première semaine de traitement et diminuaient en quelques jours ou semaines lors de la poursuite du traitement.
Chez les patients de ≥65 ans, des troubles gastro-intestinaux peuvent être plus fréquents au cours du traitement par Saxenda.
Les patients dont la fonction rénale est légèrement ou modérément diminuée (clairance de la créatinine ≥30 ml/min) peuvent présenter plus fréquemment des troubles gastro-intestinaux sous traitement par Saxenda.
Affections du rein et des voies urinaires
Rares – Insuffisance rénale aiguë
Une insuffisance rénale aigüe a été rapportée chez des patients traités par des agonistes du récepteur du GLP-1. La plupart des évènements rapportés apparaissaient chez des patients ayant présenté des nausées, des vomissements ou des diarrhées entraînant une déplétion volumique (voir sous «Mises en garde et précautions»).
Troubles généraux et anomalies au site d'administration
Fréquents – Réactions au site d'injection
Des réactions au site d'injection ont été rapportées chez des patients traités par Saxenda. Ces réactions étaient généralement légères et passagères, et la plupart ont disparu au cours du traitement.
Expériences de post-marketing
Après l'obtention de l'autorisation, les effets indésirables ci-après ont été rapportés lors de l'utilisation du liraglutide, le principe actif de Saxenda. Ces effets indésirables ont été enregistrés sur la base de rapports volontaires fournis par une population dont la taille n'est pas connue. Il n'est donc pas toujours possible d'estimer clairement leur fréquence ou d'établir un lien causal avec l'exposition au principe actif.
Néoplasmes: carcinome médullaire de la thyroïde.
Affections gastro-intestinales: pancréatite aiguë, pancréatite hémorragique et nécrosante, dont certaines d'issue fatale.
Troubles du métabolisme et de la nutrition: déshydratation consécutive à des nausées, vomissements et diarrhées.
Affections du rein et des voies urinaires: augmentation de la créatininémie, insuffisance rénale aiguë ou aggravation d'une insuffisance rénale chronique, nécessitant parfois une hémodialyse.
Troubles généraux et anomalies au site d'administration: réactions allergiques: éruption cutanée et prurit.
Affections du système immunitaire: angio-œdème et réactions anaphylactiques.
Affections hépatobiliaires: élévation du taux d'enzymes hépatiques, hyperbilirubinémie, cholestase, hépatite.
Enfants et adolescents
Lors d'une étude clinique menée sur des aolescents atteints d'obésité et âgés d'entre 12 et 18 ans, 125 patients ont été traités pendant 56 semaines avec Saxenda.
La fréquence, le type et la gravité des effets secondaires ont été dans l'ensemble comparable à ceux observées pour les effets secondaires chez les adultes. Les vomissements surviennent deux fois plus fréquemment chez les adolescents que chez les adultes
Le taux de patients ayant signalé au moins un épisode d'hypoglycémie cliniquement pertinente était plus élevé chez les patients traités par Liraglutide (1.6 %) que chez ceux traités par placebo (0.8 %). Aucun épisode hypoglycémique sévère n'est survenu au cours de l'étude.
Aucun effet sur la croissance et le développement pubaire n'a été relevé après un traitement de 56 semaines.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou sévère via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDes surdosages, pouvant aller jusqu'à 72 mg (24 fois la dose recommandée pour la réduction pondérale), ont été rapportés lors d'études cliniques et lors de l'emploi du liraglutide après la mise sur le marché.
Signes et symptômes
Les évènements rapportés incluent des nausées sévères, des vomissements sévères et des diarrhées, correspondant aussi aux symptômes attendus d'un surdosage de liraglutide. Des cas d'hypoglycémie grave ont été rapportés. Tous les patients ont récupéré sans présenter de complication.
Traitement
En cas de surdosage, un traitement symptomatique approprié doit être initié en fonction des signes et des symptômes cliniques du patient. Les signes cliniques de déshydratation doivent être recherchés chez le patient et sa glycémie doit être surveillée.
Propriétés/EffetsCode ATC
A10BJ02
Mécanisme d'action
Le liraglutide est un analogue acylé du glucagon-like peptide 1 humain (GLP-1) présentant 97 % d'homologie en termes de séquence d'acides aminés avec le GLP-1 humain, qui se lie au récepteur du GLP-1 et l'active.
Le GLP-1 est un régulateur physiologique de l'appétit et de la prise de nourriture, mais le mécanisme d'action exact n'est pas encore complètement connu. Au cours d'études d'expérimentation animale, l'administration périphérique de liraglutide a entraîné une assimilation dans des régions cérébrales spécifiques impliquées dans la régulation de l'appétit. En augmentant l'activité spécifique du GLP-1R, le liraglutide a provoqué une augmentation des signes principaux de satiété et une diminution des principaux signaux de faim, ce qui a entraîné une perte de poids.
