CompositionPrincipes actifs
Rurioctocogum alfa pegolum (Factor VIII coagulationis humanus (ADNr)). La substance de départ, facteur VIII de coagulation recombinant, est produite dans des cellules d'ovaire d'hamster chinois (CHO).
Excipients
Trometamolum, Calcii chloridum dihydricum, Mannitolum, Natrii chloridum (correspond à 10.4 mg sodium par flacon), Trehalosum dihydricum, Histidinum, Polysorbatum 80, L-Glutathionum
Solvant: Aqua ad iniectabile
Indications/Possibilités d’emploiTraitement et prophylaxie des épisodes hémorragiques chez les patients atteints d'hémophilie A (déficit congénital en facteur VIII).
ADYNOVI ne contient pas de facteur de von Willebrand en quantité pharmacologiquement active. Par conséquent, il n'est pas indiqué dans le traitement de la maladie de von Willebrand.
Posologie/Mode d’emploiLe traitement doit être instauré sous la surveillance d'un médecin expérimenté dans le traitement de l'hémophilie.
Posologie usuelle
La dose et la durée du traitement substitutif dépendent de la sévérité du déficit en facteur VIII, de la localisation et de l'importance de l'hémorragie ainsi que de l'état clinique du patient.
Le nombre d'unités de facteur VIII est exprimé en Unités Internationales (UI), ramenées au standard de l'OMS pour les produits de facteur VIII. L'activité en facteur VIII dans le plasma est exprimée soit en pourcentage (par rapport au plasma humain normal), soit en UI (par rapport au Standard international du facteur VIII plasmatique).
Une Unité internationale (UI) d'activité de facteur VIII correspond à la quantité de facteur VIII contenue dans un ml de plasma humain normal.
Traitement à la demande
Le calcul de la dose nécessaire de facteur VIII est basé sur le résultat empirique que 1 UI de facteur VIII par kg de poids corporel augmente l'activité plasmatique du facteur VIII de 2 UI/dl.
La dose nécessaire est déterminée à l'aide de la formule suivante:
Nombre d'unités (UI) nécessaires = poids corporel (kg) x augmentation souhaitée du taux de facteur VIII (%) x 0.5
La dose et la fréquence d'administration doivent toujours être adaptées à chaque cas individuel en fonction de l'efficacité clinique du produit.
En cas de survenue de l'un des événements hémorragiques suivants, l'activité de facteur VIII ne doit pas chuter en dessous du taux d'activité plasmatique indiqué (en % de la normale ou UI/dl) pendant la durée mentionnée.
Le tableau 1 ci-dessous peut servir de guide pour la détermination des posologies lors d'épisodes hémorragiques et lors de chirurgie:
Dans le cas des épisodes hémorragiques sévères ou mettant en jeu le pronostic vital, une surveillance soigneuse du traitement substitutif est notamment nécessaire.
Tableau 1
|
Degré de l'hémorragie/type d'intervention chirurgicale
|
Niveau plasmatique de facteur VIII nécessaire (% ou UI/dl)
|
Fréquence des doses (heures)/durée du traitement (jours)
| |
Hémorragie
| |
Début d'hémarthrose, de saignement musculaire ou buccal
|
20 - 40
|
Répéter les injections toutes les 12 à 24 heures pendant au moins 1 jour, jusqu'à la fin de l'épisode hémorragique, indiquée par la disparition de la douleur ou l'atteinte d'une guérison
| |
Hémarthrose plus étendue, saignements musculaires ou hématome
|
30 - 60
|
Répéter les injections toutes le 12 – 24 heures pendant 3-4 jours ou plus longtemps jusqu'à disparition de la douleur et de l'invalidité aiguë.
| |
Hémorragie mettant en jeu le pronostic vital
|
60 - 100
|
Répéter les injections toutes les 8 à 24 heures jusqu'à ce que le danger pour le patient soit éliminé.
| |
Chirurgie
| |
Mineure dont extraction dentaire
|
30 - 60
|
Toutes les 12 heures, au moins 1 jour, jusqu'à l'obtention d'une cicatrisation.
| |
Majeure
|
80 - 100
(pré- et postopératoire)
|
Répéter les injections toutes les 8-24 heures jusqu' à cicatrisation satisfaisante de la plaie, puis poursuivre le traitement pendant au moins 7 jours supplémentaires pour maintenir un taux de facteur VIII entre 30% et 60% (UI/dl).
|
Prophylaxie
ADYNOVI est administré moins fréquemment que le factor VIII recombinant. La dose recommandée d'ADYNOVI est de 40 – 50 UI/kg de poids corporel 2 fois par semaine chez les adolescents et les adultes (≥12 ans) et de 40 – 60 UI/kg de poids corporel 2 fois par semaine chez les enfants (<12 ans). Afin d'offrir la protection maximale pour l'activité physique, administrez ADYNOVI au prochain jour prévu de la perfusion et adaptez la fréquence conformément.
Posologie individualisée
Jusqu'à 80 IU/kg peuvent être administré afin d'obtenir un taux cible de facteur VIII ≥1%. La dose doit être adaptée en tenant compte de la réponse clinique du patient.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Instructions posologiques particulières
Enfants et adolescents
La sécurité et l'efficacité chez les enfants, adolescents et adultes étaient comparables dans les études cliniques (traitement à la demande, prophylaxie).
Les études pharmacocinétiques ont montré une clairance supérieure, une demi-vie et une récupération incrémentielle inférieure du facteur VIII chez les enfants de moins de 12 ans, en comparaison aux adultes.
Mode d'administration
ADYNOVI doit être administré par voie intraveineuse. Si ADYNOVI n'est pas administré par un personnel médical, il est nécessaire d'effectuer un entraînement correspondant.
La vitesse d'administration sera déterminée en fonction du niveau de confort du patient et ne devra pas être supérieure à 10 ml/min.
Pour des instructions concernant la reconstitution du médicament avant l'administration, voir sous «Remarques concernant la manipulation».
Contre-indicationsHypersensibilité connue mettant en jeu le pronostic vital, inclus anaphylaxie, à l'ADYNOVI, à la substance de départ ADVATE, aux protéines de souris ou de hamster, ainsi qu'aux autres composants d'ADYNOVI.
