CompositionPrincipes actifs
Ixazomib sous forme de citrate d'ixazomib.
Excipients
Contenu de la gélule: cellulose microcristalline, stéarate de magnésium, talc.
Enveloppe de la gélule: gélatine, dioxyde de titane (E 171), oxyde de fer (E 172) rouge (Ninlaro 2,3 mg et 4,0 mg), jaune (Ninlaro 4,0 mg), resp. noir (Ninlaro 3,0 mg).
Encre d'impression: gomme-laque, propylène glycol, hydroxyde de potassium, oxyde de fer noir (E 172), solution d'ammoniaque à 30%.
Indications/Possibilités d’emploiNINLARO en association avec le lénalidomide et la dexaméthasone est indiqué dans le traitement des patients adultes atteints de myélome multiple
·qui ont reçu au moins un traitement antérieur et présentent des caractéristiques à haut risque ou
·qui ont reçu au moins deux traitements antérieurs.
En dehors des études cliniques contrôlées, NINLARO n'est pas recommandé pour le traitement d'entretien du myélome multiple.
Voir rubrique «Propriétés/Effets».
Posologie/Mode d’emploiLe traitement doit être instauré et suivi sous la surveillance de médecins expérimentés dans le traitement du myélome multiple.
Posologie
NINLARO en association avec le lénalidomide et la dexaméthasone
La dose initiale recommandée de NINLARO est de 4 mg (une gélule), administrée par voie orale une fois par semaine aux jours 1, 8 et 15 d'un cycle de traitement de 28 jours.
La dose initiale recommandée de lénalidomide est de 25 mg, administrée une fois par jour aux jours 1 à 21 d'un cycle de traitement de 28 jours.
La dose initiale recommandée de dexaméthasone est de 40 mg, administrée aux jours 1, 8, 15 et 22 d'un cycle de traitement de 28 jours.
Schéma posologique: NINLARO en association avec le lénalidomide et la dexaméthasone
+ Prise du médicament
|
Cycle de 28 jours (cycle de 4 semaines)
| |
|
Semaine 1
|
Semaine 2
|
Semaine 3
|
Semaine 4
| |
|
Jour
1
|
Jours
2 à 7
|
Jour
8
|
Jours
9 à 14
|
Jour
15
|
Jours
16 à 21
|
Jour
22
|
Jours
23 à 28
| |
NINLARO
|
+
|
|
+
|
|
+
|
|
|
| |
Lénalidomide
|
+
|
+ Tous les jours
|
+
|
+ Tous les jours
|
+
|
+ Tous les jours
|
|
| |
Dexaméthasone
|
+
|
|
+
|
|
+
|
|
+
|
|
Pour de plus amples informations sur le lénalidomide et la dexaméthasone, voir les informations professionnelles correspondantes.
Avant de commencer un nouveau cycle de traitement:
·Le nombre absolu de neutrophiles doit être ≥1000/mm3
·Le nombre de plaquettes doit être ≥75'000/mm3
·Les toxicités non hématologiques doivent de manière générale être revenues à l'état initial du patient ou à un grade ≤1 selon l'appréciation du médecin.
Le traitement doit être poursuivi jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable.
Retard ou oubli de dose
En cas de retard ou d'oubli d'une dose de NINLARO, la dose ne doit être prise que si le délai jusqu'à la dose suivante prévue est ≥72 heures. La dose oubliée ne doit pas être prise si le délai jusqu'à la dose suivante prévue est inférieur à 72 heures. Le patient ne doit pas prendre deux doses pour compenser une dose oubliée.
En cas de vomissements après la prise d'une dose, le patient ne doit pas prendre une nouvelle dose, mais prendre la dose suivante au moment prévu.
Ajustements posologiques
Les paliers de réduction de la dose de NINLARO figurent dans le tableau 1. Les directives relatives à l'ajustement posologique sont mentionnées dans le tableau 2.
Tableau 1: Paliers de réduction de la dose de NINLARO
|
Dose initiale recommandée*
|
Première réduction à
|
Deuxième réduction à
|
Arrêt
| |
4 mg
|
3 mg
|
2,3 mg
|
* Dose initiale recommandée de 3 mg chez les patients atteints d'une insuffisance hépatique modérée à sévère, d'une insuffisance rénale sévère ou d'une insuffisance rénale terminale (IRT) nécessitant une dialyse.
Il est recommandé d'ajuster en alternance les doses de NINLARO et de lénalidomide en raison du chevauchement des toxicités de type thrombocytopénie, neutropénie et rash. En présence de ces toxicités, la première étape de l'ajustement posologique consiste à arrêter ou à réduire le lénalidomide. Concernant les paliers de réduction de la dose lors de ces toxicités, voir l'information professionnelle du lénalidomide, rubrique «Posologie/Mode d'emploi».
Tableau 2: Directives relatives à l'ajustement posologique de NINLARO en association avec le lénalidomide et la dexaméthasone
|
Toxicités hématologiques
|
Mesures recommandées
| |
Thrombocytopénie (nombre de plaquettes)
| |
Nombre de plaquettes <30'000/mm3
|
·Arrêter NINLARO et le lénalidomide, jusqu'à ce que le nombre de plaquettes soit ≥30'000/mm3.
·En cas de normalisation, reprendre le lénalidomide à la dose immédiatement inférieure selon l'information professionnelle de celui-ci et reprendre NINLARO à la dernière dose utilisée.
·Si le nombre de plaquettes baisse à nouveau à <30'000/mm3, arrêter NINLARO et le lénalidomide, jusqu'à ce que le nombre de plaquettes soit ≥30'000/mm3.
·En cas de normalisation, reprendre NINLARO à la dose immédiatement inférieure et le lénalidomide à la dernière dose utilisée.*
| |
Neutropénie (nombre de neutrophiles absolu)
| |
Nombre de neutrophiles absolu <500/mm3
|
·Arrêter NINLARO et le lénalidomide jusqu'à ce que le nombre de neutrophiles absolu soit ≥500/mm3.
·Conformément aux lignes directrices cliniques, il est aussi possible d'envisager l'ajout de G-CSF.
·Après le retour à la normale, poursuivre avec le lénalidomide à la dose inférieure la plus proche, conformément à l'information professionnelle correspondante, et avec NINLARO à la dernière dose.
·Si le nombre de neutrophiles absolu baisse à nouveau à <500/mm3, arrêter NINLARO et le lénalidomide jusqu'à ce que le nombre de neutrophiles absolu revienne à ≥500/mm3.
·Après le retour à la normale, poursuivre avec NINLARO à la dose inférieure la plus proche et avec le lénalidomide à la dernière dose.*
| |
Toxicités non hématologiques
|
Mesures recommandées
| |
Rash
| |
Grade† 2 ou 3
|
·Arrêter le lénalidomide jusqu'à ce que le rash soit de grade ≤1.
·Après le retour à la normale, reprendre le lénalidomide à la dose immédiatement inférieure selon l'information professionnelle de celui-ci.
