Propriétés/EffetsCode ATC
L04AC13
Mécanisme d'action
L'ixekizumab est un anticorps monoclonal recombinant humanisé sélectif pour l'interleukine 17-A. L'ixekizumab, produit par des cellules ovariennes de hamster chinois (CHO), est un anticorps monoclonal IgG4 modifié qui se lie spécifiquement et avec une affinité élevée (< 3 pM) à la cytokine pro-inflammatoire interleukine 17A (à la fois à l'IL-17A et à l'hétérodimère IL-17A/F). Des concentrations élevées d'IL-17A ont été associées à la pathogenèse de diverses maladies autoimmunes. Dans le psoriasis, le ligand IL-17A joue un rôle important dans la prolifération et l'activation excessives des kératinocytes.
La neutralisation de l'IL-17A par l'ixekizumab inhibe ces phénomènes. L'IL-17A joue un rôle clé dans la pathogenèse du psoriasis en plaques et de l'arthrite psoriasique et est régulée à la hausse dans la peau affectée par les lésions par opposition à la peau non lésionnelle des patients atteints de psoriasis en plaques.
Dans la spondylarthrite axiale, le ligand IL-17A joue un rôle important de par l'augmentation de l'inflammation, qui entraîne des lésions osseuses érosives et une nouvelle formation osseuse pathologique.
L'ixekizumab ne se lie pas aux ligands IL-17B, IL-17C, IL-17D, IL-17E ni IL-17F.
L'ixekizumab a une faible capacité de liaison aux récepteurs Fcγ ou aux composants du complément. Les essais de liaison in vitro ont confirmé que l'ixekizumab ne se lie pas aux récepteurs humains Fcγ I, IIa et IIIa ni au composant du complément C1q.
Pharmacodynamique
Sur la base des données des biopsies de peau psoriasique issues d'une étude de phase I, on observe une tendance dose-dépendante en faveur d'une diminution de l'épaisseur de l'épiderme, du nombre de kératinocytes, de lymphocytes T et de cellules dendritiques en prolifération, ainsi que des diminutions des marqueurs d'inflammation locale entre le stade initial et le jour 43. En conséquence, le traitement par ixekizumab réduit l'érythème, l'induration et la desquamation dans les lésions de psoriasis en plaques.
Il a été observé que Taltz réduisait (après 1 semaine de traitement) les taux de protéine C réactive.
Efficacité clinique
Psoriasis en plaques
L'efficacité et la sécurité de Taltz ont été évaluées dans le cadre de trois études de phase III randomisées, en double-aveugle, contrôlées versus placebo chez des patients adultes atteints de psoriasis en plaques modéré à sévère, candidats à la photothérapie ou à un traitement systémique (UNCOVER-1, UNCOVER-2 et UNCOVER-3). L'efficacité et la sécurité de Taltz ont également été évaluées versus étanercept (UNCOVER-2 et UNCOVER-3). Les patients randomisés dans le groupe Taltz qui, à la 12ème semaine, étaient répondeurs selon le score sPGA (static Physician Global Assessment ou Evaluation Globale du Médecin) de 0 ou de 1 (0,1) ont été à nouveau randomisés dans le groupe placebo ou Taltz pendant 48 semaines supplémentaires (UNCOVER-1 et UNCOVER-2). Les patients randomisés dans le groupe placebo, étanercept ou Taltz, non-répondeurs selon le score sPGA (0,1), ont été traités par Taltz jusqu'à 48 semaines. De plus, l’efficacité et la sécurité à long terme ont été évaluées pour les trois études sur une durée totale de 5 ans chez les patients ayant participé à l’intégralité des études.
Sur les 3866 patients inclus dans ces études contrôlées versus placebo, 64 % avaient reçu un traitement systémique antérieur (biologique, systémique conventionnel ou psoralène et ultraviolets A (PUVA)), 43.5 % avaient déjà été traités par photothérapie, 49.3 % avaient déjà reçu un traitement systémique conventionnel et 26.4 % avaient déjà reçu une biothérapie pour le traitement du psoriasis. Sur l'ensemble de ces patients, 14.9 % avaient reçu au moins un agent anti-TNF alfa et 8.7 % un anti-IL-12/IL-23. A l'inclusion 23.4 % des patients avaient des antécédents d'arthrite psoriasique.
Critères d'exclusion: les critères d'exclusion essentiels qui s'opposaient à la participation dans UNCOVER-1, UNCOVER-2 et UNCOVER-3 étaient l'utilisation concomitante de traitements anti-psoriasiques systémiques ou biologiques ou la photothérapie, tout trouble médical grave ou maladie instables, sauf le psoriasis en plaques, et toute infection grave actuelle ou récente. Dans UNCOVER-2 et UNCOVER-3, l'utilisation précédente d'étanercept était un critère d'exclusion.
Dans les trois études, les co-critères d'évaluation principaux étaient la proportion de patients ayant atteint une réponse PASI 75 (Psoriasis Area and Severity Index; PASI 75 = amélioration du PASI d'au-moins 75 %) et une réponse sPGA 0 («blanchi») ou 1 («minimal») à la 12ème semaine par rapport au placebo. A l'inclusion, les patients de tous les groupes de traitement présentaient un score PASI médian compris entre 17.4 et 18.3; 48.3 % à 51.2 % des patients présentaient un score sPGA initial «sévère» ou «très sévère» et un score de prurit moyen compris entre 6.3 et 7.1 sur l'échelle d'évaluation numérique des démangeaisons (itch NRS, itch Numeric Rating Scale).
Tableau 1: réponse clinique après 12 semaines (NRI) dans les études UNCOVER-1, -2 et -3
|
|
Placebo
|
Taltz
80 mg 1x/4 sem.
|
Taltz
80 mg 1x/2 sem.
|
Etanercept
50 mg deux fois par semaine
| |
UNCOVER 1
| |
Nombre de patients (N)
|
431
|
432
|
433
|
NA
| |
sPGA de «0» (blanchi) ou de «1» (minimal), n (%)
|
14
(3.2%)
|
330
(76.4%)a
|
354
(81.8%)a
|
NA
| |
sPGA de «0» (blanchi), n (%)
|
0
|
149
(34.5%)a
|
160
(37.0%)a
|
NA
| |
PASI 75, n (%)
|
17
(3.9%)
|
357
(82.6%)a
|
386
(89.1%)a
|
NA
| |
PASI 90, n (%)
|
2
(0.5%)
|
279
(64.6%)a
|
307
(70.9%)a
|
NA
| |
PASI 100, n (%)
|
0
|
145
(33.6%)a
|
153
(35.3%)a
|
NA
| |
UNCOVER 2
| |
Nombre de patients (N)
|
168
|
347
|
351
|
358
| |
sPGA de «0» (blanchi) ou de «1» (minimal), n (%)
|
4
(2.4%)
|
253
(72.9%)a
|
292
(83.2%)a
|
129
(36.0%)a
| |
sPGA de «0» (blanchi), n (%)
|
1
(0.6%)
|
112
(32.3%)a,b
|
147
(41.9%)a,b
|
21
(5.9%)c
| |
PASI 75, n (%)
|
4
(2.4%)
|
269
(77.5%)a
|
315
(89.7%)a
|
149
(41.6%)a
| |
PASI 90, n (%)
|
1
(0.6%)
|
207
(59.7%)a,b
|
248
(70.7%)a,b
|
67
(18.7%)a
| |
PASI 100, n (%)
|
1
(0.6%)
|
107
(30.8%)a,b
|
142
(40.5%)a,b
|
19
(5.3%)c
| |
UNCOVER 3
| |
Nombre de patients (N)
|
193
|
386
|
385
|
382
| |
sPGA de «0» (blanchi) ou de «1» (minimal), n (%)
|
13
(6.7%)
|
291
(75.4%)a,b
|
310
(80.5%)a,b
|
159
(41.6%)a
| |
sPGA de «0» (blanchi), n (%)
|
0
|
139
(36.0%)a,b
|
155
(40.3%)a,b
|
33
(8.6%)a
| |
PASI 75, n (%)
|
14
(7.3%)
|
325
(84.2%)a,b
|
336
(87.3%)a,b
|
204
(53.4%)a
| |
PASI 90, n (%)
|
6
(3.1%)
|
252
(65.3%)a,b
|
262
(68.1%)a,b
|
98
(25.7%)a
| |
PASI 100, n (%)
|
0
|
135
(35.0%)a,b
|
145
(37.7%)a,b
|
28
(7.3%)a
|
Abréviations: n = nombre de patients dans cette catégorie; N = nombre de patients dans la population en intention de traiter: NA = non applicable; NRI = Non-Responder Imputation; NRS = numeric rating scale; PASI = Psoriasis Area and Severity Index; 1x/2 sem. = toutes les 2 semaines; 1x/4 sem. = toutes les 4 semaines; sPGA = static Physician Global Assessment.
