CompositionPrincipes actifs
Lumacaftor et ivacaftor.
Excipients
Comprimés
Noyau du comprimé:
Cellulose microcristalline, croscarmellose sodique, succinate d'acétate d'hypromellose, povidone (K30), laurilsulfate de sodium, stéarate de magnésium.
Pelliculage:
Alcool polyvinylique, dioxyde de titane, macrogol 3350, talc, acide carminique, bleu brillant FCF, indigotine.
Encre d'impression:
Hydroxyde d'ammonium, oxyde de fer noir, propylène glycol, gomme laque.
1 comprimé pelliculé Orkambi 100 mg/125 mg contient 2,56 mg de sodium.
1 comprimé pelliculé Orkambi 200 mg/125 mg contient 3,51 mg de sodium.
Granulés
Cellulose microcristalline, croscarmellose sodique, succinate d'acétate d'hypromellose, povidone (K30), laurilsulfate de sodium.
1 sachet de granulés Orkambi 75 mg/94 mg contient au maximum 0,67 mg de sodium.
1 sachet de granulés Orkambi 100 mg/125 mg contient au maximum 0,89 mg de sodium.
1 sachet de granulés Orkambi 150 mg/188 mg contient au maximum 1,34 mg de sodium.
Indications/Possibilités d’emploiComprimés
Orkambi comprimés est indiqué dans le traitement de la mucoviscidose chez les patients âgés de 6 ans et plus, homozygotes pour la mutation F508del du gène CFTR (voir «Posologie/Mode d'emploi», «Mises en garde et précautions» et «Propriétés/Effets»).
Granulés
Orkambi granulés est indiqué dans le traitement de la mucoviscidose chez les enfants âgés d'un an et plus, homozygotes pour la mutation F508del du gène CFTR (voir «Posologie/Mode d'emploi», «Mises en garde et précautions» et «Propriétés/Effets»).
Posologie/Mode d’emploiLa prescription d'Orkambi est réservée aux médecins expérimentés dans le traitement de la mucoviscidose. Si le génotype du patient n'est pas connu, un génotypage par une méthode fiable et validée devra être réalisé pour confirmer la présence de la mutation F508del sur les deux allèles du gène CFTR.
Posologie usuelle
Pour les recommandations posologiques standards, voir le tableau 1.
|
Tableau 1: Dose d'Orkambi recommandée chez les patients âgés d'un an et plus
| |
Âge
|
Poids
|
Dosage
|
Dose (toutes les 12 heures)
| |
|
|
|
Matin
|
Soir
| |
1 à < 2 ans
|
7 kg à < 9 kg*
|
lumacaftor 75 mg/ivacaftor 94 mg
|
1 sachet
|
1 sachet
| |
|
9 kg à < 14 kg
|
lumacaftor 100 mg/ivacaftor 125 mg
|
1 sachet
|
1 sachet
| |
|
≥14 kg
|
lumacaftor 150 mg/ivacaftor 188 mg
|
1 sachet
|
1 sachet
| |
2 à 5 ans
|
< 14 kg
|
lumacaftor 100 mg/ivacaftor 125 mg
|
1 sachet
|
1 sachet
| |
|
≥14 kg
|
lumacaftor 150 mg/ivacaftor 188 mg
|
1 sachet
|
1 sachet
| |
6 à < 12 ans
|
-
|
lumacaftor 100 mg/ivacaftor 125 mg
|
2 comprimés
|
2 comprimés
| |
12 ans et plus
|
-
|
lumacaftor 200 mg/ivacaftor 125 mg
|
2 comprimés
|
2 comprimés
|
* Pour les patients âgés de 1 à moins de 2 ans pesant de 7 kg à moins de 9 kg traités avec le dosage lumacaftor 75 mg/ivacaftor 94 mg deux fois par jour, les données ne sont disponibles que pour un nombre limité de patients (voir «Propriétés/Effets» et «Pharmacocinétique»).
Orkambi doit être pris avec des aliments contenant des graisses. Les comprimés doivent être pris immédiatement avant ou après un repas ou une collation contenant des graisses (voir «Pharmacocinétique»).
Prise retardée
En cas d'oubli d'une prise, s'il s'est écoulé moins de 6 heures depuis l'heure de prise habituelle, la dose prévue d'Orkambi doit être prise avec un repas ou une collation contenant des graisses. Si un délai de plus de 6 heures s'est écoulé, le patient doit attendre et prendre la dose suivante à l'heure habituelle. Les patients ne doivent pas prendre de dose double pour compenser la dose oubliée.
Administration concomitante d'inhibiteurs du CYP3A
Aucune adaptation de la posologie n'est nécessaire en cas d'instauration d'un traitement par des inhibiteurs du CYP3A chez les patients en cours de traitement par Orkambi. Cependant, en cas d'instauration du traitement chez des patients traités par des inhibiteurs puissants du CYP3A, la posologie d'Orkambi doit être réduite à un comprimé par jour chez les patients âgés de 6 ans et plus et à un sachet un jour sur deux chez les patients âgés de 1 à 5 ans pendant la première semaine de traitement afin que l'effet d'induction du lumacaftor atteigne son état d'équilibre. Après cette période, le traitement sera poursuivi à la dose quotidienne recommandée (voir tableau 2).
|
Tableau 2: Instauration du traitement chez les patients recevant des inhibiteurs puissants du CYP3A
| |
Âge
|
Poids
|
Dosage
|
1re semaine de traitement
|
À partir de la 2e semaine
| |
1 à < 2 ans
|
7 kg à < 9 kg
|
lumacaftor 75 mg/ivacaftor 94 mg
|
1 sachet un jour sur deux, c'est-à-dire les jours 1, 3, 5, 7
|
À partir du jour 8, le traitement doit être administré à la dose quotidienne recommandée
| |
|
9 kg à < 14 kg
|
lumacaftor 100 mg/ivacaftor 125 mg
|
|
| |
|
≥14 kg
|
lumacaftor 150 mg/ivacaftor 188 mg
|
|
| |
2 à 5 ans
|
<14 kg
|
lumacaftor 100 mg/ivacaftor 125 mg
|
|
| |
|
≥14 kg
|
lumacaftor 150 mg/ivacaftor 188 mg
|
|
| |
6 à < 12 ans
|
-
|
lumacaftor 100 mg/ivacaftor 125 mg
|
1 comprimé par jour
|
À partir du jour 8, le traitement doit être administré à la dose quotidienne recommandée
| |
12 ans et plus
|
-
|
lumacaftor 200 mg/ivacaftor 125 mg
|
|
|
Si le traitement par Orkambi est interrompu pendant plus d'une semaine puis repris pendant un traitement en cours par des inhibiteurs puissants du CYP3A, la dose d'Orkambi doit être réduite à un comprimé par jour chez les patients âgés de 6 ans et plus et à un sachet un jour sur deux chez les patients âgés de 1 à 5 ans pendant la première semaine suivant la reprise du traitement (voir tableau 2). Après cette période, le traitement sera poursuivi à la dose quotidienne recommandée (voir «Interactions»).
Mode d'administration
Comprimés
Voie orale. Les patients doivent être informés que les comprimés doivent être avalés entiers (par exemple, les patients ne doivent pas croquer, fractionner ou dissoudre les comprimés).
Granulés
Voie orale. Chaque sachet est à usage unique.
Le contenu de chaque sachet de granulés doit être entièrement mélangé avec une cuillère à café (5 ml) d'aliment semi-liquide ou de liquide adapté à l'âge de l'enfant et ingéré en totalité. Les aliments semi-liquides ou les liquides sont par exemple les compotes de fruits, les purées de légumes, les yaourts, la compote de pommes, l'eau, le lait, le lait maternel, le lait infantile ou le jus de fruit. L'aliment ou le liquide servant à la préparation du mélange doit être à température ambiante ou inférieure. Si le mélange n'est pas consommé immédiatement, il reste stable pendant une heure et doit donc être ingéré dans ce délai.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucune adaptation de la posologie n'est nécessaire en cas d'insuffisance hépatique légère (Child-Pugh classe A). En cas d'insuffisance hépatique modérée (Child-Pugh classe B), une réduction de la posologie est recommandée. Il n'y a pas d'expérience de l'utilisation d'Orkambi en cas d'insuffisance hépatique sévère (Child-Pugh classe C), mais une exposition systémique plus élevée que chez les patients présentant une insuffisance hépatique modérée est attendue. Par conséquent, Orkambi doit être utilisé avec précaution à une dose réduite chez les patients présentant une insuffisance hépatique sévère, après évaluation du rapport bénéfice/risque (voir «Mises en garde et précautions», «Effets indésirables» et «Pharmacocinétique»).
Pour les adaptations de la posologie chez les patients âgés de 1 à 5 ans présentant une insuffisance hépatique modérée ou sévère, voir le tableau 3.
|
Tableau 3: Recommandations pour les adaptations de la posologie chez les patients âgés de 1 à 5 ans présentant une insuffisance hépatique modérée ou sévère
| |
Âge
|
Poids
|
Dosage
|
Modérée
(Child-Pugh classe B)
|
Sévère
(Child-Pugh classe C)
| |
|
|
|
Matin
|
Soir
|
Matin
|
Soir
| |
1 à < 2 ans
|
7 kg à < 9 kg
|
lumacaftor 75 mg/ivacaftor 94 mg
|
1 sachet de granulés par jour
|
1 sachet de granulés un jour sur deux
|
1 sachet de granulés par jour ou moins fréquemment*
|
Pas de prise
| |
|
9 kg à < 14 kg
|
lumacaftor 100 mg/ivacaftor 125 mg
|
|
|
|
| |
|
≥14 kg
|
lumacaftor 150 mg/ivacaftor 188 mg
|
|
|
|
| |
2 à 5 ans
|
< 14 kg
|
lumacaftor 100 mg/ivacaftor 125 mg
|
|
|
|
| |
|
≥14 kg
|
lumacaftor 150 mg/ivacaftor 188 mg
|
|
|
|
|
* L'intervalle entre chaque prise doit être modifié en fonction de la réponse clinique et de la tolérance.
Pour les adaptations de la posologie chez les patients âgés de 6 ans et plus présentant une insuffisance hépatique modérée ou sévère, voir le tableau 4.
|
Tableau 4: Recommandations pour les adaptations de la posologie chez les patients âgés de 6 ans et plus présentant une insuffisance hépatique modérée ou sévère
| |
Âge
|
Dosage
|
Dose quotidienne totale
| |
|
|
Modérée
(Child-Pugh classe B)
|
Sévère
(Child-Pugh classe C)
| |
6 à < 12 ans
|
lumacaftor 100 mg/ivacaftor 125 mg
|
2 comprimés le matin +
1 comprimé le soir (à 12 heures d'intervalle)
|
1 comprimé le matin +
1 comprimé le soir ou moins fréquemment*
| |
12 ans et plus
|
lumacaftor 200 mg/ivacaftor 125 mg
|
|
|
* L'intervalle entre chaque prise doit être modifié en fonction de la réponse clinique et de la tolérance.
Patients présentant des troubles de la fonction rénale
Aucune adaptation de la posologie n'est nécessaire chez les patients présentant une insuffisance rénale légère à modérée. La prudence est recommandée en cas d'insuffisance rénale sévère (clairance de la créatinine ≤30 ml/min) ou d'insuffisance rénale terminale (voir «Mises en garde et précautions» et «Pharmacocinétique»).
Patients âgés
La sécurité et l'efficacité d'Orkambi chez les patients âgés de 65 ans et plus n'ont pas été évaluées.
Enfants et adolescents
La sécurité et l'efficacité d'Orkambi chez les enfants âgés de moins de 1 an n'ont pas encore été établies. Aucune donnée n'est disponible.
Contre-indicationsHypersensibilité aux principes actifs ou à l'un des excipients (voir «Composition»).
Mises en garde et précautionsPatients hétérozygotes pour la mutation F508del du gène CFTR
Le lumacaftor/ivacaftor n'est pas efficace chez les patients atteints de mucoviscidose porteurs de la mutation F508del sur un seul allèle avec, sur le second allèle, une mutation susceptible d'entraîner l'absence de synthèse de la protéine CFTR ou la synthèse d'une protéine CFTR ne répondant pas à l'ivacaftor in vitro (voir «Propriétés/Effets»).
Patients porteurs d'une mutation de défaut de régulation (classe III) du gène CFTR
L'association lumacaftor/ivacaftor n'a pas été étudiée chez les patients atteints de mucoviscidose porteurs d'une mutation de défaut de régulation (classe III) du gène CFTR sur un allèle, avec ou sans la mutation F508del sur l'autre allèle. L'exposition systémique à l'ivacaftor étant significativement diminuée lorsque celuici est administré en association avec le lumacaftor, l'association lumacaftor/ivacaftor ne doit pas être utilisée chez ces patients.
Effets indésirables respiratoires
Les effets indésirables respiratoires (tels que: gêne thoracique, dyspnée, bronchospasme et respiration anormale) ont été plus fréquents au début du traitement par l'association lumacaftor/ivacaftor. Ces événements ont entraîné l'arrêt du traitement et peuvent être graves, en particulier chez les patients ayant un VEMS < 40 % de la valeur théorique.
L'expérience clinique chez les patients ayant un VEMS < 40 % de la valeur théorique est limitée et une surveillance supplémentaire de ces patients est recommandée en début de traitement (voir «Effets indésirables»). Une diminution transitoire du VEMS a également été observée chez certains patients au début du traitement par l'association lumacaftor/ivacaftor. En l'absence de données, l'instauration du traitement par l'association lumacaftor/ivacaftor chez des patients présentant une exacerbation pulmonaire n'est pas recommandée.
Effet sur la pression artérielle
Une augmentation de la pression artérielle a été observée chez certains patients traités par l'association lumacaftor/ivacaftor. La pression artérielle doit être surveillée régulièrement chez tous les patients pendant le traitement (voir «Effets indésirables»).
Patients présentant une atteinte hépatique avancée
Les patients atteints de mucoviscidose peuvent présenter des anomalies de la fonction hépatique, avec éventuellement une atteinte hépatique avancée. Une détérioration de la fonction hépatique a été rapportée chez des patients présentant une atteinte hépatique avancée. Des cas de décompensation hépatique, y compris d'insuffisance hépatique d'issue fatale, ont été rapportés chez des patients atteints de mucoviscidose présentant une cirrhose préexistante avec hypertension portale et traités par l'association lumacaftor/ivacaftor. Chez ces patients, l'association lumacaftor/ivacaftor doit être utilisée avec précaution, et seulement si le bénéfice attendu du traitement surpasse le risque encouru. La dose doit être réduite et une surveillance étroite est requise après l'instauration du traitement (voir «Posologie/Mode d'emploi», «Effets indésirables» et «Pharmacocinétique»).
Effets indésirables hépatobiliaires
Une augmentation des transaminases hépatiques, associée dans certains cas à une augmentation de la bilirubine totale, a été rapportée fréquemment chez des patients atteints de mucoviscidose recevant l'association lumacaftor/ivacaftor. Des augmentations des transaminases ont été observées plus fréquemment chez les patients pédiatriques que chez les patients adultes. Dans les cohortes pédiatriques de patients de différents âges, des augmentations des transaminases ont été observées plus fréquemment chez les patients âgés de 2 à 5 ans que chez les patients âgés de 6 à moins de 12 ans (voir «Effets indésirables»).