Mises en garde et précautionsRéactions d'hypersensibilité
Des réactions d'hypersensibilité de type allergiques peuvent apparaître avec ADYNOVI. Des réactions d'hypersensibilité de type allergique, inclus l'anaphylaxie, sont connues après l'administration de préparations de facteur VIII de coagulation recombinant, y compris ADYNOVI et sa substance mère ADVATE. En cas de survenue de réactions d'hypersensibilité, l'administration doit être interrompue immédiatement et un traitement approprié doit être entamé.
Les patients doivent être informés des signes précoces des réactions d'hypersensibilité, y compris rash, urticaire généralisée, oppression thoracique, sibilances, hypotension et anaphylaxie.
En cas de choc anaphylactique, une thérapie de choc basée sur les règles actuelles de la médecine doit être appliquée.
Au cours des études cliniques aucun cas de réactions d'hypersensibilité n'a été rapporté.
Inhibiteurs
L'apparition d'anticorps neutralisants (inhibiteurs) du facteur VIII est une complication connue dans le traitement des patients atteints d'hémophilie A. Ces inhibiteurs sont habituellement des immunoglobulines G dirigées contre l'activité procoagulante du facteur VIII et sont mesurées en Unités Bethesda (UB) par ml de plasma par le test de Bethesda modifié.
Le risque de développer des inhibiteurs est corrélé à la durée de l'exposition au facteur VIII, le risque étant le plus élevé au cours des 20 premiers jours d'exposition. Rarement, les inhibiteurs peuvent apparaître après les 100 premiers jours d'exposition.
Des cas de réapparition d'inhibiteurs (faible titre) ont été observés après le changement d'un facteur VIII recombinant pour un autre, chez des patients préalablement traités (PTPs) ayant plus de 100 jours d'exposition et qui avaient des antécédents de développement d'inhibiteur.
Par conséquent, il est recommandé de surveiller attentivement l'apparition d'inhibiteurs chez tous les patients après tout changement de produit.
D'une manière générale, tous les patients traités avec un facteur VIII de coagulation doivent faire l'objet d'une surveillance étroite clinique et au moyen d'examens de laboratoire appropriés afin de détecter l'apparition éventuelle d'inhibiteurs. Si le taux d'activité plasmatique du facteur VIII attendu n'est pas atteint ou si l'épisode hémorragique n'est pas contrôlé malgré l'administration d'une dose appropriée, une recherche de la présence d'inhibiteurs du facteur VIII doit être réalisée. Chez les patients présentant des taux élevés d'inhibiteurs, le traitement par facteur VIII peut être inefficace et d'autres options thérapeutiques doivent être envisagées. La prise en charge de ces patients doit être effectuée par des médecins expérimentés dans la prise en charge de l'hémophilie et des inhibiteurs du facteur VIII.
Complications liées au cathéter lors du traitement
Si un dispositif d'accès veineux central est requis, le risque de complications liées au dispositif d'accès veineux central, telles que des infections locales, une bactériémie et une thrombose au site du cathéter, doit être pris en compte.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c.-à-d. qu'il est essentiellement « sans sodium ».
InteractionsAucune étude d'interaction d'ADYNOVI avec d'autres produits n'a été réalisée.
Grossesse, allaitementADYNOVI n'a pas fait l'objet d'études de reproduction chez l'animal. En raison de la rareté de l'hémophilie A chez les femmes, il n'y a pas de données sur l'utilisation d'ADYNOVI pendant la grossesse et l'allaitement. Par conséquent, il faut peser l'intérêt de l'utilisation d'ADYNOVI au cours de la grossesse et de l'allaitement par rapport au risque pour la mère et l'enfant. ADYNOVI ne doit être prescrit qu'en cas de nécessité absolue.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune information n'est disponible sur l'effet de ADYNOVI sur l'aptitude à la conduite et l'utilisation de machines.
Effets indésirablesUne hypersensibilité ou des réactions allergiques (incluant angiooedème, sensation de brûlure et de piqûre au site de perfusion, frissons, bouffées congestives, urticaire généralisée, céphalées, rash, hypotension, léthargie, nausées, agitations nerveuses, tachycardie, oppression thoracique, paresthésie, vomissements, sibilances) ont été observées dans de rares cas après traitement avec facteur VIII et peuvent, dans certains cas, évoluer vers une anaphylaxie sévère (y compris choc).
Les patients atteints d'hémophilie A peuvent développer des anticorps neutralisants (inhibiteurs) du facteur VIII. L'apparition de tels inhibiteurs peut se manifester par une réponse clinique insuffisante. Il est alors recommandé de contacter un centre spécialisé en hémophilie.
La sécurité d'ADYNOVI a été évaluée chez 365 patients préalablement traités ou non préalablement traités, présentant une hémophilie sévère et ayant reçu au moins une dose d'ADYNOVI dans le cadre de 6 études cliniques multicentriques, prospectives, en ouvert et de 1 étude clinique toujours en cours.
L'effet indésirable qui a été observé fréquemment (≥1% des patients) lors des études cliniques était la céphalée.
La fréquence a été définie selon les critères suivants: très fréquent (≥10%), fréquent (≥1% à <10%), occasionnel (≥0.1% à <1%), rare (≥0.01% à <0.1%), très rare (<0.01%)
Les effets indésirables suivants ont été décrits dans des études cliniques:
Affections gastro-intestinales
Fréquent: diarrhée, nausée
Affections oculaires
Occasionnel: hyperémie oculaire
Affections du système immunitaire
Occasionnel: hypersensibilité
Affections du système nerveux
Très fréquent: céphalées (11.2%)
Fréquent: sensation de vertiges
Affections de la peau et du tissu sous-cutané
Fréquent: rash
Occasionnel: Eruption cutanée prurigineuse
Fréquent: urticaire
Affections vasculaires
Occasionnel: bouffée de chaleur
Investigations
Occasionnel: augmentation du nombre d'éosinophiles
Lésions, intoxications et complications liées aux procédures
Occasionnel: réaction liée à une perfusion
Effets indésirables identifiés après la mise sur le marché
Affections du système immunitaire
Inconnue: réactions anaphylactiques
Description de certains effets indésirables
Développement d'inhibiteurs
Après l'administration d'ADYNOVI des anticorps neutralisants (inhibiteurs) contre le facteur VIII peuvent se développer.