Les récepteurs du GLP-1 agissent à des endroits spécifiques du cœur, du système vasculaire, du système immunitaire et des reins. Des études chez l'homme et chez l'animal ont montré que ces récepteurs pouvaient atténuer les effets cardiovasculaires du liraglutide, ainsi qu'entraîner une diminution de l'inflammation et une progression retardée de l'athérosclérose.
Pharmacodynamique
Le liraglutide entraîne une perte de poids chez l'homme essentiellement par la perte de masse adipeuse, la réduction relative de la graisse viscérale étant supérieure à celle de la graisse sous-cutanée. Le liraglutide régule l'appétit en augmentant la sensation de réplétion et de satiété tout en réduisant la sensation de faim et l'envie de manger, ce qui diminue la prise alimentaire. Le liraglutide n'augmente pas la dépense énergétique par rapport au placebo.
Le liraglutide stimule la sécrétion d'insuline et diminue la sécrétion de glucagon par un mécanisme glucose-dépendant, ce qui provoque une diminution de la glycémie postprandiale et de la glycémie à jeun. L'effet hypoglycémiant est plus marqué chez les patients pré-diabétiques et diabétiques que chez les patients dont les glycémies se situent dans des fourchettes normales. Des études cliniques suggèrent que le liraglutide améliore et prolonge la fonction bêta-cellulaire, selon le modèle HOMA-B, et le rapport pro-insuline/insuline.
Efficacité clinique
L'efficacité et la sécurité du liraglutide sur la régulation pondérale en association à une réduction de l'apport calorique et une augmentation de l'activité physique ont été évaluées au cours de quatre études randomisées en double aveugle et contrôlées par placebo, ayant inclus au total 5 358 patients.
Étude 1 (SCALE Obesity & Pre-Diabetes - 1839): au total, 3'731 patients obèses (IMC ≥30 kg/m2) ou en surpoids (IMC ≥27 kg/m2) présentant une dyslipidémie et/ou une hypertension artérielle ont été stratifiés selon leur stade prédiabétique au moment de l'inclusion et de leur IMC au départ (≥30 kg/m² ou <30 kg/m²). Les 3'731 patients randomisés ont reçu un traitement pendant 56 semaines et 2'254 patients randomisés prédiabétiques au moment de l'inclusion ont reçu un traitement pendant 160 semaines. Les deux périodes de traitement ont été suivies d'une période de suivi de 12 semaines sans médicament ni placebo. La thérapie de base pour tous les patients consistait en une intervention axée sur le mode de vie sous la forme d'un régime à valeur énergétique réduite et en conseils sur l'activité physique.La perte de poids a été évaluée pendant 56 semaines au cours de l'étude 1 chez les 3'731 patients randomisés (2'590 patients ont terminé l'étude).Dans la partie de l'étude 1 d'une durée de 160 semaines, le délai d'apparition du diabète sucré de type 2 a été évalué auprès de 2'254 patients randomisés atteints de pré-diabète (1'128 patients ont terminé l'étude).
Étude 2 (SCALE Diabetes - 1922): une étude randomisée de 56 semaines évaluant la perte de poids comme critère d'évaluation principal chez 846 patients obèses ou en surpoids (dont 628 ont terminé l'étude) présentant un diabète de type 2 insuffisamment contrôlé (HbA1c compris entre 7 et 10 %). Le traitement de fond au début de l'étude était soit un régime alimentaire et de l'activité physique seuls, ou en association avec un traitement par la metformine, une sulfonylurée ou une glitazone en monothérapie, soit une association de ces traitements.
Étude 3 (SCALE Maintenance - 1923): une étude de 56 semaines évaluant la perte de poids et la stabilisation du poids comme critère d'évaluation principal chez 422 patients randomisés obèses ou en surpoids (dont 305 ont terminé l'étude) présentant une hypertension artérielle ou une dyslipidémie après une perte de poids antérieure ≥5 % obtenue par un régime hypocalorique.
Étude 4 (SCALE Sleep Apnoe - 3970): une étude de 32 semaines évaluant la sévérité de l'apnée du sommeil comme critère d'évaluation principal et la perte de poids comme critère d'évaluation secondaire chez 359 patients obèses randomisés (dont 276 ont terminé l'étude) présentant une apnée obstructive du sommeil modérée ou sévère.
Poids corporel
Les données concernant la perte de poids, la réponse au traitement, l'évolution et la distribution cumulative des modifications de poids (en %) des études 1 à 3 sont rapportées dans le Tableau 3 et les schémas 1, 2, 3 et 4.