Le risque d'atteinte hépatique liée au traitement ne pouvant être exclu, il est recommandé de réaliser un bilan biologique hépatique (ALAT, ASAT et bilirubine) avant le début du traitement par l'association lumacaftor/ivacaftor, tous les trois mois pendant la première année de traitement puis une fois par an. Chez les patients ayant des antécédents d'augmentations des ALAT, des ASAT ou de la bilirubine, une surveillance plus fréquente doit être envisagée.
En cas d'augmentation significative des ALAT ou des ASAT, avec ou sans augmentation de la bilirubine (ALAT ou ASAT > 5 fois la limite supérieure de la normale [LSN] ou ALAT ou ASAT > 3 x LSN avec bilirubine > 2 x LSN et/ou ictère clinique), le traitement par l'association lumacaftor/ivacaftor doit être arrêté et le bilan hépatique doit être étroitement surveillé jusqu'à normalisation. Un examen approfondi sur les causes possibles doit être effectué et les patients doivent être étroitement surveillés afin de détecter une éventuelle progression clinique. La décision d'une éventuelle reprise du traitement devra tenir compte des risques encourus par rapport au bénéfice attendu (voir «Posologie/Mode d'emploi», «Effets indésirables» et «Pharmacocinétique»).
Dépression
Des cas de dépression (incluant idées suicidaires et tentatives de suicide), apparaissant généralement au cours des trois mois suivant l'instauration du traitement, ont été rapportés chez des patients traités par Orkambi et ayant des antécédents de troubles psychiatriques. Dans certains cas, une amélioration des symptômes a été observée après une réduction de la dose ou l'arrêt du traitement. Les patients (et aidants) doivent être avertis de la nécessité d'être attentifs à l'apparition d'une humeur dépressive, de pensées suicidaires ou de modifications inhabituelles du comportement et de prendre immédiatement avis auprès du médecin en cas de survenue de ces symptômes.
Interactions avec d'autres médicaments
Substrats du CYP3A
Le lumacaftor est un inducteur puissant du CYP3A. L'association avec des substrats du CYP3A à forte affinité ou ayant une marge thérapeutique étroite n'est pas recommandée (voir «Interactions»).
Les contraceptifs hormonaux, qu'ils soient administrés par voie orale, injectable, transdermique ou par des dispositifs implantables, ne doivent pas être considérés comme une méthode de contraception efficace en cas d'administration concomitante avec Orkambi (voir «Interactions»).
Inducteurs puissants du CYP3A
L'ivacaftor est un substrat du CYP3A4 et du CYP3A5. Par conséquent, l'administration concomitante avec des inducteurs puissants du CYP3A (par exemple rifampicine, millepertuis [Hypericum perforatum]) n'est pas recommandée (voir «Interactions»).
Insuffisance rénale
La prudence est recommandée en cas d'utilisation du lumacaftor/ivacaftor chez des patients présentant une insuffisance rénale sévère ou en phase terminale (voir «Posologie/Mode d'emploi» et «Pharmacocinétique»).
Cataractes
Des cas d'opacités du cristallin non congénitales sans répercussions sur la vision ont été rapportés chez des enfants et adolescents traités par l'association lumacaftor/ivacaftor et par l'ivacaftor en monothérapie. Bien que d'autres facteurs de risque aient été présents dans certains cas (tels que: une corticothérapie, une exposition à des rayonnements), un risque possible imputable à l'ivacaftor ne peut être exclu (voir «Données précliniques»). Des examens ophtalmologiques avant et pendant le traitement sont recommandés chez les patients pédiatriques recevant un traitement par l'association lumacaftor/ivacaftor.
Patients greffés
L'association lumacaftor/ivacaftor n'a pas été étudiée chez les patients atteints de mucoviscidose ayant reçu une transplantation d'organe. Par conséquent, son utilisation chez ces patients n'est donc pas recommandée. Voir la «Interactions» pour les interactions avec les immunosuppresseurs.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c'est-à-dire qu'il est essentiellement «sans sodium».
InteractionsCompte tenu de la biodisponibilité et des doses indiquées, le risque d'interactions est considéré comme similaire pour tous les dosages et formes galéniques.
Le lumacaftor est un inducteur puissant du CYP3A. L'ivacaftor est un faible inhibiteur du CYP3A lorsqu'il est administré en monothérapie. D'autres médicaments administrés de façon concomitante peuvent avoir des effets sur l'association lumacaftor/ivacaftor et vice-versa.
Effets potentiels d'autres médicaments sur l'association lumacaftor/ivacaftor
Inhibiteurs du CYP3A
L'administration concomitante de l'association lumacaftor/ivacaftor et d'itraconazole, un inhibiteur puissant du CYP3A, n'a pas eu d'effet sur l'exposition systémique au lumacaftor, mais a augmenté de 4,3 fois l'exposition systémique à l'ivacaftor. Du fait de l'effet inducteur du lumacaftor sur le CYP3A, l'administration concomitante d'un inhibiteur du CYP3A ne devrait pas entraîner d'exposition systémique plus élevée de l'ivacaftor comparativement à celle obtenue lorsque l'ivacaftor est administré sans lumacaftor et à la dose recommandée en monothérapie de 150 mg toutes les 12 heures.
Aucune adaptation de la posologie n'est nécessaire lors de la mise en route d'un traitement par des médicaments inhibiteurs du CYP3A chez les patients en cours de traitement par l'association lumacaftor/ivacaftor. Cependant, en cas d'instauration du traitement par l'association lumacaftor/ivacaftor chez des patients recevant des inhibiteurs puissants du CYP3A, la posologie doit être adaptée (voir «Posologie/Mode d'emploi» et «Mises en garde et précautions»).
Aucune adaptation de la posologie n'est nécessaire en cas d'utilisation avec des inhibiteurs faibles ou modérés du CYP3A.
Inducteurs du CYP3A
L'administration concomitante de l'association lumacaftor/ivacaftor et de rifampicine, un inducteur puissant du CYP3A, n'a eu qu'un effet minimal sur l'exposition systémique au lumacaftor, mais l'aire sous la courbe (ASC) de l'ivacaftor a diminué de 57 %. Par conséquent, l'administration concomitante de l'association lumacaftor/ivacaftor avec des inducteurs puissants du CYP3A n'est pas recommandée (voir «Posologie/Mode d'emploi» et «Mises en garde et précautions»).
Aucune adaptation de la posologie n'est nécessaire en cas d'utilisation avec des inducteurs faibles ou modérés du CYP3A.
Effets potentiels de l'association lumacaftor/ivacaftor sur d'autres médicaments
Substrats du CYP3A
Le lumacaftor est un inducteur puissant du CYP3A. L'ivacaftor est un faible inhibiteur du CYP3A lorsqu'il est administré en monothérapie. Un effet inducteur puissant sur le CYP3A est attendu avec l'association lumacaftor/ivacaftor. Par conséquent, l'administration concomitante de l'association lumacaftor/ivacaftor avec des substrats du CYP3A peut diminuer leur exposition systémique (voir «Mises en garde et précautions»).
Substrats de la Pgp
Des études in vitro ont mis en évidence des effets à la fois inhibiteurs et inducteurs du lumacaftor sur la Pgp. De plus, une étude clinique menée avec l'ivacaftor en monothérapie a montré que l'ivacaftor est un faible inhibiteur de la Pgp. Par conséquent, l'administration concomitante de l'association lumacaftor/ivacaftor avec des substrats de la Pgp (par exemple digoxine) peut modifier leur exposition systémique.
Substrats du CYP2B6 et du CYP2C
Les interactions avec les substrats du CYP2B6 et du CYP2C n'ont pas été étudiées in vivo. Des études in vitro semblent indiquer que le lumacaftor peut avoir un effet inducteur sur les isoenzymes CYP2B6, CYP2C8, CYP2C9 et CYP2C19; cependant, une inhibition du CYP2C8 et du CYP2C9 a également été observée in vitro. De plus, des études in vitro semblent indiquer que l'ivacaftor peut inhiber le CYP2C9. Par conséquent, l'administration concomitante de l'association lumacaftor/ivacaftor peut modifier (augmenter ou diminuer) l'exposition systémique aux substrats du CYP2C8 et du CYP2C9, diminuer l'exposition systémique aux substrats du CYP2C19 et diminuer de façon importante l'exposition systémique aux substrats du CYP2B6.
Interactions potentielles de l'association lumacaftor/ivacaftor avec les transporteurs
Les études in vitro montrent que le lumacaftor est un substrat de la protéine de résistance du cancer du sein (BCRP – Breast Cancer Resistance Protein). L'administration concomitante d'Orkambi avec des médicaments qui inhibent la BCRP peut augmenter la concentration plasmatique de lumacaftor. Le lumacaftor inhibe les transporteurs d'anions organiques (OAT) 1 et 3. Le lumacaftor et l'ivacaftor sont des inhibiteurs de la BCRP. L'administration concomitante d'Orkambi avec des médicaments qui sont des substrats des transporteurs OAT1/3 et BCRP peut augmenter les concentrations plasmatiques de ces médicaments. Le lumacaftor et l'ivacaftor ne sont pas des inhibiteurs d'OATP1B1, d'OATP1B3 et des transporteurs de cations organiques (OCT) 1 et 2. L'ivacaftor n'est pas un inhibiteur d'OAT1 et d'OAT3.
Interactions établies et autres interactions potentiellement significatives
Le tableau 5 ci-dessous présente l'effet établi ou attendu de l'association lumacaftor/ivacaftor sur d'autres médicaments ou l'effet d'autres médicaments sur l'association lumacaftor/ivacaftor. Les données présentées dans le tableau sont issues principalement des études in vitro. Les recommandations figurant dans la colonne intitulée «Commentaire clinique» du tableau 5 sont basées sur les études d'interactions, la pertinence clinique ou les interactions attendues en relation avec les voies d'élimination. Les interactions les plus cliniquement pertinentes sont présentées en premier.
|
Tableau 5: Interactions établies et autres interactions potentiellement significatives – Recommandations posologiques en cas d'association du lumacaftor/ivacaftor avec d'autres médicaments
| |
Classe thérapeutique du médicament concomitant:
Nom du principe actif
|
Effet
|
Commentaire clinique
| |
Médicaments concomitants présentant les interactions cliniquement les plus pertinentes
| |
Antiallergiques:
montélukast
|
↔ LUM, IVA
↓ montélukast
liée à l'effet inducteur du LUM sur les CYP3A/2C8/2C9
|
Aucune adaptation de la dose de montélukast n'est nécessaire. Seule une surveillance clinique appropriée est préconisée en cas d'administration concomitante avec le lumacaftor/ivacaftor. Le lumacaftor/ivacaftor peut diminuer l'exposition systémique au montélukast, ce qui peut entraîner une diminution de son efficacité.
| |
fexofénadine
|
↔ LUM, IVA
↑ ou ↓ fexofénadine
liée à l'effet potentiel inducteur ou inhibiteur sur la Pgp
|
Une adaptation de la dose de fexofénadine peut être nécessaire pour obtenir l'effet clinique attendu. Le lumacaftor/ivacaftor peut modifier l'exposition systémique à la fexofénadine.
| |
Antibiotiques:
clarithromycine, télithromycine
|
↔ LUM
↑ IVA
liée à l'effet inhibiteur de la clarithromycine, la télithromycine sur le CYP3A
↓ clarithromycine, télithromycine
liée à l'effet inducteur du LUM sur le CYP3A
|
Aucune adaptation de la dose de lumacaftor/ivacaftor n'est nécessaire en cas de mise en route d'un traitement par la clarithromycine ou la télithromycine chez des patients en cours de traitement par l'association lumacaftor/ivacaftor.
La dose de lumacaftor/ivacaftor doit être réduite à 1 comprimé par jour ou à 1 sachet tous les deux jours pendant la première semaine en cas d'instauration du traitement par le lumacaftor/ivacaftor chez des patients en cours de traitement par la clarithromycine ou la télithromycine.
L'utilisation d'un autre antibiotique, par exemple l'azithromycine, doit être envisagée. Le lumacaftor/ivacaftor peut diminuer l'exposition systémique à la clarithromycine et à la télithromycine, ce qui peut entraîner une diminution de leur efficacité.
| |
érythromycine
|
↔ LUM
↑ IVA
liée à l'effet inhibiteur de l'érythromycine sur le CYP3A.
↓ érythromycine
liée à l'effet inducteur du LUM sur le CYP3A.
|
Aucune adaptation de la posologie de lumacaftor/ivacaftor n'est nécessaire en cas d'administration concomitante avec l'érythromycine.
L'utilisation d'un autre antibiotique, par exemple l'azithromycine, doit être envisagée. Le lumacaftor/ivacaftor peut diminuer l'exposition systémique à l'érythromycine, ce qui peut entraîner une diminution de son efficacité.
| |
Antiépileptiques:
carbamazépine, phénobarbital, phénytoïne
|
↔ LUM
↓ IVA
liée à l'effet inducteur de ces antiépileptiques sur le CYP3A
↓ carbamazépine, phénobarbital, phénytoïne
liée à l'effet inducteur du LUM sur le CYP3A
|
L'administration concomitante de lumacaftor/ivacaftor avec ces antiépileptiques n'est pas recommandée. Les expositions à l'ivacaftor et à l'antiépileptique peuvent être significativement diminuées, ce qui peut entraîner une diminution de l'efficacité de chacune des substances actives.
| |
Antifongiques:
itraconazole*, kétoconazole, posaconazole, voriconazole
|
↔ LUM
↑ IVA
liée à l'effet inhibition de ces antifongiques sur le CYP3A
↓ itraconazole, kétoconazole, voriconazole
liée à l'effet inducteur du LUM sur le CYP3A
↓ posaconazole
liée à l'effet inducteur du LUM sur les UGT
|
Aucune adaptation de la dose de lumacaftor/ivacaftor n'est nécessaire en cas d'instauration d'un traitement par ces antifongiques chez des patients en cours de traitement par lumacaftor/ivacaftor.
La dose de lumacaftor/ivacaftor doit être réduite à 1 comprimé par jour ou à 1 sachet tous les deux jours pendant la première semaine en cas d'instauration du traitement par le lumacaftor/ivacaftor chez des patients en cours de traitement par ces antifongiques.
L'administration concomitante de lumacaftor/ivacaftor avec ces antifongiques n'est pas recommandée. Si l'utilisation de ces médicaments est nécessaire, les patients doivent être surveillés étroitement afin de détecter l'apparition d'infections fongiques sous traitement. Le lumacaftor/ivacaftor peut diminuer l'exposition à ces antifongiques, ce qui peut entraîner une diminution de leur efficacité.
| |
fluconazole
|
↔ LUM
↑ IVA
liée à l'effet inhibiteur du fluconazole sur le CYP3A.
↓ fluconazole
liée à l'effet inducteur du LUM; le fluconazole est éliminé essentiellement par excrétion rénale sous forme inchangée; cependant, une légère diminution de l'exposition au fluconazole a été observée avec les inducteurs puissants.
|
Aucune adaptation de la dose de lumacaftor/ivacaftor n'est nécessaire en cas d'administration concomitante avec le fluconazole.