Aucun des patients ayant participé à une ou plusieurs des 6 études achevées et réalisées sur des patients prétraités (PTP) n'a développé des anticorps neutralisants (inhibiteurs) persistants dirigés contre le FVIII à ≥0.6 UB/ml (sur la base du test Bethesda-Nijmegen). Pendant une prophylaxie personnalisée avec une valeur cible de FVIII de 8-12%, un patient a développé transitoirement des inhibiteurs du FVIII à la limite basse de détection positive (0.6 UB).
On dispose de rapports provisoires relatifs à 9 cas de développement d'inhibiteurs du FVIII lié au traitement par ADYNOVI, issus d'une étude en cours menée sur des patients non prétraités âgés de < 6 ans atteints d'hémophilie A sévère.
L'identification des anticorps réactifs au facteur VIII dépend fortement de facteurs différents, y compris la sensitivité et spécificité des tests, manipulation des échantillons, moment du prélèvement, médicament administrés simultanément et maladie sous-jacente.
Effet de classe
Effets indésirables associés avec la substance de départ, incluent: réactions anaphylactiques, hypersensibilité, inhibition du facteur VIII.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucun symptôme de surdosage avec du facteur VIII de coagulation humain recombinant n'a été rapporté.
Propriétés/EffetsCode ATC
B02BD02 (facteur VIII de coagulation)
Mécanisme d'action
Le complexe facteur VIII/facteur von Willebrand se compose de deux molécules (facteur VIII et facteur von Willebrand) aux fonctions physiologiques différentes.
Lorsque le facteur VIII est perfusé à un patient hémophile, il se lie dans la circulation sanguine au facteur von Willebrand endogène. Le facteur VIII activé agit comme Cofacteur du facteur IX activé, accélérant la conversion du facteur X en facteur X activé. Le facteur X activé convertit la prothrombine en thrombine. La thrombine convertit ensuite le fibrinogène en fibrine, ce qui aboutit à la formation d'un caillot.
L'hémophilie A est une maladie de la coagulation sanguine, héréditaire liée au sexe (liée au chromosome X), due à la diminution de l'activité du facteur VIII provoquant des accidents hémorragiques fortes au niveau des articulations, des muscles ou des organes internes, spontanés ou provoqués par des traumatismes accidentels ou chirurgicaux. Le taux plasmatique en facteur VIII est augmenté grâce au traitement substitutif, ce qui permet de corriger temporairement le déficit en facteur VIII et les tendances hémorragiques.
ADYNOVI (facteur VIII humain recombinant pégylé) contient le principe active ADVATE (Octacogum alfa, facteur VIII humain recombinant) in extenso, constitué de 2332 acides aminés (poids moléculaire (PM) approximatif de 280 kD), conjugué avec le polyéthylène glycol (PEG) (PM 20 kD).
L'activité thérapeutique d'ADYNOVI est déduite d'ADVATE. ADVATE est produit par la technologie de l'ADN recombinant à partir de cellules d'ovaire d'hamster chinois (CHO).
La molécule ADVATE est ensuite liée de manière covalente au polyéthylène glycol, qui cible le résidu lysine. Le groupe fonctionnel PEG est lié au principe actif d'ADVATE, afin d'augmenter le temps de demi-vie plasmatique en réduisant la clairance de la molécule de facteur VIII médiée par le récepteur LRP-1.
Pharmacodynamique
Efficacité clinique
L'efficacité clinique d'ADYNOVI a été mise en évidence par une étude ouverte prospective, multicentrique de phase 2/3. 137 patients préalablement traités âgés entre 12 et 65 ans ont reçu ADYNOVI soit de manière prophylactique (deux fois par semaine à la dose de 40-50 UI/kg; n = 120), soit en traitement aigu des hémorragies (10-60 UI/kg; n = 17). Tous les patients étaient atteints d'une hémophilie A sévère (taux de facteur VIII <1%). L'intervalle posologique moyen était de 3,6 jours. La fréquence d'administration a pu être réduite (médiane: 33.7%) chez 91 patients sur 98 traités de manière prophylactique avant le début de l'étude.
Le taux annuel médian d'hémorragies dans le groupe de traitement prophylactique se montait à 1.9 (min. 0.0, max. 18.4) et à 0.0 (hémorragies spontanées: min. 0.0, max. 18.2; hémorragies articulaires: min. 0.0, max. 13.1) pour les hémorragies spontanées et pour les hémorragies articulaires. 40 % des patients n'ont enregistré aucun épisode hémorragique.
Au total, 518 épisodes hémorragiques ont été traités par ADYNOVI, dont 361 dans le groupe de traitement aigu et 157 dans le groupe de traitement prophylactique. Dans 95.9% des cas, les hémorragies ont été traitées par 1 à 2 injections d'ADYNOVI.
Pour 96.1% de l'ensemble des épisodes hémorragiques traités, l'efficacité hémostatique a été qualifiée d'excellente ou de bonne.
La sécurité et l'efficacité d'ADYNOVI dans le cadre d'une utilisation périopératoire ont été évaluées par une étude de phase 3 ouverte, multicentrique, non contrôlée, prospective sur des patients masculins préalablement traités présentant une hémophilie A sévère. Au total, 15 interventions (11 majeures et 4 mineures) réalisées chez 15 patients (âgés de 19 à 52 ans) ont été incluses dans l'analyse. L'efficacité hémostatique périopératoire a été jugée excellente pour l'ensemble des 15 interventions (perte de sang attendue inférieure ou comparable à celle subie par un patient non hémophile lors d'une intervention similaire).
Sécurité et efficacité en pédiatrie
Population < 12 ans
Au total, 66 patients atteints d'hémophilie A sévère préalablement traités ont été inclus dans l'étude pédiatrique (32 patients âgés de moins de 6 ans et 34 patients âgés de 6 à moins de 12 ans).
La posologie du schéma prophylactique était de 40 à 60 UI/kg d'ADYNOVI deux fois par semaine.
