CompositionPrincipes actifs
INN = lonoctocog alfa.
Facteur VIII antihémophilique recombinant à chaîne unique (rVIII-SingleChain) produit dans des cellules ovariennes de hamster chinois (CHO).
Excipients
Poudre: L-histidine, polysorbate 80, chlorure de calcium dihydraté, chlorure de sodium (correspond à 0,7 mmol ou 16,1 mg de sodium par flacon de 500 U.I. et 1000 U.I. et 1,4 mmol ou 32,3 mg par flacon de 2000 U.I.), saccharose.
Solvant: eau pour préparations injectables.
Indications/Possibilités d’emploiTraitement et prophylaxie d'épisodes hémorragiques chez des patients atteints d'hémophilie A (déficit congénital en facteur VIII) traités préalablement.
AFSTYLA n'est pas indiqué pour le traitement de la maladie de von Willebrand (vWD).
Posologie/Mode d’emploiPosologie pour adultes et enfants
Le traitement par AFSTYLA doit être instauré sous la surveillance d'un médecin qui a de l'expérience dans le traitement de l'hémophilie.
La décision relative à une utilisation à domicile du produit pour le traitement d'hémorragies ou pour la prophylaxie d'hémorragies chez des patients atteints d'hémophilie A doit être prise individuellement pour chaque patient par le médecin traitant. Le médecin traitant doit s'assurer que le patient reçoit une formation correspondante et doit évaluer l'utilisation à intervalles réguliers.
Posologie
La posologie et la durée du traitement dépendent de la gravité du déficit en facteur VIII, du site et de l'étendue de l'hémorragie, ainsi que de l'état clinique du patient.
Le nombre d'unités de facteur VIII administrées est indiqué en unités internationales (U.I.), conformément à la norme de l'OMS actuellement en vigueur relative aux produits contenant le facteur VIII. L'activité plasmatique en facteur VIII s'exprime soit en pourcentage (par rapport au plasma humain normal), soit en unités internationales (par rapport à la norme internationale pour le facteur VIII dans le plasma).
L'étiquette de chaque flacon d'AFSTYLA indique l'activité en facteur VIII en unités internationales (U.I.). Une U.I. correspond à l'activité en facteur VIII qui est contenue dans 1 ml de plasma humain normal.
La détermination de l'activité indiquée est réalisée à l'aide d'un dosage chromogénique.
La surveillance du taux plasmatique en facteur VIII peut être réalisée à l'aide d'un dosage chromogénique ou d'un test de coagulation en une étape (cf. «Mises en garde et précautions»).
Traitement en fonction des besoins
Le calcul de la dose de facteur VIII nécessaire se base sur un constat empirique, à savoir que 1 U.I. de facteur VIII par kg de poids corporel accroît le taux plasmatique de facteur VIII de 2 U.I./dl. In vivo, la hausse maximale attendue du taux de facteur VIII, exprimé en U.I./dl (ou en % de la normale), est estimée au moyen de la formule suivante:
Hausse estimée du taux de facteur VIII (U.I./dl ou % de la normale) = [dose totale (U.I.)/poids corporel (kg)] × 2 (U.I./dl par U.I./kg)
La dose nécessaire pour atteindre la hausse maximale souhaitée in vivo du taux de facteur VIII peut être calculée au moyen de la formule suivante:
Dose (U.I.) = poids corporel (kg) × hausse souhaitée du FVIII (U.I./dl ou % de la normale) × 0,5 (U.I./kg par U.I./dl)
La posologie et la fréquence d'administration doivent toujours s'orienter en fonction de l'efficacité clinique dans chaque cas particulier.
Le tableau suivant sert de recommandation pour la posologie d'AFSTYLA en vue du contrôle et de la prévention d'épisodes hémorragiques. Le taux de facteur VIII doit être maintenu dans la zone cible ou au-dessus de celle-ci.
|
Sévérité de l'hémorragie/ nature de l'intervention chirurgicale
|
Taux de facteur VIII nécessaire (% ou U.I./dl)
|
Fréquence des doses (heures)/durée du traitement (jours)
| |
Hémorragie
| |
Hémarthrose naissante, hémorragies musculaires ou buccales
|
20 à 40
|
Répéter l'injection toutes les 12 à 24 heures jusqu'à l'arrêt de l'hémorragie
| |
Hémarthroses plus étendues, hémorragies musculaires ou hématomes
|
30 à 60
|
Répéter l'injection toutes les 12 à 24 heures jusqu'à l'arrêt de l'hémorragie
| |
Hémorragies menaçant le pronostic vital
|
60 à 100
|
Répéter l'injection toutes les 8 à 24 heures jusqu'à l'arrêt de l'hémorragie
| |
Interventions chirurgicales
| |
Interventions mineures
y compris extractions dentaires
|
30 à 60
|
Répéter l'injection toutes les 24 heures pendant au moins 1 journée jusqu'à cicatrisation de la plaie
| |
Interventions majeures
|
80 à 100 (pré- et post-opératoire)
|
Répéter l'injection toutes les 8-24 heures jusqu'à une cicatrisation adéquate de la plaie puis poursuivre le traitement pendant au moins 7 jours pour maintenir un taux d'activité en facteur VIII de 30 à 60% (U.I./dl)
|
Prophylaxie
La posologie initiale recommandée est de 20 à 50 U.I. d'AFSTYLA/kg 2 ou 3 fois par semaine.