·En cas de réapparition d'un rash de grade 2 ou 3, arrêter NINLARO et le lénalidomide jusqu'à ce que le rash soit de grade ≤1.
·Après le retour à la normale, reprendre NINLARO à la dose immédiatement inférieure et le lénalidomide à la dernière dose utilisée.*
| |
Grade 4
|
Arrêter le traitement.
| |
Autres toxicités non hématologiques
| |
Neuropathie périphérique de grade 1 avec des douleurs ou neuropathie périphérique de grade 2
|
·Arrêter NINLARO jusqu'à ce que la neuropathie périphérique soit de grade ≤1 sans douleur ou jusqu'au retour à l'état initial du patient.
·En cas de normalisation, reprendre NINLARO à la dernière dose utilisée.
| |
Neuropathie périphérique de grade 2 avec des douleurs ou neuropathie périphérique de grade 3
Autres toxicités non hématologiques de grades 3 ou 4
|
·Arrêter NINLARO. Avant de reprendre le traitement par NINLARO, les toxicités doivent de manière générale être revenues à l'état initial du patient ou à un grade ≤1, selon l'appréciation du médecin.
·En cas de normalisation, reprendre NINLARO à la dose immédiatement inférieure.
| |
Neuropathie périphérique de grade 4
|
Arrêter le traitement.
|
* En cas d'autres événements, ajuster en alternance les doses de lénalidomide et de NINLARO
† Classification reposant sur les critères de toxicité de l'Institut national du cancer des USA (CTCAE) version 4.03
Traitements concomitants
Chez les patients traités par NINLARO, il faut envisager une prophylaxie antivirale pour diminuer le risque de réactivation du zona. Dans les études portant sur l'ixazomib, la fréquence du zona était plus faible chez les patients ayant reçu une prophylaxie antivirale que chez les patients qui n'en avaient pas reçu.
Instructions spéciales pour la posologie
Patients âgés
Aucun ajustement de la dose de NINLARO n'est nécessaire chez les patients de plus de 65 ans selon les résultats d'une analyse pharmacocinétique de population. Dans les études portant sur NINLARO, aucune différence cliniquement significative concernant la sécurité et l'efficacité n'a été observée entre les patients de moins de 65 ans et ceux de plus de 65 ans.
Enfants et adolescents
La sécurité et l'efficacité de NINLARO pour les enfants et les adolescents de moins de 18 ans ne sont pas établies. Aucune donnée n'est disponible.
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la dose de NINLARO n'est nécessaire chez les patients atteints d'une insuffisance hépatique légère (bilirubine totale ≤ limite supérieure de la normale (LSN) et aspartate aminotransférase (ASAT) >LSN ou bilirubine totale >1-1,5 x LSN et tout taux d'ASAT), selon les résultats d'une analyse pharmacocinétique de population. Une dose initiale plus faible (3 mg) est recommandée chez les patients atteints d'une insuffisance hépatique modérée (bilirubine totale >1,5-3 x LSN) ou sévère (bilirubine totale >3 x LSN) selon les résultats d'une analyse pharmacocinétique de population (voir rubrique «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la dose de NINLARO n'est nécessaire chez les patients atteints d'une insuffisance rénale légère ou modérée (clairance de la créatinine ≥30 ml/min), selon les résultats d'une analyse pharmacocinétique de population. Une dose initiale plus faible (3 mg) est recommandée chez les patients atteints d'une insuffisance rénale sévère (clairance de la créatinine <30 ml/min) ou d'une insuffisance rénale terminale (IRT) nécessitant une dialyse, selon les résultats d'une analyse pharmacocinétique. NINLARO n'est pas dialysable et peut donc être pris indépendamment du moment de la dialyse (voir rubrique «Pharmacocinétique»).
Concernant les recommandations posologiques chez les patients atteints d'une insuffisance rénale, voir aussi l'information professionnelle du lénalidomide.
Origine ethnique
La sécurité et l'efficacité d'une dose initiale de 4 mg d'ixazomib en association avec le lénalidomide et la dexaméthasone ont été démontrées dans une étude clinique globale menée auprès de patients asiatiques. Les ASC de l'ixazomib ont été concordantes chez les patients asiatiques et chez les patients caucasiens; l'ASC moyenne était cependant supérieure de 35% chez les patients asiatiques. Une surveillance étroite de ces patients est recommandée. Voir paragraphe «Ajustements posologiques».
Mode d'administration
Voie orale.
NINLARO doit être pris à peu près à la même heure de la journée, aux jours 1, 8 et 15, et au moins 1 heure avant ou 2 heures après un repas (voir rubrique «Pharmacocinétique»). Les gélules doivent être avalées entières avec de l'eau. Les gélules ne doivent pas être broyées, mâchées ou ouvertes (voir rubrique «Remarques particulières, Remarques concernant la manipulation»).
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients selon la composition.
Mises en garde et précautionsThrombocytopénie
Des thrombocytopénies ont été observées au cours du traitement par NINLARO (voir rubrique «Effets indésirables»). Le nadir du nombre de plaquettes est généralement survenu entre le 14ème et le 21ème jour chaque cycle de 28 jours. Le taux était revenu au taux initial au début du cycle suivant. La thrombocytopénie n'a entraîné aucune augmentation des événements hémorragiques ou des transfusions de plaquettes.
La numération plaquettaire doit être contrôlée au moins une fois par mois au cours du traitement par NINLARO. Selon l'information professionnelle du lénalidomide, une surveillance plus fréquente doit être envisagée au cours des trois premiers cycles. Une thrombocytopénie peut être traitée par des ajustements posologiques (voir rubrique «Posologie/Mode d'emploi») et des transfusions de plaquettes, conformément aux directives médicales standard.
Toxicités gastro-intestinales
Des nausées, des vomissements et diarrhées ont été rapportés au cours du traitement par NINLARO (voir rubrique «Effets indésirables»). L'administration d'antiémétiques et d'antidiarrhéiques ainsi que des mesures de soutien ont occasionnellement été nécessaires. La dose doit être ajustée en cas de symptômes sévères (grades 3-4) (voir rubrique «Posologie/Mode d'emploi»).
Neuropathie périphérique
Des cas de neuropathie périphérique ont été rapportés chez des patients traités par NINLARO (voir rubrique «Effets indésirables»). Il convient de surveiller les patients afin de détecter tout symptôme de neuropathie. Un ajustement posologique peut s'avérer nécessaire en cas d'apparition ou d'aggravation d'une neuropathie périphérique chez un patient (voir rubrique «Posologie/Mode d'emploi»).
Œdèmes périphériques
Des œdèmes périphériques ont été rapportés sous traitement par NINLARO (voir rubrique «Effets indésirables»). Les causes sous-jacentes doivent être déterminées et des mesures de soutien doivent être mises en œuvre si nécessaire. Il convient d'ajuster la posologie de la dexaméthasone conformément à l'information professionnelle et d'ajuster la posologie de NINLARO conformément aux instructions posologiques en présence de symptômes de grades 3 ou 4 (voir rubrique «Posologie/Mode d'emploi»).