a p < 0.001 versus au placebo
b p < 0.001 versus étanercept
c p < 0.01 versus placebo
L'étude UNCOVER-1 incluait 1296 patients. Les patients ont été randomisés (1:1:1) pour recevoir pendant 12 semaines le placebo ou Taltz (80 mg toutes les deux ou quatre semaines [1x/2 sem. ou 1x/4 sem.] après une dose initiale de 160 mg).
L'étude UNCOVER-2 incluait 1224 patients. Les patients ont été randomisés (1:2:2:2) pour recevoir pendant 12 semaines le placebo ou Taltz (80 mg toutes les deux ou quatre semaines [1x/2 sem. ou 1x/4 sem.] après une dose initiale de 160 mg) ou de l'étanercept 50 mg deux fois par semaine.
L'étude UNCOVER-3 incluait 1346 patients. Les patients ont été randomisés (1:2:2:2) pour recevoir pendant 12 semaines le placebo ou Taltz (80 mg toutes les deux ou quatre semaines [1x/2 sem. ou 1x/4 sem.] après une dose initiale de 160 mg) ou de l'étanercept 50 mg deux fois par semaine.
Le dosage de 80 mg 1x/2 sem. a démontré dans toutes les études pour tous les critères d'évaluation une efficacité supérieure (voir tableau ci-dessus), en particulier dans les degrés élevés d'amélioration de la peau (PASI 90, PASI 100, sPGA 0) et dans la réduction des démangeaisons. En comparaison avec le placebo et l'étanercept, à la semaine 12, des améliorations significativement supérieures par rapport aux valeurs initiales ont été observées dans le psoriasis des ongles (mesuré par le Nail Psoriasis Severity Index [NAPSI]), dans le psoriasis du cuir chevelu (mesuré par le Psoriasis Scalp Severity Index [PSSI]) et le psoriasis palmoplantaire (mesuré par le Psoriasis Palmoplantar Severity Index [PPASI]).
Taltz a été associé à une efficacité précoce avec une diminution > 50 % du score PASI moyen à la semaine 2 (Figure 1). Le pourcentage de patients ayant obtenu une réponse PASI 75 était significativement supérieur dans le groupe Taltz comparé aux groupes placebo et étanercept dès la semaine 1 dans les trois études. Environ 25 % des patients traités par Taltz ont obtenu un score PASI < 5 à la semaine 2, plus de 55 % ont obtenu un score PASI < 5 à la semaine 4 et jusqu'à 85 % à la semaine 12 (contre respectivement 3 %, 14 % et 50 % dans le groupe étanercept). Une amélioration significative de l'intensité des démangeaisons a été observée à la semaine 1 chez les patients traités par Taltz.
Taltz a également montré un effet favorable sur le prurit, qui était significativement supérieur au placebo.
Figure 1. Score PASI, amélioration en pourcentage à chaque visite suivant la visite initiale (mBOCF) dans la population en intention de traiter pendant la phase d'induction (UNCOVER-2 et UNCOVER-3)

L'efficacité et la sécurité de Taltz ont été démontrées quels que soient l'âge, le sexe, l'origine ethnique, le poids corporel, le score de gravité PASI initial, et la prise d'un traitement antérieur biologique. Les taux de réponse après 12 semaines chez les patients traités par Taltz 80 mg 1x/2 sem étaient pour les sous-groupes < 100 kg et ≥100kg respectivement PASI 75 (90.1% et 85.7%) et sPGA 0,1 (84.5% et 75.6%). Les taux de réponse après 12 semaines chez les patients traités par Taltz 80 mg 1x/4 sem étaient pour les sous-groupes < 100 kg et ≥100kg respectivement PASI 75 (85.6% et 74.2%) et sPGA 0,1 (79.0% et 67.4%). La réponse n'a pas été différente chez les patients avec psoriasis des ongles, psoriasis du visage ou psoriasis du cuir chevelu au début de l'étude. Taltz s'est montré efficace chez les patients ayant reçu précédemment un traitement systémique, avec ou sans exposition biologique/anti-TNF préalable, y compris des patients en échec aux traitements biologiques/anti-TNF. Les améliorations des critères d'évaluation sPGA et PASI chez les patients présentant une arthrite psoriasique concomitante au départ de l'étude étaient semblables à ceux de l'ensemble de la population avec psoriasis en plaques modéré à sévère. Environ 45% des patients présentaient un psoriasis dans la zone du visage au départ de l'étude. Parmi ces patients, 80,4% des patients traités avec Taltz étaient débarrassées du psoriasis dans la zone du visage à la semaine 12.
Efficacité chez les non-répondeurs à l'étanercept: chez les patients identifiés comme non-répondeurs à l'étanercept selon le score sPGA (0,1) à la semaine 12 dans l'étude UNCOVER-2 (N = 200) puis traités par Taltz 80 mg 1x/4 sem. après une période de sevrage thérapeutique de 4 semaines, 73 % ont obtenu un score sPGA (0,1) et 83.5 % un PASI 75 après 12 semaines de traitement.
Maintien de la réponse à la semaine 60 et jusqu’à 5 ans
Afin d'évaluer le maintien de la réponse, les patients initialement randomisés dans le groupe Taltz et considérés comme répondeurs à la semaine 12 (c'est-à-dire, score sPGA de 0,1) dans les études UNCOVER-1 et UNCOVER-2 ont été re-randomisés pour 48 semaines supplémentaires dans l'un des groupes de traitement suivants: placebo ou Taltz (80 mg toutes les quatre ou douze semaines ([1x/4 sem. ou 1x/12 sem.]).
Les patients qui n'ont pas atteint un sPGA (0,1) à la semaine 12 ou qui ont subi une rechute pendant la phase de maintenance (sPGA ≥3), ont reçu par la suite Taltz 1x/4 sem.
Chez les répondeurs sPGA (0,1) à la semaine 12 (dans les études combinées UNCOVER-1 et UNCOVER-2), la proportion de patients qui ont maintenu cette réponse à la semaine 60 était significativement plus élevée chez les patients traités avec Taltz 80 mg 1x/4 sem. (71 %) comparé à ceux traités par Taltz 80 mg 1x/12 sem. (35.5 %) ou le placebo (7 %).
Le tableau 2 montre les taux de réponse chez les patients re-randomisés pour recevoir la dose de maintenance recommandée de Taltz 80 mg toutes les 4 semaines, en fonction de la dose randomisée à l'inclusion.
Tableau 2. Maintien de la réponse et de l'efficacité au bout de 60 semaines (résultats combinés des études UNCOVER-1 et UNCOVER-2, NRI)
|
Critère d'évaluation
|
80 mg 1x/2 sem.
(induction)/Placebo (maintien)
(N = 211)
|
80 mg 1x/4 sem.