Tableau 3. Modifications du poids à la semaine 56 et à la semaine 160 par rapport à l'état initial – Études 1, 2 et 3
|
|
Étude 1
(semaine 56)
|
Étude 1
(semaine 160)
|
Étude 2
|
Étude 3
| |
|
Saxenda
n=2'437
|
Placebo
n=1'225
|
Saxenda
(n=1'472)
|
Placebo
(n=738)
|
Saxenda
n=412
|
Placebo
n=211
|
Saxenda
n=207
|
Placebo
n=206
| |
Poids corporel
| |
Valeur initiale moyenne, en kg (ET)
|
106.3
(21.2)
|
106.3
(21.7)
|
107.6
(21.6)
|
108.0
(21.8)
|
105.6
(21.9)
|
106.7
(21.2)
|
100.7
(20.8)
|
98.9
(21.2)
| |
Modification par rapport à la valeur initiale, en %
|
-8.0
|
-2.6
|
-6.2
|
-1.8
|
-5.9
|
-2.0
|
-6.3
|
-0.2
| |
Saxenda versus
placebo, en % (IC à 95 %)
|
-5.4*
(-5.8; -5.0)
|
-4.3**
(-4.9; -3.7)
|
-4.0**
(-4.8; -3.1)
|
-6.1**
(-7.5; -4.6)
| |
Modification par rapport à la valeur initiale, en kg
|
-8.4
|
-2.8
|
-6.5
|
-2.0
|
-6.2
|
-2.2
|
-6.0
|
-0.2
| |
Saxenda versus
placebo, en kg (IC à 95 %)
|
-5.6**
(-6.0; -5.1)
|
-4.6**
(-5.3; -3.9)
|
-4.1**
(-5.0; -3.1)
|
-5.9**
(-7.3; -4.4)
| |
% de patients ayant perdu ≥5 % du poids corporel
|
63.5
|
26.6
|
49.6
|
23.4
|
49.8
|
13.5
|
50.7
|
21.3
| |
Saxenda versus
placebo, kg (IC à 95 %)
|
4.8**
(4.1; 5.6)
|
3.2**
(2.6; 3.9)
|
6.4**
(4.1; 10.0)
|
3.8**
(2.4; 6.0)
| |
% de patients ayant perdu >10 % de leur poids corporel
|
32.8
|
10.1
|
24.4
|
9.5
|
22.9
|
4.2
|
27.4
|
6.8
| |
Saxenda versus
placebo, kg (IC à 95 %)
|
4.3**
(3.5; 5.3)
|
3.1**
(2.3; 4.1)
|
6.8**
(3.4; 13.8)
|
5.1**
(2.7; 9.7)
|
Analyse complète (FAS=Full Analysis Set). Concernant le poids, les valeurs initiales sont des moyennes, les modifications à la semaine 56 et à la semaine 160 par rapport aux valeurs initiales sont des moyennes estimées (LSMeans = moindres carrés) et les différences de traitement à la semaine 56 et à la semaine 160 sont des différences de traitement estimées. Pour les proportions de patients (%) ayant perdu ≥5/>10% de leur poids, les odds ratios estimés sont présentés. Les valeurs post-inclusion manquantes ont été imputées à l'aide de la dernière observation reportée (Last Observation Carried Forward, LOCF). * p <0.05. ** p <0.0001. ET = écart-type. IC = intervalle de confiance.
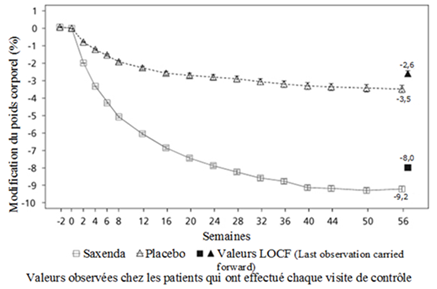
Schéma 1. Modification du poids corporel (%) par rapport à la valeur initiale dans l'étude 1 (de 0 à 56 semaines) en fonction du temps

Schéma 2. Modification du poids corporel (%) par rapport à la valeur initiale dans l'étude 1 (de 0 à 160 semaines) en fonction du temps
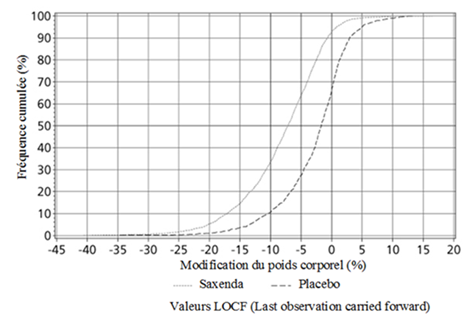
Schéma 3. Distribution cumulative de la modification de poids (%) après 56 semaines de traitement dans l'étude 1
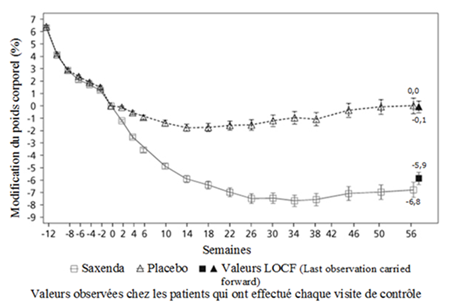
Schéma 4. Modification du poids (%) en fonction du temps dans l'étude 3 par rapport à la randomisation (semaine 0)
Avant la semaine 0, les patients avaient pour seul traitement un régime hypocalorique et de l'activité physique. Au cours de la semaine 0, les patients ont été randomisés pour recevoir Saxenda ou le placebo.
