Propriétés/EffetsCode ATC
L01XG03
Mécanisme d'action
Le citrate d'ixazomib, une prodrogue, est un principe actif qui, dans des conditions physiologiques, s'hydrolyse rapidement en ixazomib, sa forme biologiquement active. Le nom chimique du citrate d'ixazomib est l'acide 2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acétyl]amino]-3- méthylbutyl]-5-oxo-1,3,2- dioxaborolane-4,4-diacétique.
L'ixazomib est un inhibiteur hautement sélectif et réversible du protéasome, administré par voie orale. L'ixazomib se lie de façon préférentielle et inhibe l'activité chymotrypsique de la sous-unité bêta 5 du protéasome 20S.
In vitro, l'ixazomib a induit l'apoptose de différents types de cellules tumorales. In vitro, l'ixazomib a entraîné une cytotoxicité contre les cellules du myélome de patients ayant présenté une récidive après plusieurs traitements antérieurs, dont le bortézomib, le lénalidomide et la dexaméthasone. L'association d'ixazomib et de lénalidomide a eu des effets cytotoxiques synergiques dans plusieurs lignées de cellules myélomateuses. In vivo, l'ixazomib a présenté une activité antitumorale dans divers modèles de xénogreffe de tumeur, y compris des modèles de myélome multiple.
L'ixazomib altère aussi le micro-environnement médullaire. In vitro, l'ixazomib a inhibé la prolifération de cellules myélomateuses mises en coculture avec des cellules stromales de la moelle osseuse. L'ixazomib a présenté un effet antiangiogénique dans un test in vitro sur la formation de nouveaux capillaires. In vitro, l'ixazomib a stimulé l'ostéoblastogenèse et l'activité ostéoblastique et a inhibé l'ostéoclastogenèse et la résorption ostéoclastique. En outre, l'ixazomib a prévenu la perte osseuse dans un modèle murin de myélome multiple in vivo.
Pharmacodynamique
Electrophysiologie cardiaque
D'après les résultats d'une analyse pharmacocinétique et pharmacodynamique de données de 245 patients, NINLARO n'entraîne pas d'allongement de l'intervalle QTc à des expositions cliniquement significatives. A la dose de 4 mg, la variation moyenne du QTcF par rapport à la valeur initiale, déterminée selon l'analyse basée sur des modèles, a été estimée à 0,07 ms (IC à 90%; -0,22, 0,36).
Aucun lien perceptible entre la concentration d'ixazomib et l'intervalle RR n'a été établi, ce qui suggère que NINLARO n'a pas d'effet cliniquement significatif sur la fréquence cardiaque.
Efficacité clinique
L'efficacité et la sécurité de NINLARO en association avec le lénalidomide et la dexaméthasone ont été évaluées dans une étude de supériorité de phase 3, multicentrique, internationale, randomisée, en double aveugle, contrôlée contre placebo, menée chez des patients atteints d'un myélome multiple récidivant et/ou réfractaire et ayant reçu au moins un traitement antérieur.
Au total, 722 patients (population en intention de traiter [ITT]) ont été randomisés selon un ratio 1:1 afin de recevoir soit l'association NINLARO-lénalidomide-dexaméthasone (N=360; groupe NINLARO) soit l'association placebo-lénalidomide-dexaméthasone (N=362; groupe placebo) jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable. La randomisation a été stratifiée selon le nombre de lignes de traitement antérieures (1 versus 2 ou 3), le système international de stratification (International Myeloma Staging System, ISS) (stade I ou II versus III) et les traitements antérieurs par un inhibiteur du protéasome (patients exposés ou naïfs).
Les patients inclus dans l'étude avaient un myélome multiple qui était mesurable au moyen du taux de paraprotéine dans le sérum ou l'urine ou par le dosage des chaînes légères libres, étaient atteints d'une maladie réfractaire, y compris réfractaire primaire (c.-à-d. aucune réponse aux traitements antérieurs), avaient présenté une récidive après un traitement antérieur, ou une récidive et une maladie réfractaire à tous les traitements antérieurs.
Les patients ayant changé de traitement avant la progression de la maladie, de même que les patients présentant des maladies cardiovasculaires contrôlées ont également été inclus. Les patients qui étaient réfractaires au lénalidomide ou aux inhibiteurs du protéasome ou qui avaient reçu plus de trois traitements antérieurs ont été exclus de l'étude de phase 3. Comme les données disponibles chez ces patients sont limitées, une évaluation soigneuse du rapport bénéfice/risque est recommandée avant l'instauration du traitement.
Conformément à l'information professionnelle du lénalidomide, une prophylaxie antithrombotique a été recommandée pour tous les patients, dans les deux groupes de traitement. Des traitements associés tels qu'antiémétiques, antiviraux et antihistaminiques ont été administrés aux patients à titre prophylactique et/ou pour traiter leurs symptômes, selon l'appréciation du médecin.