(induction)/Placebo (maintien)
(N = 191)
|
80 mg 1x/2 sem
(induction)/80 mg 1x/4 sem (maintien)
(N = 221)
|
80 mg 1x/4 sem.
(induction)/80 mg 1x/4 sem. (maintien)
(N = 195)
| |
Score sPGA de «0» (blanchi) ou de «1» (minimal) conservé
|
7.6 %
|
6.3 %
|
78.3 %
|
68.7 %
| |
Score sPGA de «0» (blanchi) conservé ou atteint
|
2.8 %
|
1.6 %
|
58.8 %
|
49.2 %
| |
PASI 75 conservé ou atteint
|
9.0 %
|
7.9 %
|
83.3 %
|
74.4 %
| |
PASI 90 conservé ou atteint t
|
4.7 %
|
4.7 %
|
76.5 %
|
66.7 %
| |
PASI 100 conservé ou atteint t
|
2.8 %
|
1.6 %
|
57.5 %
|
49.7 %
|
Abréviations: N = nombre de patients dans la population d'analyse; NRI = Non-Responder Imputation
76.4% des répondeurs sPGA (0,1) à la semaine 12, qui ont été assignés au traitement de maintenance Taltz 80 mg 1x/4 sem., avaient un PASI <5 maintenu ou atteint à la semaine 60. L'amélioration de la gravité de la démangeaison a été maintenue jusqu'à la semaine 60 chez les patients traités avec Taltz qui étaient répondeurs sPGA (0,1) à la semaine 12. En ce qui concerne le maintien de la réponse jusqu'à la semaine 60, Taltz a permis de maintenir la réponse chez les patients ayant précédemment reçu un traitement systémique, avec ou sans exposition préalable à des traitements biologiques/anti-TNF, y compris les patients en échec aux traitements biologiques/anti-TNF.
Pour les patients ayant obtenu un score sPGA de répondeurs (0,1) à la semaine 12 et re-randomisés dans le groupe placebo, le délai moyen de rechute (sPGA ≥3) a été de 164 (95% CI [143, 169]) jours en regroupant les études UNCOVER-1 et UNCOVER-2. Parmi ces patients, 71.5 % ont de nouveau obtenu un score sPGA de 0,1 dans les 12 semaines suivant la reprise du traitement par Taltz 80 mg 1x/4 sem.
Chez les patients ayant obtenu un score sPGA de répondeurs (0,1) à la semaine 12, les améliorations du psoriasis des ongles, du cuir chevelu et palmoplantaire persistaient aussi jusqu'à la semaine 60.
Sur 591 patients ayant été traités par Taltz 1x/2 sem. pendant la période d’induction, puis 1x/4 sem. par la suite dans les études UNCOVER-1, UNCOVER-2 et UNCOVER-3, 427 ont achevé 5 ans de traitement par Taltz. Parmi eux, 101 ont eu besoin d’une augmentation de la dose. Parmi les patients ayant terminé l’évaluation à la semaine 264 (N=427), 295 (69 %), 289 (68 %) et 205 (48 %) ont respectivement atteint une réponse sPGA (0,1), PASI 90 et PASI 100 à la semaine 264. Les scores DLQI ont été collectés après la période d’induction dans UNCOVER-1 et UNCOVER-2, 113 patients (66 %) ont présenté une réponse DLQI (0,1).
Qualité de vie/Résultats rapportés par les patients
Des améliorations statistiquement significatives du score DLQI (Dermatology Life Quality Index ou indice de qualité de vie en dermatologie) ont été démontrées entre l'inclusion et la 12e semaine (études 1 à 3). Ces améliorations se sont maintenues pendant 60 semaines. Taltz était associé à des améliorations significativement supérieures des douleurs cutanées (mesurées au moyen de l'échelle analogique visuelle, visual analogic scale ou VAS).
Etudes de comparaison directe après la mise sur le marché
L'efficacité et la sécurité de l'ixekizumab ont également été étudiées dans l'étude en double-aveugle RHBS (IXORA-S), en comparaison à l'ustekinumab, avec une supériorité de l'ixekizumab sur le critère principal de l'étude (réponse PASI 90 à la semaine 12, tableau 3). La supériorité versus ustekinumab a été démontrée rapidement dans les 3 catégories de réponse PASI. La supériorité de l'ixekizumab sur l'ustekinumab a également été démontrée dans les sous-groupes stratifiés par poids.
Tableau 3: Taux de réponse PASI dans l'étude comparative ixekizumab versus ustekinumab
|
|
Semaine 12
|
Semaine 52
| |
|
ixekizumab*
|
ustekinumab**
|
ixekizumab*
|
ustekinumab**
| |
Patients (n)
|
136
|
166
|
136
|
166
| |
PASI 75,
n (%)
|
120 (88.2 %)
|
114 (68.7 %)
|
120 (88.2 %)
|
126 (75.9 %)
| |
PASI 90,
n (%)
|
99 (72.8 %)§
|
70 (42.2 %)
|
104 (76.5 %)
|
98 (59.0 %)
| |
PASI 100,
n (%)
|
49 (36.0 %)
|
24 (14.5 %)
|
71 (52.2 %)
|
59 (35.5 %)
|
* 160 mg d'ixekizumab a été administrée en dose initiale suivie de 80 mg aux semaines 2, 4, 6, 8, 10 et 12, et 80 mg 1x/4 sem. par la suite
** Posologie en fonction du poids: les patients traités par ustekinumab ont eu une dose de 45 mg ou 90 mg aux semaines 0 et 4, puis toutes les 12 semaines jusqu'à la semaine 52 (dose en fonction du poids conformément à la posologie approuvée)
§ p < 0.001 versus ustekinumab (valeur de p mentionnée uniquement pour le critère principal)
L’efficacité et la sécurité de l’ixekizumab ont également été étudiées dans l’étude RHCR (IXORA-R) sur 24 semaines, randomisée, en double aveugle, en groupes parallèles, comparant l’ixekizumab au guselkumab. L’ixekizumab a été supérieur dès la semaine 4 dans l’obtention d’un blanchiment cutané complet, ainsi que sur le critère principal de l’étude PASI 100 à la semaine 12 [ixekizumab 41.3% vs guselkumab 24.9% (p<0.001)], et a été non-inférieur sur le PASI 100 à la semaine 24 [ixekizumab 50.0% vs guselkumab 52.3% (p=0.414)].
Efficacité dans le psoriasis génital
Une étude randomisée en double-aveugle, contrôlée versus placebo (IXORA-Q) a été conduite chez 149 patients adultes (24 % de femmes) présentant un psoriasis génital modéré à sévère (score du sPGA-Génital ≥3), avec une atteinte d'au moins 1 % de la surface corporelle (body surface area, BSA) (60.4 % avaient une BSA ≥10 %) et un antécédent d'échec ou d'intolérance à au moins un traitement topique pour un psoriasis génital. Les patients présentaient un psoriasis en plaques au-moins modéré (défini par un score sPGA ≥3 et candidats à une photothérapie et/ou à un traitement systémique) depuis au moins 6 mois.
Les participants à l'étude randomisés dans le groupe Taltz ont reçu une dose initiale de 160 mg suivie de 80 mg toutes les 2 semaines pendant 12 semaines. Le critère principal était la proportion de patients ayant atteint une réponse du sPGA-Génital de «0» (blanchi) ou «1» (minimal) (sPGA-G 0/1). A la semaine 12, il y a eu significativement plus de patients dans le groupe Taltz comparé au groupe placebo qui ont atteint un sPGA-G 0/1 et un sPGA 0/1 indépendamment de la surface corporelle à l'inclusion (surface corporelle à l'inclusion BSA 1 % à <10 %, resp. BSA ≥10 %: réponse sPGA-G de «0» ou «1»: Taltz 71 % resp. 75 %; placebo 0 % resp. 13 %).