La posologie sera adaptée en fonction de la réponse du patient.
Patients non traités précédemment (PUP)
Globalement, le profil de sécurité et d'efficacité d'AFSTYLA chez les PUP correspondait au profil risques-bénéfices chez les patients traités précédemment (previously treated patients, PTP) à l'exception de l'apparition d'anticorps neutralisants qui représente un risque reconnu associé au traitement de substitution du FVIII dans cette population de patients (voir «Mises en garde et précautions»).
Patients âgés
Les études cliniques portant sur AFSTYLA ne comportaient pas de participants âgés de plus de 65 ans.
Enfants et adolescents
La posologie initiale recommandée chez les enfants (de 0 à 12 ans) est de 30 à 50 U.I d'AFSTYLA/kg 2 à 3 fois par semaine. Une posologie plus fréquente ou plus élevée peut être nécessaire pour les enfants de moins de 12 ans car il y a lieu de tenir compte d'une clairance plus élevée dans cette catégorie d'âge.
Pour les adolescents de plus de 12 ans, la posologie recommandée est la même que pour adultes.
Les données actuellement disponibles sont mentionnées dans la rubrique «Pharmacocinétique».
Surveillance des inhibiteurs
Les patients doivent être suivis pour surveiller le développement d'inhibiteurs du facteur VIII, cf. également la rubrique «Mises en garde et précautions».
Mode d'administration
Vitesse d'injection
Pour utilisation intraveineuse.
Pour les instructions portant sur la reconstitution du médicament avant l'administration, cf. «Remarques concernant la manipulation» dans la rubrique «Remarques particulières». La préparation reconstituée doit être injectée lentement et à une vitesse confortable pour le patient.
Le patient doit être observé pour déceler des réactions immédiates. En cas d'apparition d'une réaction qui pourrait être associée à l'administration d'AFSTYLA, la vitesse d'injection doit être diminuée ou l'administration arrêtée en fonction de l'état clinique du patient (cf. également «Mises en garde et précautions»).
Afin de garantir la traçabilité des produits pharmaceutiques issus de la biotechnologie, il est recommandé de documenter le nom commercial et le numéro de lot pour chaque traitement.
Contre-indicationsAFSTYLA est contre-indiqué chez les patients ayant eu des réactions d'hypersensibilité menaçant le pronostic vital, y compris une anaphylaxie, au principe actif, à l'un des excipients selon la composition ou aux protéines de hamster.
Mises en garde et précautionsHypersensibilité
AFSTYLA peut provoquer des réactions d'hypersensibilité allergiques, y compris une anaphylaxie. Les patients doivent être informés sur les signes précoces de réactions d'hypersensibilité, tels qu'une éruption cutanée papuleuse, une urticaire généralisée, une sensation d'oppression au niveau du thorax, un stridor, une hypotension et un prurit. En cas d'apparition de réactions d'hypersensibilité, l'administration doit être arrêtée immédiatement et un traitement approprié doit être instauré.
Il y a lieu de recommander aux patients d'interrompre l'administration d'AFSTYLA et de contacter leur médecin.
Chez les patients qui ont déjà eu une réaction d'hypersensibilité, l'administration préalable d'antihistaminiques peut être envisagée.
Anticorps neutralisants
La formation d'anticorps neutralisants (inhibiteurs) anti-facteur VIII a été observée après l'administration de produits contenant du facteur VIII, y compris AFSTYLA.
Le développement d'anticorps neutralisants (inhibiteurs) doit être surveillé chez les patients, au moyen d'examens cliniques et biologiques adéquats. Si le taux plasmatique en facteur VIII attendu après l'administration d'AFSTYLA n'est pas atteint ou si l'hémorragie n'a pas pu être arrêtée, la présence d'un inhibiteur (anticorps neutralisant) doit être soupçonnée.
Si un patient développe un inhibiteur, un centre d'hémophilie doit être contacté.
Un test de recherche d'inhibiteur par la méthode Bethesda doit être réalisé lorsque les taux plasmatiques en facteur VIII attendus ne sont pas atteints ou si l'hémorragie ne peut pas être arrêtée avec la dose attendue d'AFSTYLA. Des unités Bethesda (UB) doivent être utilisées pour exprimer le taux d'inhibiteurs.