Les patients ont reçu 4 mg de NINLARO ou un placebo aux jours 1, 8 et 15 plus le lénalidomide (25 mg) aux jours 1 à 21 et la dexaméthasone (40 mg) aux jours 1, 8, 15 et 22 d'un cycle de 28 jours. Les patients atteints d'une insuffisance rénale ont reçu une dose initiale de lénalidomide correspondant à celle figurant dans l'information professionnelle. Le traitement a été poursuivi jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable.
Les données démographiques initiales et les caractéristiques de la maladie étaient équilibrées et comparables entre les groupes de l'étude. L'âge médian était de 66 ans (intervalle: 38-91 ans); 58% des patients étaient âgés de plus de 65 ans. 57% des patients étaient des hommes. 85% des patients de la population étudiée étaient caucasiens, 9% asiatiques et 2% noirs. 93% des patients avaient un indice de performance ECOG de 0-1 et 12% avaient un stade ISS III au début de l'étude (N=90). 25% des patients avaient une clairance de la créatinine <60 ml/min. 23% des patients présentaient un myélome à chaînes légères et 12% des patients une maladie mesurable uniquement par le dosage des chaînes légères libres. 43% des patients de la population totale (N=309) avaient un risque cytogénétique accru (risque élevé [del(17), t(4;14), t(14;16)] ou amplification 1q [1q21]). 19% (N=137) des patients présentaient des anomalies cytogénétiques à haut risque et 10% (N=69) étaient porteurs de la délétion del(17). Les patients avaient reçu une à trois lignes de traitement antérieures (médiane: 1) incluant un traitement antérieur par le bortézomib (69%), le carfilzomib (<1%), le thalidomide (45%), le lénalidomide (12%) et le melphalan (81%). 57% des patients avaient reçu préalablement une greffe de cellules souches. 77% des patients avaient présenté une récidive après les lignes de traitement antérieures et 11% des patients étaient réfractaires aux lignes de traitement antérieures. Une maladie réfractaire primaire (c.-à-d. que la meilleure réponse à toutes les lignes de traitement antérieures était une maladie stable ou une progression de la maladie) a été documentée chez 6% des patients.
Le critère d'évaluation principal était la survie sans progression (PFS) établie conformément au consensus de l'International Myeloma Working Group 2011 (IMWG) sur les critères de réponse uniforme, après évaluation en aveugle par un comité d'experts indépendants (IRC) sur la base des résultats d'un laboratoire central. La réponse a été évaluée toutes les 4 semaines jusqu'à la progression de la maladie.
Lors des analyses primaire et finale réalisées pour l'évaluation statistique (période de suivi médiane de 14,7 mois et un nombre médian de 13 cycles), des résultats significativement meilleurs ont été mis en évidence dans le groupe traité par NINLARO, la PFS médiane ayant augmenté d'environ 6 mois. 129 (36%) événements de PFS sont survenus dans le groupe NINLARO et 157 (43%) dans le groupe placebo. La PFS médiane a été de 20,6 mois dans le groupe NINLARO et de 14,7 mois dans le groupe placebo (HR=0,74 [IC à 95% (0,587, 0,939)], p=0,012).
Remarque: Le hazard ratio repose sur le modèle de régression de Cox stratifié; un hazard ratio inférieur à 1 indique une supériorité du traitement par NINLARO. La valeur de p repose sur le test du log-rank stratifié.
L'amélioration de la PFS dans le groupe NINLARO a été appuyée par des améliorations du taux de réponse globale.
Après un suivi médian de 23 mois, une analyse intermédiaire planifiée de la survie globale (OS) a été réalisée avec 35% du nombre de décès requis pour l'analyse finale de l'OS dans la population ITT. 81 décès sont survenus dans le groupe NINLARO et 90 dans le groupe placebo. L'OS médiane n'a été atteinte dans aucun des deux bras de traitement. Une analyse exploratoire noninférentielle de la PFS a parallèlement été réalisée. La PFS médiane estimée dans la population ITT a été de 20 mois dans le bras NINLARO et de 15,9 mois dans le bras placebo (HR=0,82 [IC à 95% (0,67, 1,0)]).
Lors de l'analyse finale de l'OS au bout d'une période de suivi médiane d'env. 85 mois, l'OS médiane dans la population ITT s'élevait à 53,6 mois pour les patients du groupe NINLARO et 51,6 mois pour les patients du groupe placebo (HR=0,94 [IC à 95% (0,78, 1,13)]).