Réactions cutanées
Un rash a été rapporté sous NINLARO (voir rubrique «Effets indésirables»). Il doit être traité par un traitement adjuvant ou, s'il atteint un grade 2 ou plus, par un ajustement posologique (voir rubrique «Posologie/Mode d'emploi»). Des cas de syndrome de Stevens-Johnson et de nécrolyse épidermique toxique ont également été rapportés sous NINLARO, y compris des cas d'issue fatale (voir rubrique «Effets indésirables»). En cas de survenue d'un syndrome de Stevens-Johnson ou d'une nécrolyse épidermique toxique, NINLARO doit être arrêté.
Grossesse
Compte tenu du mécanisme d'action et des résultats des études chez l'animal, il est possible que l'ixazomib puisse être nocif pour le fœtus lorsqu'il est administré à une femme enceinte. Il convient de recommander aux femmes en âge de procréer d'éviter de tomber enceintes pendant le traitement par l'ixazomib. Si l'ixazomib est administré pendant la grossesse ou si une grossesse survient pendant le traitement par l'ixazomib, la patiente doit être informée des risques potentiels pour le fœtus. Les femmes en âge de procréer doivent être informées de la nécessité d'utiliser une méthode de contraception efficace pendant le traitement par l'ixazomib et pendant les 90 jours suivant la dernière dose. Les femmes utilisant des contraceptifs hormonaux doivent utiliser une méthode barrière de contraception supplémentaire. (Voir rubriques «Grossesse, Allaitement» et «Données précliniques»).
Inducteurs puissants du CYP3A
Les inducteurs puissants peuvent diminuer l'efficacité de NINLARO. L'administration concomitante d'inducteurs puissants du CYP3A tels que la carbamazépine, la phénytoïne, la rifampicine et le millepertuis (Hypericum perforatum) doit donc être évitée (voir «Interactions» et «Pharmacocinétique»). Si l'administration concomitante d'un inducteur puissant du CYP3A ne peut être évitée, il faut surveiller étroitement le patient afin d'assurer un bon contrôle de la maladie.
InteractionsInducteurs puissants du CYP3A
L'administration concomitante d'inducteurs puissants du CYP3A et de NINLARO n'est pas recommandée. Lors de l'administration concomitante de NINLARO et de rifampine, la Cmax de l'ixazomib a diminué 54% et l'ASC de 74%.
Inhibiteurs puissants du CYP3A
L'administration concomitante de NINLARO et de clarithromycine n'a pas entraîné de modification cliniquement significative de l'exposition systémique à l'ixazomib. La Cmax de l'ixazomib a diminué de 4% et l'ASC a augmenté de 11%. Dans une étude séparée menée avec le kétoconazole, dans laquelle un protocole à séquence fixe a été utilisé, les taux d'exposition à l'ixazomib ont été plus élevés lors de l'administration conjointe avec le kétoconazole durant la deuxième période. L'effet réel du kétoconazole n'a pas été évalué, car il a été faussé par un effet de période. La prudence est donc recommandée lors de l'association d'inhibiteurs puissants du CYP3A et de NINLARO.
En revanche, l'administration concomitante de NINLARO et de clarithromycine ne nécessite pas d'ajustement posologique.
Inhibiteurs puissants du CYP1A2
Aucun ajustement posologique n'est nécessaire lors de l'utilisation concomitante d'inhibiteurs puissants du CYP1A2 et de NINLARO. L'administration concomitante de NINLARO et d'inhibiteurs puissants du CYP1A2 n'a pas entraîné de modification cliniquement significative de l'exposition systémique à l'ixazomib selon les résultats d'une analyse pharmacocinétique de population.
Effets de NINLARO sur d'autres médicaments
NINLARO ne devrait pas provoquer d'interactions médicamenteuses liées à une inhibition ou à une induction du CYP.
L'ixazomib n'est pas un inhibiteur réversible ou temps-dépendant des CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6 ou 3A4/5. L'ixazomib n'a pas stimulé l'activité des CYP1A2, CYP2B6 et CYP3A4/5 ni des protéines immunoréactives correspondantes.
Interactions impliquant des transporteurs
NINLARO ne devrait pas provoquer d'interactions médicamenteuses médiées par les transporteurs. L'ixazomib est un substrat de la gp-P à faible affinité. L'ixazomib n'est pas un substrat de la BCRP, de la MRP2 et des OATP hépatiques. L'ixazomib n'est pas un inhibiteur de la gp-P, de la BCRP, de la MRP2, de l'OATP1B1, de l'OATP1B3, du transporteur cationique organique (OCT)2, ou des transporteurs anioniques organiques (OAT)1, OAT3, du MATE1 ou du MATE2-K.
Grossesse, allaitementGrossesse
Il n'existe aucune donnée chez l'être humain concernant les effets potentiels de l'ixazomib sur la grossesse et le développement embryonnaire ou fœtal. Les expérimentations animales sur le développement embryonnaire et fœtal ont cependant montré que l'ixazomib a un effet létal potentiel chez l'embryon et le fœtus (voir rubrique «Données précliniques»).
NINLARO n'est pas recommandé pendant la grossesse et chez les femmes en âge de procréer qui n'utilisent pas de méthode contraceptive.
Les hommes et les femmes en âge de procréer doivent utiliser des méthodes de contraception efficaces pendant le traitement et les 90 jours suivant l'arrêt de celui-ci.
Lorsque l'ixazomib est administré en association avec la dexaméthasone qui est connue pour être un inhibiteur faible à modéré du CYP3A4 et d'autres enzymes et protéines de transport, il convient de tenir compte du risque de réduction de l'efficacité des contraceptifs oraux. Les femmes qui utilisent des contraceptifs hormonaux oraux doivent utiliser une méthode barrière de contraception supplémentaire.
Allaitement
On ignore si NINLARO ou ses métabolites passent dans le lait maternel.
Un risque pour le nouveau-né/nourrisson ne peut pas être exclu.
Il est recommandé d'évaluer le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la mère et en conséquence, soit d'interrompre l'allaitement soit d'arrêter ou de s'abstenir du traitement par NINLARO.
Effet sur l’aptitude à la conduite et l’utilisation de machinesIl n'existe aucune donnée sur les effets sur l'aptitude à la conduite et l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
La population de sécurité de l'étude clinique de phase 3, randomisée, en double aveugle et contrôlée contre placebo a compris 720 patients atteints d'un myélome multiple récidivant et/ou réfractaire qui ont reçu soit NINLARO en association avec le lénalidomide et la dexaméthasone (groupe NINLARO; N=361) soit un placebo en association avec le lénalidomide et la dexaméthasone (groupe placebo; N=359).