Analyses de laboratoire pour la surveillance
L'activité plasmatique du facteur VIII doit être surveillée par l'utilisation du dosage chromogénique ou du test de coagulation en une étape chez les patients traités par AFSTYLA.
Les résultats d'efficacité d'une étude clinique pivot à grande échelle ont confirmé que les résultats du dosage chromogénique reflètent le plus exactement le potentiel hémostatique clinique. Le dosage chromogénique doit donc être utilisé s'il est disponible pour déterminer l'activité en facteur VIII dans des échantillons de patients. Si le test de coagulation en une étape est utilisé pour déterminer l'activité en facteur VIII dans des échantillons de patients, il convient de prendre en compte le fait que les résultats de ce test sont inférieurs d'environ 45% à ceux du dosage chromogénique et qu'ils peuvent être ajustés par multiplication par un facteur 2 (cf. ci-dessus).
Complications liées à un cathéter
Si le recours à un cathéter veineux central est nécessaire, il faut envisager la possibilité de complications associées, notamment des infections locales, une bactériémie et des thromboses liées au cathéter.
Enfants et adolescents
Les précautions et mises en garde indiquées s'appliquent de la même manière aux adultes et aux enfants.
AFSTYLA 500 U.I. et 1000 U.I. contiennent moins de 1 mmol de sodium (23 mg) par flacon, ce qui signifie qu'ils sont pratiquement «sans sodium».
AFSTYLA 2000 U.I. contient ≤1,4 mmol (32,3 mg) de sodium par flacon, ce qui correspond à 1,6 % de l'apport quotidien maximum en sodium de 2 g recommandé par l'OMS pour un adulte dans le cadre de son régime alimentaire.
InteractionsAucune interaction entre AFSTYLA et d'autres médicaments n'a été rapportée.
Grossesse, allaitementGrossesse
Aucune étude sur la toxicité d'AFSTYLA pour la reproduction et le développement chez l'animal n'a été réalisée. Le risque d'atteinte au fœtus ou de troubles reproductifs en cas d'administration d'AFSTYLA à des femmes enceintes est inconnu. Par conséquent, AFSTYLA ne doit être administré pendant la grossesse qu'en cas de nécessité absolue.
Allaitement
On ignore si AFSTYLA passe dans le lait maternel humain. Étant donné que de nombreux médicaments passent dans le lait maternel humain, AFSTYLA ne doit pas être administré aux femmes qui allaitent.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAFSTYLA n'a aucune influence sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
Une hypersensibilité ou des réactions allergiques, pouvant inclure un angioœdème, une sensation de brûlure et de picotement au point d'injection, des frissons, une sensation de chaleur, une urticaire généralisée, des céphalées, une éruption cutanée papuleuse, une hypotension, une léthargie, des nausées, une agitation, de la tachycardie, une sensation d'oppression au niveau du thorax, des picotements (fourmillements), des vomissements ou un stridor, ont été rarement observés lors de l'utilisation de produits contenant du facteur VIII. Dans certains cas, ces réactions ont évolué vers une anaphylaxie sévère (y compris un choc anaphylactique) (cf. également «Mises en garde et précautions»). Des réactions d'hypersensibilité ont été observées dans les études cliniques portant sur AFSTYLA (cf. tableau ADR ci-dessous), mais aucune réaction anaphylactique n'a été signalée.
Les patients atteints d'hémophilie A peuvent développer des anticorps neutralisants (inhibiteurs) contre le facteur VIII. L'apparition de tels inhibiteurs se manifeste par une réponse clinique insuffisante. Dans ces cas, il est recommandé de contacter un centre d'hémophilie spécialisé. Aucune réaction de ce type n'a été signalée lors des études cliniques portant sur AFSTYLA. La formation d'anticorps neutralisants (inhibiteurs) contre le facteur VIII a été observée durant l'étude clinique chez des patients non traités auparavant par AFSTYLA (voir la liste suivante des effets indésirables).
Liste des effets indésirables
La liste ci-dessous correspond à la classification MedDRA par classe de système d'organes (SOC et termes privilégiés).
Les fréquences des effets indésirables ont été calculées par patient, sur la base des données issues des études cliniques achevées, et classées selon la convention suivante: fréquents (≥1/100 à <1/10); occasionnels (≥1/1000 à <1/100); rares (≥1/10 000 à <1/1000) et très rares (<1/10 000), fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Affections hématologiques et du système lymphatique
Occasionnels (chez les patients traités auparavant, PTP), fréquence de l'inhibition du facteur VIII basée sur la fréquence de classes pour les produits à base de FVIII), très fréquents (chez les patients non traités auparavant (previously untreated patients, PUP): inhibition du facteur VIII.