Perte de poids après 12 semaines de traitement par liraglutide (3.0 mg)
Les répondeurs précoces (early responders) étaient définis comme les patients ayant obtenu une perte de poids ≥5 % après 12 semaines avec une dose thérapeutique de liraglutide (4 semaines d'augmentation de la dose et 12 semaines de traitement). Dans le cadre de l'étude 1, 67.5 % des patients étaient des répondeurs précoces et dans le cadre de l'étude 2, 50.4 % des patients. En poursuivant le traitement au liraglutide, 86.2 % de ces répondeurs précoces devraient atteindre une réduction de poids ≥5 % après 1 an de traitement et 51 % une perte de poids ≥10 %. La perte de poids moyenne prévue chez les répondeurs précoces qui ont suivi le traitement pendant 1 an est de 11.2 % de leur poids corporel initial (9.7 % chez les hommes et 11.6 % chez les femmes). Dans le groupe de patients ayant atteint une perte de poids <5 % après un traitement de 12 semaines de 3 mg de liraglutide par jour, 6.6 % des patients ont encore présenté une perte de poids ≥10 % après 1 an.
Glycémie et paramètres cardiométaboliques
Les données concernant la glycémie et les paramètres cardiométaboliques issues des études 1 et 2 sont rapportées dans le Tableau 4.
Tableau 4. Modification de la glycémie et des paramètres cardiométaboliques par rapport à la valeur initiale, après 56 semaines (études 1 et 2) et 160 semaines (étude1)
|
|
Saxenda (n=2437)
|
Placebo (n=1225)
|
Saxenda versus placebo
| |
Étude 1 (semaine 56)
|
Valeur initiale
|
Modification
|
Valeur initiale
|
Modification
|
| |
HbA1c, %
|
5.6
|
-0.3
|
5.6
|
-0.1
|
-0.23**
(0.25; -0.21)
| |
Glycémie à jeun, mmol/l
|
5.3
|
-0.4
|
5.3
|
-0.01
|
-0.38**
(0.42; -0.35)
| |
Pression artérielle systolique, mmHg
|
123.0
|
-4.3
|
123.3
|
-1.5
|
-2.8**
(-3.6; -2.1)
| |
Pression artérielle diastolique, mmHg
|
78.7
|
-2.7
|
78.9
|
-1.8
|
-0.9*
(-1.4; - 0.4)
| |
Tour de taille, cm
|
115.0
|
-8.2
|
114.5
|
-4.0
|
-4.2**
(-4.7; -3.7)
| |
|
Saxenda
(n=1472)
|
Placebo
(n=738)
|
Saxenda versus placebo
| |
Étude 1 (semaine 160)
|
Valeur initiale
|
Modification
|
Valeur initiale
|
Modification
|
| |
HbA1c, %
|
5.75
|
-0.35
|
5.74
|
-0.14
|
-0.21**
(-0.24; -0.18)
| |
Glycémie à jeun,
mmol/l
|
5.50
|
-0.37
|
5.46
|
0.04
|
-0.41**
(-0.46; -0.36)
| |
Pression artérielle systolique, mmHg
|
124.80
|
-3.24
|
125.01
|
-0.44
|
-2.80**
(-3.81; -1.79)
| |
Pression artérielle diastolique, mmHg
|
79.40
|
-2.36
|
79.83
|
-1.74
|
-0.62
(-1.33; 0.09)
| |
Tour de taille, cm
|
116.64
|
-6.88
|
116.74
|
-3.35
|
-3.53**
(-4.23; -2.83)
| |
Étude 2
|
(n=412)
|
(n=211)
|
| |
HbA1c, %
|
7.9
|
-1.3
|
7.9
|
-0.4
|
-0.9**
(-1.1; -0.8)
| |
Glycémie à jeun, mmol/l
|
8.8
|
-1.9
|
8.6
|
-0.1
|
-1.8**
(-2.1; -1.4)
| |
Pression artérielle systolique, mmHg
|
128.9
|
-3.0
|
129.2
|
-0.4
|
-2.6*
(-4.6; - 0.6)
| |
Pression artérielle diastolique, mmHg
|
79.0
|
-1.0
|
79.3
|
-0.6
|
-0.4
(1.7; 1.0)
| |
Tour de taille, cm
|
118.1
|
-6.0
|
117.3
|
-2.8
|
-3.2**
(-4.2; -2.2)
| |
Lors de l'analyse statistique des paramètres glycémiques et cardiométaboliques, le test multiple n'a pas été évalué et les résultats ne doivent donc pas être considérés comme statistiquement confirmatoires. Analyse complète (FAS = Full Analyse Set). Pour l'HbA1c, la glycémie à jeun, la pression artérielle et le tour de taille, les valeurs initiales sont des moyennes, les modifications observées à la semaine 56 et à la semaine 160 par rapport aux valeurs initiales sont des moyennes estimées (LSmeans, moindres carrés) et les différences de traitement à la semaine 56 et à la semaine 160 sont des différences de traitement estimées. Les valeurs post-inclusion manquantes ont été calculées à l'aide de la dernière observation reportée (Last Observation Carried Forward – LOCF). * p <0.05. ** p <0.0001.