Les effets indésirables les plus fréquemment rapportés (≥20%) dans le groupe NINLARO ont été: diarrhée, thrombocytopénie, constipation, neuropathie périphérique, nausées, infection des voies respiratoires supérieures, œdèmes périphériques, douleurs dorsales, rash, vomissements et bronchite. Les effets indésirables sévères observés chez ≥2% des patients ont été: diarrhée (3%), thrombocytopénie (2%) et bronchite (2%).
Effets indésirables
Les effets indésirables sont répertoriés ci-dessous par classes de systèmes d'organes et par fréquence. Les fréquences sont définies comme suit: «très fréquents» (≥1/10); «fréquents» (< 1/10, ≥1/100); «occasionnels» (<1/100, ≥1/1000); «rares» (<1/1000, ≥1/10'000); «très rares» (<1/10'000); fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Effets indésirables observés chez les patients traités par NINLARO en association avec le lénalidomide et la dexaméthasone (tous les grades, grade 3 et grade 4).
Infections et infestations
Très fréquents (tous les grades): infection des voies respiratoires supérieures (27%), bronchite (22%).
Fréquents (tous les grades): zona.
Fréquents (grade 3): infection des voies respiratoires supérieures, bronchite.
Occasionnels (grade 3): zona.
Affections hématologiques et du système lymphatique
Très fréquents (tous les grades): thrombocytopénie* (37%).
Très fréquents (grade 3): thrombocytopénie* (13%).
Fréquents (grade 4): thrombocytopénie*.
Affections du système immunitaire
Rares (tous les grades): réaction anaphylactique†.
Très rares (grade 3): réaction anaphylactique†.
Très rares (grade 4): réaction anaphylactique†.
Affections du système nerveux
Très fréquents (tous les grades): neuropathies périphériques* (32%).
Fréquents (grade 3): neuropathies périphériques*.
Affections gastro-intestinales
Très fréquents (tous les grades): diarrhée (52%), constipation (35%), nausées (32%), vomissements (26%).
Très fréquents (grade 3): diarrhée (10%).
Fréquents (grade 3): nausées, vomissements.
Occasionnels (grade 3): constipation
Affections de la peau et du tissu sous-cutané
Très fréquents (tous les grades): rash* (27%).
Fréquents (grade 3): rash*.
Rares (tous les grades): syndrome de Stevens-Johnson†, nécrolyse épidermique toxique†, angioœdème†.
Rares (grade 3): syndrome de Stevens-Johnson†, angioœdème†.
Rares (grade 4): nécrolyse épidermique toxique†.
Affections musculo-squelettiques et du tissu conjonctif
Très fréquents (tous les grades): douleurs dorsales (27%).
Occasionnels (grade 3): douleurs dorsales.
Troubles généraux et anomalies au site d'administration
Très fréquents (tous les grades): œdèmes périphériques (27%).
Fréquents (grade 3): œdèmes périphériques.
* Représente un résumé des termes préférentiels.
† Rapportés en dehors de l'étude de phase III.
Description d'effets indésirables sélectionnés
Rash
Un rash est survenu chez 27% des patients du groupe NINLARO. La plupart des événements de type rash étaient de grade 1 ou 2. Un rash de grade 3 a été observé chez 3% des patients du groupe NINLARO. Aucun événement de grade 4 de type rash n'a été observé. Des réactions indésirables sévères de type rash ont été observées chez <1% des patients du groupe NINLARO. Les types de rash les plus fréquemment observés étaient des exanthèmes maculopapuleux et maculeux. Un rash a entraîné l'arrêt d'au moins un des trois médicaments chez <1% des patients.
En dehors de l'étude de phase 3, l'effet indésirable sévère observé dans de rares cas, pour lequel le lien de causalité n'est pas établi, a été le suivant: dermatose neutrophile fébrile aiguë (syndrome de Sweet). De rares cas de syndrome de Stevens-Johnson, dont un d'issue fatale, ont également été rapportés sous NINLARO.
Des réactions dermatologiques ont été observées sous lénalidomide et dexaméthasone.
Neuropathie périphérique
La plupart des réactions indésirables de neuropathie périphérique étaient de grade 1 (18% dans le groupe NINLARO et 16% dans le groupe placebo) et de grade 2 (11% dans le groupe NINLARO et 6% dans le groupe placebo). Des réactions indésirables de type neuropathie périphérique de grade 3 ont été observées chez 2% des patients des deux groupes; aucun événement de grade 4 ni aucun effet indésirable sévère ne sont survenus.
La réaction la plus fréquemment rapportée a été une neuropathie sensitive périphérique (24% dans le groupe NINLARO et 17% dans le groupe placebo). Des neuropathies motrices périphériques ont été observées occasionnellement dans les deux groupes (<1%). Des neuropathies périphériques ont entraîné l'arrêt d'un ou de plusieurs des trois médicaments chez 4% des patients du groupe NINLARO et <1% des patients du groupe placebo.
Thrombocytopénie
Deux pour cent des patients du groupe NINLARO comme du groupe placebo ont présenté un nombre de thrombocytes ≤10 000/mm3 pendant le traitement. Moins de 1% des patients sous les deux traitements ont présenté un nombre de thrombocytes ≤5000/mm3 pendant le traitement. La thrombocytopénie a entraîné l'arrêt d'un ou de plusieurs des trois médicaments chez 2% des patients du groupe NINLARO et 3% des patients du groupe placebo.
Toxicité gastro-intestinale
La diarrhée a entraîné l'arrêt d'un ou de plusieurs des trois médicaments chez 3% des patients du groupe NINLARO et 2% des patients du groupe placebo.
Œdèmes périphériques
Des œdèmes périphériques ont été rapportés chez 27% des patients du groupe NINLARO et 21% des patients du groupe placebo. La plupart des réactions indésirables sous forme d'œdèmes périphériques étaient de grade 1 (17% dans le groupe NINLARO et 14% dans le groupe placebo) et de grade 2 (7% dans le groupe NINLARO et 6% dans le groupe placebo).
Des œdèmes périphériques de grade 3 ont été rapportés chez 2% des patients du groupe NINLARO et 1% des patients du groupe placebo. Aucun œdème périphérique de grade 4 n'a été rapporté. Des œdèmes périphériques ont entraîné l'arrêt d'un ou de plusieurs des trois médicaments chez <1% des patients des deux groupes.
Autres effets indésirables
En dehors de l'étude de phase 3, les événements indésirables sévères observés dans de rares cas, pour lesquels un lien de causalité n'est pas établi, ont été les suivants: myélite transverse, syndrome d'encéphalopathie postérieure réversible, syndrome de lyse tumorale et purpura thrombocytopénique thrombotique.
Effets indésirables après commercialisation
Les effets indésirables cliniquement notables sont cités ici s'ils ne sont pas décrits plus haut.