Affections du système immunitaire:
Fréquents: hypersensibilité.
Affections du système nerveux:
Fréquents: vertiges, paresthésie.
Affections de la peau et du tissu sous-cutané
Fréquents: éruption cutanée.
Occasionnels: érythème, prurit.
Troubles généraux et anomalies au site d'administration
Fréquents: fièvre.
Occasionnels: douleur au point d'injection, frissons, sensation de chaleur.
Effets indésirables identifiés après la mise sur le marché
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucun cas de surdosage n'a été signalé.
Le cas d'un patient ayant reçu plus du double de la dose prescrite a été rapporté. Aucun effet indésirable associé à ce surdosage n'a été rapporté.
Propriétés/EffetsCode ATC:
B02BD02
Groupe pharmacothérapeutique: antihémorragique: facteur VIII de coagulation
Mécanisme d'action
AFSTYLA (INN: Lonoctocog alfa) contient une protéine recombinante qui remplace le facteur VIII manquant nécessaire à une hémostase efficace. Le lonoctocog alfa est un facteur VIII recombinant, à chaîne unique dans lequel la plus grande partie du domaine B présent dans le facteur VIII de type sauvage complet a été supprimée. Après activation, la molécule de lonoctocog alfa formée contient une séquence d'acides aminés identique à celle du facteur VIIIa, formé à partir du facteur VIII endogène complet. En outre, la structure à chaîne unique conduit à une meilleure affinité du lonoctocog alfa au facteur de von Willebrand.
Pharmacodynamique
L'hémophilie A est un trouble héréditaire, lié au chromosome X, de la coagulation du sang provoqué par un taux réduit en facteur VIII. Elle provoque des hémorragies importantes dans les articulations, les muscles ou les organes internes, spontanément ou suite à des lésions dues à un accident ou à une intervention chirurgicale. Le traitement de substitution augmente le taux plasmatique de facteur VIII et entraîne donc une correction limitée dans le temps du déficit en facteur VIII et une correction de la tendance hémorragique.
Efficacité clinique
Études ouvertes menées auprès de patients déjà traités auparavant (previously treated patients, PTP): étude 1001 auprès d'adultes et d'adolescents, étude 3002 dans une population pédiatrique
La pharmacocinétique, l'innocuité et l'efficacité clinique d'AFSTYLA ont été étudiées dans une étude de phase I/III avec des adultes/adolescents et dans une étude de phase III avec des enfants. La pharmacocinétique d'AFSTYLA a été caractérisée dans les études et l'efficacité hémostatique dans le traitement d'hémorragies et dans la prophylaxie de routine pour éviter les hémorragies a été déterminée. Dans l'étude de phase I/III avec des adultes/adolescents, l'efficacité hémostatique dans la prophylaxie périopératoire a également été étudiée chez des patients ayant subi une intervention chirurgicale.
L'étude de phase I/III portant sur les adultes/adolescents regroupait 175 patients masculins atteints d'une hémophilie A sévère (activité FVIII endogène <1%), qui avaient déjà reçu préalablement un traitement. Les patients avaient entre 12 et 65 ans; parmi eux, se trouvaient 14 adolescents âgés d'au moins 12 ans à moins de 18 ans. Parmi les 175 participants à l'étude, 174 ont reçu au moins 1 dose d'AFSTYLA et 173 (99%) ont pu être évalués dans l'étude d'efficacité. 161 patients (92,5%) ont complété l'étude. 120 (69,0%) de ces participants ont été soumis à au moins 50 jours de traitement, 52 (29,9%) de ces patients ont été soumis à au moins 100 jours de traitement. Les patients ont reçu au total 14'592 injections, la médiane étant de 67 injections, correspondant à une fourchette de 1 à 395 injections par patient.
L'étude de phase III portant sur les enfants regroupait 84 patients masculins atteints d'hémophilie A sévère, 35 patients étant âgés de 0 à moins de 6 ans et 49 de 6 à 12 ans. Parmi les 84 participants à l'étude, tous ont reçu au moins une dose d'AFSTYLA et 83 (99%) ont pu être évalués dans l'étude d'efficacité. 65 (77,4%) des participants ont été soumis à au moins 50 jours de traitement, 8 (9,5%) de ces patients ont été soumis à au moins 100 jours de traitement. Les patients ont reçu au total 5'313 injections, la médiane étant de 59 injections, correspondant à une fourchette de 4 à 145 injections par patient.
Traitement et prévention des épisodes hémorragiques
Dans l'étude menée portant sur les adultes/adolescents, 848 épisodes hémorragiques, principalement des hémorragies articulaires, ont été traités par AFSTYLA et l'efficacité a été évaluée chez 835 par l'investigateur.