Le taux annualisé d'hémorragies (TAH) médian global était de 2.0 (EI: 3.9) pour les 65 patients de la population per protocole, et les TAH spontanées et articulaires médians étaient tous deux de 0 (EI: 1.9). 37 % des patients n'ont présenté aucun épisode de saignement, 72% n'ont présenté aucun épisode de saignement articulaire et 66%) n'ont présenté aucun épisode de saignement spontané au cours du traitement prophylactique.
Sur les 70 épisodes de saignement observés au cours de l'étude pédiatrique, 82.9% ont été contrôlés avec 1 injection et 91.4% avec 1 ou 2 injections.
Le contrôle du saignement a été jugé excellent ou bon dans 90.0% des cas.
Population de < 6 ans
La sécurité, l'immunogénicité et l'efficacité hémostatique d'ADYNOVI ont été évaluées dans une étude de phase 3 multicentrique et prospective en ouvert chez des patients âgés de moins de 6 ans, atteints d'hémophilie A sévère (facteur VIII < 1 %) et naïfs de traitement. Les patients ont reçu soit un traitement en fonction des besoins à une dose de 10 à 50 UI/kg et jusqu'à 80 UI/kg, soit un traitement prophylactique à une dose de 25 à 50 UI/kg au moins une fois par semaine, pouvant être portée à 80 UI/kg.
L'évaluation intermédiaire comprenait 59 patients qui avaient reçu au moins une dose. Parmi les 59 patients traités, 52 ont été inclus dans l'analyse primaire du développement d'inhibiteurs et 10 de ceux-là ont développé des inhibiteurs (19.2%). Sur la base d'un modèle linéaire généralisé moyen (MLG) avec distribution binomiale négative, l'indicateur (IC à 95%) du taux annuel total d'hémorragies s'élevait respectivement à 3.206 (1.632-6.297) et 3.199 (2.026-5.049) pour le groupe traité en fonction des besoins et le groupe recevant un traitement prophylactique.
Une seule perfusion a été nécessaire pour la plupart des hémorragies (77.7%). Lors du contrôle de l'hémorragie, l'efficacité hémostatique a été jugée excellente (32.7%) ou bonne (27.1%) par les patients ou les soignants. Il convient de noter que l'efficacité hémostatique n'a pas été évaluée lors du contrôle de 36.8% des hémorragies. Ces résultats intermédiaires confirment la sécurité et l'efficacité démontrées dans les études réalisées sur des patients pédiatriques, des adolescents, ainsi que chez des adultes prétraités.
Prophylaxie au long cours chez les patients pédiatriques et adultes
La sécurité et l'efficacité à long terme d'ADYNOVI pour la prophylaxie et le traitement en fonction des besoins des épisodes hémorragiques ont été étudiées chez 216 patients prétraités pédiatriques et adultes atteints d'hémophilie A sévère qui avaient participé antérieurement à d'autres études portant sur ADYNOVI ou qui étaient naïfs de traitement par ADYNOVI jusque-là. Les participants de la population traitée recevaient une dose fixe de 40 à 50 UI/kg (participants âgés de ≥12 ans) ou 40 à 60 UI/kg (participants âgés de < 12 ans) deux fois par semaine. La dose a été ajustée jusqu'à 80 UI/kg deux fois par semaine lorsque cela était nécessaire pour maintenir un taux résiduel de FVIII de > 1%. Les participants qui avaient décidé de suivre un schéma prophylactique personnalisé (individualisé du point de vue de la pharmacocinétique) recevaient au moins deux fois par semaine des doses allant jusqu'à 80 UI/kg par perfusion, visant un taux résiduel de FVIII de ≥3%. Les participants âgés de ≥12 ans qui recevaient un régime à deux administrations par semaine et qui ne présentaient pas d'hémorragies spontanées (zéro) durant une période de 6 mois consécutifs pouvaient passer à une prophylaxie avec dose fixe tous les 5 jours. Si ces sujets continuaient à ne pas avoir d'hémorragies spontanées (zéro) pendant les 6 mois suivants, ils pouvaient recevoir un régime à une administration tous les 7 jours. Au total, l'exposition au long cours durait en moyenne (ET) 195.4 (101.57) jours d'exposition à la prophylaxie (ED) par sujet. Le TSA est représenté en fonction du schéma prophylactique, du site de l'hémorragie et de l'étiologie dans le Tableau 2.
|
Tableau 2: Taux de saignements annualisé (TSA) selon le schéma prophylactique (population ITT)
| |
Localisation et étiologie du saignement
|
Deux fois par semaine
(n = 186)
|
Tous les 5 jours
(n = 56)
|
Tous les 7 jours
(n = 15)
|
Sur la base de la PCa
(n = 25)
| |
Valeur moyenne
[Estimation ponctuelle – intervalle de confiance 95%]
| |
Total
|
2.2 [1.85-2.69]
|
2.1 [1.54-2.86]
|
2.7 [1.44-5.20]
|
2.6 [1.70-4.08]
| |
Articulation
|
1.2 [0.96-1.58]
|
1.1 [0.81-1.55]
|
2.0 [0.90-4.62]
|
1.4 [0.91-2.17]
| |
Spontané
|
1.2 [0.92-1.56]
|
1.3 [0.87-2.01]
|
1.8 [0.78-4.06]
|
1.0 [0.54-1.71]
| |
Traumatique
|
1.0 [0.83-1.28]
|
0.7 [0.45-0.99]
|
0.9 [0.41-1.91]
|
1.6 [1.03-2.50]
| |
Estimation ponctuelle et intervalle de confiance à 95% issus d'un modèle linéaire généralisé suivant une répartition binomiale négative avec fonction de lien logarithmique.
Les participants qui recevaient des doses dans plusieurs schémas sont pris en considération dans les récapitulations de plusieurs schémas.
n = nombre de patients inclus dans l'analyse
a Valeurs résiduelles visées pour l'activité FVIII de ≥3% de la valeur normale
|
L'effet hémostatique à long terme a été étudié sur 910 épisodes hémorragiques traités par ADYNOVI et classé comme excellent ou bon pour 88.5% de ces épisodes hémorragiques. Toutes catégories d'âge confondues, > 85% des traitements d'hémorragies ont été classés comme excellents ou bons, tant pour le schéma posologique à dose fixe que pour la posologie basée sur la PC. La plupart des épisodes hémorragiques ont été contrôlés avec une (74.0%) ou deux (15.4%) perfusions. Au total, pendant une exposition au long cours d'en moyenne (ET) 195.4 (101.57) jours d'exposition à la prophylaxie par patient (médiane de 208.5 ED), 16.7% (36/216) des patients n'ont pas eu d'épisodes hémorragiques dans cette période élargie.