Une dose plus élevée de fluconazole peut être nécessaire pour obtenir l'effet clinique attendu. Le lumacaftor/ivacaftor peut diminuer l'exposition systémique au fluconazole, ce qui peut entraîner une diminution de son efficacité.
| |
Antiinflammatoires:
ibuprofène
|
↔ LUM, IVA
↓ ibuprofène
liée à l'effet inducteur du LUM sur les CYP3A/2C8/2C9
|
Une dose plus élevée d'ibuprofène peut être nécessaire pour obtenir l'effet clinique attendu. Le lumacaftor/ivacaftor peut diminuer l'exposition à l'ibuprofène, ce qui peut entraîner une diminution de son efficacité.
| |
Antimycobactériens:
rifabutine, rifampicine*, rifapentine
|
↔ LUM
↓ IVA
liée à l'effet inducteur des antimycobactériens sur le CYP3A
↓ rifabutine
liée à l'effet inducteur du LUM sur le CYP3A
↔ rifampicine, rifapentine
|
L'administration concomitante de lumacaftor/ivacaftor avec ces antimycobactériens n'est pas recommandée. Diminution de l'exposition systémique à l'ivacaftor, ce qui peut entraîner une diminution de l'efficacité de lumacaftor/ivacaftor.
Une dose plus élevée de rifabutine peut être nécessaire pour obtenir l'effet clinique attendu. Le lumacaftor/ivacaftor peut diminuer l'exposition systémique à la rifabutine, ce qui peut entraîner une diminution de son efficacité.
| |
Benzodiazépines:
midazolam, triazolam
|
↔ LUM, IVA
↓ midazolam, triazolam
liée à l'effet inducteur du LUM sur le CYP3A
|
L'administration concomitante de lumacaftor/ivacaftor avec ces benzodiazépines n'est pas recommandée. Diminution de l'exposition systémique au midazolam et au triazolam, ce qui entraînera une diminution de leur efficacité.
| |
Contraceptifs hormonaux:
éthinylestradiol, noréthistérone et autres progestatifs
|
↓ éthinylestradiol, noréthistérone et autres progestatifs
liée à l'effet inducteur du LUM sur le CYP3A et les UGT
|
Les contraceptifs hormonaux, qu'ils soient administrés par voie orale, injectable, transdermique ou par dispositif implantable, ne doivent pas être considérés comme une méthode de contraception efficace en cas d'administration concomitante avec le lumacaftor/ivacaftor. Le lumacaftor/ivacaftor peut diminuer l'exposition systémique aux contraceptifs hormonaux, ce qui peut entraîner une diminution de leur efficacité.
| |
Immunosuppresseurs:
ciclosporine, évérolimus, sirolimus, tacrolimus (utilisés après une transplantation d'organe)
|
↔ LUM, IVA
↓ ciclosporine, évérolimus, sirolimus, tacrolimus
liée à l'effet inducteur du LUM sur le CYP3A.
|
L'administration concomitante de lumacaftor/ivacaftor avec ces immunosuppresseurs n'est pas recommandée. Le lumacaftor/ivacaftor diminuera l'exposition systémique à ces immunosuppresseurs, ce qui peut entraîner une diminution de leur efficacité. L'utilisation du lumacaftor/ivacaftor chez des patients greffés n'a pas été étudiée.
| |
Inhibiteurs de la pompe à protons:
ésoméprazole, lansoprazole, oméprazole
|
↔ LUM, IVA
↓ ésoméprazole, lansoprazole, oméprazole
liée à l'effet inducteur du LUM sur les CYP3A/2C19
|
Une dose plus élevée de ces inhibiteurs de la pompe à protons peut être nécessaire pour obtenir l'effet clinique attendu. Le lumacaftor/ivacaftor peut diminuer l'exposition systémique à ces inhibiteurs de la pompe à protons, ce qui peut entraîner une diminution de leur efficacité.
| |
Produits de phytothérapie:
millepertuis (Hypericum perforatum)
|
↔ LUM
↓ IVA
liée à l'effet inducteur du millepertuis sur le CYP3A
|
L'administration concomitante de lumacaftor/ivacaftor avec le millepertuis n'est pas recommandée.
Diminution de l'exposition systémique à l'ivacaftor pouvant entraîner une diminution de l'efficacité du lumacaftor/ivacaftor.
| |
Autres médicaments concomitants cliniquement pertinents
| |
Antiarythmiques:
digoxine
|
↔ LUM, IVA
↑ ou ↓ digoxine
liée à l'effet inducteur ou inhibiteur potentiel de la Pgp
|
Les concentrations sériques de digoxine doivent être surveillées et la dose doit être adaptée pour obtenir l'effet clinique attendu. Le lumacaftor/ivacaftor peut modifier l'exposition systémique à la digoxine.
| |
Anticoagulants:
dabigatran
|
↔ LUM, IVA
↑ ou ↓ dabigatran
liée à l'effet inducteur ou inhibiteur potentiel sur la Pgp
|
Une surveillance clinique appropriée est requise en cas d'administration concomitante avec le lumacaftor/ivacaftor. Une adaptation de la dose de dabigatran peut être nécessaire pour obtenir l'effet clinique attendu. Le lumacaftor/ivacaftor peut modifier l'exposition systémique au dabigatran.
| |
warfarine
|
↔ LUM, IVA
↑ ou ↓ warfarine
liée à l'effet inducteur ou inhibiteur potentiel du LUM sur le CYP2C9
|
Le rapport normalisé international (INR) doit être surveillé si l'administration concomitante de warfarine et de lumacaftor/ivacaftor est nécessaire. Le lumacaftor/ivacaftor peut modifier l'exposition systémique à la warfarine.
| |
Antidépresseurs:
citalopram, escitalopram, sertraline
|
↔ LUM, IVA
↓ citalopram, escitalopram, sertraline
liée à l'effet inducteur du LUM sur les CYP3A/2C19
|
Une dose plus élevée de ces antidépresseurs peut être nécessaire pour obtenir l'effet clinique attendu. Le lumacaftor/ivacaftor peut diminuer l'exposition systémique à ces antidépresseurs, ce qui peut entraîner une diminution de leur efficacité.
| |
Bupropion
|
↔ LUM, IVA
↓ bupropion
liée à l'effet inducteur du LUM sur le CYP2B6
|
Une dose plus élevée de bupropion peut être nécessaire pour obtenir l'effet clinique attendu. Le lumacaftor/ivacaftor peut diminuer l'exposition systémique au bupropion, ce qui peut entraîner une diminution de son efficacité.
| |
Corticoïdes systémiques:
méthylprednisolone, prednisone
|
↔ LUM, IVA
↓ méthylprednisolone, prednisone
lié à l'effet induction du LUM sur le CYP3A
|
Une dose plus élevée de ces corticoïdes systémiques peut être nécessaire pour obtenir l'effet clinique attendu. Le lumacaftor/ivacaftor peut diminuer l'exposition systémique à la méthylprednisolone et à la prednisone, ce qui peut entraîner une diminution de leur efficacité.
| |
Antagonistes des récepteurs H2 :
ranitidine
|
↔ LUM, IVA
↑ ou ↓ ranitidine
liée à l'effet inducteur ou inhibiteur potentiel sur la Pgp
|
Une adaptation de la dose de ranitidine peut être nécessaire pour obtenir l'effet clinique attendu. Le lumacaftor/ivacaftor peut modifier l'exposition systémique à la ranitidine.
| |
Antidiabétiques oraux:
répaglinide
|
↔ LUM, IVA
↓ répaglinide
liée à l'effet inducteur du LUM sur les CYP3A/2C8
|
Une dose plus élevée de répaglinide peut être nécessaire pour obtenir l'effet clinique attendu. Le lumacaftor/ivacaftor peut diminuer l'exposition systémique au répaglinide, ce qui peut entraîner une diminution de son efficacité.
| |
Légende: ↑ = augmentation, ↓ = diminution, ↔ = pas de modification; LUM = lumacaftor; IVA = ivacaftor.
* Sur la base des études cliniques d'interactions. Toutes les autres interactions présentées sont les interactions attendues.
|
Résultats faussement positifs pour les analyses de dépistage urinaire du THC
Des résultats faussement positifs pour les analyses de dépistage urinaire du tétrahydrocannabinol (THC) ont été rapportés chez des patients recevant Orkambi. Une autre méthode de confirmation doit être envisagée pour vérifier les résultats.
Enfants et adolescents
Les études d'interaction n'ont été réalisées que chez l'adulte.
Grossesse, allaitementGrossesse
Les données disponibles sur l'utilisation de l'association lumacaftor/ivacaftor chez la femme enceinte sont limitées (moins de 300 grossesses). Les études effectuées chez l'animal avec le lumacaftor et l'ivacaftor n'ont pas mis en évidence d'effets délétères directs ou indirects sur le développement et la reproduction, tandis que des effets ont été observés avec l'ivacaftor à des doses maternotoxiques (voir «Données précliniques»). Par mesure de précaution, il est préférable d'éviter l'utilisation du lumacaftor/ivacaftor pendant la grossesse, sauf si l'état clinique de la femme justifie le traitement avec le lumacaftor/ivacaftor.
Allaitement
Des données limitées montrent une excrétion de l'ivacaftor et du lumacaftor dans le lait maternel humain. Un risque pour les nouveaunés/nourrissons ne peut être exclu. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre/de s'abstenir du traitement avec l'association lumacaftor/ivacaftor en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement attendu pour la femme.
Fertilité
Il n'existe pas de données concernant les effets du lumacaftor et/ou de l'ivacaftor sur la fertilité humaine. Le lumacaftor n'a pas eu d'effets sur la fertilité et les indices de performance de reproduction chez des rats mâles et femelles. L'ivacaftor a diminué la fertilité et les indices de performance de reproduction chez les rats mâles et femelles (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesL'effet d'Orkambi sur l'aptitude à la conduite et à l'utilisation de machines n'a pas été étudié.
L'ivacaftor peut provoquer des sensations vertigineuses (voir «Effets indésirables»).
Il doit être recommandé aux patients qui présentent des sensations vertigineuses pendant le traitement par Orkambi de ne pas conduire des véhicules ni utiliser des machines jusqu'à la disparition des symptômes.
Effets indésirablesRésumé du profil de sécurité
Les effets indésirables les plus fréquents sont: dyspnée (14,0 %), diarrhée (11,0 %) et nausées (10,2 %).
Les effets indésirables graves étaient des événements hépatobiliaires, tels que: augmentations des transaminases (0,5 %), hépatite cholestatique (0,3 %) et encéphalopathie hépatique (0,1 %).
Liste tabulée des effets indésirables
Les effets indésirables identifiés dans les études de phase III contrôlées contre placebo de 24 semaines (étude VX12-809-103 et étude VX12-809-104) menées chez des patients âgés de 12 ans et plus et dans l'étude contrôlée contre placebo de 24 semaines menée chez des patients âgés de 6 à moins de 12 ans (étude VX-14-809-109), homozygotes pour la mutation F508del du gène CFTR, sont présentés ci-dessous dans le tableau 6 par classe de systèmes d'organes et fréquence. Les effets indésirables observés avec l'ivacaftor en monothérapie sont également présentés dans le tableau 6. Les effets indésirables sont classés par fréquence selon la définition MedDRA: très fréquent (≥1/10), fréquent (≥1/100, < 1/10), peu fréquent (≥1/1 000, < 1/100), rare (≥1/10 000, < 1/1 000), très rare (< 1/10 000) et fréquence inconnue (ne peut être estimée sur la base des données disponibles).
|
Tableau 6: Effets indésirables chez les patients traités par le lumacaftor/ivacaftor et chez les patients traités par l'ivacaftor en monothérapie
| |
Classe de systèmes d'organes
|
Fréquence de survenue
|
Effets indésirables
| |
Infections et infestations
|
Très fréquent
|
Rhinopharyngite*
| |
|
Fréquent
|
Infections des voies respiratoires supérieures, rhinite
| |
Affections vasculaires
|
Peu fréquent
|
Hypertension artérielle
| |
Affections psychiatriques
|
Inconnue
|
Dépression
| |
Affections du système nerveux
|
Très fréquent
|
Céphalées*, sensations vertigineuses*
| |
|
Peu fréquent
|
Encéphalopathie hépatique†
| |
Affections de l'oreille et du labyrinthe
|
Fréquent
|
Otalgie*, sensation anormale au niveau de l'oreille*, acouphènes*, hyperhémie du tympan*, trouble vestibulaire*
| |
|
Peu fréquent
|
Congestion de l'oreille*
| |
Affections respiratoires, thoraciques et médiastinales
|
Très fréquent
|
Congestion nasale*, dyspnée, toux productive, augmentation des expectorations
| |
|
Fréquent
|
Respiration anormale, douleur oropharyngée, congestion des sinus*, rhinorrhée, érythème pharyngé*, bronchospasme
| |
Affections gastrointestinales
|
Très fréquent
|
Douleur abdominale*, douleur abdominale haute, diarrhée, nausées
| |
|
Fréquent
|
Flatulences, vomissements
| |
Affections hépatobiliaires
|
Fréquent
|
Augmentations des transaminases
| |
|
Peu fréquent
|
Hépatite cholestatique‡
| |
Affections de la peau et du tissu souscutané
|
Fréquent
|
Rash
| |
Affections des organes de reproduction et du sein
|
Fréquent
|
Règles irrégulières, dysménorrhée, métrorragie, masse au niveau du sein*
| |
|
Peu fréquent
|
Ménorragie, aménorrhée, polyménorrhée, inflammation du sein*, gynécomastie*, affection du mamelon*, douleur au niveau du mamelon*, oligoménorrhée
| |
Investigations
|
Très fréquent
|
Contamination bactérienne de l'expectoration*
| |
|
Fréquent
|
Augmentation de la créatine kinase sanguine
| |
|
Peu fréquent
|
Augmentation de la pression artérielle
|
* Effets indésirables et fréquences observés chez les patients inclus dans les études cliniques menées avec l'ivacaftor (un des composants d'Orkambi) en monothérapie.
† 1 patient sur 738.
‡ 2 patients sur 738.
Les données de sécurité chez 1 029 patients âgés de 12 ans et plus homozygotes pour la mutation F508del du gène CFTR traités par le lumacaftor/ivacaftor pendant une durée allant jusqu'à 96 semaines supplémentaires dans l'étude d'extension de l'efficacité et de la sécurité à long terme (étude VX12-809-105) étaient comparables à celles issues des études contrôlées contre placebo de 24 semaines (voir «Propriétés/Effets»).
Description de certains effets indésirables
Effets indésirables hépatobiliaires
Pendant les études VX12-809-103 et VX12-809-104, les valeurs maximales des transaminases (ALAT ou ASAT) étaient > 8 x LSN, > 5 x LSN et > 3 x LSN chez respectivement 0,8 %, 2,0 % et 5,2 % chez les patients traités par le lumacaftor/ivacaftor et de 0,5 %, 1,9 % et 5,1 % chez les patients recevant le placebo. L'incidence des augmentations des transaminases était de 5,1 % chez les patients traités par le lumacaftor/ivacaftor et de 4,6 % chez les patients recevant le placebo. Sept patients qui recevaient le lumacaftor/ivacaftor ont présenté des effets indésirables hépatiques graves avec des transaminases élevées, associées pour 3 d'entre eux, à une augmentation de la bilirubine totale. Après l'arrêt du traitement par le lumacaftor/ivacaftor, le bilan hépatique est revenu à son état initial ou s'est nettement amélioré chez tous les patients (voir «Mises en garde et précautions»).