La médiane des doses d'injection pour le traitement des épisodes hémorragiques était de 31,7 U.I./kg. 686 (81%) de cette totalité de 848 épisodes hémorragiques ont été traités par une seule dose d'AFSTYLA, 107 autres épisodes (13%) par 2 doses. 55 (6%) des 848 épisodes hémorragiques ont nécessité 3 injections ou plus.
L'efficacité hémostatique a été notée comme excellente ou bonne pour 94% des épisodes hémorragiques par l'investigateur.
Dans l'étude pédiatrique, un total de 347 épisodes hémorragiques, principalement des saignements articulaires, ont été traités par AFSTYLA et l'efficacité évaluée par l'investigateur.
La médiane des doses pour le traitement des épisodes hémorragiques était de 27,3 U.I./kg. 298 (86%) de cette totalité de 347 épisodes hémorragiques ont été traités par une seule dose d'AFSTYLA, 34 autres épisodes (10%) par 2 doses d'injection. 15 (4%) des 347 épisodes hémorragiques ont nécessité 3 injections ou plus.
L'efficacité hémostatique a été notée comme excellente ou bonne pour 96% des épisodes hémorragiques par l'investigateur.
Prophylaxie de routine
Dans l'étude portant sur les adultes/adolescents et l'étude pédiatrique, le schéma posologique pour la prophylaxie de routine était déterminé par l'investigateur. Ce plan a été établi en tenant compte du schéma de traitement par le FVIII que chaque patient suivait avant le début de l'étude et en tenant compte de son phénotype hémorragique.
Dans l'étude portant sur les adultes/adolescents, 54% des 146 patients sous traitement prophylactique ont reçu de l'AFSTYLA 3 fois par semaine. 32% ont reçu AFSTYLA 2 fois par semaine. 6% des patients sous traitement prophylactique ont reçu AFSTYLA tous les 2 jours et 8% des patients sous traitement prophylactique ont suivi un autre schéma posologique.
Pendant la prophylaxie de routine, 63 (43%) des 146 patients n'ont présenté aucune hémorragie.
La médiane du taux d'hémorragie annuel (ABR) sous une prophylaxie de routine était de 1,14 (écart interquartile 0,0 à 4,2) et était donc, de manière statistiquement significative, inférieure (p <0,0001) à celle chez les patients sous un traitement selon les besoins (médiane de 19,64, écart interquartile 6,2 à 46,5). L'ABR de patients traités 3 fois par semaine (médiane de 1,53, écart interquartile 0,0 à 4,4) était comparable à l'ABR de patients traités 2 fois par semaine (médiane de 0,00, écart interquartile 0,00 à 3,3). Le taux annuel d'hémorragies spontanées (AsBR) chez ces patients était identique, avec un AsBR médian de 0,00, tant dans le cadre du traitement 3 fois par semaine (écart interquartile 0,00 à 3,5) que pour le traitement 2 fois par semaine (écart interquartile 0,00 à 1,1).
La médiane de la dose prescrite pour des patients traités 3 fois par semaine était de 30 U.I./kg par injection et de 35 U.I./kg pour des patients traités 2 fois par semaine.
Dans l'étude pédiatrique, 54% des 80 patients sous traitement prophylactique ont reçu AFSTYLA 2 fois par semaine. 30% des patients sous traitement prophylactique ont reçu AFSTYLA 3 fois par semaine. 4% des patients sous traitement prophylactique ont reçu AFSTYLA tous les 2 jours et 12% des patients sous traitement prophylactique ont suivi un autre schéma posologique.
Pendant la prophylaxie de routine, 21 (26%) des 80 patients n'ont présenté aucune hémorragie.
L'ABR de patients sous traitement prophylactique était de 3,69 (écart interquartile 0,00 à 7,20) avec une médiane de 2,30 (écart interquartile 0,00 à 11,58) pour les patients traités 3 fois par semaine et de 4,37 (écart interquartile 2,31 à 7,24) pour les patients traités 2 fois par semaine. Le taux annuel d'hémorragies spontanées (AsBR) chez ces patients était identique, avec un AsBR médian de 0,00 dans le cadre du traitement 3 fois par semaine ainsi que pour le traitement 2 fois par semaine (écart interquartile 0,00 à 3,03 et 0,00 à 2,08).
La médiane de la dose prescrite pour les patients traités 3 fois par semaine était de 32 U.I./kg par injection et de 35 U.I./kg pour les patients traités 2 fois par semaine.