Microangiopathie thrombotique
Des cas de microangiopathie thrombotique (MAT), y compris de purpura thrombotique thrombocytopénique (PTT), ont été signalés chez des patients ayant reçu de l'ixazomib. Certains de ces cas ont connu une issue fatale. Il convient de surveiller la survenue des signes et symptômes d'une MAT. Si ce diagnostic est suspecté, arrêter l'ixazomib et examiner les patients afin de déceler une éventuelle MAT. Si l'examen exclut une MAT, l'administration d'ixazomib peut reprendre. L'innocuité de la reprise d'un traitement par l'ixazomib chez les patients qui ont déjà eu une MAT est inconnue.
Hépatotoxicité
Des lésions hépatiques d'origine médicamenteuse, des lésions hépatocellulaires, une stéatose hépatique, une hépatite cholestatique et une hépatotoxicité ont été signalées occasionnellement chez des patients traités par l'ixazomib (voir rubrique «Effets indésirables»). Les valeurs des enzymes hépatiques doivent faire l'objet d'une surveillance régulière et la dose doit être adaptée en cas de survenue de symptômes de degré 3 ou 4 (voir «Posologie/Mode d'emploi»).
Syndrome d'encéphalopathie postérieure réversible (SEPR)
Un syndrome d'encéphalopathie postérieure réversible (SEPR) est survenu chez des patients traités par l'ixazomib. Le SEPR est un trouble neurologique rare et réversible qui peut se manifester par des crises d'épilepsie, une hypertension artérielle, des céphalées, des modifications de l'état de conscience et des troubles visuels. Une tomographie cérébrale, de préférence par résonance magnétique, sert à confirmer le diagnostic. Le traitement par l'ixazomib doit être arrêté si le patient développe un SEPR.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDes cas surdosage ont été signalés chez des patients traités par NINLARO. Les symptômes de surdosage sont généralement compatibles avec les risques connus de NINLARO (voir «Effets indésirables»). Les rapports de surdosage accidentel ont été associés à des effets indésirables sévères comme des nausées sévères, la pneumonie par aspiration, une défaillance multiorganique et la mort.
Il n'existe pas d'antidote spécifique en cas de surdosage de NINLARO. En cas de surdosage, il convient de surveiller étroitement le patient à la recherche de réactions indésirables et mettre en œuvre des mesures de soutien appropriées.
Les professionnels de la santé doivent aviser les patients et les soignants de ne prendre qu'une seule dose de NINLARO à la fois et uniquement à l'intervalle prescrit (une gélule, une fois par semaine, aux jours 1, 8 et 15 de chaque cycle de 28 jours). Il y a lieu de discuter de l'importance de suivre scrupuleusement toutes les instructions de dosage avec les patients qui commencent le traitement.
Propriétés/EffetsCode ATC
L01XG03
Mécanisme d'action
Le citrate d'ixazomib, une prodrogue, est un principe actif qui, dans des conditions physiologiques, s'hydrolyse rapidement en ixazomib, sa forme biologiquement active. Le nom chimique du citrate d'ixazomib est l'acide 2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acétyl]amino]-3- méthylbutyl]-5-oxo-1,3,2- dioxaborolane-4,4-diacétique.
L'ixazomib est un inhibiteur hautement sélectif et réversible du protéasome, administré par voie orale. L'ixazomib se lie de façon préférentielle et inhibe l'activité chymotrypsique de la sous-unité bêta 5 du protéasome 20S.
In vitro, l'ixazomib a induit l'apoptose de différents types de cellules tumorales. In vitro, l'ixazomib a entraîné une cytotoxicité contre les cellules du myélome de patients ayant présenté une récidive après plusieurs traitements antérieurs, dont le bortézomib, le lénalidomide et la dexaméthasone. L'association d'ixazomib et de lénalidomide a eu des effets cytotoxiques synergiques dans plusieurs lignées de cellules myélomateuses. In vivo, l'ixazomib a présenté une activité antitumorale dans divers modèles de xénogreffe de tumeur, y compris des modèles de myélome multiple.
L'ixazomib altère aussi le micro-environnement médullaire. In vitro, l'ixazomib a inhibé la prolifération de cellules myélomateuses mises en coculture avec des cellules stromales de la moelle osseuse. L'ixazomib a présenté un effet antiangiogénique dans un test in vitro sur la formation de nouveaux capillaires. In vitro, l'ixazomib a stimulé l'ostéoblastogenèse et l'activité ostéoblastique et a inhibé l'ostéoclastogenèse et la résorption ostéoclastique. En outre, l'ixazomib a prévenu la perte osseuse dans un modèle murin de myélome multiple in vivo.
Pharmacodynamique
Electrophysiologie cardiaque
D'après les résultats d'une analyse pharmacocinétique et pharmacodynamique de données de 245 patients, NINLARO n'entraîne pas d'allongement de l'intervalle QTc à des expositions cliniquement significatives. A la dose de 4 mg, la variation moyenne du QTcF par rapport à la valeur initiale, déterminée selon l'analyse basée sur des modèles, a été estimée à 0,07 ms (IC à 90%; -0,22, 0,36).
Aucun lien perceptible entre la concentration d'ixazomib et l'intervalle RR n'a été établi, ce qui suggère que NINLARO n'a pas d'effet cliniquement significatif sur la fréquence cardiaque.
Efficacité clinique
L'efficacité et la sécurité de NINLARO en association avec le lénalidomide et la dexaméthasone ont été évaluées dans une étude de supériorité de phase 3, multicentrique, internationale, randomisée, en double aveugle, contrôlée contre placebo, menée chez des patients atteints d'un myélome multiple récidivant et/ou réfractaire et ayant reçu au moins un traitement antérieur.
Au total, 722 patients (population en intention de traiter [ITT]) ont été randomisés selon un ratio 1:1 afin de recevoir soit l'association NINLARO-lénalidomide-dexaméthasone (N=360; groupe NINLARO) soit l'association placebo-lénalidomide-dexaméthasone (N=362; groupe placebo) jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable. La randomisation a été stratifiée selon le nombre de lignes de traitement antérieures (1 versus 2 ou 3), le système international de stratification (International Myeloma Staging System, ISS) (stade I ou II versus III) et les traitements antérieurs par un inhibiteur du protéasome (patients exposés ou naïfs).
Les patients inclus dans l'étude avaient un myélome multiple qui était mesurable au moyen du taux de paraprotéine dans le sérum ou l'urine ou par le dosage des chaînes légères libres, étaient atteints d'une maladie réfractaire, y compris réfractaire primaire (c.-à-d. aucune réponse aux traitements antérieurs), avaient présenté une récidive après un traitement antérieur, ou une récidive et une maladie réfractaire à tous les traitements antérieurs.
Les patients ayant changé de traitement avant la progression de la maladie, de même que les patients présentant des maladies cardiovasculaires contrôlées ont également été inclus. Les patients qui étaient réfractaires au lénalidomide ou aux inhibiteurs du protéasome ou qui avaient reçu plus de trois traitements antérieurs ont été exclus de l'étude de phase 3. Comme les données disponibles chez ces patients sont limitées, une évaluation soigneuse du rapport bénéfice/risque est recommandée avant l'instauration du traitement.