ET = écart-type. IC = intervalle de confiance.
|
Immunogénicité
Les patients traités par Saxenda peuvent développer des anticorps anti-liraglutide. Lors d'une analyse effectuée après le début du traitement, 42 (2.8 %) des 1'505 patients traités par Saxenda présentaient des anticorps anti-liraglutide. Des anticorps dotés d'un effet neutralisant au cours d'un test effectué in vitro sont apparus chez 18 (1.2 %) des 1'505 patients traités par Saxenda. La présence d'anticorps pourrait être en rapport avec l'apparition accrue de réactions au site d'injection et d'hypoglycémies rapportées. Au cours d'études cliniques, ces résultats ont été généralement d'intensité légère et disparaissaient lors de la poursuite du traitement. La détection de formation d'anticorps est fortement dépendante de la sensibilité et de la spécificité du test. En outre, l'incidence observée des anticorps détectés (y compris des anticorps neutralisants) dans un test peut être influencée par divers facteurs, comme la méthode de test, la gestion des échantillons, le déroulement du recueil des échantillons, la comédication ainsi que les maladies sous-jacentes. Cela explique pourquoi la survenue d'anticorps contre Saxenda ne peut être directement comparée à la survenue d'anticorps d'autres produits.
Évaluation cardiovasculaire
Les événements indésirables cardiovasculaires majeurs (MACE) ont été évalués par un groupe d'experts indépendants externes et définis comme infarctus du myocarde non fatal, accident vasculaire cérébral non fatal et le décès cardiovasculaire. Dans l'ensemble des études menées avec Saxenda, 6 MACE ont été observés chez les patients traités par liraglutide et 10 chez les patients recevant un placebo. Le rapport de risque et l'IC à 95 % est de 0.33 [0.12; 0.90] pour le liraglutide par rapport au placebo. Une élévation moyenne de la fréquence cardiaque de 2.5 battements par minute par rapport à la valeur initiale (allant de 1.6 à 3.6 battements par minute suivant l'étude) a été observée avec le liraglutide dans les études de phase 3, qui a atteint son maximum après environ 6 semaines et était réversible après l'arrêt du liraglutide (voir «Mises en garde et précautions»).
L'étude LEADER (Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcome Results) a étudié l'incidence des événements cardiovasculaires sévères (MACE: décès cardiovasculaire, infarctus du myocarde non mortel, accident vasculaire cérébral non mortel) chez 9'340 patients présentant un diabète sucré de type 2 important et un risque cardiovasculaire accru. Après randomisation (1:1), les patients ont été traités (durée moyenne du traitement environ 3.5 ans), en plus du traitement standard, soit avec jusqu'à 1.8 mg de liraglutide par jour (4'668 patients) ou un placebo (4'672 patients). Le critère d'évaluation principal était le délai d'apparition d'un MACE. Le liraglutide a significativement réduit ce risque à la dose testée (rapport de risque 0.87 [0.78; 0,97] IC à 95 %).
Enfants et adolescents
Lors d'une étude en double-aveugle, sur l'efficacité et la sûreté de Saxenda par rapport à un placebo en ce qui concerne la perte de poids chez l'adolescent dès 12 ans souffrant d'obésité, Saxenda s'est avéré supérieur au placebo en ce qui concerne la réduction de la masse corporelle après un traitement de 56 semaines (mesuré par le IMC-SDS; voir tableau 5).
Une plus grande proportion de patients a obtenu une réduction de ≥5 % et de ≥10 % de l'IMC qu'avec le placebo, ainsi qu'une diminution moyenne de l'IMC et une perde de poids plus importante (voir tableau 5).
La modification de la composition corporelle n'a pas été étudiée.
Tous les patients doivent faire l'objet d'une surveillance étroite (voir également la règle d'arrêt du traitement à la rubrique «Indication»).
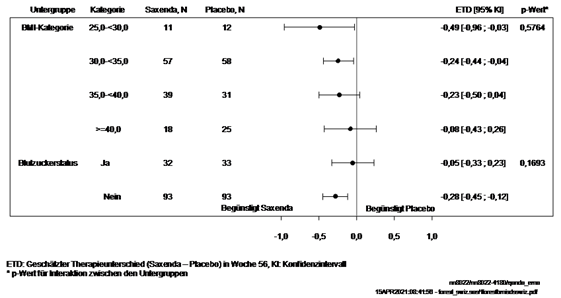
Figure 5.