Prophylaxie personnalisée, étude PROPEL chez des adolescents et des adultes
La sécurité et l'efficacité d'ADYNOVI ont été examinées dans une étude multicentrique ouverte, randomisée, prospective menée sur 121 (115 randomisés) PTP adolescents (âge 12-18 ans) et adultes atteints d'hémophilie sévère durant une période de traitement de 12 mois. Dans l'étude, deux schémas posologiques prophylactiques d'ADYNOVI guidés par la PC ont été comparés, visant un taux résiduel de facteur VIII de 1 à 3% (n = 57) pour l'administration deux fois par semaine et de 8 à 12% pour l'administration tous les deux jours (n = 58). La comparaison a été réalisée par l'évaluation de la proportion de patients qui avaient atteint un TSA total de 0 au cours de la seconde période d'étude de 6 mois.
En moyenne, des doses prophylactiques de 3866.1 UI kg par an ont été administrées dans le bras visant un taux résiduel de 1 à 3% (moyenne [ET] des perfusions/semaine = 2.3 [058]) et de 7532.8 UI/kg par an dans le bras visant un taux résiduel de 8 à 12% (moyenne [ET] des perfusions/semaine = 3.6 [1.18]). Après ajustement de la dose au cours des 6 premiers mois de la prophylaxie, les taux résiduels médians se situaient, dans la seconde période de 6 mois, entre 2.10 UI/dl et 3.00 UI/dl dans le bras visant un taux résiduel de 1 à 3% et entre 10.70 UI/dl et 11.70 UI/dl dans le bras visant un taux résiduel de 8 à 12% (taux résiduel calculé sur la base du test de coagulation en une étape, à la fin de l'intervalle de perfusion planifié). Par conséquent, la posologie permettait généralement d'atteindre et de maintenir les taux résiduels de FVIII souhaités dans les deux schémas prophylactiques.
Le critère d'évaluation principal de l'étude (proportion de sujets qui avaient un TSA total de 0 pendant la seconde période de 6 mois) n'a pas été atteint dans la population de patients ITT (p = 0.545), mais l'a été dans la population per protocole (p = 0.0154).
Au total, 242 épisodes hémorragiques ont été traités par ADYNOVI chez 66 participants, dont 155 hémorragies chez 40 participants dans le bras de taux résiduel de 1 à 3% et 87 hémorragies chez 26 participants dans le bras de taux résiduel de 8 à 12%. La plupart des hémorragies (86.0%, 208/242) ont été contrôlées avec 1 ou 2 perfusions. Dans 84.7% (205/242) des cas d'hémorragies, le contrôle de l'hémorragie a été classé comme excellent ou bon à la fin de l'épisode hémorragique.
PharmacocinétiqueLa pharmacocinétique d'ADYNOVI a été déterminée dans le cadre d'un essai croisé avec ADVATE mené chez 26 patients (18 adultes et 8 adolescents) et chez 22 patients (16 adultes et 6 adolescents) après un traitement de 6 mois par ADYNOVI. Les deux produits ont été utilisés à la dose unitaire de 45 ± 5 UI/kg. Les paramètres PC, répertoriés par groupe d'âge (adultes et adolescents) dans le tableau ci-dessous, reposent sur l'activité plasmatique du facteur VIII mesurée à l'aide du dosage chronométrique en un temps.
ADYNOVI a une demi-vie 1.4 à 1.5 fois plus longue, comme l'ont montré le dosage chronométrique en un temps et le dosage chromogénique. Une augmentation de l'AUC et une baisse de la clairance par rapport à la substance de départ ADVATE ont également été observées. La récupération incrémentielle était similaire avec les deux produits.
L'évolution des paramètres pharmacocinétiques était semblable dans les deux groupes, adultes et adolescents, ainsi qu'entre le dosage chronométrique en un temps et le dosage de l'activité du substrat chromogène.
|
Paramètres pharmacocinétiques chez les adultes (18 ans et plus)
| |
Paramètre PC (moyenne ± (ET)
|
ADVATE lors de dose initiale
n = 18
|
ADYNOVI lors de dose initiale
n = 18
|
ADYNOVI ≥50 EDs
n = 16
| |
Demi-vie terminale (h)
|
10.83 ± 2.08
|
14.69 ± 3.79
|
16.39 ± 5.28
| |
TRM (h)
|
13.41 ± 3.00
|
20.27 ± 5.23
|
21.09 ± 4.73
| |
Clairance (dl/kg*h)
|
0.0388 ± 0.0124
|
0.0227 ± 0.0084
|
0.0237 ± 0.0077
| |
Récupération incrémentielle (I.E./dl/I.E./kg)*
|
2.57 ± 0.43
|
2.66 ± 0.68
|
2.33 ± 0.55
| |
AUC0-Inf (I.E. * h/dl)
|
1286 ± 390
|
2264 ± 729
|
2062 ± 575
| |
Volume de distribution à l'état d'équilibre (dl/kg)
|
0.50 ± 0.11
|
0.43 ± 0.11
|
0.49 ± 0.17
| |
Cmax (U.I./dl)
|
117 ± 20
|
122 ± 29
|
105 ± 25
| |
Tmax (h)
|
0.033 ± 0.19
|
0.46 ± 0.29
|
0.38 ± 0.18
|
|
Paramètres pharmacocinétiques chez les adolescents (12 jusqu'à moins de 18 ans)
| |
Paramètre PC (moyenne ± (ET)
|
ADVATE lors de dose initiale
(IC à 95%)
n = 8
|
ADYNOVI lors de dose initiale (IC à 95%)
n = 8
|
ADYNOVI ≥50 EDs
(IC à 95%)
N = 6
| |
Demi-vie terminale (h)
|
9.45 ± 2.45
|
13.43 ± 4.05
|
15.06 ± 4.08
| |
TRM (h)
|
11.63 ± 2.94
|
17.96 ± 5.49
|
19.47 ± 5.32
| |
Clairance (dl/kg*h)
|
0.0607 ± 0.0305
|
0.0387 ± 0.0331
|
0.0275 ± 0.0096
| |
Récupération incrémentielle (U.I./dl/U.I./kg)*
|
1.94 ± 0.52
|
2.12 ± 0.60
|
2.22 ± 0.88
| |
AUC0-Inf (U.I. * h/dl)
|
902 ± 400
|
1642 ± 752
|
1868 ± 807
| |
Volume de distribution à l'état d'équilibre (dl/kg)
|
0.67 ± 0.31
|
0.56 ± 0.18
|
0.51 ± 0.13
| |
Cmax (U.I./dl)
|
89 ± 29
|
95 ± 25
|
100 ± 42
| |
Tmax (h)
|
0.21 ± 0.04
|
0.26 ± 0.10
|
0.71 ± 1.16
|
Cinétique pour certains groupes de patients
Enfants et adolescents
Les paramètres pharmacocinétiques ont été calculés chez 39 patients de moins de 18 ans (analyse en intention de traiter): 14 enfants (âge compris entre 2 et moins de 6 ans), 17 enfants plus âgés (âge compris entre 6 et moins de 12 ans) et 8 adolescents (âge compris entre 12 et moins de 18 ans) (voir tableau ci-dessous).