Conformément à l'information professionnelle du lénalidomide, une prophylaxie antithrombotique a été recommandée pour tous les patients, dans les deux groupes de traitement. Des traitements associés tels qu'antiémétiques, antiviraux et antihistaminiques ont été administrés aux patients à titre prophylactique et/ou pour traiter leurs symptômes, selon l'appréciation du médecin.
Les patients ont reçu 4 mg de NINLARO ou un placebo aux jours 1, 8 et 15 plus le lénalidomide (25 mg) aux jours 1 à 21 et la dexaméthasone (40 mg) aux jours 1, 8, 15 et 22 d'un cycle de 28 jours. Les patients atteints d'une insuffisance rénale ont reçu une dose initiale de lénalidomide correspondant à celle figurant dans l'information professionnelle. Le traitement a été poursuivi jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable.
Les données démographiques initiales et les caractéristiques de la maladie étaient équilibrées et comparables entre les groupes de l'étude. L'âge médian était de 66 ans (intervalle: 38-91 ans); 58% des patients étaient âgés de plus de 65 ans. 57% des patients étaient des hommes. 85% des patients de la population étudiée étaient caucasiens, 9% asiatiques et 2% noirs. 93% des patients avaient un indice de performance ECOG de 0-1 et 12% avaient un stade ISS III au début de l'étude (N=90). 25% des patients avaient une clairance de la créatinine <60 ml/min. 23% des patients présentaient un myélome à chaînes légères et 12% des patients une maladie mesurable uniquement par le dosage des chaînes légères libres. 43% des patients de la population totale (N=309) avaient un risque cytogénétique accru (risque élevé [del(17), t(4;14), t(14;16)] ou amplification 1q [1q21]). 19% (N=137) des patients présentaient des anomalies cytogénétiques à haut risque et 10% (N=69) étaient porteurs de la délétion del(17). Les patients avaient reçu une à trois lignes de traitement antérieures (médiane: 1) incluant un traitement antérieur par le bortézomib (69%), le carfilzomib (<1%), le thalidomide (45%), le lénalidomide (12%) et le melphalan (81%). 57% des patients avaient reçu préalablement une greffe de cellules souches. 77% des patients avaient présenté une récidive après les lignes de traitement antérieures et 11% des patients étaient réfractaires aux lignes de traitement antérieures. Une maladie réfractaire primaire (c.-à-d. que la meilleure réponse à toutes les lignes de traitement antérieures était une maladie stable ou une progression de la maladie) a été documentée chez 6% des patients.
Le critère d'évaluation principal était la survie sans progression (PFS) établie conformément au consensus de l'International Myeloma Working Group 2011 (IMWG) sur les critères de réponse uniforme, après évaluation en aveugle par un comité d'experts indépendants (IRC) sur la base des résultats d'un laboratoire central. La réponse a été évaluée toutes les 4 semaines jusqu'à la progression de la maladie.
Lors des analyses primaire et finale réalisées pour l'évaluation statistique (période de suivi médiane de 14,7 mois et un nombre médian de 13 cycles), des résultats significativement meilleurs ont été mis en évidence dans le groupe traité par NINLARO, la PFS médiane ayant augmenté d'environ 6 mois. 129 (36%) événements de PFS sont survenus dans le groupe NINLARO et 157 (43%) dans le groupe placebo. La PFS médiane a été de 20,6 mois dans le groupe NINLARO et de 14,7 mois dans le groupe placebo (HR=0,74 [IC à 95% (0,587, 0,939)], p=0,012).
Remarque: Le hazard ratio repose sur le modèle de régression de Cox stratifié; un hazard ratio inférieur à 1 indique une supériorité du traitement par NINLARO. La valeur de p repose sur le test du log-rank stratifié.
L'amélioration de la PFS dans le groupe NINLARO a été appuyée par des améliorations du taux de réponse globale.
Après un suivi médian de 23 mois, une analyse intermédiaire planifiée de la survie globale (OS) a été réalisée avec 35% du nombre de décès requis pour l'analyse finale de l'OS dans la population ITT. 81 décès sont survenus dans le groupe NINLARO et 90 dans le groupe placebo. L'OS médiane n'a été atteinte dans aucun des deux bras de traitement. Une analyse exploratoire noninférentielle de la PFS a parallèlement été réalisée. La PFS médiane estimée dans la population ITT a été de 20 mois dans le bras NINLARO et de 15,9 mois dans le bras placebo (HR=0,82 [IC à 95% (0,67, 1,0)]).
Lors de l'analyse finale de l'OS au bout d'une période de suivi médiane d'env. 85 mois, l'OS médiane dans la population ITT s'élevait à 53,6 mois pour les patients du groupe NINLARO et 51,6 mois pour les patients du groupe placebo (HR=0,94 [IC à 95% (0,78, 1,13)]).
Lors de l'analyse intermédiaire de l'OS dans le sous-groupe des patients à haut risque dont le myélome multiple a été analysé de manière centralisée dans un laboratoire certifié CLIA à la recherche d'une délétion du chromosome 17 (del[17]), moins de décès sont survenus chez les patients porteurs de la délétion del(17) dans le bras NINLARO (11,1%) que dans le bras placebo (27,3%). Lors de l'analyse finale, l'OS médiane dans cette sous-population était de 42,2 mois pour les patients du groupe NINLARO et 29,4 mois pour les patients du groupe placebo (HR=0,92 [IC à 95% (0,52, 1,63)]).
Chez les patients présentant un risque cytogénétique élevé, l'OS médiane dans l'analyse finale était de 46,9 mois pour le groupe NINLARO et 30,9 mois dans le groupe placebo (HR=0,87 [IC à 95% (0,58, 1,31)]) et chez les patients présentant un risque cytogénétique accru, l'OS médiane était de 44,6 mois pour le groupe NINLARO et 33,4 mois dans le groupe placebo (HR=0,86 [IC à 95% (0,66, 1,12)]).
Le myélome multiple étant une maladie hétérogène, le bénéfice peut varier dans les différents sous-groupes (voir Figure 1).