Le diagramme indique les différences estimées (Saxenda vs. placebo) dans la modification de l'IMC-SDS entre le début de l'étude jusqu'à la semaine 56, en fonction de la valeur de l'IMC de départ ([kg/m2]: ≥25 - <30, ≥30 - <35, ≥35 - <40 et ≥40) et du statut glycémique au départ de l'étude (normoglycémie vs prédiabète ou diabète de type 2), y compris les valeurs p des test d'interaction.
Après une période de suivi de 26 semaines sans prise du médicament étudié ou du placebo, une reprise de poids a été observée dans le groupe Saxenda par rapport au groupe placebo (voir tableau 5):
Tableau 5. NN8022-4180 Modification du poids et de l'IMC en semaine 56 et modification de l'IMC-SDS de la semaine 56 à la semaine 82
|
|
Saxenda (N=125)
|
Placebo
(N=126)
|
Saxenda vs. Placebo
| |
IMC-SDS
|
|
|
| |
Valeur de base, IMC-SDS (SD)
|
3.14 (0.65)
|
3.20 (0.77)
|
| |
Modification moyenne en semaine 56 (95%-KI)
|
-0.23
|
-0.00
|
-0.22*
(-0.37; -0.08)
| |
Semaine 56, IMC-SDS (SD)
|
2.88 (0.94)
|
3.14 (0.98)
|
| |
Modification moyenne de la semaine 56 à la semaine 82, IMC-SDS (95%-KI)
|
0.22
|
0.07
|
0.15**
(0.07; 0.23)
| |
Poids corporel
|
|
|
| |
Valeur de base, kg (SD)
|
99.3 (19.7)
|
102.2 (21.6)
|
-
| |
Modification moyenne en semaine 56, % (95%-KI)
|
-2.65
|
2.37
|
-5.01**
(-7.63; -2.39)
| |
Modification moyenne en semaine 56, kg (95%-KI)
|
-2.26
|
2.25
|
-4.50**
(-7.17; -1.84)
| |
IMC
|
|
|
| |
Valeur de base, kg/m2 (SD)
|
35.3 (5.1)
|
35.8 (5.7)
|
-
| |
Modification moyenne en semaine 56, kg/m2 (95%-KI)
|
-1.39
|
0.19
|
-1.58**
(-2.47; -0.69)
| |
Proportion de patients avec une réduction de la valeur de base de l'IMC de ≥5% en semaine 56, % (95%-KI)
|
43.25
|
18.73
|
3.31**
(1.78; 6.16)
| |
Proportion de patients avec une rédution de la valeur de base de l'IMC de ≥10% en semaine 56, % (95%-KI)
|
26.08
|
8.11
|
4.00**
(1.81; 8.83)
| |
Groupe total (FAS=Full Analysis Set). Pour l'IMC-SDS, le poids, et l'IMC, les valeurs de base sont des moyennes, les modifications par rapport aux valeurs de base en semaine 56 sont des moyennes estimées (LSMeans) et les contrastes de traitements en semaine 56 sont des différences de traitement estimées. Les valeurs IMC -SDS en semaine 56 sont des valeurs moyennes, les modification entre la semaine 56 et la semaine 82 sont des moyennes estimées (LSMeans) et les contrastes de traitement en semaine 82 sont des différences de traitement estimées. Pour la proportion de patients pour lequel la valeur de base de l'IMC a été réduite de ≥5%/≥10%, le rapport de cotes estimé est indiqué. Les observations manquantes ont été complétées par une imputation multiple (approche de saut à a référence, x100) basée sur le groupe placebo.
*p<0.01, **p<0.001. KI=Intervalle de confiance. SD=Déviation standard.
|
En fonction de la tolérance, la dose a été augmentée à 3.0 mg chez 103 patients (82.4 %), à 2.4 mg chez 11 patients (8.8 %), à 1.8mg chez 4 patients (3.2 %), à 1.2 mg chez 4 patients (3.2 %) et 3 patients (2.4 %) sont restés à 0.6 mg.
L'efficacité et la sécurité de Saxenda chez des patients pédiatriques atteints du syndrome de Prader-Willi et d'obésité ont été évaluées dans une étude en double aveugle (partie A) pendant 16 semaines, puis en ouvert durant 36 semaines. Dans la partie A ont été inclus en tout 32 adolescents âgés de ≥12 à <18 ans (stade Tanner 2 à 5), randomisés pour recevoir soit du liraglutide 3 mg (n = 20), soit un placebo (n = 12), dont 18 et 12 respectivement sont arrivés au terme de la phase en double aveugle (semaine 16), et 17 et 12 respectivement au terme de la phase d'extension en ouvert (semaine 52). Dans la partie B, 24 enfants entre ≥6 et <12 ans (stade Tanner inférieur à 2) au total ont été randomisés pour recevoir soit du liraglutide 3 mg (n = 17), soit un placebo (n = 7), dont 16 et 7 respectivement sont arrivés au terme de la phase en double aveugle (semaine 16), et 14 et 7 respectivement au terme de la phase d'extension en ouvert (semaine 52).