Prophylaxie périopératoire (prophylaxie chirurgicale)
13 patients parmi ceux qui participaient à l'étude portant sur les adultes/adolescents ont subi au total 16 interventions chirurgicales. Pour ces interventions, il s'agissait d'une extraction des dents de sagesse, de chirurgies d'hernies abdominales, d'une prothèse totale de coude, de cinq arthroplasties du genou d'une cholécystectomie, d'un allongement du tendon d'Achille combiné à un redressement, de trois circoncisions et d'une procédure d'ostéosynthèse à l'articulation du pied droit avec une reposition ouverte et une fixation interne et une élimination consécutive de cette fixation. Au total, l'efficacité hémostatique d'AFSTYLA dans 15 parmi les 16 interventions a été notée comme excellente par les investigateurs et comme bonne dans 1 parmi les 16 interventions (une arthroplastie du genou). La médiane de consommation du FVIII le jour de l'opération était de 89,4 U.I./kg (fourchette 40,5-108,6 U.I./kg).
Enfants, adolescents et adultes (étude de prolongation 3001)
Dans l'étude 3001, 222 PTP (67 participants <12 ans) ont été inclus, dont 204 PTP issus des études lead-in 1001 et 3002. Le nombre moyen (ET) de jours d'exposition (Ed) chez les PTP au cours de cette étude était de 341,9 (135,48). Au total, 212 participants (95,5 %) ont atteint >100 Ed. Aucun patient n'a développé un inhibiteur ou n'a été victime d'une réaction anaphylactique. Aucun nouveau problème de sécurité n'a été constaté au cours de cette étude de prolongation. Les résultats relatifs à la prophylaxie individuelle et au traitement des hémorragies ont été comparables avec ceux des études antérieures.
Prophylaxie individuelle: parmi les 211 patients traités prophylactiquement (ABR médian 1,21 [écart interquartile: 0,33; 3,03]), 85 (40 %) ont été attribués à un régime de deux fois par semaine et 97 (46 %) à un régime de trois fois par semaine. Les patients sous traitement prophylactique 2 et 3 fois par semaine ont reçu des doses médianes de 35 et 32 U.I./kg par injection, respectivement, avec une consommation annuelle médiane de 4,603 U.I./kg par an, tous régimes prophylactiques confondus.
Traitement des hémorragies: parmi les 2413 épisodes hémorragiques apparus et traités au cours de l'étude 3001, 86,3 % ont été contrôlés avec 2 injections ou moins.
Patients non traités auparavant (PUP)
Population pédiatrique <12 ans (étude 3001)
Au total, 24 PUP avec un âge moyen de 1,0 an (intervalle: 0 à 5 ans) ont été inclus dans cette étude. Les participants ont totalisé 5909 Ed au rVIII-SingleChain.
Prophylaxie individuelle: au total, 23 PUP ont suivi un régime de traitement prophylactique pendant l'étude (11 après changement à partir du traitement en fonction des besoins). L'ABR médian a été de 1,84 (intervalle: 0,0 à 23,6), l'AsBR médian a été de 0,88 (intervalle: 0,0 à 19,7).
Traitement des hémorragies: parmi les 315 épisodes hémorragiques traités, 88,9 % ont été contrôlés avec 2 injections ou moins. La dose médiane utilisée pour traiter les épisodes hémorragiques était de 66,2 U.I./kg.
Globalement, le profil risques-bénéfices d'AFSTYLA chez les PUP correspondait à celui rapporté chez les PTP, à l'exception de l'apparition d'anticorps neutralisants qui représente un risque reconnu associé au traitement de substitution par le FVIII chez les PUP.
Des données sur l'induction de l'immunotolérance (ITI) ont été recueillies auprès de patients avec hémophilie A ayant développé des inhibiteurs contre le FVIII. Pendant l'étude clinique menée auprès des PUP, 11 patients ont reçu un traitement ITI par AFSTYLA et 9 patients ont atteint une immunotolérance objectivée par un résultat négatif au test des anticorps à la fin de l'étude.
Il convient de noter que le taux d'hémorragie annuel (ABR) n'est pas comparable entre différents concentrés de facteurs et différentes études cliniques.
PharmacocinétiqueAdultes
La pharmacocinétique (PC) d'AFSTYLA a été étudiée chez 81 sujets adultes après injection par voie intraveineuse d'une dose unique de 50 UI/kg.
Les paramètres PC s'appuient sur l'activité plasmatique du facteur VIII mesurée par le dosage chromogénique. Le profil PC obtenu 3 à 6 mois après l'évaluation de la PC initiale était comparable au profil obtenu après la première dose.