Figure 1: Forest plot de la survie sans progression dans les sous-groupes
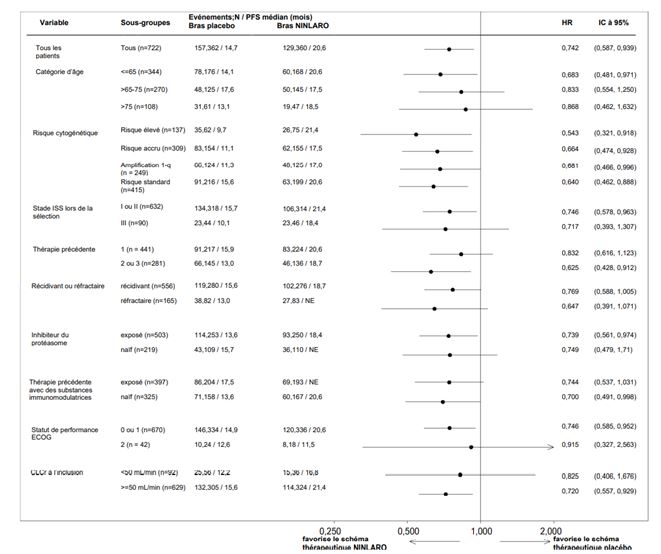
Population indiquée
Une analyse a été réalisée pour déterminer les résultats d'efficacité clinique à l'aide de caractéristiques de risque cytogénétique fixées et de caractéristiques de risque clinique défavorable stratifiées. Dans la population ITT, une amélioration plus importante des résultats d'efficacité a été observée chez 70% des patients (N=505, «population indiquée») qui avaient reçu au moins un traitement antérieur et présentaient des caractéristiques à haut risque (définies comme un risque cytogénétique accru [risque élevé (del[17], t[4;14], t[14;16]) ou 1q21] ou stade ISS III) ou qui avaient reçu au moins deux traitements antérieurs. Lors de l'analyse primaire, 98 (39%) événements de PFS sont survenus dans le bras NINLARO et 122 (49%) dans le bras placebo. La PFS médiane a été de 18,4 mois dans le bras NINLARO et de 12,2 mois dans le bras placebo (HR=0,681 [IC à 95% (0,520, 0,892)], p=0,005).
L'amélioration de la PFS dans le bras NINLARO a été appuyée par des améliorations du taux de réponse global dans la population indiquée.
Au moment de l'analyse intermédiaire planifiée de l'OS, 58 décès étaient survenus dans le bras NINLARO et 76 décès dans le bras placebo, dans la population indiquée. L'OS médiane n'a pas été atteinte dans le bras NINLARO et a été de 30,9 mois dans le bras placebo (HR=0,706 [IC À 95% (0,499, 0,997)]). Les résultats de l'analyse exploratoire noninférentielle de la PFS ont montré dans la population indiquée une PFS médiane estimée de 19,1 mois dans le bras NINLARO et de 12,6 mois dans le bras placebo (HR=0,728 [IC à 95% (0,573, 0,924)]). Les résultats sont concordants avec la conclusion de l'analyse primaire de la PFS, à savoir que le traitement a un effet positif.
Au moment de l'analyse finale de l'OS, 181 décès ont été dénombrés dans la population indiquée dans le groupe NINLARO et 179 décès dans le groupe placebo. L'OS médiane était de 50,4 mois dans le groupe NINLARO et 42,0 mois dans le groupe placebo (HR=0,912 [IC à 95% (0,74, 1,125)]), p=0,005).
Pédiatrie
L'Agence européenne des médicaments a supprimé l'obligation de soumettre les résultats des études réalisées avec NINLARO dans tous les sous-groupes de la population pédiatrique (voir rubrique «Posologie/Mode d'emploi, Enfants et adolescents»).
PharmacocinétiqueAbsorption
Les concentrations plasmatiques maximales de l'ixazomib ont été atteintes environ une heure après l'administration orale. La biodisponibilité absolue moyenne est de 58%. L'ASC de l'ixazomib augmente proportionnellement à la dose dans un intervalle posologique compris entre 0,2 et 10,6 mg.
La prise avec un repas riche en graisses a réduit de 28% l'ASC de l'ixazomib par rapport à l'administration à jeun (voir rubrique «Posologie/Mode d'emploi»).
Distribution
L'ixazomib se lie à 99% aux protéines plasmatiques et est distribué dans les globules rouges selon un rapport ASC sang/plasma de 10. Le volume de distribution à l'état d'équilibre est de 543 l.
Métabolisme
Pas de données disponibles.
Élimination
L'ixazomib présente un profil d'élimination multiexponentiel. Selon une analyse pharmacocinétique de population, la clairance (CL) systémique était d'environ 1,86 l/h, avec une variabilité interindividuelle de 44%. La demi-vie des phases d'élimination (terminale) alpha, bêta et gamma du profil pharmacocinétique était de 0,272 heure, 17,8 heures et 9,5 jours.
Lors d'une administration orale hebdomadaire, une accumulation environ 2 fois plus élevée de l'ASC a été observée au jour 15.
Biotransformation
Après administration orale d'une dose radiomarquée, 70% de la substance liée au médicament ixazomib était présente dans le plasma. L'ixazomib est métabolisé en plusieurs métabolites déboronés qui sont pharmacologiquement inactifs et sont éliminés dans l'urine. Le métabolisme par plusieurs enzymes du cytochrome (CYP) et des protéines non-CYP devrait être le principal mécanisme de clairance de l'ixazomib. Des études in vitro réalisées avec des isoenzymes du cytochrome P450 issues de l'ADNc humain, menées à des concentrations cliniquement significatives d'ixazomib, indiquent qu'aucune isoenzyme spécifique du CYP ne contribue de façon prédominante au métabolisme de l'ixazomib et que des protéines non-CYP contribuent au métabolisme global.
Excrétion
Après administration orale d'une dose unique de 14C-ixazomib à 5 patients atteints d'un cancer avancé, 62% de la radioactivité administrée a été excrétée dans l'urine et 22% dans les selles. L'ixazomib sous forme inchangée a représenté <3,5% de la dose administrée retrouvée dans l'urine.
Cinétique pour certains groupes de patients
Insuffisance hépatique
Selon les résultats d'une analyse pharmacocinétique de population, les propriétés pharmacocinétiques de l'ixazomib sont comparables chez les patients présentant une fonction hépatique normale et chez les patients atteints d'une insuffisance hépatique légère (bilirubine totale ≤LSN et ASAT > LSN ou bilirubine totale >1-1,5 x LSN et tout taux d'ASAT).
Les propriétés pharmacocinétiques de l'ixazomib ont été évaluées à la dose de 4 mg chez les patients présentant une fonction hépatique normale (N=12), à la dose de 2,3 mg en cas d'insuffisance hépatique modérée (bilirubine totale >1,5-3 x LSN, N=13) et à la dose de 1,5 mg en cas d'insuffisance hépatique sévère (bilirubine totale >3 x LSN, N=18). L'ASC libre normalisée en fonction de la dose était supérieure de 27% chez les patients atteints d'insuffisance hépatique modérée ou sévère par rapport aux patients présentant une fonction hépatique normale (voir rubrique «Posologie/Mode d'emploi»).
Insuffisance rénale
Selon les résultats d'une analyse pharmacocinétique de population, les propriétés pharmacocinétiques de l'ixazomib sont comparables chez les patients présentant une fonction rénale normale et chez les patients atteints d'une insuffisance rénale légère ou modérée (clairance de la créatinine ≥30 ml/min).