Les patients ayant un poids corporel inférieur à 45 kg ont débuté l'augmentation de la dose à une dose plus faible de 0.3 mg au lieu de 0.6 mg, jusqu'à une dose maximale de 2.4 mg.
La modification estimée de l'IMC-SDS moyen était comparable dans les deux bras de traitement à la semaine 16 (différence entre le liraglutide 3 mg et le placebo [IC à 95 %]: -0.07 [-0.23; 0.09] dans la partie A et -0.06 [-1.06; 0.93] dans la partie B) comme à la semaine 52 (différence entre le liraglutide 3 mg et le placebo [IC à 95 %]: -0.14 [-0.62; 0.34] dans la partie A et -0.07 [-0.89; 0.76] dans la partie B).
PharmacocinétiqueAbsorption
L'absorption du liraglutide administré par voie sous-cutanée est lente; la concentration sérique maximale est atteinte environ 11 heures après l'injection. Après administration d'une dose unique de 3.0 mg de liraglutide à un patient obèse (IMC 30-40 kg/m2), la concentration moyenne à l'état d'équilibre (AUCτ/24) du liraglutide était d'environ 31 nmol/l. L'exposition au liraglutide s'élevait dans la fourchette entre 0.6 mg et 3.0 mg de manière proportionnelle à la dose. La biodisponibilité absolue du liraglutide après administration sous-cutanée est d'environ 55 %.
Distribution
Après administration sous-cutanée de 3.0 mg de liraglutide, le volume de distribution moyen apparent est de 20-25 l (chez une personne pesant environ 100 kg). Le liraglutide se lie largement aux protéines plasmatiques (>98 %).
Métabolisme
Dans les 24 heures suivant l'administration d'une dose unique de [3H]-liraglutide à des sujets sains, on trouvait principalement du liraglutide intact dans le plasma. Deux métabolites ont été détectés dans le plasma (≤9% et ≤5% de la radioactivité plasmatique totale).
Élimination
Le liraglutide est métabolisé par voie endogène de la même manière que les grosses protéines et aucun organe en particulier n'a été identifié comme voie d'élimination principale. Après administration d'une dose de [3H]-liraglutide, le liraglutide intact n'a été détecté ni dans les urines ni dans les fèces. Seule une petite proportion de la radioactivité administrée a été éliminée dans les urines ou dans les fèces sous forme de métabolites issus du liraglutide (6 % et 5 % respectivement). L'élimination de la radioactivité par les urines et les fèces a principalement eu lieu dans les 6 à 8 premiers jours, sous la forme des trois métabolites mineurs.
Après administration sous-cutanée du liraglutide, la clairance moyenne est d'environ 0.9-1.4 l/h avec une demi-vie d'élimination d'environ 13 heures.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
La pharmacocinétique du liraglutide a été évaluée chez des sujets présentant un degré variable d'insuffisance hépatique dans une étude avec des doses uniques (0.75 mg). L'exposition au liraglutide a diminué de 13-23 % chez les sujets qui présentaient une insuffisance hépatique légère à modérée par rapport aux sujets sains. L'exposition avait nettement diminuée (44 %) chez les patients atteints d'une insuffisance hépatique sévère (score de Child Pugh >9).
Troubles de la fonction rénale
Au cours de l'étude à doses uniques (0.75 mg), l'exposition au liraglutide s'est révélée plus faible chez les sujets qui présentaient une insuffisance rénale par rapport à ceux ayant une fonction rénale normale. L'exposition au liraglutide a diminué de respectivement 33%, 14 %, 27 % et 26 % chez les sujets présentant respectivement une insuffisance rénale légère (clairance de la créatinine ClCr 50-80 ml/min), modérée (ClCr 30-50 ml/min), sévère (ClCr <30 ml/min) et une insuffisance rénale terminale nécessitant une dialyse.
Patients âgés
Les résultats des analyses pharmacocinétiques de population des patients en surpoids et obèses (18 à 82 ans) n'ont pas montré d'effet cliniquement significatif de l'âge sur la pharmacocinétique du liraglutide. Aucun ajustement posologique n'est nécessaire chez les patients âgés.
Enfants et adolescents
Des analyses de population ont été réalisées pour le liraglutide en utilisant les données des essais cliniques pédiatriques.
Les propriétés pharmacocinétiques du liraglutide 3.0 mg ont été évaluées dans des études cliniques chez des adolescents obèses âgés de 12 à 18 ans (134 patients, poids corporel 62-178 kg). L'exposition au liraglutide chez les adolescents (12-18 ans) était comparable à celle des adultes atteints d'obésité.
Les propriétés pharmacocinétiques ont également été évaluées dans une étude de pharmacologie clinique chez des enfants obèses âgés de 7 à 11 ans (13 patients, poids corporel 54-87 kg). L'exposition associée à 3.0 mg de liraglutide a été estimée comparable chez les enfants âgés de 7 à 11 ans et chez les adultes après correction en fonction du poids corporel.