Lors de l'analyse intermédiaire de l'OS dans le sous-groupe des patients à haut risque dont le myélome multiple a été analysé de manière centralisée dans un laboratoire certifié CLIA à la recherche d'une délétion du chromosome 17 (del[17]), moins de décès sont survenus chez les patients porteurs de la délétion del(17) dans le bras NINLARO (11,1%) que dans le bras placebo (27,3%). Lors de l'analyse finale, l'OS médiane dans cette sous-population était de 42,2 mois pour les patients du groupe NINLARO et 29,4 mois pour les patients du groupe placebo (HR=0,92 [IC à 95% (0,52, 1,63)]).
Chez les patients présentant un risque cytogénétique élevé, l'OS médiane dans l'analyse finale était de 46,9 mois pour le groupe NINLARO et 30,9 mois dans le groupe placebo (HR=0,87 [IC à 95% (0,58, 1,31)]) et chez les patients présentant un risque cytogénétique accru, l'OS médiane était de 44,6 mois pour le groupe NINLARO et 33,4 mois dans le groupe placebo (HR=0,86 [IC à 95% (0,66, 1,12)]).
Le myélome multiple étant une maladie hétérogène, le bénéfice peut varier dans les différents sous-groupes (voir Figure 1).
Figure 1: Forest plot de la survie sans progression dans les sous-groupes
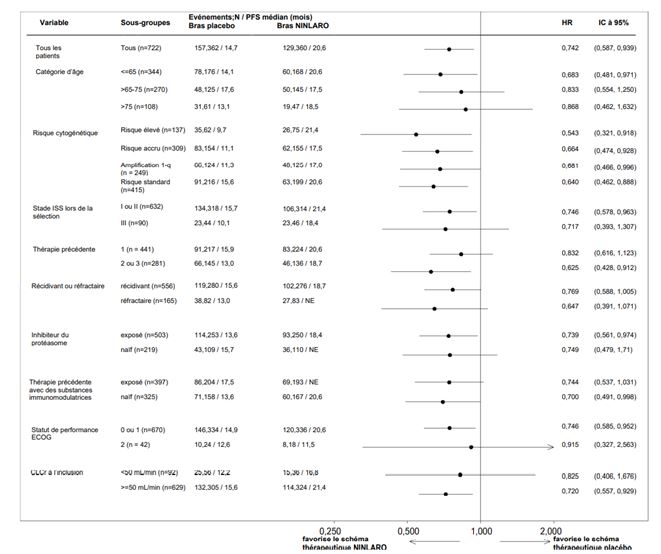
Population indiquée
Une analyse a été réalisée pour déterminer les résultats d'efficacité clinique à l'aide de caractéristiques de risque cytogénétique fixées et de caractéristiques de risque clinique défavorable stratifiées. Dans la population ITT, une amélioration plus importante des résultats d'efficacité a été observée chez 70% des patients (N=505, «population indiquée») qui avaient reçu au moins un traitement antérieur et présentaient des caractéristiques à haut risque (définies comme un risque cytogénétique accru [risque élevé (del[17], t[4;14], t[14;16]) ou 1q21] ou stade ISS III) ou qui avaient reçu au moins deux traitements antérieurs. Lors de l'analyse primaire, 98 (39%) événements de PFS sont survenus dans le bras NINLARO et 122 (49%) dans le bras placebo. La PFS médiane a été de 18,4 mois dans le bras NINLARO et de 12,2 mois dans le bras placebo (HR=0,681 [IC à 95% (0,520, 0,892)], p=0,005).
L'amélioration de la PFS dans le bras NINLARO a été appuyée par des améliorations du taux de réponse global dans la population indiquée.
Au moment de l'analyse intermédiaire planifiée de l'OS, 58 décès étaient survenus dans le bras NINLARO et 76 décès dans le bras placebo, dans la population indiquée. L'OS médiane n'a pas été atteinte dans le bras NINLARO et a été de 30,9 mois dans le bras placebo (HR=0,706 [IC À 95% (0,499, 0,997)]). Les résultats de l'analyse exploratoire noninférentielle de la PFS ont montré dans la population indiquée une PFS médiane estimée de 19,1 mois dans le bras NINLARO et de 12,6 mois dans le bras placebo (HR=0,728 [IC à 95% (0,573, 0,924)]). Les résultats sont concordants avec la conclusion de l'analyse primaire de la PFS, à savoir que le traitement a un effet positif.
Au moment de l'analyse finale de l'OS, 181 décès ont été dénombrés dans la population indiquée dans le groupe NINLARO et 179 décès dans le groupe placebo. L'OS médiane était de 50,4 mois dans le groupe NINLARO et 42,0 mois dans le groupe placebo (HR=0,912 [IC à 95% (0,74, 1,125)]), p=0,005).
Pédiatrie
L'Agence européenne des médicaments a supprimé l'obligation de soumettre les résultats des études réalisées avec NINLARO dans tous les sous-groupes de la population pédiatrique (voir rubrique «Posologie/Mode d'emploi, Enfants et adolescents»).
|