Paramètres pharmacocinétiques (moyenne arithmétique, %CV) après une injection unique de 50 UI/kg d'AFSTYLA - Dosage chromogénique:
|
Paramètre pharmacocinétique
|
rVIII-SingleChain
50 UI/kg
(N = 81)
| |
IR (UI/dl)/(UI/kg)
|
2,00 (20,8)
| |
Cmax (UI/dl)
|
106 (18,1)
| |
AUC0-inf (UI*h/dl)
|
1960 (33,1)
| |
t½(h)
|
14,2 (26,0)
| |
MRT (h)
|
20,4 (25,8)
| |
CL (ml/h/kg)
|
2,90 (34,4)
| |
Vss (ml/kg)
|
55,2 (20,8)
|
IR = recouvrement incrémentiel, enregistré 30 minutes après injection; Cmax = concentration maximale; AUC0-inf = surface sous la courbe activité du facteur VIII - temps extrapolée à l'infini; t½= durée de demi-vie; MRT = durée moyenne de séjour; CL = clairance corrigée par le poids corporel; Vss = volume de répartition corrigé par le poids corporel à l'état stationnaire.
Enfants et adolescents
La pharmacocinétique (PC) d'AFSTYLA a été étudiée chez 10 adolescents (âge 12 à <18 ans) et 39 enfants (âge 0 à <12 ans) après injection par voie intraveineuse d'une dose unique de 50 UI/kg.
Les paramètres PC s'appuient sur l'activité plasmatique du facteur VIII mesurée par le dosage chromogénique.
Comparaison des paramètres pharmacocinétiques en fonction de la classe d'âge (moyenne arithmétique, %CV) après une injection unique de 50 UI/kg d'AFSTYLA - Dosage chromogénique:
|
Paramètre pharmacocinétique
|
0 à <6 ans
(N = 20)
|
6 à <12 ans
(N = 19)
|
12 à <18 ans
(N = 10)
| |
IR (UI/dl)/(UI/kg)
|
1,60 (21,1)
|
1,66 (19,7)
|
1,69 (24,8)
| |
Cmax (UI/dl)
|
80,2 (20,6)
|
83,5 (19,5)
|
89,7 (24,8)
| |
AUC0-inf (UI*h/dl)
|
1080 (31,0)
|
1170 (26,3)
|
1540 (36,5)
| |
t½ (h)
|
10,4 (28,7)
|
10,2 (19,4)
|
14,3 (33,3)
| |
MRT (h)
|
12,4 (25,0)
|
12,3 (16,8)
|
20,0 (32,2)
| |
CL (ml/h/kg)
|
5,07 (29,6)
|
4,63 (29,5)
|
3,80 (46,9)
| |
Vss (ml/kg)
|
71,0 (11,8)
|
67,1 (22,3)
|
68,5 (29,9)
|
IR = recouvrement incrémentiel, enregistré 30 minutes après injection sur des sujets de l'âge de 12 à <18 ans et 60 minutes après injection sur des sujets de l'âge de 1 à <12 ans; Cmax = concentration maximale; AUC0-inf = surface sous la courbe activité du facteur VIII - temps extrapolée à l'infini; t½ = durée de demi-vie; MRT = durée moyenne de séjour; CL = clairance corrigée par le poids corporel; Vss = volume de répartition corrigé par le poids corporel à l'état stationnaire
Absorption
Sans objet.
Distribution
Sans objet.
Métabolisme
Sans objet.
Élimination
Sans objet.
Données précliniquesLes données précliniques issues des études conventionnelles sur la pharmacologie de sécurité, la toxicité en administration unique et répétée, ainsi que les tests portant sur la tolérance locale et la thrombogénicité n'ont pas révélé de risque particulier pour l'homme.
Aucune étude pour examiner la génotoxicité, la carcinogénicité et la toxicité pour la reproduction avec le principe actif d'AFSTYLA n'a été réalisée.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments, diluants ou solvants, à l'exception du solvant indiqué dans la rubrique «Composition».
Stabilité
AFSTYLA ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Stabilité après ouverture:
La stabilité chimique et physique d'administration après reconstitution a été prouvée pour 48 heures à température ambiante (maximum 25 °C). D'un point de vue microbiologique, le produit doit être utilisé immédiatement. S'il n'est pas utilisé immédiatement, une conservation de 4 heures à température ambiante (à maximum 25 °C) ne doit pas être dépassée.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C)
Ne pas congeler.
Conserver hors de portée des enfants.
Conserver le récipient dans son carton pour protéger le contenu de la lumière.
AFSTYLA peut être conservé à température ambiante, mais à une température ne dépassant pas 25 °C, exceptionnellement pendant une période pouvant aller jusqu'à 3 mois dans le délai de conservation indiqué sur le carton et l'étiquette du flacon.
Remarques concernant la manipulation
Remarques générales
·La solution doit être pratiquement incolore, transparente ou légèrement opalescente. Après filtration/prélèvement (cf. ci-dessus), le produit reconstitué doit être inspecté visuellement pour exclure la présence de particules et tout changement de coloration avant administration
·Ne pas utiliser de solutions visiblement troubles ou qui contiennent encore des flocons ou des particules.