Parmi les sept patients présentant une cirrhose et/ou une hypertension portale préexistantes et qui recevaient le lumacaftor/ivacaftor dans les études de phase III contrôlées contre placebo, une détérioration de la fonction hépatique avec augmentation des ALAT, des ASAT, de la bilirubine et encéphalopathie hépatique a été observée chez un patient. L'événement est survenu dans les cinq jours suivant le début du traitement et les anomalies se sont résolues après l'arrêt du lumacaftor/ivacaftor (voir «Mises en garde et précautions»).
Des cas de décompensation hépatique, y compris d'insuffisance hépatique d'issue fatale, ont été rapportés depuis la commercialisation chez des patients atteints de mucoviscidose présentant une cirrhose préexistante avec hypertension portale et qui étaient traités par l'association lumacaftor/ivacaftor (voir «Mises en garde et précautions»).
Effets indésirables respiratoires
Pendant les études VX12-809-103 et VX12-809-104, l'incidence d'effets indésirables respiratoires (tels que: gêne thoracique, dyspnée, bronchospasme et respiration anormale) était de 26,3 % chez les patients traités par le lumacaftor/ivacaftor et de 17,0 % chez les patients qui recevaient le placebo. La survenue de ces effets indésirables était plus fréquente chez les patients ayant un VEMS plus faible avant le traitement. Environ trois quarts des événements ont débuté au cours de la première semaine de traitement et ont régressé sans interruption du traitement chez la majorité des patients. Dans la majorité des cas, les événements ont été d'intensité légère ou modérée, sans gravité, et n'ont pas entraîné l'arrêt du traitement (voir «Mises en garde et précautions»).
Pendant une étude clinique de phase IIIb en ouvert d'une durée de 24 semaines (étude VX14-809-106) menée chez 46 patients âgés de 12 ans et plus présentant une pneumopathie avancée (VEMS < 40 % de la valeur théorique) (VEMS moyen initial: 29,1 % de la valeur théorique [valeurs extrêmes: 18,3 % à 42,0 %), l'incidence d'effets indésirables respiratoires était de 65,2 %. L'incidence était de 71,4 % dans le sousgroupe de 28 patients chez lesquels le traitement avait été instauré à la pleine dose de lumacaftor/ivacaftor (2 comprimés toutes les 12 heures) et de 55,6 % chez les 18 patients ayant commencé le traitement à une dose réduite de lumacaftor/ivacaftor (1 comprimé toutes les 12 heures pendant une durée allant jusqu'à 2 semaines, avec augmentation ensuite à la pleine dose). Chez les patients ayant commencé le traitement par l'association lumacaftor/ivacaftor à la pleine dose, un patient a présenté un effet indésirable respiratoire grave, la dose a été réduite par la suite chez trois patients et trois patients ont arrêté le traitement. Il n'a pas été observé d'effets indésirables respiratoires graves, de réductions de dose ou d'arrêts du traitement chez les patients qui avaient commencé le traitement à la demi-dose (voir «Mises en garde et précautions»).
Troubles menstruels
Pendant les études VX12-809-103 et VX12-809-104, l'incidence de troubles menstruels (aménorrhée, dysménorrhée, ménorragie, règles irrégulières, métrorragie, oligoménorrhée et polyménorrhée) était de 9,9 % chez les patientes traitées par le lumacaftor/ivacaftor et de 1,7 % chez les patientes recevant le placebo. Ces troubles menstruels sont survenus plus fréquemment dans le sousgroupe de patientes qui utilisaient des contraceptifs hormonaux (25,0 %) que chez celles qui n'en utilisaient pas (3,5 %) (voir «Interactions»). Dans la majorité des cas, ces effets ont été d'intensité légère ou modérée et sans gravité. Chez les patientes traitées par le lumacaftor/ivacaftor, ces effets indésirables se sont résolus dans environ deux tiers des cas et leur durée médiane était de 10 jours.
Augmentation de la pression artérielle
Au cours des études VX12-809-103 et VX12-809-104, des effets indésirables en relation avec une augmentation de la pression artérielle (hypertension, pression artérielle augmentée) ont été rapportés chez 0,9 % (7/738) des patients traités par l'association lumacaftor/ivacaftor mais pas chez les patients qui recevaient le placebo.
Chez les patients traités par l'association lumacaftor/ivacaftor (valeurs initiales moyennes: systolique 114 mmHg et diastolique 69 mmHg), l'augmentation maximale de la pression artérielle systolique moyenne et de la pression diastolique moyenne était respectivement de 3,1 mmHg et 1,8 mmHg par rapport aux valeurs moyennes à l'inclusion. Chez les patients qui recevaient le placebo (valeurs moyennes à l'inclusion: pression systolique 114 mmHg et pression diastolique 69 mmHg), l'augmentation maximale de la pression artérielle systolique moyenne et de la pression diastolique moyenne était respectivement de 0,9 mmHg et 0,9 mmHg par rapport aux valeurs moyennes à l'inclusion.
Les pourcentages de patients présentant à deux reprises au moins une valeur de la pression artérielle systolique > 140 mmHg ou de la pression artérielle diastolique > 90 mmHg étaient respectivement de 3,4 % et 1,5 % chez les patients traités par l'association lumacaftor/ivacaftor alors qu'elles étaient de respectivement de 1,6 % et 0,5 % des patients qui recevaient le placebo (voir «Mises en garde et précautions»).
Enfants et adolescents
Les données de sécurité de l'association lumacaftor/ivacaftor ont été évaluées chez 46 patients âgés de 1 à moins de 2 ans (étude VX16-809-122), 60 patients âgés de 2 à 5 ans (étude VX15-809-115), 161 enfants âgés de 6 à moins de 12 ans (études VX13-809-011B, VX14-809-109 et VX15-809-115) et 194 patients âgés de 12 à 17 ans atteints de mucoviscidose, homozygotes pour la mutation F508del et traités par le lumacaftor/ivacaftor dans les études cliniques. La sécurité de l'association lumacaftor/ivacaftor chez les patients âgés de 1 an et plus qui étaient homozygotes pour la mutation F508del du gène CFTR a également été évaluée dans le cadre d'une étude en ouvert d'une durée de 96 semaines (étude VX19-809-124) chez 52 patients (39 qui ont participé à l'étude VX16-809-122 et 13 patients naïfs de traitement par Orkambi). Les patients âgés de 12 à 17 ans étaient inclus dans les études VX12-809-103 et VX12-809-104.
Le profil de sécurité global chez ces patients pédiatriques est généralement similaire à celui observé chez les patients adultes. Peu d'effets indésirables d'intérêt particulier sont rapportés spécifiquement dans la population pédiatrique.
Les effets indésirables identifiés dans l'étude VX13-809-011B sont inclus dans le tableau 6.
Description de certains effets indésirables chez les patients âgés de 1 à moins de 12 ans
Effets indésirables hépatobiliaires
Pendant l'étude clinique de phase III en ouvert d'une durée de 24 semaines menée chez 58 patients âgés de 6 à moins de 12 ans (étude VX13-809-011B), les valeurs maximales des transaminases (ALAT ou ASAT) étaient > 8 x LSN, > 5 x LSN et > 3 x LSN chez respectivement 5,3 %, 8,8 % et 19,3 % des patients. Aucun patient n'a présenté un taux de bilirubine totale > 2 x LSN. Le traitement par le lumacaftor/ivacaftor a été maintenu ou repris avec succès après une interruption chez tous les patients présentant des élévations des transaminases, à l'exception d'un patient chez lequel le traitement a été arrêté.
Pendant l'étude clinique de phase III contrôlée contre placebo d'une durée de 24 semaines menée chez 204 patients âgés de 6 à moins de 12 ans (étude VX14-809-109), les valeurs maximales des transaminases (ALAT ou ASAT) étaient > 8 x LSN, > 5 x LSN et > 3 x LSN chez respectivement 1,0 %, 4,9 % et 12,6 % des patients traités par l'association lumacaftor/ivacaftor et chez 2,0 %, 3,0 % et 7,9 % des patients recevant le placebo. Aucun patient n'a présenté un taux de bilirubine totale > 2 x LSN. Le traitement a été arrêté en raison d'élévations des transaminases chez deux patients du groupe lumacaftor/ivacaftor et deux patients du groupe placebo.
Au cours de l'étude clinique de phase III en ouvert d'une durée de 24 semaines menée chez 60 patients âgés de 2 à 5 ans (étude VX15-809-115), les valeurs maximales des transaminases (ALAT ou ASAT) étaient > 8 x LSN, > 5 x LSN et > 3 x LSN chez respectivement 8,3 % (5/60), 11,7 % (7/60) et 15,0 % (9/60) des patients. Aucun patient n'a présenté un taux de bilirubine totale > 2 x LSN. Le traitement par l'association lumacaftor/ivacaftor a été arrêté en raison d'élévations des transaminases chez trois patients.
Au cours de l'étude clinique de phase III en ouvert d'une durée de 24 semaines menée chez 46 patients âgés de 1 à moins de 2 ans (étude VX16-809-122), les valeurs maximales des transaminases (ALAT ou ASAT) étaient > 8 x LSN, > 5 x LSN et > 3 x LSN chez respectivement 2,2 % (1/46), 4,3 % (2/46) et 10,9 % (5/46) des patients. Aucun patient n'a présenté un taux de bilirubine totale > 2 x LSN. Le traitement par l'association lumacaftor/ivacaftor a été arrêté en raison d'élévations des transaminases chez un patient.
Les données de sécurité à long terme issues d'une étude d'extension de 96 semaines (étude VX15-809-110) portant sur 239 patients âgés de 6 ans et plus qui étaient homozygotes pour la mutation F508del du gène CFTR étaient globalement cohérentes avec celles des études principales de 24 semaines portant sur des patients âgés de 6 à moins de 12 ans (études VX13-809-011B et VX14-809-109). Dans l'étude VX15-809-110, des augmentations des ALAT et des ASAT sont survenues chez 18,8 % et 13,4 % des patients respectivement. L'incidence des taux maximaux de transaminases (ASAT ou ALAT) > 8, > 5 et > 3 x LSN était respectivement de 3,8 %, 6,7 % et 18,4 %. Aucun patient n'a présenté un taux de bilirubine totale > 2 x LSN. Quatre patients (1,7 %) ont terminé le traitement par Orkambi en raison d'une augmentation des transaminases, et 9 patients (3,8 %) ont interrompu le traitement en raison d'une augmentation des transaminases. Chez 2 patients (0,8 %), l'augmentation des transaminases était sévère, dont un cas a été jugé potentiellement lié au médicament et le traitement a été interrompu, tandis que dans l'autre cas, l'événement a été jugé non lié au médicament et le traitement a été poursuivi. Une hépatite a été signalée chez deux patients, dont l'une a été considérée comme liée au médicament. Dans les deux cas, des augmentations initiales des transaminases étaient présentes et, dans un cas d'hépatite auto-immune classée comme liée au médicament, d'autres facteurs de risque étaient présents en raison de l'utilisation d'un médicament concomitant.
Les données de sécurité à long terme issues d'une étude d'extension de 96 semaines (étude VX15-809-116) portant sur 57 patients âgés de 2 ans et plus, homozygotes pour la mutation F508del du gène CFTR, concordaient pour la plupart avec celles de l'étude principale de 24 semaines portant sur des patients âgés de 2 à 5 ans (étude VX15-809-115).
Dans l'étude VX16-809-116, des augmentations des ALAT et des ASAT ont été observées comme effets indésirables chez 17,5 % et 8,8 % des patients respectivement. L'incidence des augmentations maximales des transaminases (ALAT ou ASAT) > 8, > 5 et > 3 x LSN était respectivement de 3,5 %, 10,5 % et 19,3 %. Aucun patient n'a présenté une augmentation du taux de bilirubine totale > 2 x LSN.
Effets indésirables respiratoires
Pendant l'étude clinique de phase III en ouvert d'une durée de 24 semaines (étude VX13-809-011B) menée chez 58 patients âgés de 6 à moins de 12 ans (VEMS moyen de 91,4 % de la valeur théorique lors de l'inclusion), l'incidence d'effets indésirables respiratoires était de 6,9 % (4/58).
Pendant l'étude clinique de phase III contrôlée contre placebo d'une durée de 24 semaines (étude VX14-809-109) menée chez des patients âgés de 6 à moins de 12 ans (VEMS moyen de 89,8 % de la valeur théorique lors de l'inclusion), l'incidence d'effets indésirables respiratoires était de 18,4 % chez les patients traités par l'association lumacaftor/ivacaftor et de 12,9 % chez les patients recevant le placebo. Une diminution du VEMS exprimé en pourcentage de la valeur théorique a été observée en début de traitement lors des tests de spirométrie en série postdose. La variation absolue entre la valeur prédose et la valeur 4 à 6 heures postdose était de -7,7 le jour 1 et de -1,3 le jour 15 chez les patients du groupe lumacaftor/ivacaftor. La diminution postdose était résolue à la semaine 16.
Exacerbations pulmonaires
Des exacerbations pulmonaires ont été observées dans l'étude ouverte et non contrôlée d'observation de la sécurité à long terme (étude VX15-809-110). L'incidence variait entre 32,3 % (taux d'événements par patient-année 0,30, IC à 95 %: 0,21; 0,43) dans la cohorte ayant reçu le placebo dans l'étude principale, puis un traitement par Orkambi pendant l'étude VX15-809-110, et 49,5 % (taux d'événements par patient-année 0,45, IC à 95 %: 0,33; 0,61) dans la cohorte qui avait été traitée par Orkambi dans les études principales et qui avait également reçu un traitement par Orkambi pendant l'étude VX15-809-110.
Dans l'étude VX16-809-115, l'incidence des exacerbations pulmonaires était de 30,0 % (taux d'événements par patient-année 0,9) chez les patients âgés de 2 à 5 ans. Dans l'étude VX16-809-122, l'incidence des exacerbations pulmonaires était de 19,6 % (taux d'événements par patient-année 0,6) chez les patients âgés de 1 à moins de 2 ans. La définition d'une exacerbation pulmonaire était différente dans les études cliniques menées chez des patients âgés de moins de 6 ans et dans les études menées chez les patients âgés de plus de 6 ans. En outre, la durée de l'étude était de 24 semaines pour les études VX16-809-115 et VX16-809-122, contre 96 semaines pour l'étude VX15-809-110. Par conséquent, les données relatives aux exacerbations pulmonaires ne doivent pas être comparées entre les différentes tranches d'âge et études.
Cataractes
Dans l'étude VX15-809-110, des anomalies oculaires ont été observées en fin d'étude chez 7,5 % des patients en bonne santé oculaire au début de l'étude. Des cataractes ou des opacités du cristallin ont été constatées dans 9 cas (3,8 %): dans un cas, la cataracte a été constatée avant le traitement par Orkambi et dans 2 cas, elle n'a plus été observée lors d'examens ophtalmologiques ultérieurs; des anomalies du cristallin ont été constatées lors de la sélection dans 2 cas; dans les 4 autres cas, il existait des facteurs de risque tels que l'utilisation de corticostéroïdes ou des antécédents familiaux.
Événements de rash
Dans la partie A de l'étude VX16-809-122, menée chez des patients âgés de 1 à moins de 2 ans, un patient (7,1 %) a présenté un événement indésirable de type rash ayant entraîné l'arrêt du traitement, qui s'est résolu sans traitement. Dans la partie B de l'étude VX16-809-122, 4 patients (8,7 %) ont présenté des événements indésirables de type rash. Tous les événements étaient de sévérité légère ou modérée et aucun n'a entraîné l'arrêt du traitement.