La clairance moyenne (basée sur le poids corporel) d'ADYNOVI était supérieure et la demi-vie moyenne était inférieure chez les enfants de moins de 12 ans, en comparaison aux adultes. L'administration d'une dose plus forte peut s'avérer nécessaire chez les enfants de moins de 12 ans.
|
Paramètres pharmacocinétiques chez les patients pédiatriques mesurés par le test de coagulation en une étape
| |
Paramètre PC (moyenne ± (ET)
|
Etude pédiatrique
|
Etude pivotale chez les adolescents et enfants
| |
< 6 ans
n = 14
|
6 à < 12 ans
n = 17
|
12 à < 18 ans
n = 8
| |
Schéma
|
PC de population déterminée à partir d'un échantillon de prélèvementsa
|
PC individuelle déterminée à partir de l'ensemble des prélèvementsb
| |
Demi-vie terminale (h)
|
11.8 ± 2.43
|
12.4 ± 1.67
|
13.43 ± 4.05
| |
TRM (h)
|
17.0 ± 3.50
|
17.8 ± 2.42
|
17.96 ± 5.49
| |
Clairance (dl/kg*h)
|
3.53 ± 1.29
|
3.11 ± 0.76
|
3.87 ± 3.31
(2.73 ± 0.93)*
| |
Récupération incrémentielle (U.I./dl/U.I./kg)*
|
s.o.c
(1.89 ± 0.49)
|
s.o.c
(1.95 ± 0.47)
|
2.12 ± 0.60
| |
AUC0-Inf (U.I. * h/dl)
|
1947 ± 757
|
2012 ± 45
|
1642 ± 752
| |
Volume de distribution à l'état d'équilibre (dl/kg)
|
0.56 ± 0.12
|
0.54 ± 0.09
|
0.56 ± 0.18
| |
Cmax (U.I./dl)
|
s.o.c
(115 ± 30)
|
s.o.c
(115 ± 33)
|
95 ± 25
| |
Tmax (h)
|
d
|
d
|
0.26 ± 0.10
|
* moyenne estimée et ET calculées sans un patient dont la clairance estimée était de 11.8 ml/kg*h. Médiane de tous les patients est 2.78 ml/kg*h.
a Modèle de PC de population mesurée à partir de 3 échantillons prélevés post-injection selon un calendrier de prélèvements aléatoire
b PC individuelle mesurée à partir de 12 échantillons prélevés post-injection.
c s.o.: sans objet, car la récupération progressive et la Cmax chez les enfants reposent sur la PC individuelle. Pour la récupération progressive et la Cmax, les résultats reposent sur la PC individuelle indiquée entre parenthèses.
d Tmax n'a pas pu être calculé pour les patients de l'étude pédiatrique, parce que seulement un échantillon a été pris dans les 3 premières heures après la perfusion (15-30 minutes post-injection)
|
Paramètres pharmacocinétiques chez les patients pédiatriques mesurés par le par dosage chromogénique
| |
Paramètre PC (moyenne ± (ET)
|
Etude pédiatrique
|
Etude pivotale chez les adolescents et enfants
| |
< 6 ans
n = 14
|
6 à < 12 ans
n = 17
|
12 à < 18 ans
n = 8
| |
Schéma
|
PK de population déterminée à partir d'un échantillon de prélèvementsa
|
PK individuelle déterminée à partir de l'ensemble des prélèvementsb
| |
Demi-vie terminale (h)
|
13.0 ± 8.74
|
11.9 ± 2.58
|
13.80 ± 4.00
| |
TRM (h)
|
18.7 ± 12.6
|
17.2 ± 3.72
|
17.73 ± 5.44
| |
Clairance (dl/kg*h)
|
|
|
3.41 ± 3.14
| |
Récupération incrémentielle (U.I./dl/U.I./kg)*
|
s.o.c
1.90 ± 0.27
|
s.o.c
2.20 ± 0.38
|
2.60 ± 0.69
| |
AUC0-Inf (U.I. * h/dl)
|
2190 ± 1590
|
2260 ± 514
|
1900 ± 841
| |
Volume de distribution à l'état d'équilibre (dl/kg)
|
0.91 ± 0.12
|
1.33 ± 0.23
|
0.49 ± 0.20
| |
Cmax (U.I./dl)
|
s.o.c
117 ± 16
|
s.o.c
130 ± 24
|
117 ± 28
| |
Tmax (h)
|
d
|
d
|
0.26 ± 0.14
|
a Modèle de PC de population mesurée à partir de 3 échantillons prélevés post-injection selon un calendrier de prélèvements aléatoire
b PC individuelle mesurée à partir de 12 échantillons prélevés post-injection.
c s.o.: sans objet, car la récupération progressive et la Cmax chez les enfants reposent sur la PC individuelle. Pour la récupération progressive et la Cmax, les résultats reposent sur la PC individuelle indiquée entre parenthèses.
d Tmax n'a pas pu être calculé pour les patients de l'étude pédiatrique, parce-que seulement un échantillon a été pris dans les 3 premières heures après la perfusion (15-30 minutes post-injection)
Les données de pharmacocinétique ont mis en évidence qu' ADYNOVI à une demi-vie circulante allongée.