Les propriétés pharmacocinétiques de l'ixazomib ont été évaluées à la dose de 3 mg chez des patients présentant une fonction rénale normale (clairance de la créatinine ≥90 ml/min, N=18), une insuffisance rénale sévère (clairance de la créatinine <30 ml/min, N=14) ou une insuffisance rénale terminale (IRT) nécessitant une dialyse (N=6). L'ASC libre était supérieure de 38% chez les patients atteints d'une insuffisance rénale sévère ou d'une IRT nécessitant une dialyse par rapport aux patients présentant une fonction rénale normale. Les concentrations prédialyse et postdialyse mesurées au cours de l'hémodialyse étaient comparables, ce qui indique que l'ixazomib n'est pas dialysable (voir rubrique «Posologie/Mode d'emploi»).
Sexe, âge
Selon les résultats d'une analyse pharmacocinétique de population, l'âge (23-91 ans), le sexe ou la surface corporelle (1,2-2,7 m2) n'ont eu aucun effet cliniquement significatif sur la clairance de l'ixazomib.
Données précliniquesCarcinogenèse et mutagenèse
L'ixazomib ne s'est avéré ni mutagène dans un test de mutation bactérienne inverse (test d'Ames) ni clastogène dans un test du micronoyau sur moelle osseuse de souris. L'ixazomib s'est avéré positif au test de clastogénicité in vitro sur des lymphocytes de sang humain périphérique. D'autre part, l'ixazomib s'est avéré négatif au test des comètes in vivo chez la souris, lors duquel la proportion d'ADN présent dans la queue a été mesurée dans l'estomac et le foie. Les données probantes permettent donc de conclure que NINLARO n'est pas associé à un risque génotoxique. Aucune étude de carcinogénicité n'a été réalisée avec NINLARO.
Reproduction et développement embryonnaire et fœtal
Les études de toxicité sur le développement, menées chez le rat et le lapin, n'ont pas montré de toxicité embryonnaire et fœtale directe en dessous des doses d'ixazomib toxiques pour la mère. Dans des études de recherche de doses menées chez des rates (0,6 mg/kg; 3,6 mg/m2) et des lapines (1,0 mg/kg; 12 mg/m2) gestantes, une réduction du poids corporel des fœtus, une tendance à une plus faible viabilité fœtale et/ou une augmentation des pertes postimplantatoires ont été observées; ces événements n'ont cependant pas été reproduits clairement dans les études définitives et n'ont été observés qu'à des doses toxiques pour la mère (des doses qui ont entraîné une réduction du poids corporel et/ou de la prise alimentaire). Dans l'étude définitive menée chez le lapin, une augmentation de la fréquence des modifications/anomalies du squelette fœtal (vertèbres caudales, nombre de vertèbres lombaires et côtes surnuméraires complètes), également associées à la toxicité maternelle, a été observée à des doses ≥0,3 mg/kg (3,6 mg/m2). Aux doses également toxiques pour la mère, l'ixazomib doit être considéré comme toxique pour l'embryon et le fœtus. Une dose de 0,1 mg/kg (1,2 mg/m2) n'a pas induit de toxicité maternelle ni d'effets embryonnaires et fœtaux.
Aucune étude sur la fertilité, le développement embryonnaire précoce et la toxicité prénatale et postnatale n'a été menée avec l'ixazomib. Mais une évaluation des tissus reproducteurs a été réalisée dans les études de toxicité générale. Le traitement par l'ixazomib n'a eu aucun effet sur les organes reproducteurs mâles et femelles dans des études d'une durée maximale de 6 mois chez le rat et de 9 mois chez le chien.
Propriétés toxicologiques et/ou pharmacologiques chez l'animal
Dans des études de toxicité générale conduites sur plusieurs cycles chez le rat et le chien, les principaux organes cibles étaient le tractus gastro-intestinal (GI), le tissu lymphoïde et le système nerveux. Les effets GI comportaient des vomissements et/ou diarrhées, une augmentation des leucocytes et des modifications microscopiques (inflammation, hyperplasie épithéliale, infiltration neutrophile, nécrose de cellules isolées et érosion/ulcération). La toxicité sur le système lymphatique était caractérisée par une déplétion/nécrose lymphoïde (y compris de la moelle osseuse), une infiltration neutrophile et une nécrose de cellules isolées. Des effets sur le système nerveux sont survenus essentiellement chez le chien à des doses ≥0,1 mg/kg (2 mg/m2) et ont compris des modifications microscopiques d'une dégénérescence neuronale minime à légère des ganglions sympathiques, spinaux, autonomes périphériques (glandes salivaires) et des organes terminaux, et une dégénérescence axonale secondaire minime des nerfs périphériques et des faisceaux ascendants dans les cornes postérieures de la moelle épinière (neurotoxicité). Dans l'étude de 9 mois (comprenant 10 cycles) menée chez le chien, dans laquelle le schéma posologique correspondait au schéma clinique (cycle de 28 jours), les effets neuronaux microscopiques ont généralement été minimes et n'ont été observés qu'à la dose de 0,2 mg/kg (4 mg/m2; ASC168 1940 h*ng/ml). La majorité des modifications des organes cibles ont été partiellement ou totalement réversibles à l'arrêt du traitement, à l'exception des modifications neuronales des ganglions spinaux lombaires et de la corne postérieure. L'absence de dégénérescence neuronale progressive dans les ganglions périphériques et la présence de modifications seulement périphériques secondaires dans les fibres nerveuses et les axones indiquent l'absence de toxicité persistante.
D'après les expérimentations animales, l'ixazomib ne traverse pas la barrière hémato-encéphalique. En outre, des études précliniques et des études sur la pharmacologie de sécurité in vitro (sur les canaux hERG) et in vivo ont montré que l'ixazomib n'a pas d'effet sur la fonction cardiovasculaire.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver hors de portée des enfants. Ne pas conserver au-dessus de 30 °C. Protéger de l'humidité. Ne pas congeler.
La gélule ne doit être sortie de la plaquette qu'immédiatement avant son utilisation.
Remarques concernant la manipulation
NINLARO est cytotoxique. Les gélules ne doivent pas être ouvertes ou broyées. Eviter tout contact direct avec le contenu de la gélule. En cas de rupture de la gélule, éviter de soulever la poussière lors du nettoyage. En cas de contact, laver abondamment avec de l'eau et du savon.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation locale en vigueur.
Numéro d’autorisation65959 (Swissmedic).
PrésentationNINLARO 4 mg: emballage multiple de 3 plaquettes en étui portefeuille contenant chacune une gélule (A).
NINLARO 3 mg: emballage multiple de 3 plaquettes en étui portefeuille contenant chacune une gélule (A).
NINLARO 2.3 mg: emballage multiple avec 3 plaquettes en étui portefeuille contenant chacune une gélule (A).
Titulaire de l’autorisationTakeda Pharma AG, 8152 Opfikon
Mise à jour de l’informationFévrier 2024
|