Augmentations de la créatine kinase
Dans la partie A de l'étude VX16-809-122, menée chez des patients âgés de 1 à moins de 2 ans, aucun patient n'a présenté d'événement indésirable d'augmentation de la créatine kinase. Dans la partie B de l'étude VX16-809-122, un patient (2,2 %) a présenté un événement indésirable d'augmentation de la créatine kinase qui était de sévérité légère et n'a pas entraîné l'interruption ou l'arrêt du traitement par le médicament expérimental. Aucun patient dans la partie A ni la partie B n'a présenté un taux de CK > 3 x LSN.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageTraitement
Il n'existe pas d'antidote spécifique en cas de surdosage de lumacaftor/ivacaftor. La conduite à tenir en cas de surdosage est la surveillance des signes vitaux et de l'état clinique du patient et de mesures adaptées.
Signes et symptômes
Les effets indésirables survenus avec une incidence plus élevée (≥5 %) avec une dose supérieure à la dose thérapeutique comparativement à l'administration d'une dose thérapeutique étaient: des céphalées, un rash généralisé et une augmentation des transaminases.
Propriétés/EffetsCode ATC
R07AX30
Mécanisme d'action
Classe pharmacothérapeutique: autres produits pour le système respiratoire
La protéine CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) est un canal chlorure présent à la surface des cellules épithéliales de nombreux organes. La mutation F508del affecte la protéine CFTR de différentes façons, principalement en entraînant un défaut de maturation (repliement incorrect) et de trafic intracellulaires qui diminue la quantité de protéines CFTR à la surface des cellules. La petite quantité de protéines F508del-CFTR qui atteint la surface cellulaire présente une probabilité faible d'ouverture du canal (régulation défectueuse du canal). Le lumacaftor est un correcteur de CFTR qui agit directement sur la protéine F508del-CFTR pour améliorer sa maturation et son trafic intracellulaires, en augmentant ainsi la quantité de protéines CFTR fonctionnelles à la surface cellulaire. L'ivacaftor potentialise l'activité de la protéine CFTR et améliore le transport des ions chlorures en augmentant la probabilité d'ouverture (ou de régulation) du canal CFTR à la surface cellulaire. L'effet combiné du lumacaftor et de l'ivacaftor est une augmentation de la quantité et de l'activité des protéines F508del-CFTR à la surface cellulaire, ce qui entraîne une augmentation du transport des ions chlorures. Les mécanismes exacts par lesquels le lumacaftor améliore la maturation et le transport intracellulaires de la protéine F508del-CFTR et par lesquels l'ivacaftor potentialise son activité ne sont pas connus.
Pharmacodynamique
Effets sur la concentration en chlorures dans la sueur
Les variations du taux de chlorures dans la sueur en réponse au lumacaftor administré seul ou en association avec l'ivacaftor ont été évaluées dans une étude clinique de phase II en double aveugle, contrôlée contre placebo, menée chez des patients atteints de mucoviscidose âgés de 18 ans et plus. Dans cette étude, 10 patients (homozygotes pour la mutation F508del-CFTR) ont été traités par le lumacaftor en monothérapie à la dose de 400 mg toutes les 12 heures pendant 28 jours, avec ensuite l'ajout d'ivacaftor 250 mg toutes les 12 heures pendant une période supplémentaire de 28 jours; 25 patients (homozygotes ou hétérozygotes pour la mutation F508del) ont reçu le placebo. La différence entre le lumacaftor 400 mg toutes les 12 heures en monothérapie et le placebo, évaluée par la variation moyenne du taux de chlorures dans la sueur au jour 28 par rapport à la valeur initiale était statistiquement significative, avec une différence de -8,2 mmol/l (IC à 95 %: -14; -2). La différence entre l'association de lumacaftor 400 mg/ivacaftor 250 mg toutes les 12 heures et le placebo, évaluée par la variation moyenne du taux de chlorures dans la sueur au jour 56 par rapport à la valeur initiale était statistiquement significative, avec une différence de -11 mmol/l (IC à 95 %: -18; -4).
Les variations du taux de chlorures dans la sueur en réponse à l'association lumacaftor/ivacaftor ont été évaluées dans le cadre d'une étude clinique de phase III contrôlée contre placebo d'une durée de 24 semaines (étude VX14-809-109) menée chez 204 patients atteints de mucoviscidose âgés de 6 à moins de 12 ans homozygotes pour la mutation F508del du gène CFTR (103 patients recevaient l'association lumacaftor 200 mg/ivacaftor 250 mg toutes les 12 heures et 101 patients recevaient le placebo). Par rapport au placebo, le traitement par le lumacaftor/ivacaftor a induit une diminution statistiquement significative du taux de chlorures dans la sueur, qui s'est maintenue pendant les 24 semaines de traitement. Par rapport au placebo, la différence entre les traitements (moyenne des moindres carrés [MC]) de la variation absolue moyenne du taux de chlorures dans la sueur au jour 15 et à la semaine 4 était de -20,8 mmol/l (IC à 95 %: -23,4; -18,2; P < 0,0001). Par rapport au placebo, la différence entre les traitements (moyenne des MC) de la variation absolue du taux de chlorures dans la sueur à la semaine 24 était de -24,9 mmol/l (P < 0,0001).
Dans l'étude VX15-809-115 menée chez des patients âgés de 2 à 5 ans homozygotes pour la mutation F508del-CFTR, la variation intragroupe absolue moyenne du taux de chlorures dans la sueur à la semaine 24 par rapport à la valeur initiale était de -31,7 mmol/l (IC à 95 %: -35,7; -27,6). De plus, la variation absolue moyenne du taux de chlorures dans la sueur de la semaine 24 à la semaine 26 après la période de wash-out de 2 semaines (destinée à évaluer la réponse sans traitement) était une augmentation de 33,0 mmol/l (IC à 95 %: 28,9; 37,1; valeur P nominale < 0,0001), ce qui représente un retour à la valeur initiale après le sevrage thérapeutique. À la semaine 24, le taux de chlorures dans la sueur avait diminué en dessous de 60 mmol/l chez 16 % des enfants, aucun d'entre eux n'étant descendu en dessous de 30 mmol/l.
Dans l'étude VX16-809-122 menée chez des patients âgés de 1 à moins de 2 ans homozygotes pour la mutation F508del-CFTR, le traitement par l'association lumacaftor/ivacaftor avait induit à la semaine 4 une réduction du taux de chlorures dans la sueur qui s'est maintenue jusqu'à la semaine 24. La variation absolue moyenne du taux de chlorures dans la sueur à la semaine 24 par rapport à la valeur initiale était de -29,1 (écarttype [ET]: 13,5) mmol/l (IC à 95 %: -34,8; -23,4). De plus, la variation absolue moyenne du taux de chlorures dans la sueur de la semaine 24 à la semaine 26 après la période de wash-out de deux semaines était de 27,3 (ET: 11,1) mmol/l (IC à 95 %: 22,3; 32,3). Cette variation représente un retour vers la valeur initiale après le sevrage thérapeutique.
Variations du VEMS
Les variations du VEMS en pourcentage de la valeur théorique en réponse au lumacaftor administré seul ou en association avec l'ivacaftor ont également été évaluées dans l'étude de phase II en double aveugle, contrôlée contre placebo, menée chez des patients atteints de mucoviscidose âgés de 18 ans et plus. La différence entre la variation absolue moyenne du VEMS en pourcentage de la valeur théorique mesurée dans le groupe lumacaftor 400 mg administré seul toutes les 12 heures et dans le groupe placebo, était de -4,6 % (IC à 95 %: -9,6; 0,4) au jour 28 par rapport à la valeur initiale et de 4,2 % (IC à 95 %: -1,3; 9,7) au jour 56 par rapport à la valeur initiale et de 7,7 % (IC à 95 %: 2,6; 12,8) au jour 56 par rapport au jour 28 (après l'ajout de l'ivacaftor à la monothérapie par le lumacaftor).
Diminution de la fréquence cardiaque
Pendant les études de phase III contrôlées contre placebo de 24 semaines, une diminution maximale de la fréquence cardiaque moyenne de 6 battements par minute (bpm) par rapport à la valeur initiale a été observée le jour 1 et le jour 15, 4 à 6 heures environ après l'administration. Après le jour 15, la fréquence cardiaque n'était pas contrôlée pendant la période post-dose dans ces études. À partir de la semaine 4, la variation de la fréquence cardiaque moyenne mesurée avant administration de la dose était de 1 à 2 bpm en dessous de la valeur initiale chez les patients traités par l'association lumacaftor/ivacaftor. Les pourcentages de patients ayant des valeurs de fréquence cardiaque < 50 bpm sous traitement étaient de 11 % chez ceux qui recevaient l'association lumacaftor/ivacaftor
versus 4,9 % chez ceux qui recevaient le placebo.
Électrophysiologie cardiaque
Aucune modification significative de l'intervalle QTc ou de la pression artérielle n'a été observée dans le cadre d'une étude clinique approfondie sur le QT évaluant le lumacaftor 600 mg une fois par jour/ivacaftor 250 mg toutes les 12 heures et le lumacaftor 1000 mg une fois par jour/ivacaftor 450 mg toutes les 12 heures.
Efficacité clinique
Efficacité et sécurité cliniques
Études chez des patients âgés de 12 ans et plus atteints de mucoviscidose homozygotes pour la mutation F508del du gène CFTR
L'efficacité de l'association lumacaftor/ivacaftor chez les patients atteints de mucoviscidose homozygotes pour la mutation F508del du gène CFTR a été évaluée dans deux études cliniques randomisées en double aveugle, contrôlées contre placebo, menées chez 1 108 patients cliniquement stables, dans lesquelles 737 patients ont été randomisés pour être traités par l'association lumacaftor/ivacaftor. Dans les deux études, les patients ont été randomisés (1:1:1) pour recevoir le lumacaftor 600 mg une fois par jour + ivacaftor 250 mg toutes les 12 heures, le lumacaftor 400 mg toutes les 12 heures + ivacaftor 250 mg toutes les 12 heures ou le placebo. Les patients ont pris le médicament expérimental avec un repas ou une collation contenant des graisses pendant 24 semaines, en plus de leurs traitements prescrits pour la mucoviscidose (tels que: bronchodilatateurs, antibiotiques inhalés, dornase alfa et nébulisation de solution saline hypertonique). Les patients de ces études étaient éligibles pour entrer dans une étude d'extension en aveugle.
Il n'a pas été mené d'étude clinique comparative évaluant directement la supériorité de l'association par rapport aux composants en monothérapie.
L'étude VX12-809-103 a évalué 549 patients atteints de mucoviscidose âgés de 12 ans et plus (âge moyen: 25,1 ans) ayant un VEMS allant de 40 % à 90 % de la valeur théorique lors de la sélection (VEMS moyen initial: 60,7 % de la valeur théorique [valeurs extrêmes: 31,1 % à 94,0 %]). L'étude VX12-809-104 a évalué 559 patients âgés de 12 ans et plus (âge moyen: 25,0 ans) ayant un VEMS allant de 40 % à 90 % de la valeur théorique lors de la sélection (VEMS moyen initial: 60,5 % de la valeur théorique [valeurs extrêmes: 31,3 % à 99,8 %]). Les patients ayant des antécédents de colonisation par des agents pathogènes tels que Burkholderia cenocepacia, Burkholderia dolosa ou Mycobacterium abscessus ou qui présentaient des anomalies de trois paramètres hépatiques ou plus (ALAT, ASAT, PA, GGT ≥3 x LSN ou bilirubine totale ≥2 x LSN) étaient exclus.
Le critère d'évaluation principal de l'efficacité dans les deux études était la variation absolue du VEMS en pourcentage de la valeur théorique à la semaine 24 par rapport à la valeur initiale. Les autres critères d'évaluation de l'efficacité étaient: la variation relative du VEMS en pourcentage de la valeur théorique à la semaine 24 par rapport à la valeur initiale, la variation absolue de l'indice de masse corporel (IMC) à la semaine 24 par rapport à la valeur initiale, la variation absolue du score du domaine respiratoire du questionnaire CFQ-R (Cystic Fibrosis Questionnaire-Revised) à la semaine 24 par rapport à la valeur initiale, le pourcentage de patients obtenant une variation relative ≥5 % du VEMS en pourcentage de la valeur théorique à la semaine 24 par rapport à la valeur initiale et le nombre d'exacerbations pulmonaires (incluant celles nécessitant une hospitalisation ou une antibiothérapie par voie intraveineuse) jusqu'à la semaine 24.
Dans les deux études, une amélioration statistiquement significative du VEMS exprimé en pourcentage de la valeur théorique a été observée dans le groupe traité par le lumacaftor/ivacaftor (voir tableau 7). L'amélioration moyenne du VEMS en pourcentage de la valeur théorique est apparue rapidement (jour 15) et s'est maintenue pendant toute la période de traitement de 24 semaines. Dans l'analyse regroupant les valeurs des études VX12-809-103 et VX12-809-104, la différence observée au jour 15 entre la variation absolue moyenne (IC à 95 %) du VEMS exprimé en pourcentage de la valeur théorique, par rapport à la valeur initiale entre le groupe lumacaftor 400 mg/ivacaftor 250 mg administré toutes les 12 heures et le groupe placebo était de 2,51 % (P < 0,0001). Les améliorations observées sur le VEMS exprimé en pourcentage de la valeur théorique, n'étaient pas influencées par l'âge, la sévérité de la maladie, le sexe et la région géographique. Les études de phase III du lumacaftor/ivacaftor ont inclus 81 patients ayant un VEMS < 40 % de la valeur théorique lors de l'inclusion. La différence entre les traitements dans ce sousgroupe de patients a été comparable à celle observée chez les patients ayant un VEMS ≥40 % de la valeur théorique. Dans l'analyse regroupant les valeurs des études VX12-809-103 et VX12-809-104, la différence entre la variation absolue moyenne (IC à 95 %) du VEMS en pourcentage de la valeur théorique à la semaine 24 par rapport à la valeur initiale observée dans le groupe lumacaftor 400 mg/ivacaftor 250 mg administré toutes les 12 heures et dans le groupe placebo était de 3,39 % (P = 0,0382) chez les patients ayant un VEMS < 40 % de la valeur théorique et de 2,47 % (P < 0,0001) chez ceux qui avaient un VEMS ≥40 % de la valeur théorique.