Psoriasis en plaques pédiatrique
Une étude clinique multicentrique, randomisée, en double-aveugle, contrôlée contre placebo (IXORA-Peds) a inclus 201 patients pédiatriques âgés de 6 ans à moins de 18 ans, atteints de psoriasis en plaques modéré à sévère (défini par un score sPGA ≥3 avec une atteinte ≥10 % de la surface corporelle et un score PASI ≥12) qui étaient candidats à la photothérapie ou à un traitement systémique, ou qui étaient insuffisamment contrôlés par un traitement topique.
Les principaux critères d'exclusion étaient similaires à ceux des études mentionnées plus haut chez l'adulte, en particulier les infections sévères actives ou récentes ainsi que les traitements concomitants du psoriasis systémiques conventionnels ou biologiques ou la photothérapie.
Durant la phase de 12 semaines en double aveugle contrôlée contre placebo et la phase de contrôle actif, les patients ont été traités par placebo (n = 56), étanercept (n = 30) ou Taltz (n = 115), la posologie adaptée au poids étant ajustée comme suit:
Taltz:
<25 kg: 40 mg à la semaine 0, suivi de 20 mg 1x/4 sem.
de 25 kg à 50 kg: 80 mg à la semaine 0, suivi de 40 mg 1x/4 sem.
>50 kg: 160 mg à la semaine 0, suivi de 80 mg 1x/4 sem.
Etanercept:
0.8 mg/kg, maximum 50 mg par dose 1x/ sem.
La réponse au traitement a été évaluée après 12 semaines de traitement et a été définie par la proportion de patients ayant atteint le cocritère d'évaluation principal, par un score sPGA de «0» (blanchi) ou «1» (minimal) avec une amélioration d'au moins 2 points par rapport au score à l'inclusion, ainsi que par la proportion de patients ayant atteint une réduction du score PASI d'au moins 75 % (PASI 75) par rapport au score à l'inclusion.
Les autres résultats évalués à la semaine 12 comprenaient la proportion de patients ayant atteint un score PASI 90, PASI 100, sPGA de «0» et une amélioration de la sévérité des démangeaisons, mesurée par une réduction d'au moins 4 points sur une échelle d'évaluation numérique des démangeaisons de 11 points (itch Numeric Rating Scale, itch NRS).
Les patients avaient un score PASI médian de 17 à l'inclusion, les scores allant de 12 à 49. Le score sPGA à l'inclusion était sévère ou très sévère dans 49 % des cas. Sur l'ensemble des patients, 22 % avaient préalablement reçu une photothérapie, 32 % un traitement systémique conventionnel et 4% un traitement biologique pour le traitement du psoriasis.
Les données de réponse clinique sont présentées dans le Tableau 4.
Tableau 4: Résultats d'efficacité à 12 semaines chez les patients pédiatriques avec psoriasis en plaques, NRI
|
Critères d'évaluation
|
Taltza
(N=115)
n (%)
|
Placebo
(N=56)
n (%)
|
Différence vs placebo
(95% CI)
| |
sPGA «0» (blanchi) ou «1» (minimal)b
|
93 (81)
|
6 (11)
|
70.2 (59.3, 81.0) d
| |
sPGA «0» (blanchi)
|
60 (52)
|
1 (2)
|
50.4 (40.6, 60.2) d
| |
PASI 75c
|
102 (89)
|
14 (25)
|
63.7 (51.0, 76.4)d
| |
PASI 90
|
90 (78)
|
3 (5)
|
72.9 (63.3, 82.5)d
| |
PASI 100
|
57 (50)
|
1 (2)
|
47.8 (38.0, 57.6)d
| |
Score Itch NRS (amélioration ≥4 points) c
|
59 (71)
|
8 (20)
|
51.1 (35.3, 66.9) d
|
Abréviations: N = Nombre de patients dans la population en intention de traiter (ITT); NRI = Non-Responder Imputation ou imputation d'absence de réponse.
a À la semaine 0, les patients ont reçu une dose de 160 mg (poids coroporel [PC] >50kg),, 80 mg (PC 25–50 kg), ou de 40 mg (PC < 50 kg) de Taltz, suivie de 80 mg (PC >50kg), 40 mg (PC 25–50 kg), ou 20 mg (< 25 kg) toutes les 4 semaines pendant 12 semaines.
b Co-critères d'évaluation principaux.
c Score Itch NRS (amélioration ≥4) chez les patients ayant un score Itch NRS ≥4 à l'inclusion. Le nombre de patients en ITT avec un score Itch NRS ≥4 à l'inclusion est le suivant: Taltz, n = 83; PBO, n = 40.
d p<0.001
Un total de 87 patients pédiatriques atteints de psoriasis en plaques sévère (PASI ≥20 ou sPGA ≥4) ont été randomisés pour recevoir de l'ixekizumab 1x/4 sem. (38 patients), de l'étanercept 1x/ sem. (30 patients) ou un placebo (19 patients).
À la semaine 12, des améliorations ont été observées pour le groupe ixekizumab 1x/4 sem. par rapport au groupe étanercept 1x/ sem. et au groupe placebo, mesurées par PASI 75 (84.2%, 63.3%, 26.3%) et sPGA (0,1) (76.3%, 53.3% und 5.3%).
Les critères secondaires les plus importants ont montré des améliorations statistiquement significatives dans le groupe ixekizumab Q4W par rapport au groupe étanercept Q1W et au groupe placebo, mesurées par: PASI 90 (76.3%, 40.0%, 0), PASI 100 (60.5%, 16.7%, 0) et sPGA (0) (63.2%, 16.7%, 0).
Les patients du groupe de traitement ixékizumab ont présenté une réponse CDLQI (children Dermatology Life Quality Index)/DLQI (0,1) à la semaine 12 (NRI) plus élevée comparé au placebo. La différence entre les groupes de traitement était observée dès la semaine 4.
La phase de traitement en double aveugle était de 12 semaines. Elle a été suivie par la phase d'entretien en ouvert de 48 semaines (phase 3) et une phase d'extension de 48 semaines (phase 4) pour les patients de pays hors UE, indépendamment de la réponse, et les non-répondeurs de l'UE (définis comme ceux qui ont sPGA 0,1 non atteint).
Sur les 115 patients qui ont reçu Taltz à la semaine 0, 94 patients ont continué à recevoir Taltz jusqu'à la semaine 108 et ont été inclus dans la population primaire d'efficacité de l'ixekizumab. Parmi les patients qui ont terminé l'évaluation à la semaine 108, 64 patients (68.1 %) ont atteint le PASI 90, 72 patients (76.6 %) le PASI 75 et 64 patients (68.1 %) ont atteint le sPGA (0,1) à la semaine 108.
Il n'y a pas suffisamment de données pour évaluer le risque possible de poussées de la maladie après l'arrêt de Taltz chez les enfants.
Arthrite psoriasique
La sécurité et l'efficacité de Taltz ont été évaluées dans deux études de phase III randomisées, en double-aveugle, contrôlées versus placebo chez 780 patients atteints d'arthrite psoriasique active (≥3 articulations gonflées et ≥3 articulations douloureuses). Le diagnostic d'arthrite psoriasique (selon les critères de classification de l'arthrite psoriasique CASPAR, Classification Criteria for Psoriatic Arthritis) a été posé chez les patients dans une médiane de 5.33 ans avant l'inclusion dans l'étude. Les patients randomisés présentaient des lésions cutanées de psoriasis en plaques actuelles (94.0 %) ou des antécédents documentés de psoriasis en plaques. A l'inclusion, 12.1 % des patients étaient atteints de psoriasis en plaques modéré à sévère. A l'inclusion, plus de 58.9 % des patients atteints d'arthrite psoriasique avaient des enthésites et 22.3 % des dactylites. Dans les deux études, le critère principal était le taux de réponse American College of Rheumatology (ACR) 20 à la semaine 24, suivi d’une période d’extension à long terme de la semaine 24 à la semaine 156 (3 ans).