·La reconstitution et le prélèvement doivent être réalisés dans des conditions aseptiques.
Préparation
Porter le solvant à température ambiante (15-25 °C). Avant d'ouvrir l'emballage du dispositif Mix2Vial™, retirer les capsules amovibles des flacons de solvant et d'AFSTYLA, désinfecter les bouchons avec une solution antiseptique, puis laisser les sécher.
|
|
|
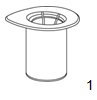
1. Ouvrez l'emballage du dispositif Mix2Vial en retirant l'opercule. Ne pas retirer le Mix2Vial de son emballage thermoformé!
| |
|
|
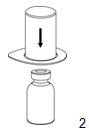
2. Placer le flacon de solvant sur une surface plane et propre et maintenez-le fermement. Saisir le dispositif Mix2Vial avec l'emballage thermoformé et enfoncer la pointe de l'extrémité bleue de l'adaptateur tout droit vers le bas dans le bouchon du flacon de solvant.
| |
|
|
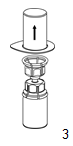
3. Retirer avec précaution l'emballage thermoformé du dispositif Mix2Vial en le tenant par les rebords et en le tirant verticalement vers le haut. Bien s'assurer que seul l'emballage thermoformé est retiré et non le dispositif Mix2Vial.
| |
|
|
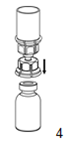
4. Poser le flacon d'AFSTYLA sur une surface plane et stable. Retourner le flacon de solvant muni du dispositif Mix2Vial et enfoncer la pointe de l'extrémité transparente de l'adaptateur verticalement vers le bas dans le bouchon du flacon d'AFSTYLA. Le solvant s'écoule automatiquement dans le flacon d'AFSTYLA.
| |
|
|
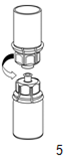
5. Saisir d'une main le côté AFSTYLA et de l'autre le côté solvant du dispositif Mix2Vial et séparer les flacons en dévissant le dispositif dans le sens inverse des aiguilles d'une montre, avec précaution afin d'éviter une formation excessive de mousse lors de la dissolution du produit.
Jeter le flacon de solvant muni de la partie bleue de l'adaptateur Mix2Vial.
| |
|
|
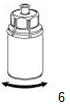
6. Tourner avec précaution le flacon d'AFSTYLA muni de la partie transparente de l'adaptateur jusqu'à dissolution complète du produit. Ne pas secouer!
| |
|
|
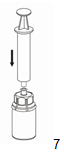
7. Aspirer de l'air dans une seringue stérile vide. Maintenir verticalement le flacon d'AFSTYLA, raccorder la seringue à l'embout Luer-Lock du dispositif Mix2Vial en vissant dans le sens des aiguilles d'une montre et injecter l'air dans le flacon d'AFSTYLA.
|
Prélèvement et administration
|
|

|
8. Maintenir le piston de la seringue enfoncé, retourner l'ensemble du système et prélever lentement le produit dans la seringue en tirant le piston.
| |
|
|
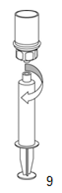
9. Une fois la solution entièrement transférée dans la seringue, saisir le corps de la seringue (en laissant le piston dirigé vers le bas) et séparer de la seringue l'adaptateur transparent du dispositif Mix2Vial en dévissant dans le sens inverse des aiguilles d'une montre.
|
Il est recommandé d'utiliser le nécessaire fourni pour l'injection d'AFSTYLA car l'absorption du facteur VIII sur la surface interne de certains dispositifs d'injection, peut entraîner un échec du traitement.
Veiller à ce qu'il n'y ait pas de sang dans la seringue remplie de produit, car le sang risque de coaguler dans la seringue et des caillots de fibrine peuvent ainsi être administrés au patient.
La solution d'AFSTYLA ne doit pas être diluée.
La solution reconstituée doit être injectée dans une voie d'injection/perfusion distincte, en injection intraveineuse lente à une vitesse confortable pour le patient.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation locale en vigueur.
Numéro d’autorisation66030 (Swissmedic)
PrésentationPour chaque flacon à:
500 U.I. de poudre et solvant (2,5 ml) (B)
1000 U.I. de poudre et solvant (2,5 ml) (B)
2000 U.I. de poudre et solvant (5 ml) (B)
sont fournis:
1 filtre de transfert 20/20
1 kit d'administration (carton intérieur):
1 kit de ponction veineuse,
2 tampons imbibés d'alcool,
1 pansement non stérile,
1 seringue à usage unique
Titulaire de l’autorisationCSL Behring Lengnau AG,
Lengnau
Mise à jour de l’informationSeptembre 2023
|