|
Tableau 7: Synthèse des résultats du critère d'évaluation principal et des principaux critères secondaires dans l'étude VX12-809-103 et l'étude VX12-809-104*
| |
|
Étude VX12-809-103
|
Étude VX12-809-104
|
Données combinées
(étude VX-12-809-103 et étude VX-12-809-104)
| |
|
Placebo
(n = 184)
|
LUM 400 mg ttes les 12 h/ IVA 250 mg ttes les 12 h
(n = 182)
|
Placebo
(n = 187)
|
LUM 400 mg ttes les 12 h/IVA 250 mg ttes les 12 h
(n = 187)
|
Placebo
(n = 371)
|
LUM 400 mg ttes les 12 h/IVA 250 mg ttes les 12 h
(n = 369)
| |
Variation absolue du VEMS en pourcentage de la valeur théorique à la semaine 24 (%)
|
Différence entre les traitements
|
–
|
2,41
(P = 0,0003) †
|
–
|
2,65
(P = 0,0011) †
|
–
|
2,55
(P < 0,0001)
| |
|
Variation intragroupe
|
-0,73
(P = 0,2168)
|
1,68
(P = 0,0051)
|
-0,02
(P = 0,9730)
|
2,63
(P < 0,0001)
|
-0,39
(P < 0,3494)
|
2,16
(P < 0,0001)
| |
Variation relative du VEMS en pourcentage de la valeur théorique à la semaine 24 (%)
|
Différence entre les traitements
|
–
|
4,15
(P = 0,0028)†
|
–
|
4,69
(P = 0,0009)†
|
–
|
4,4
(P < 0,0001)
| |
|
Variation intragroupe
|
-0,85
(P = 0,3934)
|
3,3
(P = 0,0011)
|
0,16
(P = 0,8793)
|
4,85
(P < 0,0001)
|
-0,34
(P = 0,6375)
|
4,1
(P < 0,0001)
| |
Variation absolue de l'IMC à la semaine 24 (kg/m2)
|
Différence entre les traitements
|
–
|
0,13
(P = 0,1938)
|
–
|
0,36
(P < 0,0001)†
|
–
|
0,24
(P = 0,0004)
| |
|
Variation intragroupe
|
0,19
(P = 0,0065)
|
0,32
(P < 0,0001)
|
0,07
(P = 0,2892)
|
0,43
(P < 0,0001)
|
0,13
(P = 0,0066)
|
0,37
(P < 0,0001)
| |
Variation absolue du score du domaine respiratoire du questionnaire CFQ-R à la semaine 24 (points)
|
Différence entre les traitements
|
–
|
1,5
(P = 0,3569)
|
–
|
2,9
(P = 0,0736)
|
–
|
2,2
(P = 0,0512)
| |
|
Variation intragroupe
|
1,1
(P = 0,3423)
|
2,6
(P = 0,0295)
|
2,8
(P = 0,0152)
|
5,7
(P < 0,0001)
|
1,9
(P = 0,0213)
|
4,1
(P < 0,0001)
| |
Pourcentage de patients présentant une variation relative ≥5 % du VEMS en pourcentage de la valeur théorique à la semaine 24
|
%
|
25 %
|
32 %
|
26 %
|
41 %
|
26 %
|
37 %
| |
|
Odds ratio
|
–
|
1,43
(P = 0,1208)
|
–
|
1,90
(P = 0,0032)
|
–
|
1,66
(P = 0,0013)
| |
Nombre d'exacerbations pulmonaires jusqu'à la semaine 24
|
Nombre d'événements (taux par période de 48 semaines)
|
112 (1,07)
|
73 (0,71)
|
139 (1,18)
|
79 (0,67)
|
251 (1,14)
|
152 (0,70)
| |
|
Rapport des taux
|
–
|
0,66
(P = 0,0169)
|
–
|
0,57
(P = 0,0002)
|
–
|
0,61
(P < 0,0001)
| |
* Dans chaque étude, une procédure de tests hiérarchisés a été effectuée dans chaque groupe de traitement actif pour les critères d'évaluation principal et secondaires versus placebo; à chaque étape, une valeur de P ≤0,0250 avec tous les tests antérieurs satisfaisant également ce seuil de significativité étaient requis pour déclarer la significativité statistique.
† Indique la significativité statistique confirmée dans la procédure de tests hiérarchisés.
|
À la semaine 24, le pourcentage de patients qui restaient sans exacerbations pulmonaires était significativement plus élevé chez les patients traités par l'association lumacaftor/ivacaftor que chez ceux qui recevaient le placebo. Dans l'analyse regroupant les 2 études, le rapport des taux d'exacerbations jusqu'à la semaine 24 chez les patients traités par le lumacaftor/ivacaftor (lumacaftor 400 mg/ivacaftor 250 mg toutes les 12 heures; n = 369) était de 0,61 (P < 0,0001), soit une réduction de 39 % par rapport au placebo. Le taux d'événements, annualisé à la semaine 48, était de 0,70 par an dans le groupe lumacaftor/ivacaftor et de 1,14 par an dans le groupe placebo. Comparativement au placebo, le traitement par l'association lumacaftor/ivacaftor a entraîné une diminution significative de 61 % du risque d'exacerbations nécessitant une hospitalisation (rapport des taux = 0,39, P < 0,0001; taux d'événements par période de 48 semaines: 0,17 dans le groupe lumacaftor/ivacaftor et 0,45 dans le groupe placebo) et une réduction de 56 % des exacerbations nécessitant une antibiothérapie par voie intraveineuse (rapport des taux = 0,44, P < 0,0001; taux d'événements par période de 48 semaines: 0,25 avec le lumacaftor/ivacaftor et 0,58 avec le placebo). Ces résultats n'ont pas été considérés comme statistiquement significatifs dans le cadre de la hiérarchie de tests pour chaque étude.
Étude d'extension de l'efficacité et de la sécurité à long terme
L'étude VX12-809-105 était une étude d'extension de phase III multicentrique, en groupes parallèles, menée chez des patients atteints de mucoviscidose âgés de 12 ans et plus ayant participé aux études VX12-809-103 et VX12-809-104. Cette étude d'extension avait pour objectif l'évaluation de la sécurité et de l'efficacité d'un traitement à long terme par l'association lumacaftor/ivacaftor. Sur les 1 108 patients ayant reçu un des traitements dans l'étude VX12-809-103 ou l'étude VX12-809-104, 1 029 patients (93 %) ont été traités dans l'étude VX12-809-105 et ont reçu le traitement actif (lumacaftor 600 mg une fois par jour/ivacaftor 250 mg q12h ou lumacaftor 400 mg q12h/ivacaftor 250 mg q12h) pendant une durée allant jusqu'à 96 semaines supplémentaires (c'est-à-dire jusqu'à 120 semaines au total). L'analyse principale d'efficacité de cette étude d'extension incluait les données jusqu'à la semaine 72 de l'étude VX12-809-105, avec une analyse de sensibilité incluant les données jusqu'à la semaine 96 de cette étude VX12-809-105.
Les patients traités par l'association lumacaftor/ivacaftor dans l'étude VX12-809-103 et l'étude VX12-809-104 ont présenté un effet qui était maintenu par rapport à l'inclusion après 96 semaines supplémentaires dans l'étude VX12-809-105. Chez les patients initialement traités par le placebo et chez qui le traitement actif était débuté, les variations observées par rapport à la valeur initiale étaient comparables à celles observées chez les patients initialement traités par l'association lumacaftor/ivacaftor dans l'étude VX12-809-103 et l'étude VX12-809-104 (voir tableau 7). Les résultats de l'étude VX12-809-105 sont présentés dans la figure 1 et le tableau 8.
Figure 1. Variation absolue du VEMS exprimé en pourcentage de la valeur théorique par rapport à la valeur initiale lors de chaque visite†
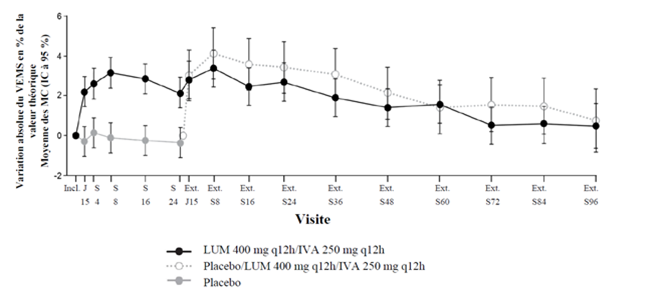
† Données des études VX12-809-103, VX12-809-104 et VX12-809-105.
|
Tableau 8: Effet à long terme de l'association lumacaftor/ivacaftor dans l'étude VX12-809-105*
|
| |
Valeur initiale et résultat
|
Relais du placebo au
lumacaftor 400 mg q12h/
ivacaftor 250 mg q12h
(N = 176)**
|
lumacaftor 400 mg q12h/
ivacaftor 250 mg q12h
(N = 369)†
|
| |
|
Moyenne (ET)
|
Moyennes des MC (IC à 95 %)
|
Valeur P
|
Moyenne (ET)
|
Moyennes des MC
(IC à 95 %)
|
Valeur P
| |
Variation absolue du VEMS en pourcentage de la valeur théorique par rapport à la valeur initiale (%)
|
| |
VEMS en pourcentage de la valeur théorique initial‡
|
60,2 (14,7)
|
|
|
60,5 (14,1)
|
|
| |
Semaine 72 de l'étude d'extension
|
|
(n = 134)
1,5
(0,2; 2,9)
|
0,0254
|
|
(n = 273)
0,5
(-0,4; 1,5)
|
0,2806
| |
Semaine 96 de l'étude d'extension
|
|
(n = 75)
0,8
(-0,8; 2,3)
|
0,3495
|
|
(n = 147)
0,5
(-0,7; 1,6)
|
0,4231
| |
Variation relative du VEMS en pourcentage de la valeur théorique par rapport à la valeur initiale (%)
|
| |
Semaine 72 de l'étude d'extension
|
|
(n = 134)
2,6
(0,2; 5,0)
|
0,0332
|
|
(n = 273)
1,4
(-0,3; 3,2)
|
0,1074
| |
Semaine 96 de l'étude d'extension
|
|
(n = 75)
1,1
(-1,7; 3,9)
|
0,4415
|
|
(n = 147)
1,2
(-0,8; 3,3)
|
0,2372
| |
IMC initial (kg/m2)‡
|
20,9 (2,8)
|
|
|
21,5 (3,0)
|
|
| |
Variation absolue de l'IMC par rapport à la valeur initiale (kg/m2)
|
| |
Semaine 72 de l'étude d'extension
|
|
(n = 145)
0,62
(0,45; 0,79)
|
< 0,0001
|
|
(n = 289)
0,69
(0,56; 0,81)
|
< 0,0001
| |
Semaine 96 de l'étude d'extension
|
|
(n = 80)
0,76
(0,56; 0,97)
|
< 0,0001
|
|
(n = 155)
0,96
(0,81; 1,11)
|
< 0,0001
| |
Score initial du domaine respiratoire du questionnaire CFQ-R (points)‡
|
70,4 (18,5)
|
|
|
68,3 (18,0)
|
|
| |
Variation absolue du score du domaine respiratoire du questionnaire CFQ-R (points)
|
| |
Semaine 72 de l'étude d'extension
|
|
(n = 135)
3,3
(0,7; 5,9)
|
0,0124
|
|
(n = 269)
5,7
(3,8; 7,5)
|
< 0,0001
| |
Semaine 96 de l'étude d'extension
|
|
(n = 81)
0,5
(-2,7; 3,6)
|
0,7665
|
|
(n = 165)
3,5
(1,3; 5,8)
|
0,0018
| |
Nombre d'exacerbations pulmonaires (événements) ** † ***
|
| |
Nombre d'événements par patient-année (IC à 95 %) (taux/48 semaines)
|
|
0,69
(0,56; 0,85)
|
|
|
0,65
(0,56; 0,75)
|
| |
Nombre d'événements nécessitant une hospitalisation par patient-année (IC à 95 %) (taux/48 semaines)
|
|
0,30
(0,22; 0,40)
|
|
|
0,24
(0,19; 0,29)
|
| |
Nombre d'événements nécessitant une antibiothérapie par voie IV par patient-année (IC à 95 %) (taux/48 semaines)
|
|
0,37
(0,29; 0,49)
|
|
|
0,32
(0,26; 0,38)
|
| |
* Au total, 82 % (421 des 516 patients éligibles) ont terminé la période de 72 semaines de cette étude; 42 % ont terminé la période de 96 semaines. Dans la majorité des cas, les patients sont sortis de l'étude pour des raisons autres que des événements indésirables.
** Chez les patients qui avaient participé aux études VX12-809-103 et VX12-809-104 (groupe avec remplacement du placebo par l'association lumacaftor/ivacaftor), l'exposition totale allait jusqu'à 96 semaines. La présentation du groupe de dose lumacaftor 400 mg q12h/ivacaftor 250 mg q12h concorde avec la posologie recommandée.
*** Le taux d'événements par patient-année était annualisé à 48 semaines.
† Chez les patients qui avaient participé aux études VX12-809-103 et VX12-809-104 (groupe traité par l'association lumacaftor/ivacaftor dans ces études), l'exposition totale allait jusqu'à 120 semaines. La posologie du groupe lumacaftor 400 mg q12h/ivacaftor 250 mg q12h concorde avec la posologie recommandée.
‡ La valeur initiale pour le groupe de patients sous placébo puis traités par l'association lumacaftor 400 mg q12h/ivacaftor 250 mg q12h était la valeur à l'inclusion dans l'étude VX12-809-105. La valeur initiale pour le groupe lumacaftor 400 mg q12h/ivacaftor 250 mg q12h était la valeur à l'inclusion dans l'étude VX12-809-103 et VX12-809-104.
|
Étude chez des patients atteints de mucoviscidose hétérozygotes pour la mutation F508del du gène CFTR
L'étude VX09-809-102 était une étude de phase II multicentrique, randomisée en double aveugle, contrôlée contre placebo, menée chez 125 patients atteints de mucoviscidose âgés de 18 ans et plus ayant un VEMS allant de 40 % à 90 % de la valeur théorique et qui étaient porteurs de la mutation F508del sur un allèle, avec sur le second allèle une mutation susceptible d'entraîner l'absence de synthèse de la protéine CFTR ou la synthèse d'une protéine CFTR ne répondant pas à l'ivacaftor in vitro.
Les patients ont reçu l'association lumacaftor/ivacaftor (n = 62) ou le placebo (n = 63) en plus de leurs traitements prescrits pour la mucoviscidose. Le critère d'évaluation principal était l'amélioration de la fonction pulmonaire, déterminée par la variation absolue moyenne du VEMS exprimé en pourcentage de la valeur théorique au jour 56 par rapport à la valeur initiale. Par rapport au placebo, le traitement par l'association lumacaftor/ivacaftor n'a pas entraîné d'amélioration significative du VEMS exprimé en pourcentage de la valeur théorique- chez les patients atteints de mucoviscidose hétérozygotes pour la mutation F508del du gène CFTR (différence entre traitements: 0,60 [P = 0,5978]), ni d'améliorations significatives de l'IMC ou du poids (voir «Mises en garde et précautions»).
Enfants et adolescents
Étude chez des patients atteints de mucoviscidose âgés de 6 à moins de 12 ans homozygotes pour la mutation F508del du gène CFTR
L'étude VX14-809-109 était une étude clinique de phase III contrôlée contre placebo d'une durée de 24 semaines menée chez 204 patients atteints de mucoviscidose âgés de 6 à moins de 12 ans (âge moyen: 8,8 ans). Cette étude a évalué des patients ayant un index de clairance pulmonaire (ICP2,5) ≥7,5 lors de la première visite de sélection (ICP2,5 moyen de 10,28 lors de l'inclusion [valeurs extrêmes: 6,55 à 16,38]) et un VEMS ≥70 % de la valeur théorique lors de la sélection (VEMS moyen de 89,8 % de la valeur théorique lors de l'inclusion [valeurs extrêmes: 48,6 à 119,6]). Les patients ont reçu l'association lumacaftor 200 mg/ivacaftor 250 mg toutes les 12 heures (n = 103) ou le placebo (n = 101) en plus de leurs traitements prescrits pour la mucoviscidose. Les patients qui présentaient des anomalies de deux paramètres hépatiques ou plus (ALAT, ASAT, PA, GGT ≥3 x LSN) ou des taux d'ALAT ou d'ASAT > 5 x LSN ou de bilirubine totale > 2 x LSN étaient exclus.