Dans l'étude 1 dans l'arthrite psoriasique (SPIRIT-P1), des patients atteints d'arthrite psoriasique active naïfs de traitement biologique ont été randomisés dans les groupes de traitement suivants: injections sous-cutanées de placebo, adalimumab 40 mg toutes les 2 semaines (groupe de contrôle actif), Taltz 80 mg toutes les 2 semaines (1x/2 sem.) ou Taltz 80 mg toutes les 4 semaines (1x/4 sem.). Les deux schémas posologiques de Taltz comprenaient une dose initiale était de 160 mg. Dans cette étude, 85.3 % des patients avaient déjà été traités par au moins un DMARD conventionnel. 53 % des patients étaient traités de façon concomitante par MTX à une dose hebdomadaire moyenne de 15.8 mg. 67 % des patients qui étaient traités de façon concomitante par MTX recevaient une dose supérieure ou égale à 15 mg. Dans tous les groupes de traitement, les patients ayant une réponse insuffisante à la semaine 16 ont reçu un traitement de secours (modification du traitement de fond). Les patients traités par Taltz 1x/2 sem. ou 1x/4 sem. ont conservé la dose initiale de Taltz. Les patients recevant adalimumab ou le placebo ont été re-randomisés selon un rapport 1:1 pour recevoir Taltz 1x/2 sem. ou 1x/4 sem. à la semaine 16 ou 24 en fonction du statut de répondeur. 243 patients ont terminé la période d'extension de 3 ans sous Taltz.
L'étude 2 dans l'arthrite psoriasique (SPIRIT-P2) incluait des patients ayant déjà été traités par un anti-TNF et ayant arrêté le traitement par anti-TNF en raison d'un manque d'efficacité ou d'une intolérance (patients anti-TNF-IR). Les patients ont été randomisés dans les groupes de traitement suivants: injections sous-cutanées de placebo, Taltz 80 mg toutes les 2 semaines (1x/2 sem.) ou Taltz 80 mg toutes les 4 semaines (1x/4 sem.). Les deux schémas posologiques de Taltz comprenaient une dose initiale était de 160 mg. 56 % et 35 % des patients ne répondaient pas de façon adéquate à respectivement 1 anti-TNF et ≥2 anti-TNF. L'étude SPIRIT-P2 a évalué 363 patients, parmi lesquels 41 % recevaient un traitement concomitant par MTX à une dose hebdomadaire moyenne de 16.1 mg. 73.2 % des patients traités de façon concomitante par MTX avaient une dose supérieure ou égale à 15 mg. Dans tous les groupes de traitement, les patients ayant eu une réponse insuffisante à la semaine 16 ont reçu un traitement de secours (modification du traitement de fond). Les patients traités par Taltz 1x/2 sem. ou 1x/4 sem. ont conservé la dose initiale de Taltz. Les patients recevant le placebo ont été re-randomisés selon un rapport 1:1 pour recevoir Taltz 1x/2 sem. ou 1x/4 sem. à la semaine 16 ou 24 en fonction du statut de répondeur. 168 patients ont terminé la période d'extension de 3 ans sous Taltz.
Signes et symptômes
Le traitement par Taltz a montré une amélioration significative des mesures de l'activité de la maladie par rapport au placebo à la semaine 24 (voir Tableau 5.
Tableau 5 Résultats d'efficacité des études SPIRIT-P1 et SPIRIT-P2 à la semaine 24
|
|
SPIRIT-P1
|
SPIRIT-P2
| |
Critères d'évalua-tion
|
|
|
|
Différence de taux de réponse versus placebo (CI 95 %)
|
|
|
|
Différence de taux de réponse versus placebo (CI 95 %)
| |
|
PBO
(N = 106)
|
Taltz
1x/4 sem. (N = 107)
|
Taltz
1x/2sem. (N = 103)
|
Taltz
1x/4 sem.
|
Taltz
1x/2 sem.
|
PBO
(N = 118)
|
Taltz
1x/4 sem. (N = 122)
|
Taltz
1x/2 sem. (N = 123)
|
Taltz
1x/4 sem.
|
Taltz
1x/2 sem.
| |
Taux de réponse ACR 20, n (%)
| |
Semaine 24
|
32 (30.2)
|
62 (57.9)
|
64 (62.1)
|
27,8 (15.0; 40.6)*
|
31,9 (19.1; 44.8)*
|
23 (19.5)
|
65 (53.3)
|
59 (48.0)
|
33.8 (22.4; 45.2)*
|
28.5 (17.1; 39.8)*
| |
Taux de réponse ACR 50, n (%)
| |
Semaine 24
|
16 (15.1)
|
43 (40.2)
|
48 (46.6)
|
25.1 (13.6; 36.6)*
|
31.5 (19.7; 43.3)*
|
6 (5.1)
|
43 (35.2)
|
41 (33.3)
|
30.2 (20.8; 39.5)*
|
28.3 (19.0; 37.5)*
| |
Taux de réponse ACR 70, n (%)
| |
Semaine 24
|
6 (5.7)
|
25 (23.4)
|
35 (34.0)
|
17.7 (8.6; 26.8)*
|
28.3 (18.2; 38.5)*
|
0
|
27 (22.1)
|
15 (12.2)
|
22.1 (14.8; 29.5)*
|
12.2 (6.4; 18.0)*
|
Abréviations: ACR 20/50/70 = Taux de réponse de 20 %/50 %/70 % de l'American College of Rheumatology; CI = intervalle de confiance; 1x/4 sem. = Taltz 80 mg une fois toutes les 4 semaines; 1x/2 sem. = Taltz 80 mg une fois toutes les 2 semaines; N = nombre de patients dans la population analysée; n = nombre de patients dans une catégorie précise; NRI = non-responder imputation; PBO = placebo.
Note: les patients ayant reçu un traitement de secours à la semaine 16 ou ayant arrêté le traitement ou pour lesquels des données manquaient ont été imputés comme non-répondeurs pour les analyses à la semaine 24.
Les DMARDs conventionnels concomitants incluaient MTX, léflunomide et sulfasalazine.
* p<0.001 par rapport au placebo.
Chez les patients avec des dactylites ou des enthésites pré-existantes, le traitement par Taltz 1x/4 sem. a entraîné une amélioration des dactylites et des enthésites à la semaine 24 comparé au placebo (p<0.01).
Chez les patients atteints à la fois d'un psoriasis en plaques (≥3% de surface corporelle atteinte) et d'arthrite psoriasique, le traitement avec Taltz 1x/4 sem. a entraîné une amélioration des lésions cutanées psoriatiques, comme démontré par les réponses PASI 75, PASI 90 et PASI 100 à la semaine 24 (p<0.001).
Chez les patients atteints à la fois d'un psoriasis en plaques modéré à sévère et d'arthrite psoriasique, le schéma posologique de Taltz 1x/2 sem. a montré un taux de réponse significativement plus élevé pour PASI 75, PASI 90 et PASI 100 par rapport au placebo (p<0.001) et a démontré un bénéfice cliniquement significatif par rapport au schéma posologique de Taltz 1x/4 sem.
Les réponses au traitement avec Taltz étaient significativement plus élevées que celles avec placebo dès la semaine 1 pour l'ACR 20, dès la semaine 4 pour l'ACR 50 et dès la semaine 8 pour l'ACR 70, et ces réponses ont persisté jusqu'à la semaine 24; ces effets se sont maintenus pendant 3 ans pour les patients qui sont restés dans l'étude.