Données précliniquesPharmacologie de sécurité
Des études de pharmacologie de sécurité n'ont pas relevée des signes d'un potentiel thrombogène ou d'effets secondaires sur les fonctions respiratoires ou cardiovasculaires.
Toxicité à long terme (ou toxicité en cas d'administration répétée)
Des doses uniques ou répétées n'ont pas relevée des signes de toxicité d'ADYNOVI chez les animaux de laboratoire (souris, rats, lapins et singes cynomolgus).
Une administration répétée de la portion 20 kDa PEG n'a pas relevée des signes de toxicité avec la plus haute dose testée de 65 mg/kg.
Mutagénicité et carcinogénicité
Aucune étude pour juger le potentiel mutagène ou cancérogène n'a été réalisé avec le principe actif d'ADYNOVI.
Toxicité sur la reproduction
Aucune étude de toxicologie chez l'animal n'a été effectuée avec ADYNOVI pour évaluer son effet sur la reproduction ou le développement.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ADYNOVI ne doit pas être mélangé avec d'autres médicaments.
Pour la reconstitution, utiliser uniquement l'eau pour préparations injectables stérilisée et le nécessaire de reconstitution fournis dans le coffret.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Stabilité après ouverture
La stabilité chimique et physique «in-use» a été démontrée pendant 3 heures à une température de 25 °C. Pour des raisons microbiologiques, le produit doit être utilisé immédiatement après la reconstitution. Si cela n'est pas possible, le délai d'utilisation et les conditions de stockage relèvent de la responsabilité de l'utilisateur, sauf si la reconstitution se déroule dans des conditions aseptiques contrôlées et validées. Ne pas mettre au réfrigérateur après la reconstitution.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C). Ne pas congeler. Pendant la durée de conservation, le produit peut être sorti du réfrigérateur, dans le cas d'un traitement ambulatoire, pour une période maximale de 3 mois avant emploi et à une température ne devant pas dépasser 30 °C. Après prélèvement du réfrigérateur, ADYNOVI ne peut pas être remis au réfrigérateur, mais doit être éliminé.
ADYNOVI avec le dispositif BAXJECT II: Conserver les flacons contenant le lyophilisat dans l'emballage extérieur à l'abri de la lumière.
ADYNOVI dans le système BAXJECT III: conserver la plaquette scellée dans l'emballage original à l'abri de la lumière.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
La solution reconstituée doit être inspectée visuellement pour mettre en évidence la présence de particules en suspension et/ou d'une coloration anormale.
Après la reconstitution, la solution est limpide ou légèrement opalescente.
N'utilisez pas de solutions troubles ou présentant des dépôts.
Après reconstitution, la solution est limpide, incolore et exempte de particules. Elle présente un pH de 6.7 à 7.3. L'osmolalité est ≥380 mOsmol/kg.
ADYNOVI est fourni ou avec le dispositif pour la reconstitution BAXJECT II ou avec un système BAXJECT III prêt à l'emploi dans une plaquette scellée (le flacon contenant le lyophilisat et le flacon contenant l'eau pour préparations injectables sont préassemblés avec le système pour reconstitution).
Reconstitution avec le dispositif BAXJECT II
Pour la reconstitution, utiliser uniquement l'eau pour préparations injectables stérilisée et le nécessaire de reconstitution fournis dans le coffret.
1.Respectez une méthode de travail aseptique et l'utilisation d'une surface de travail plane pendant la reconstitution
2.Amener les flacons d'ADYNOVI et de solvant (Aqua ad iniectabile) à température ambiante
3.Enlever les capsules de protection du flacon d'ADYNOVI et du solvant.
4.Nettoyez les bouchons en caoutchouc à l'aide de tampons imbibés d'alcool et laissez-les sécher avant l'utilisation.
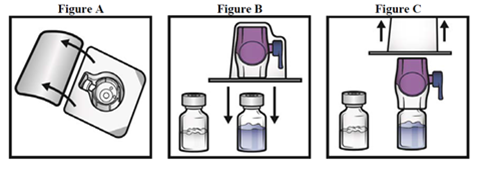
5.Ouvrez l'emballage du dispositif BAXJECT II en détachant la protection de papier sans toucher l'intérieur (fig. A). Ne sortez pas le dispositif de l'emballage.
6.Retournez l'emballage et insérez le perforateur en plastique transparent à travers le bouchon de solvant (fig. B).
7.Tenez l'emballage par les bords et retirez l'emballage de BAXJECT II (fig. C). Ne retirez pas le capuchon bleu du dispositif BAXJECT II. Ne touchez pas la pointe en plastique violet dégagée.
8.Tournez le système sur lui-même de sorte que le flacon de solvant se trouve en haut. Insérer vite la pointe en plastique violet dans le bouchon du flacon d'ADYNOVI en le pressant directement vers le bas (Fig. D). Le vide entraînera le solvant vers le flacon de poudre ADYNOVI
9.Agitez doucement jusqu'à ce que toute ADYNOVI soit dissout complètement. Ne réfrigérez pas la préparation après reconstitution
Reconstitution avec le système BAXJECT III
Ne pas utiliser si le l'opercule de la plaquette n'est pas complètement scellé
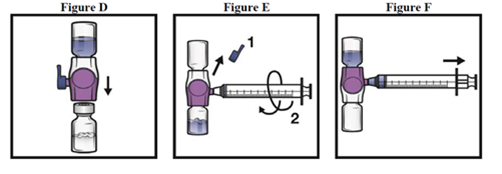
1.Laisser revenir la plaquette scellé (contient les flacons de lyophilisat et d'eau pour préparations injectables préassemblés avec le système pour reconstitution) à 15 °C – 25 °C.