Le critère d'évaluation principal de l'efficacité était la variation absolue de l'ICP2,5 jusqu'à la semaine 24 par rapport à la valeur initiale. Les principaux critères secondaires étaient la variation absolue moyenne du taux de chlorures dans la sueur au jour 15, à la semaine 4 et à la semaine 24 par rapport à la valeur initiale (voir «Effets pharmacodynamiques»), la variation absolue de l'IMC à la semaine 24 par rapport à la valeur initiale, la variation absolue du score du domaine respiratoire du questionnaire CFQ-R jusqu'à la semaine 24 par rapport au score initial. Ces résultats sont présentés dans le tableau 9 cidessous:
|
Tableau 9: Synthèse des résultats du critère d'évaluation principal et des principaux critères secondaires dans l'étude VX14-809-109
| |
|
Placebo
(n = 101)
|
LUM 200 mg/IVA 250 mg toutes les 12 h
(n = 103)
| |
Critère principal
| |
Variation absolue de l'index de clairance pulmonaire (ICP2,5) jusqu'à la semaine 24 par rapport à la valeur initiale
|
Différence entre les traitements
|
–
|
-1,09
(P < 0,0001)
| |
|
Variation intragroupe
|
0,08
(P = 0,5390)
|
-1,01 (P < 0,0001)
| |
Principaux critères secondaires*
| |
Variation absolue de l'IMC à la semaine 24 (kg/m2)
|
Différence entre les traitements
|
–
|
0,11
(P = 0,2522)
| |
|
Variation intragroupe
|
0,27
(P = 0,0002)
|
0,38
(P < 0,0001)
| |
Variation absolue du score du domaine respiratoire CFQ-R jusqu'à la semaine 24 (points)
|
Différence entre les traitements
|
–
|
2,5
(P = 0,0628)
| |
|
Variation intragroupe
|
3,0
(P = 0,0035)
|
5,5
(P < 0,0001)
| |
* L'étude comportait les principaux critères secondaires et d'autres critères secondaires.
|
Le VEMS exprimé en pourcentage de la valeur théorique était également évalué en tant qu'autre critère secondaire cliniquement significatif. Chez les patients du groupe lumacaftor/ivacaftor, la différence entre les traitements de la variation absolue du VEMS en pourcentage de la valeur théorique entre l'inclusion et la semaine 24 était de 2,4 (P = 0,0182).
Étude VX15-809-115: étude de la sécurité et de la tolérance chez des enfants atteints de mucoviscidose âgés de 2 à 5 ans homozygotes pour la mutation F508del du gène CFTR
L'étude VX15-809-115 a été menée chez 60 patients âgés de 2 à 5 ans lors de la sélection (âge moyen à l'inclusion: 3,7 ans). En fonction de leur poids lors de la sélection, les patients ont reçu les granulés mélangés avec des aliments toutes les 12 heures, à la dose de 100 mg de lumacaftor/125 mg d'ivacaftor chez les patients pesant moins de 14 kg (n = 19) ou de 150 mg de lumacaftor/188 mg d'ivacaftor chez les patients pesant 14 kg et plus (n = 41) pendant 24 semaines en plus de leurs traitements prescrits pour la mucoviscidose. Afin d'évaluer les effets sans médicament, une visite de suivi de la sécurité était effectuée après une période de wash-out de 2 semaines.
Les critères d'évaluation secondaires étaient la variation absolue du taux de chlorures dans la sueur à la semaine 24 par rapport à la valeur initiale et la variation absolue du taux de chlorures dans la sueur de la semaine 24 à la semaine 26 (voir «Effets pharmacodynamiques»). Les critères présentés dans le tableau 10 étaient en outre pris en compte. La pertinence clinique de l'ampleur de ces variations chez les enfants atteints de mucoviscidose âgés de 2 à 5 ans n'a pas été clairement établie à ce jour en cas de traitement de longue durée.
|
Tableau 10: Synthèse des résultats des critères secondaires dans l'étude VX15-809-115
| |
Critères secondaires*
|
LUM/IVA
| |
Variation absolue de l'indice de masse corporelle (IMC) par rapport à la valeur initiale
|
n = 57
0,27
IC à 95 %: 0,07; 0,47;
P = 0,0091
| |
ICP2,5
|
n = 17
-0,58
IC à 95 %: -1,17; 0,02;
P = 0,0559
|
Remarque: les valeurs P présentées dans le tableau sont les valeurs nominales.
* La variation absolue par rapport à la valeur initiale des critères présentés est la variation absolue moyenne à la semaine 24 par rapport à la valeur initiale.
Étude VX16-809-122: étude de la sécurité et de la tolérance chez des enfants atteints de mucoviscidose âgés de 1 à moins de 2 ans homozygotes pour la mutation F508del du gène CFTR
Dans la partie B de l'étude VX16-809-122, la sécurité et la tolérance (critère d'évaluation principal) ont été évaluées chez 46 patients (âge moyen à l'inclusion: 18,1 mois) pendant 24 semaines. Les critères d'évaluation secondaires étaient les paramètres pharmacocinétiques et la variation absolue du taux de chlorures dans la sueur à la semaine 24 par rapport à la valeur initiale (voir «Effets pharmacodynamiques»). En fonction de leur poids lors de la sélection, les patients ont reçu les granulés mélangés avec des aliments toutes les 12 heures, à la dose de 75 mg de lumacaftor/94 mg d'ivacaftor (patients pesant de 7 kg à moins de 9 kg), de 100 mg de lumacaftor/125 mg d'ivacaftor (patients pesant de 9 kg à < 14 kg) ou de 150 mg de lumacaftor/188 mg d'ivacaftor (patients pesant 14 kg et plus) pendant 24 semaines en plus de leurs traitements prescrits pour la mucoviscidose. Afin d'évaluer les effets sans traitement, une visite de suivi de la sécurité était effectuée après une période de wash-out de 2 semaines.
Étude VX15-809-110: étude d'extension de 96 semaines
Les patients atteints de mucoviscidose âgés de 6 ans et plus de l'étude VX13-809-011B et de l'étude VX14-809-109 ont été inclus dans une étude multicentrique d'extension de phase III (étude VX15-809-110). Cette étude d'extension non contrôlée a été conçue pour évaluer la sécurité et l'efficacité descriptive d'un traitement à long terme par lumacaftor/ivacaftor. Sur les 262 patients ayant reçu l'un des traitements dans l'étude VX13-809-011B ou l'étude VX14-809-109, 239 (91 %) ont été traités dans l'étude d'extension et ont tous reçu le traitement par lumacaftor/ivacaftor (les patients âgés de 6 à < 12 ans ont reçu lumacaftor 200 mg toutes les 12 heures/ivacaftor 250 mg toutes les 12 heures; les patients âgés de ≥12 ans ont reçu lumacaftor 400 mg toutes les 12 heures/ivacaftor 250 mg toutes les 12 heures) pendant une durée supplémentaire pouvant aller jusqu'à 96 semaines (c'est-à-dire pendant un maximum de 120 semaines au total) (voir «Effets indésirables»). Les résultats descriptifs secondaires d'efficacité et le nombre d'exacerbations pulmonaires par patient-année sont résumés dans le tableau 11.
|
Tableau 11: Effet à long terme de l'association lumacaftor/ivacaftor dans l'étude VX15-809-110
| |
Valeur initiale et résultat
|
Relais du placebo au
lumacaftor / ivacaftor
(P-L/I)
(N = 96)*
|
lumacaftor / ivacaftor –
lumacaftor / ivacaftor
(L/I-L/I)
(N = 143)*
| |
|
Moyenne (ET)
|
Moyenne des MC
(IC à 95 %)
|
Moyenne (ET)
|
Moyenne des MC
(IC à 95 %)
| |
Variation absolue de l'ICP2,5 par rapport à la valeur initiale
| |
|
n = 101
|
|
n = 128
|
| |
ICP2,5 initial‡**
|
10,26
(2,24)
|
|
10,24
(2,42)
|
| |
Semaine 96 de l'étude d'extension
|
|
(n = 69)
-0,86
(-1,33; -0,38)
|
|
(n = 88)
-0,85
(-1,25; -0,45)
| |
|
n = 101
|
|
n = 159
|
| |
VEMS en pourcentage de la valeur théorique initiale (%)‡
|
90,7
(10,8)
|
|
89,7
(13,8)
|
| |
Variation absolue du VEMS en pourcentage de la valeur théorique par rapport à la valeur initiale (%)
| |
Semaine 96 de l'étude d'extension
|
|
n = 79
0,0
(-2,7; 2,7)
|
|
n = 123
3,1
(1,0; 5,1)
| |
|
n = 96
|
|
n = 143
|
| |
IMC initial pour l'âge, Z-score (kg/m2)‡
|
-0,16
(0,90)
|
|
-0,09
(0,88)
|
| |
Variation absolue de l'IMC par rapport à la valeur initiale pour l'âge, Z-score (kg/m2)
| |
Semaine 96 de l'étude d'extension
|
|
(n = 83)
0,31
(0,19; 0,42)
|
|
(n = 130)
0,17
(0,08; 0,26)
| |
|
n = 78
|
|
n = 135
|
| |
Score initial du domaine respiratoire du questionnaire CFQ-R (points)‡
|
77,1
(15,5)
|
|
78,5
(14,3)
|
| |
Variation absolue du score du domaine respiratoire du questionnaire CFQ-R (points)
| |
Semaine 96 de l'étude d'extension
|
|
(n = 65)
6,6
(3,1; 10,0)
|
|
(n = 108)
7,4
(4,8; 10,0)
| |
Nombre d'exacerbations pulmonaires (événements) (étude VX14-809-109 FAS et ROS)†
| |
Nombre d'événements par patient-année (IC à 95 %)
|
|
n = 96
0,30
(0,21; 0,43)
|
|
n = 103
0,45
(0,33; 0,61)
| |
*Participants à l'étude traités par placebo dans l'étude VX14-809-109 (n = 96) et qui sont passés à un traitement actif par LUM/IVA dans l'étude d'extension (P-L/I).
Participants à l'étude qui ont été traités par LUM/IVA dans l'une des deux études principales [étude VX13-809-011B (n = 49) ou étude VX14-809-109 (n = 94)] et qui ont continué à recevoir un traitement actif par LUM/IVA dans l'étude d'extension (L/I-L/I).
‡La valeur initiale pour les deux groupes (P-L/I et L/I-L/I) était la valeur à l'inclusion dans l'étude VX13-809-011B et dans l'étude VX14-809-109 (étude principale) et le nombre «n» correspondant se réfère aux groupes d'analyses de l'étude principale.
**La sous-étude ICP a inclus 117 participants dans le groupe L/I-L/I et 96 participants dans le groupe P-L/I.
†FAS = ensemble d'analyse complet (n = 103), comprend les participants ayant reçu L/I dans l'étude VX13-809-109 et l'étude VX15-809-110 et qui ont été évalués pendant la durée de l'étude cumulée pour L/I; ROS = groupe d'extension (n = 96), comprend les participants ayant reçu un placebo dans l'étude VX14-809-109 et L/I dans l'étude VX15-809-110 et qui ont été évalués pendant toute la durée de l'étude VX15-809-110.
|
Étude VX16-809-116: étude d'extension de 96 semaines
Les patients atteints de mucoviscidose âgés de 2 ans et plus de l'étude VX15-809-115 ont été inclus dans une étude multicentrique d'extension de phase III (étude VX16-809-116). Cette étude d'extension non contrôlée a été conçue pour évaluer la sécurité et l'efficacité descriptive d'un traitement à long terme par lumacaftor/ivacaftor. Sur les 60 patients qui ont reçu l'un des traitements dans l'étude VX15-809-115, 57 (95 %) ont continué à recevoir le traitement dans l'étude d'extension. 33,3 % des patients étaient âgés de < 3 ans et 66, 7 % des patients étaient âgés de ≥3 ans. Dans les études d'efficacité descriptives non contrôlées, le traitement par lumacaftor/ivacaftor était associé à une amélioration des paramètres de poids et de taille (IMC, poids, taille), y compris les Z-scores. Chez les patients ayant reçu le lumacaftor/ivacaftor dans l'étude principale VX15-809-115 avant d'être inclus dans l'étude d'extension VX16-809-116, aucun changement cliniquement significatif n'a été observé en termes d'ICP2,5 (n = 17: -0,20, (IC à 95 % -0,99; 0,60)) ou de VEMS (n = 13: 0,4, (IC à 95 % -7,5; 8,4)) après un traitement supplémentaire de 96 semaines.
PharmacocinétiqueL'aire sous la courbe des concentrations plasmatiques (ASC) de lumacaftor est environ 2 fois plus élevée chez les volontaires sains adultes que chez les patients atteints de mucoviscidose. L'exposition systémique à l'ivacaftor est similaire chez les volontaires sains adultes et les patients atteints de mucoviscidose. Après administration deux fois par jour, les concentrations plasmatiques à l'état d'équilibre du lumacaftor et de l'ivacaftor chez les volontaires sains étaient généralement atteintes en 7 jours environ, avec un taux d'accumulation d'environ 1,9 pour le lumacaftor. L'exposition à l'ivacaftor à l'état d'équilibre est plus faible que l'exposition au jour 1 en raison de l'effet d'induction du CYP3A par le lumacaftor (voir «Interactions»).
Après administration orale de l'association lumacaftor 400 mg/ivacaftor 250 mg toutes les 12 heures avec un repas, les valeurs moyennes à l'état d'équilibre de l'ASC0-12h et de la Cmax étaient respectivement de 198 (écarttype, ET: ± 64,8) µg∙h/ml et 25,0 (ET: ± 7,96) µg/ml pour le lumacaftor, et de 3,66 (± 2,25) µg∙h/ml et 0,602 (± 0,304) µg/ml pour l'ivacaftor. Après administration orale d'ivacaftor seul à la dose de 150 mg toutes les 12 heures avec un repas, les valeurs moyennes à l'état d'équilibre de l'ASC0-12h et de la Cmax étaient respectivement de 9,08 μg∙h/ml (ET : ± 3,20) et 1,12 (ET : ± 0,319) μg/ml.
Absorption
Après administrations orales répétées de lumacaftor, l'exposition au lumacaftor augmentait généralement de façon proportionnelle à la dose dans l'intervalle de doses de 50 mg à 1 000 mg administrées toutes les 24 heures. L'exposition au lumacaftor était environ 2 fois plus élevée en cas d'administration avec un repas contenant des graisses par rapport à l'administration à jeun. Le Tmax médian du lumacaftor administré avec un repas est d'environ 4,0 heures (valeurs extrêmes: 2,0; 9,0).
Après administrations orales répétées d'ivacaftor en association avec le lumacaftor, l'exposition systémique à l'ivacaftor augmentait généralement de façon proportionnelle à la dose dans l'intervalle de doses de 150 mg toutes les 12 heures à 250 mg toutes les 12 heures. Chez des volontaires sains, l'exposition systémique à l'ivacaftor administré en association avec le lumacaftor était environ 3 fois plus élevée en cas d'administration avec un repas contenant des graisses. Par conséquent, le lumacaftor/ivacaftor doit être administré avec un repas ou une collation contenant des graisses. Le Tmax médian de l'ivacaftor administré avec un repas est d'environ 4,0 heures (valeurs extrêmes: 2,0; 6,0).