Figure 2: Taux de réponse ACR 20 dans l'étude SPIRIT-P1 jusqu'à la semaine 24
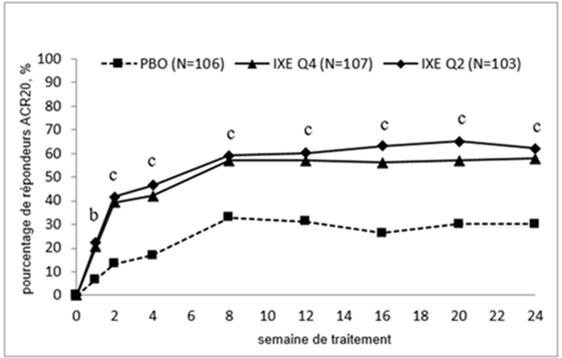
Pour Taltz 1x/2 sem. et 1x/4 sem.: b p<0.01 et c p<0.001 par rapport au placebo.
Dans SPIRIT-P1 et SPIRIT-P2, des taux de réponses similaires pour les ACR 20/50/70 ont été observés chez les patients atteints d'arthrite psoriasique indépendamment du fait qu'ils recevaient ou non un traitement concomitant par cDMARDs, y compris MTX.
Dans SPIRIT-P1 et SPIRIT-P2, des améliorations ont été observées pour toutes les composantes des scores ACR, y compris pour le critère d'évaluation de la douleur par le patient.
Dans SPIRIT-P1, l'efficacité a été maintenue jusqu'à la semaine 52 comme démontré par les taux de réponses de l'ACR 20/50/70, l'amélioration des enthésites, l'amélioration des dactilytes et le PASI 75/90/100.
L'efficacité et la sécurité de Taltz ont été démontrées quels que soient l'âge, le sexe, l'origine ethnique, la durée de la maladie, le poids corporel à l'inclusion dans l'étude, la présence de psoriasis à l'inclusion dans l'étude, la CRP initiale, la DAS28-CRP initiale, la prise concomitante de corticoïdes et un traitement antérieur par un biologique. Taltz a été efficace chez les patients naïfs de traitement biologique, ayant déjà été exposés à un traitement biologique et n'ayant pas répondu à un traitement biologique.
Il y avait trop peu de patients atteints d'arthrite mutilante, d'arthrite isolée avec atteinte des articulations interphalangiennes distales (IPD) et de spondylarthrite concomitante dans les études d'homologation pour obtenir des résultats significatifs dans ces sous-groupes spécifiques.
Dans SPIRIT-P1, 63 patients ont achevé 3 ans de traitement par ixekizumab 1x/4 sem. Parmi les 107 patients qui ont été randomisés dans le groupe ixekizumab 1x/4 sem. (analyse NRI dans la population en ITT), 54 (50 %), 41 (38 %), 29 (27 %) et 36 patients (34 %) ont présenté respectivement des réponses ACR20, ACR50, ACR70 et de MDA à la semaine 156.
Dans SPIRIT-P2, 70 patients ont achevé 3 ans de traitement par ixekizumab 1x/4 sem. Parmi les 122 patients qui ont été randomisés dans le groupe ixekizumab 1x/4 sem. (analyse NRI dans la population en ITT), 56 (46 %), 39 (32 %), 24 (20 %) et 33 patients (27 %) ont présenté respectivement des réponses ACR20, ACR50, ACR70 et de MDA à la semaine 156.
Réponse radiographique
Dans SPIRIT-P1, l'inhibition de la progression des atteintes structurales articulaires a été évaluée par radiographie et exprimée au moyen du Score total de Sharp modifié (mTSS) et de ses composants, le score d'érosion (ES, Erosion Score) et le score de pincement articulaire (JSN, Joint Space Narrowing) aux semaines 24 et 52, par rapport aux valeurs à l'inclusion.
La progression des atteintes articulaires radiographiques (mTSS) était inhibée par Taltz à la semaine 24 par rapport au placebo.
Le pourcentage de patients ne présentant pas de progression des atteintes articulaires radiographiques (définie par une variation du mTSS par rapport à l'inclusion ≤0.5) entre la randomisation et la semaine 24 était de 94.8 % chez les patients traités par Taltz 1x/2 sem., 89.0 % avec Taltz 1x/4 sem. et 77.4 % avec le placebo. L'inhibition des atteintes structurales a été maintenue avec le traitement par Taltz jusqu'à la semaine 52.
Capacité fonctionnelle et qualité de vie liée à la santé
Dans SPIRIT-P1 et SPIRIT-P2, les patients traités par Taltz 1x/2 sem. (p<0.001) et 1x/4 sem. (p<0.001) ont présenté une amélioration significative de leur capacité fonctionnelle, évaluée à l'aide de l'indice HAQ-DI (Health Assessment Questionnaire – Disability Index) à la semaine 24 et maintenue à la semaine 52 dans l'étude SPIRIT-P1.
Les patients traités par Taltz ont rapporté des améliorations de la qualité de vie liée à la santé, évaluée par le score Physical Component Summary of the Short Form-36 Health Survey (SF-36 PCS) (p<0.001). Une amélioration statistiquement significative de la fatigue a aussi été démontrée au moyen de l'échelle Fatigue Severity NRS.
Spondylarthrite ankylosante (spondylarthrite axiale radiographique, maladie de Bechterew)
La sécurité et l'efficacité de Taltz ont été étudiées dans 2 études randomisées, en double-aveugle, contrôlées contre placebo (COAST-V et COAST-W) chez 657 patients adultes ≥18 ans atteints de spondylarthrite ankylosante. Les patients avaient une maladie active définie selon le Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 et des douleurs rachidiennes ≥4 sur une échelle d'évaluation numérique malgré le traitement par AINS. Dans les deux études, les patients présentaient à l'inclusion des symptômes de spondylarthrite ankylosante depuis en moyenne 17 ans (médiane de 16 ans). A l'inclusion, environ 32 % des patients recevaient un traitement concomitant par cDMARD (dans les études COAST-V et COAST-W: méthotrexate 8.5 % et 13.0 %, sulfasalazine 28.5 % et 14.6 %, hydroxychloroquine 0.6 % et 0).
Dans les deux études, le critère d'évaluation principal était le pourcentage de patients ayant obtenu une réponse Assessment of Spondyloarthritis International Society 40 (ASAS40) à la semaine 16.
COAST-V a évalué 341 patients naïfs de traitement biologique traités avec Taltz 80 mg ou 160 mg à la semaine 0, suivis de 80 mg toutes les 2 semaines (1x/2 sem.) ou 4 semaines (1x/4 sem), adalimumab 40 mg toutes les 2 semaines ou placebo. Les patients traités avec le placebo ont été re-randomisés à la semaine 16 pour recevoir Taltz (dose initiale de 160 mg suivie de 80 mg 1x/2 sem. ou 1x/4 sem). Les patients traités avec adalimumab ont été re-randomisés à la semaine 16 pour recevoir Taltz (80 mg 1x/2 sem. ou 1x/4 sem.).
COAST-W a évalué 316 patients précédemment traités avec 1 ou 2 inhibiteurs du TNF (90 % ayant eu une réponse inadéquate et 10 % une intolérance aux inhibiteurs du TNF). Tous les patients ont été traités avec Taltz 80 mg ou 160 mg à la semaine 0, puis avec 80 mg 1x/2 sem. ou 1x/4 sem. ou avec placebo. Les patients traités avec le placebo ont été re-randomisés à la semaine 16 pour recevoir Taltz (dose initiale de 160 mg suivie de 80 mg 1x/2 sem. ou 1x/4 sem.).
Réponse clinique
Dans les deux études, les patients traités par Taltz 80 mg 1x/2 sem. ou 1x/4 sem. ont présenté des améliorations plus importantes de la réponse ASAS40 et ASAS20 par rapport au placebo à la semaine 16 (tableau 6). La réponse était comparable chez les patients, indépendamment des thérapies concomitantes. Dans COAST-W, la réponse était indépendante du nombre de traitements antérieurs par des inhibiteurs du TNF.