Sexe
D'après les résultats des analyses pharmacocinétiques de population, les femmes présentent une clairance du liraglutide ajustée selon le poids de 24 % inférieure à celle des hommes. D'après les données d'efficacité de l'exposition, aucun ajustement posologique n'est nécessaire en fonction du sexe.
Origine ethnique
Les résultats des analyses pharmacocinétiques réalisés sur une population incluant des sujets en surpoids et obèses, d'ethnie blanche, noire, asiatique et d'Amérique latine ou non n'ont pas montré d'effet cliniquement significatif de l'origine ethnique sur la pharmacocinétique du liraglutide.
Poids corporel
L'exposition au liraglutide diminue quand le poids initial augmente. D'après l'analyse des données d'efficacité de l'exposition obtenues dans le cadre des études cliniques, la dose quotidienne de 3 mg de liraglutide assurait une exposition systémique adéquate chez des patients d'un poids compris entre 60 et 234 kg. L'exposition au liraglutide n'a pas été étudiée chez les patients de poids >234 kg.
Données précliniquesLes données précliniques issues des études conventionnelles de pharmacologie de sécurité, de toxicité en administration répétée ou de génotoxicité n'ont pas révélé de risque particulier pour l'être humain.
Carcinogénicité
Lors d'études de carcinogénicité sur deux ans chez le rat et la souris, des tumeurs non létales des cellules C de la thyroïde ont été observées. Chez le rat, aucune «dose sans effet nocif observé» (DSENO) n'a été observée. Ces tumeurs n'ont pas été observées chez des singes traités pendant 20 mois. Ces résultats décrits chez les rongeurs sont dus à un mécanisme non génotoxique, spécifique, médié par le récepteur du GLP-1, auquel les rongeurs sont particulièrement sensibles. La pertinence de ces résultats pour l'homme est probablement faible mais ne peut pas être complètement exclue. Aucune autre tumeur liée au traitement n'a été constatée.
Toxicité sur la reproduction
Les études effectuées chez l'animal n'ont mis en évidence aucun effet délétère direct sur la fertilité, mais une légère augmentation des morts embryonnaires précoces a été observée à la dose la plus élevée. L'administration du liraglutide en milieu de gestation a entraîné une perte de poids maternelle et une diminution de la croissance fœtale, avec des effets ambigus sur la cage thoracique chez le rat et une modification du squelette chez le lapin. Chez les rats exposés au liraglutide, la croissance néonatale était ralentie, ce qui a perduré après le sevrage dans le groupe recevant des doses élevées. On ignore si le retard de croissance des jeunes rats est imputable à une consommation de lait réduite due à un effet direct du GLP-1 ou à une baisse de la production de lait maternel induite par une réduction de l'apport calorique.
Études juvéniles
Chez les rats mâles et femelles juvéniles, le liraglutide a provoqué un retard de maturation sexuelle lors d'expositions cliniquement pertinentes. Ces retards n'ont pas affecté la fertilité et la capacité de reproduction des deux sexes, ni la capacité des femelles à mener une grossesse à terme.
Remarques particulièresIncompatibilités
L'ajout d'autres substances à Saxenda peut provoquer la dégradation du liraglutide. Aucune étude de compatibilité n'a été menée. Ce médicament ne doit donc pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Stabilité après ouverture
Après la première utilisation du stylo Saxenda, le produit peut être conservé 1 mois à température ambiante (ne dépassant pas 30°C) ou au réfrigérateur (2-8°C).
Remarques particulières concernant le stockage
Conserver hors de portée des enfants.
Conserver au réfrigérateur (2-8°C).
Ne pas congeler.
Ne pas conserver à proximité d'éléments réfrigérants.
Conserver le capuchon du stylo pour le protéger de la lumière.
Remarques concernant la manipulation
La solution ne doit pas être utilisée si elle n'est pas limpide et incolore ou presque incolore.
Saxenda ne doit plus être utilisé s'il a été congelé.
Le stylo est prévu pour une utilisation avec une aiguille à usage unique NovoFine® ou NovoTwist® d'une longueur maximale de 8 mm et d'un diamètre minimal de 32 G.
Les aiguilles ne sont pas incluses.
Il faut dire au patient qu'il doit éliminer l'aiguille après chaque injection et qu'il doit conserver le stylo sans aiguille. Cela prévient les risques de contamination, d'infection et de fuite de liquide. De plus, un dosage correct est ainsi garanti.
Numéro d’autorisation65899 (Swissmedic)
PrésentationEmballages de 3 et 5 stylos préremplis. [B]
Chaque stylo contient 3 ml de solution et permet l'administration de doses de 0.6 mg, 1.2 mg, 1.8 mg, 2.4 mg et 3.0 mg.
Titulaire de l’autorisationNovo Nordisk Pharma AG, Kloten
Domicile: Zürich
Mise à jour de l’informationAvril 2025
|