Distribution
Le lumacaftor est lié aux protéines plasmatiques à 99 % environ, essentiellement à l'albumine. Après administration orale d'une dose de 400 mg toutes les 12 heures avec un repas chez des patients atteints de mucoviscidose, les volumes apparents de distribution typiques dans les compartiments central et périphérique ont été estimés à 23,5 litres (coefficient de variation, CV: 48,7 %) et 33,3 litres (CV: 30,5 %) respectivement.
L'ivacaftor est lié aux protéines plasmatiques à 99 % environ, essentiellement à l'alpha-1glycoprotéine acide et à l'albumine. Après administration orale d'ivacaftor 250 mg toutes les 12 heures en association avec le lumacaftor, les volumes apparents de distribution dans les compartiments central et périphérique ont été estimés à respectivement 95,0 litres (CV: 53,9 %) et 201 litres (CV: 26,6 %).
Les études in vitro indiquent que le lumacaftor est un substrat de la protéine de résistance du cancer du sein (BCRP).
Métabolisme
Le lumacaftor n'est pas fortement métabolisé chez l'homme, la majeure partie étant éliminée sous forme inchangée dans les fèces. Les données in vitro et in vivo indiquent que le lumacaftor est métabolisé principalement par oxydation et glucuroconjugaison.
L'ivacaftor est fortement métabolisé chez l'homme. Les données in vitro et in vivo indiquent que l'ivacaftor est métabolisé principalement par le CYP3A. M1 et M6 sont les deux principaux métabolites de l'ivacaftor chez l'homme. L'activité de M1 correspond à un sixième environ de celle de l'ivacaftor et ce métabolite est considéré comme pharmacologiquement actif. L'activité de M6 correspond à moins d'un cinquantième de celle de l'ivacaftor et ce métabolite n'est pas considéré comme pharmacologiquement actif.
Élimination
Après administration orale de lumacaftor, la majorité de la dose (51 %) a été éliminée dans les fèces sous forme inchangée. L'excrétion urinaire de lumacaftor sous forme inchangée était négligeable. La demivie terminale apparente est d'environ 26 heures. La clairance apparente (Cl/F) du lumacaftor a été estimée à 2,38 L/h (29,4 %) chez les patients atteints de mucoviscidose.
Après administration orale d'ivacaftor seul, la majorité de la dose (87,8 %) a été éliminée dans les fèces sous forme métabolisée. L'excrétion urinaire de l'ivacaftor sous forme inchangée était négligeable. Chez les volontaires sains, la demivie de l'ivacaftor administré avec le lumacaftor est d'environ 9 heures. La CL/F de l'ivacaftor administré en association avec le lumacaftor a été estimée à 25,1 L/h (CV: ± 40,5 %) chez les patients atteints de mucoviscidose.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Après administration de doses répétées de l'association lumacaftor/ivacaftor pendant 10 jours, l'exposition systémique était plus élevée chez les patients présentant une insuffisance hépatique modérée (Child-Pugh classe B, score de 7 à 9) comparativement aux volontaires sains appariés sur les critères démographiques (augmentation d'environ 50 % pour l'ASC0-12 h et d'environ 30 % pour la Cmax). L'effet de l'insuffisance hépatique légère (Child-Pugh classe A, score de 5 à 6) sur la pharmacocinétique du lumacaftor administré en association avec l'ivacaftor n'a pas été étudié, mais l'augmentation de l'exposition systémique ne devrait pas dépasser 50 %.
Il n'a pas été mené d'études chez des patients atteints d'insuffisance hépatique sévère (Child-Pugh classe C, score de 10 à 15), mais une exposition systémique plus élevée que chez les patients présentant une insuffisance hépatique modérée est attendue (voir «Posologie/Mode d'emploi», «Mises en garde et précautions» et «Effets indésirables»).
Troubles de la fonction rénale
Il n'a pas été réalisé d'études pharmacocinétiques avec l'association lumacaftor/ivacaftor chez des patients présentant une insuffisance rénale. Dans une étude pharmacocinétique chez l'homme menée avec le lumacaftor administré seul, l'élimination urinaire du lumacaftor et de ses métabolites était minime (seulement 8,6 % de la radioactivité totale ont été retrouvés dans les urines, avec 0,18 % sous forme de lumacaftor inchangé). Dans une étude pharmacocinétique chez l'homme menée avec l'ivacaftor administré seul, l'élimination urinaire de l'ivacaftor et de ses métabolites était minime (seulement 6,6 % de la radioactivité totale ont été retrouvés dans les urines). Une analyse pharmacocinétique de population en fonction de la clairance de la créatinine ne montre pas de tendance chez les patients présentant une insuffisance rénale légère ou modérée. Par conséquent, aucune adaptation de la posologie du lumacaftor/ivacaftor n'est recommandée en cas d'insuffisance rénale légère ou modérée. Cependant, la prudence est recommandée en cas d'administration de lumacaftor/ivacaftor chez des patients présentant une insuffisance rénale sévère (clairance de la créatinine ≤30 ml/min) ou une insuffisance rénale au stade terminal.
Patients âgés
La sécurité et l'efficacité de l'association lumacaftor/ivacaftor chez les patients âgés de 65 ans et plus n'ont pas été évaluées.
Sexe
L'influence du sexe sur la pharmacocinétique du lumacaftor a été évaluée à l'aide d'une analyse pharmacocinétique de population à partir des données issues des études cliniques du lumacaftor administré en association avec l'ivacaftor. Les résultats n'indiquent pas de différence cliniquement pertinente des paramètres pharmacocinétiques du lumacaftor ou de l'ivacaftor entre les patients de sexe masculin et féminin. Aucune adaptation de la posologie en fonction du sexe n'est nécessaire.
Enfants et adolescents
Selon les analyses (PK) de population présentées dans le tableau 12 cidessous, les expositions systémiques sont similaires chez les patients adultes et pédiatriques:
|
Tableau 12: Exposition systémique moyenne (ET) au lumacaftor et à l'ivacaftor, par tranche d'âge
| |
Tranche d'âge
|
Poids
|
Dose
|
ASCss moyenne
(ET) du lumacaftor
(μg/ml*h)
|
ASCss moyenne (ET) de l'ivacaftor
(μg/ml*h)
| |
Patients âgés de 1 à moins de 2 ans
|
7 kg à < 9 kg
N = 1
|
Un sachet de lumacaftor 75 mg/ivacaftor 94 mg toutes les 12 heures
|
234
|
7,98
| |
|
9 kg à < 14 kg
N = 44
|
Un sachet de lumacaftor 100 mg/ivacaftor 125 mg toutes les 12 heures
|
191 (40,6)
|
5,35 (1,61)
| |
|
≥14 kg
N = 1
|
Un sachet de lumacaftor 150 mg/ivacaftor 188 mg toutes les 12 heures
|
116
|
5,82
| |
Patients âgés de 2 à 5 ans
|
< 14 kg
N = 20
|
Un sachet de lumacaftor 100 mg/ivacaftor 125 mg toutes les 12 heures
|
180 (45,5)
|
5,92 (4,61)
| |
Patients âgés de 2 à 5 ans
|
≥14 kg
N = 42
|
Un sachet de lumacaftor 150 mg/ivacaftor 188 mg toutes les 12 heures
|
217 (48,6)
|
5,90 (1,93)
| |
Patients âgés de 6 à moins de 12 ans
|
-
N = 62
|
lumacaftor 200 mg/ivacaftor 250 mg toutes les 12 heures
|
203 (57,4)
|
5,26 (3,08)
| |
Patients âgés de 12 à moins de 18 ans
|
-
N = 98
|
lumacaftor 400 mg/ivacaftor 250 mg toutes les 12 heures
|
241 (61,4)
|
3,90 (1,56)
| |
Patients âgés de 18 ans et plus
|
-
N = 55
|
lumacaftor 400 mg/ivacaftor 250 mg toutes les 12 heures
|
198 (64,8)
|
3,66 (2,25)
| |
Remarque: les données d'exposition chez les patients âgés de moins de 18 ans sont issues des analyses PK de population. Les données d'exposition chez les patients adultes sont issues d'analyses non compartimentales.
|
Données précliniquesLumacaftor
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie après administration répétée, génotoxicité, cancérogenèse et des fonctions de la reproduction et du développement n'ont pas révélé de risque particulier pour l'homme. Il n'a pas été mené d'études spécifiques pour évaluer le potentiel phototoxique du lumacaftor; cependant, l'évaluation des données précliniques et cliniques disponibles ne semble pas indiquer de phototoxicité.
Ivacaftor
Dans les études à doses répétées, des effets n'ont été observés chez l'animal qu'à des expositions considérées comme suffisamment supérieures (> 25, > 35 et > 45 fois plus élevées chez la souris, le chien et le rat respectivement) à l'exposition maximale à l'ivacaftor observée chez l'homme après administration d'Orkambi indiquant une faible pertinence clinique. Les données non cliniques issues des études conventionnelles de génotoxicité et cancérogenèse n'ont pas révélé de risque particulier pour l'homme.
Pharmacologie de sécurité
L'ivacaftor a exercé un effet inhibiteur concentrationdépendant sur les courants des canaux hERG (human ether-à-go-go related gene) avec une CI15 de 5,5 µM, comparativement à la Cmax (1,5 µM) de l'ivacaftor observée à la dose thérapeutique de lumacaftor/ivacaftor. Cependant, aucun allongement de l'espace QT induit par l'ivacaftor n'a été observé lors de l'étude de télémétrie chez le chien à des doses uniques allant jusqu'à 60 mg/kg ou lors de mesures électrocardiographiques (ECG) dans les études à doses répétées d'une durée allant jusqu'à 1 an à la dose de 60 mg/kg/jour chez le chien (Cmax après 365 jours = 36,2 à 47,6 μM). L'ivacaftor a entraîné une augmentation dosedépendante, mais transitoire, des paramètres de la pression artérielle chez le chien à des doses orales uniques allant jusqu'à 60 mg/kg (voir «Propriétés/Effets»).
Gestation et fertilité
L'ivacaftor n'a pas été tératogène après administration orale à des rates et lapines gravides pendant la période d'organogenèse du développement fœtal, à des doses correspondant respectivement à 10 fois (exposition à l'ivacaftor et à ses métabolites) et 46 fois environ l'exposition à l'ivacaftor chez l'homme à la dose thérapeutique de lumacaftor/ivacaftor. Aux doses materno-toxiques chez la rate, l'ivacaftor a entraîné des réductions du poids des fœtus, une augmentation de l'incidence de syndromes de la côte cervicale, d'hypoplasie costale, de côtes ondulées et d'irrégularités du sternum, y compris des fusions. La pertinence de ces observations chez lֹ'homme n'est pas connue.
L'ivacaftor a diminué la fertilité et les indices de performance de reproduction chez des rats mâles et femelles à la dose de 200 mg/kg/jour, lorsque les mères étaient traitées avant la gestation et au cours des premiers stades de celleci. (Cette dose a entraîné des expositions correspondant à environ 11 et 7 fois respectivement, les expositions obtenues à la dose maximale recommandée chez l'homme d'ivacaftor, composant d'Orkambi, d'après les ASC totales de l'ivacaftor et de ses métabolites, extrapolées à partir des expositions au jour 90 avec la dose de 150 mg/kg/jour dans l'étude de toxicité à doses répétées de 6 mois, ainsi que des expositions au jour 17 de gestation dans l'étude pilote sur le développement embryofoetal dans cette espèce). Aucun effet sur la fertilité mâle ou femelle et sur les indices de performance de reproduction n'a été observé aux doses ≤100 mg/kg/jour. (Ces doses ont entraîné des expositions correspondant à environ 8 fois et 5 fois respectivement celles obtenues à la dose maximale recommandée chez l'homme d'ivacaftor, composant d'Orkambi, d'après les ASC totales de l'ivacaftor et de ses métabolites, extrapolées à partir des expositions au jour 90 à la dose de 100 mg/kg/jour dans l'étude de toxicité à doses répétées de 6 mois et des expositions au Jour 17 de gestation dans l'étude sur le développement embryofoetal dans cette espèce). Un passage transplacentaire de l'ivacaftor a été observé chez des rates et des lapines gravides.
Développement périnatal et postnatal
L'ivacaftor n'a pas provoqué d'anomalies du développement dans les portées de rates gravides recevant des doses orales de 100 mg/kg/jour pendant toute la durée de la gestation jusqu'à la parturition et le sevrage (conduisant à des expositions environ 4 fois supérieures à celles obtenues avec la dose la plus élevée recommandée du composant ivacaftor d'Orkambi pour l'être humain, sur la base des ASC cumulées pour l'ivacaftor et ses métabolites). Les doses supérieures à 100 mg/kg/jour ont entraîné des taux de survie et des indices de lactation qui étaient respectivement de 92 % et 98 % des valeurs observées chez les animaux témoins, ainsi qu'une réduction du poids des petits.
Animaux juvéniles
Des cataractes ont été observées chez des rats juvéniles ayant reçu l'ivacaftor à des doses représentant 0,32 fois la dose maximale recommandée chez l'homme sur la base de l'exposition systémique à l'ivacaftor et à ses métabolites après administration d'ivacaftor en association avec le lumacaftor sous forme d'Orkambi. Il n'a pas été observé de cataractes chez les fœtus de rates traitées pendant la période d'organogenèse du développement fœtal, chez les petits exposés dans une certaine mesure par l'intermédiaire du lait avant le sevrage ni dans les études de toxicologie après administration répétée d'ivacaftor. La pertinence éventuelle de ces observations chez l'homme n'est pas connue.
Lumacaftor et ivacaftor
Les études de toxicologie après administration répétée impliquant l'administration concomitante de lumacaftor et d'ivacaftor n'ont pas révélé de risque particulier pour l'homme en termes de potentiel de toxicités additives et/ou synergiques.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Comprimés pelliculés et granulés: ne pas conserver au-dessus de 30°C.
Conserver hors de vue et de portée des enfants.
Numéro d’autorisation65981, 67395 (Swissmedic)
PrésentationOrkambi 100 mg/125 mg comprimés pelliculés:
Boîte contenant 112 comprimés pelliculés (4 boîtes contenant chacune 28 comprimés pelliculés). [A]
Orkambi 200 mg/125 mg comprimés pelliculés:
Boîte contenant 112 comprimés pelliculés (4 boîtes contenant chacune 28 comprimés pelliculés). [A]
Orkambi 75 mg/94 mg granulés en sachet:
Boîte de 56 sachets (contenant 4 pochettes individuelles contenant chacune 14 sachets) [A]
Orkambi 100 mg/125 mg granulés en sachet:
Boîte de 56 sachets (contenant 4 pochettes individuelles contenant chacune 14 sachets) [A]
Orkambi 150 mg/188 mg granulés en sachet:
Boîte de 56 sachets (contenant 4 pochettes individuelles contenant chacune 14 sachets) [A]
Titulaire de l’autorisationVertex Pharmaceuticals (CH) GmbH
6300 Zug
Mise à jour de l’informationJuin 2025
|