Tableau 6 Résultats d'efficacité des études COAST-V et COAST-W à la semaine 16
|
|
COAST-V, naïfs de traitement biologique
|
COAST-W, expérience avec inhibiteurs du TNF
| |
Taltz
80 mg
1x/4 sem.a
(N=81)
|
Placebo
(N=87)
|
Différence par rapport au placebo
(CI 95%)
|
Adalimumab
40 mg
1x/2 sem.
(N= 90)
|
Taltz
80 mg
1x/4 sem.c
(N=114)
|
Placebo
(N=104)
|
Différence par rapport au placebo
(CI 95%)
| |
Réponse ASAS20b, n (%), NRI
|
52
(64.2%)
|
35
(40.2%)
|
24.0
(9.3, 38.6)**
|
53
(58.9%)
|
55
(48.2%)
|
31
(29.8%)
|
18.4
(5.7, 31.1)**
| |
Réponse ASAS40b,c, n (%), NRI
|
39
(48.1%)
|
16
(18.4%)
|
29.8
(16.2, 43.3)***
|
32
(35.6%)
|
29
(25.4%)
|
13
(12.5%)
|
12.9
(2.7, 23.2)*
| |
ASDAS à l'inclusion[variation vs inclusion]
|
3.7
[-1.4]
|
3.9
[-0.5]
|
-1.0
(-1.3, -0.7)***
|
3.7
[-1.3]***
|
4.2
[-1.2]
|
4.1
[-0.1]
|
-1.1
(-1.3, -0.8)***
| |
Score BASDAI à l'inclusion [variation vs inclusion]
|
6.8
[-2.9]
|
6.8
[-1.4]
|
-1.5
(-2.1, -0.9)***
|
6.7
[-2.5]***
|
7.5
[-2.2]
|
7.3
[-0.9]
|
-1.2
(-1.8, -0.7)***
| |
MRI Spine SPARCCd à l'inclusion [variation vs inclusion]
|
14.5
[-11.0]
|
15.8
[-1.5]
|
-9.5
(-12.6, -6.4)***
|
20.0
[-11.6]***
|
8.3
[-3.0]
|
6.4
[3.3]
|
-6.3
(-10.0, -2.5)**
| |
BASDAI50e (%), NRI
|
42
|
17
|
25
(11, 38)***
|
32*
|
22
|
10
|
12
(3, 22)*
| |
ASDAS <2.1 (%) (faible activité de la maladie), NRI
|
43
|
13
|
31
(18, 43)***
|
38***
|
18
|
5
|
13
(5, 21)**
| |
ASAS HI
à l'inclusion [variation vs inclusion]
|
7.5
[-2.4]
|
8.1
[-1.3]
|
-1.1
(-2.0, -0.3)*
|
8.2
[-2.3]*
|
10.0
[-1.9]
|
9.0
[-0.9]
|
-1.0
(-1.9, -0.1)*
| |
SF-36 PCS
à l'inclusion [variation vs inclusion]
|
34.0
[7.7]
|
32.0
[3.6]
|
4.1
(1.9, 6.2)***
|
33.5
[6.9]**
|
27.5
[6.6]***
|
30.6
[1.4]
|
5.2
(3.0, 7.4)***
|
Abréviations: N = nombre de patients dans la population en intention de traiter; NRI = Non-Responder Imputation ou imputation d'absence de réponse; les patients ayant des données manquantes ont été comptés comme non-répondeurs.
ASAS HI = Assessment of SpondyloArthritis International Society Health Index, Indice d'évaluation de la santé dans la spondyloarthrite; ASDAS = Ankylosing Spondylitis Disease Activity Score, Score d'activité de la spondylarthrite ankylosante; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index, Indice d'activité de la spondylarthrite ankylosante de Bath; Variation par rapport à l'inclusion = modification de la moyenne des moindres carrés à la semaine 16 par rapport à la valeur à l'inclusion; MRI Spine SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the Spine, Score IRM-rachis du Consortium canadien de recherche sur la spondyloarthrite (cotation de 23 unités discovertébrales) SF-36 PCS = Short Form health survey physical component summary.
a À la semaine 0, les patients ont reçu Taltz 80 mg ou 160 mg..
b Une réponse ASAS20 est définie comme une amélioration ≥20 % et une amélioration absolue par rapport à la valeur à l'inclusion ≥1 unité (intervalle de 0 à 10) dans au moins 3 des 4 domaines (Évaluation globale du patient, douleur rachidienne, fonction et inflammation), et aucune aggravation ≥20 % et ≥1 unité (intervalle de 0 à 10) dans le domaine restant. Une réponse ASAS40 est définie comme une amélioration ≥40 % et une amélioration absolue par rapport à la valeur à l'inclusion ≥2 unités dans au moins 3 des 4 domaines, sans aucune aggravation dans le domaine restant.
c Critère principal.
d Les nombres de patients en ITT avec des données IRM à l'inclusion sont les suivants: COAST-V: Taltz, n = 81; PBO, n = 82; ADA, n = 85. COAST-W: Taltz, n = 58; PBO, n = 51.
e Réponse BASDAI50 définie comme une amélioration ≥50 % du score BASDAI par rapport à sa valeur à l'inclusion.
* p<0.05; ** p<0.01; *** p<0.001 par rapport au placebo.
A la semaine 16, il y a eu une amélioration cliniquement significative des principaux composants du critère de réponse ASAS40 (douleurs rachidiennes, BASFI, évaluation globale du patient, raideur) et d'autres mesures d'activité de la maladie, y compris la CRP.
Le pourcentage de patients ayant atteint une réponse ASAS40 dans les études COAST-V et COAST-W est présenté dans la figure 3.
Figure 3. Réponse ASAS40 dans les études COAST-V et COAST-W jusqu'à la semaine 16, NRIa

a Les patients ayant des données manquantes ont été comptés comme non-répondeurs.
* p<0.05; ** p<0.01; *** p<0.001 par rapport au placebo
Un taux de réponse ASAS40 comparable a été observé chez les patients indépendamment des taux de CRP à l'inclusion, des scores ASDAS à l'inclusion et des scores IRM SPARCC-rachis. La réponse ASAS40 a été démontrée indépendamment de l'âge, du sexe, de l'ethnie, de la durée de la maladie, du poids corporel à l'inclusion, du score BASDAI à l'inclusion et du traitement biologique antérieur.
Dans les études COAST-V et COAST-W, l'efficacité a été maintenue jusqu'à la semaine 52, selon les critères d'évaluation présentés ci-dessus à la semaine 16, y compris les taux de réponse ASAS20, ASAS40, ASDAS, BASDAI, BASFI et ASAS HI.
Résultats liés à la santé
Des améliorations des douleurs rachidiennes par rapport au placebo ont été observées dès la semaine 1, et se sont maintenues jusqu'à la semaine 16 (Taltz vs placebo: COAST-V -3.2 vs -1.7; COAST-W -2.4 vs -1.0); des améliorations sur la fatigue et sur la mobilité rachidienne ont été observées par rapport au placebo à la semaine 16. Les améliorations sur la douleur rachidienne, sur la fatigue et sur la mobilité rachidienne se sont maintenues jusqu'à la semaine 52.