2.Se laver soigneusement les mains à l'eau chaude et au savon.
3.Ouvrir la plaquette d'ADYNOVI en retirant l'opercule. Retirer le système BAXJECT III de la plaquette.
4.Placer ADYNOVI sur une surface plane avec le flacon de solvant en haut (Fig. 1). Le flacon de solvant porte une bande bleue. Ne retirer le capuchon bleu que lorsque vous serez invité à le faire, ultérieurement.
5.Tout en tenant ADYNOVI d'une main dans le système BAXJECT III, appuyer fermement sur le flacon de solvant de l'autre jusqu'à ce que le système soit entièrement replié et que le solvant s'écoule dans le flacon ADYNOVI (Fig. 2). N'incliner le système qu'une fois que le transfert est terminé.
6.Vérifier que le transfert de solvant est terminé. Agiter doucement jusqu'à ce que tout ADYNOVI soit dissoute, puisque autrement la substance efficace est retenue par le filtre. Le produit se dissout rapidement (en général en moins de 1 minute). Après reconstitution, la solution doit être limpide, incolore et exempte de particules.
Administration
Vérifiez l'absence de particules et de décoloration avant administration. La solution doit être limpide et incolore. N'utilisez pas des solutions présentant des particules ou de la décoloration. Le produit reconstitué doit être utilisé immédiatement, mais en aucun cas plus tard que 3 heures après la reconstitution.
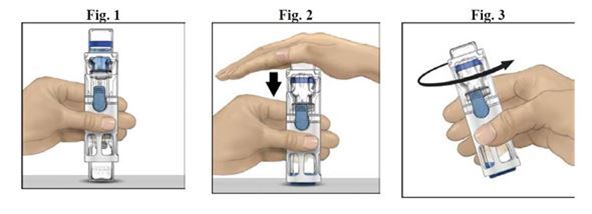
1.Retirez le capuchon bleu de BAXJECT II / BAXJECT III. Connectez la seringue à BAXJECT II / BAXJECT III (Fig. E). N'aspirez pas d'air.
2.Retournez le système – le flacon en verre contenant ADYNOVI se troue au-dessus. Aspirez la solution reconstituée dans la seringue en tirant lentement sur le piston (Fig. F).
3.Retirez la seringue, fixez une aiguille appropriée et commencez avec l'injection intraveineuse.Si plus d'un flacon d'ADYNOVI doit être injecté à un patient, la même seringue peut être utilisée pour ces flacons.Il faut faire attention au fait que BAXJECT II est prévu pour l'utilisation de seulement un flacon unique d'ADYNOVI et de solution. Pour cette raison il est nécessaire d'utiliser un deuxième dispositif BAXJECT II pour une autre reconstitution et une autre aspiration de la solution dans la seringue.
4.Administrer ADYNOVI sur une durée ne dépassant pas 5 minutes. La vitesse maximale d'injection est 10 ml/min.
Numéro d’autorisation65953 (Swissmedic)
PrésentationEmballages avec dispositif BAXJECT II
Chaque présentation d'ADYNOVI contient: 1 flacon contenant le lyophilisat, 1 flacon de solvant contenant d'aqua ad iniectabile et 1 BAXJECT II dispositif pour la reconstitution.
Emballages avec système BAXJECT III
Chaque présentation d'ADYNOVI contient: un système BAXJECT III prêt à l'emploi dans une plaquette scellée (le flacon de lyophilisat et le flacon contenant l'aqua ad iniectabile sont préassemblés avec le système pour reconstitution).
ADYNOVI 250 [B] Lyophilisat pour injection avec 250 UI de facteur VIII recombinant par flacon et un flacon de solvant contenant 2 ml d'aqua ad iniectabile.
ADYNOVI 250 [B] Lyophilisat pour injection avec 250 UI de facteur VIII recombinant par flacon et un flacon de solvant contenant 5 ml d'aqua ad iniectabile.
ADYNOVI 500 [B] Lyophilisat pour injection avec 500 UI de facteur VIII recombinant par flacon et un flacon de solvant contenant 2 ml d'aqua ad iniectabile.
ADYNOVI 500 [B] Lyophilisat pour injection avec 500 UI de facteur VIII recombinant par flacon et un flacon de solvant contenant 5 ml d'aqua ad iniectabile.
ADYNOVI 750 [B] Lyophilisat pour injection avec 750 UI de facteur VIII recombinant par flacon et un flacon de solvant contenant 2 ml d'aqua ad iniectabile.
ADYNOVI 750 [B] Lyophilisat pour injection avec 750 UI de facteur VIII recombinant par flacon et un flacon de solvant contenant 5 ml d'aqua ad iniectabile.
ADYNOVI 1000 [B] Lyophilisat pour injection avec 1000 UI de facteur VIII recombinant par flacon et un flacon de solvant contenant 2 ml d'aqua ad iniectabile.
ADYNOVI 1000 [B] Lyophilisat pour injection avec 1000 UI de facteur VIII recombinant par flacon et un flacon de solvant contenant 5 ml d'aqua ad iniectabile.
ADYNOVI 1500 [B] Lyophilisat pour injection avec 1500 UI de facteur VIII recombinant par flacon et un flacon de solvant contenant 2 ml d'aqua ad iniectabile.
ADYNOVI 1500 [B] Lyophilisat pour injection avec 1500 UI de facteur VIII recombinant par flacon et un flacon de solvant contenant 5 ml d'aqua ad iniectabile.
ADYNOVI 2000 [B] Lyophilisat pour injection avec 2000 UI de facteur VIII recombinant par flacon et un flacon de solvant contenant 5 ml d'aqua ad iniectabile.
ADYNOVI 3000 [B] Lyophilisat pour injection avec 3000 UI de facteur VIII recombinant par flacon et un flacon de solvant contenant 5 ml d'aqua ad iniectabile.
Chaque présentation d'ADYNOVI est livrée avec un set d'administration
1 mini nécessaire de perfusion
1 seringue stérile de 10 ml à usage unique
2 tampons d'alcool
2 pansements
Titulaire de l’autorisationTakeda Pharma AG, 8152 Opfikon
Mise à jour de l’informationFévrier 2024
|