Spondylarthrite axiale non radiographique
L'efficacité et la sécurité de Taltz ont été évaluées dans une étude randomisée, en double aveugle, sur une période de 52 semaines contrôlée contre placebo (COAST-X) chez 303 patients âgés de ≥18 ans ayant une spondylarthrite axiale active depuis au moins 3 mois. Les patients devaient présenter des signes objectifs d'inflammation se traduisant par des taux élevés de protéine C réactive (CRP) et/ou une sacro-iliite à l'imagerie par résonance magnétique (IRM), mais sans signe radiographique visible de lésion structurale au niveau des articulations sacro-iliaques. Les patients avaient une maladie active définie par un score BASDAI (Bath Ankylosing Spondylitis Disease Activity Index) ≥4 et une douleur rachidienne ≥4 sur une échelle d'évaluation numérique (NRS) de 0 à 10, malgré un traitement par AINS. Les patients ont été traités par Taltz 80 mg ou 160 mg à la semaine 0, suivi par Taltz 80 mg toutes les 2 semaines (1x/2 sem.) ou toutes les 4 semaines (1x/4 sem.) ou par un placebo. L'instauration et/ou l'ajustement de la dose de traitements concomitants (AINS, cDMARD, corticostéroïdes, analgésiques) étaient autorisés à partir de la semaine 16.
A l'inclusion les patients avaient des symptômes de spondylarthrite axiale non-radiographique depuis en moyenne 11 ans. Environ 39% des patients recevaient un traitement concomitant par cDMARD.
Le critère d'évaluation principal était le pourcentage de patients atteignant une réponse ASAS40 à la semaine 16.
Réponse clinique
A la semaine 16, des proportions plus élevées de patients traités par Taltz 80 mg 1x/4 sem. ont atteint une réponse ASAS40 par rapport à ceux sous placebo (Tableau 7). Les réponses étaient similaires, indépendamment des traitements concomitants.
Tableau 7 Résultats d'efficacité de l'étude COAST-X à la semaine 16, NRI
|
|
Taltz 80 mg
1x/4 sem.a
(N=96)
|
Placebo
(N=105)
|
Différence par rapport au placebo
(95% CI)f
| |
Réponse ASAS20 b, n (%), NRI
|
52
(54.2%)
|
41
(39.0%)
|
15.1
(1.5, 28.8)*
| |
Réponse ASAS40 b,c, n (%), NRI
|
34
(35.4%)
|
20
(19.0%)
|
16.4
(4.2, 28.5)**
| |
ASDAS à l'inclusion
[variation par rapport à l'inclusion]
|
3.8
[-1.1]
|
3.8
[-0.6]
|
-0.5
(-0.8, -0.3) ***
| |
Score BASDAI à l'inclusion
[variation par rapport à l'inclusion]
|
7.0
[-2.2]
|
7.2
[-1.5]
|
-0.7
(-1.3, -0.1) *
| |
MRI SIJ SPARCCd à l'inclusion
[variation par rapport à l'inclusion]
|
5.1
[-3.4]
|
6.3
[-0.3]
|
-3.1
(-4.6, -1.6) ***
| |
ASDAS <2.1, n (%)
(faible activité de la maladie), NRIe
|
26
(27.7%)
|
13
(12.4%)
|
15.3
(4.3, 26.3) **
| |
SF-36 PCS
à l'inclusion
[variation par rapport à l'inclusion]
|
33.5
[8.1]
|
32.6
[5.2]
|
2.9
(0.6, 5.1) *
|
Abréviations: N = nombre de patients dans la population en intention de traiter; NRI = Non-Responder Imputation ou Imputation d'absence de réponse. ASDAS = Ankylosing Spondylitis Disease Activity Score, Score d'activité de la spondylarthrite ankylosante; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index, Indice d'activité de la spondylarthrite ankylosante de Bath; Variation par rapport à l'inclusion = modification de la moyenne des moindres carrés à la semaine 16 par rapport à la valeur à l'inclusion; MRI SIJ SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the sacroiliac joint, Score IRM des articulations sacro-iliaques du Consortium canadien de recherche sur la spondyloarthrite, SF-36 PCS = Short Form health survey physical component summary.
Les patients ayant des données manquantes ont été comptés comme non-répondeurs.
a À la semaine 0, les patients ont reçu 80 mg ou 160 mg de Taltz.
b Une réponse ASAS20 est définie comme une amélioration ≥20 % et une amélioration absolue par rapport à la valeur à l'inclusion ≥1 unité (intervalle de 0 à 10) dans au moins 3 des 4 domaines (Évaluation globale du patient, douleurs rachidiennes, fonction et inflammation), et aucune aggravation ≥20 % et ≥1 unité (intervalle de 0 à 10) dans le domaine restant. Une réponse ASAS40 est définie comme une amélioration ≥40 % et une amélioration absolue par rapport à la valeur à l'inclusion ≥2 unités dans au moins 3 des 4 domaines, sans aucune aggravation dans le domaine restant.
c Critère principal à la semaine 16.
d Le nombre de patients en ITT avec des données IRM à l'inclusion et à la semaine 16 est le suivant: Taltz, n = 85; PBO, n = 90.
e Les patients ayant des données manquantes ont été comptés comme non-répondeurs. Les pourcentages sont fondés sur le nombre de patients dans la population en ITT ayant un score ASDAS ≥2,1 à l'inclusion.
f Les valeurs mentionnées correspondent à la différence en % (IC 95 %) pour les variables catégorielles, et la différence en LSM (IC 95 %) pour les variables continues.
* p<0.05; ** p<0.01; *** p<0.001 par rapport au placebo.
Les principales composantes du critère de réponse ASAS40 (douleur rachidienne, BASFI, évaluation globale du patient, raideur) ainsi que d'autres mesures de l'activité de la maladie présentaient une amélioration clinique significative à la semaine 16.
La figure 4 montre le pourcentage de patients ayant atteint une réponse ASAS40 lors des visites.
Figure 4. Réponse ASAS40 jusqu'à la semaine 16 dans l'étude COAST-X, NRIa

a Les patients ayant des données manquantes ont été comptés comme non-répondeurs.
** p < 0.01 par rapport au placebo.
Dans COAST-X l'efficacité a été maintenue jusqu'à la semaine 52.
Résultats à long terme dans la spondylarthrite axiale
Les patients ayant terminé l'une des trois études pivots COAST-V/W/X (52 semaines) se sont vu proposer de participer à l'étude d'extension à long terme et de retrait randomisé (COAST-Y, avec 350 patients sous Taltz 1x/4 sem et 423 patients sous 1x/2 sem). Parmi les 157/773 (20.3 %) qui ont obtenu une rémission, (défini comme Ankylosing Spondylitis Disease Activity Score [ASDAS] < 1.3 au moins une fois, et aucun score ASDAS ≥2.1 aux semaines 16 et 20), 155 patients déjà traités par Taltz jusqu'à 76 semaines (52 semaines de l'étude COAST V/W/X + 24 semaines de COAST Y) ont été randomisés à la semaine 24 de l'étude COAST-Y (population de retrait randomisé: placebo, N = 53; Taltz 1x/4 sem., N = 48 et Taltz 1x/2 sem., N = 54). Sur l'ensemble de ces patients, 148 (95.5 %) ont terminé la visite de la semaine 64 (placebo, N = 50; Taltz 1x/4 sem., N = 47 et Taltz 1x/2 sem., N = 51). Le critère principal d'évaluation était la proportion de patients dans la population de retrait randomisé qui ne présentait pas de poussée de la maladie pendant les semaines 24 à 64 (groupes Taltz 1x/2 sem. et Taltz 1x/4 sem. combinés versus placebo). Dans les groupes Taltz combinés (83.3 % (85/102), p < 0.001) et Taltz 1x/4 sem. (83.3 % (40/48), p = 0.003), une proportion significativement plus élevée de patients (non responder imputation, NRI) n'a pas présenté de poussée entre les semaines 24 et 64 comparé à ceux qui ont arrêté Taltz pour recevoir un placebo (54.7 % (29/53)).
Taltz (dans les deux groupes Taltz combinés et Taltz 1x/4 sem., respectivement) a significativement retardé le délai d'apparition des poussées (Test du Log-Rank p < 0.001 et p < 0.01, respectivement) comparé au placebo.
|