CompositionPrincipes actifs
[18F] Flutemetamolum (18F) 150 MBq à la date et à l’heure de référence.
Excipients
Ethanolum anhydricum (55,2 mg), Polysorbatum 80, Natrii chloridum (9,00 mg), Natrii dihydrogenophosphas dihydricus (0,49 mg), Dinatrii phosphas dodecahydricus (3,88 mg), Aqua ad iniectabilia.
Chaque ml de solution contient 4,1 mg de sodium.
Spécifications
Pureté radiochimique: ≥ 90 %
Pureté du radionucléide: ≥ 99,9 %
L’activité par flacon peut varier de 150 MBq à 1500 MBq.
Date et heure de calibration: voir bulletin de livraison et étiquette du flacon.
Indications/Possibilités d’emploiVIZAMYL est un médicament radiopharmaceutique indiqué pour l’imagerie en Tomographie par Émission de Positons (PET) afin d’estimer la densité des plaques β-amyloïdes neuritiques dans le cerveau de patients adultes ayant une déficience cognitive qui sont en cours d’évaluation pour la maladie d’Alzheimer (MA) et pour d’autres causes de déficience cognitive. VIZAMYL doit être utilisé en complément à l’évaluation clinique.
Un résultat négatif de l’examen PET indique une densité très faible à nulle de plaques amyloïdes, ce qui n’est pas compatible avec un diagnostic de MA. Pour les limitations d’interprétation des résultats positifs de l’examen PET, voir les rubriques «Mises en garde et précautions, limites d’utilisation».
Produit radiodiagnostique.
Posologie/Mode d’emploiUn examen PET avec flutémétamol (18F) doit être demandé par des cliniciens expérimentés dans la prise en charge clinique des maladies neurodégénératives. Les images obtenues avec VIZAMYL ne doivent être interprétées que par des évaluateurs formés à l’interprétation d’images PET avec le flutémétamol (18F). Une imagerie Tomodensitométrique (TDM) ou une imagerie par Résonance Magnétique (IRM) récente du patient est recommandée afin d’obtenir une image de fusion PET-TDM ou PET-IRM en cas d’incertitude quant à l’emplacement de la substance grise et de la limite substance grise/blanche sur l’examen PET (voir la rubrique «Mises en garde et précautions; Interprétation des images obtenues avec VIZAMYL»).
Posologie
Adultes
L’activité recommandée pour un adulte est de 185 MBq de flutémétamol (18F) administrés par voie intraveineuse (en bolus de 40 secondes environ). Le volume de l’injection ne doit pas être inférieur à 1 ml et ne doit pas excéder 10 ml.
Groupes de patients particuliers
Aucune étude étendue de détermination et d’ajustement de la dose chez des populations saines et particulières n’a été effectuée avec ce médicament. La pharmacocinétique du flutémétamol (18F) chez des patients atteints d’insuffisance rénale ou hépatique n’a pas été caractérisée.
Patients âgés
Aucune adaptation posologique n’est recommandée sur la base de l’âge.
Patients présentant des troubles de la fonction rénale ou hépatique
VIZAMYL n’a pas été étudié chez des patients présentant une insuffisance rénale ou hépatique significative. Il convient d’évaluer attentivement l’activité à administrer car ces patients sont potentiellement plus exposés aux rayonnements (voir la rubrique «Mises en garde et précautions»).
Enfants et adolescents
VIZAMYL n’est pas indiqué chez les enfants et les adolescents.
Mode d’administration
VIZAMYL doit être administré par voie intraveineuse. L’activité du flutémétamol (18F) doit être mesurée avec un activimètre immédiatement avant l’injection.
L’injection de VIZAMYL à l’aide d’un cathéter intraveineux court (environ 12,5 cm ou moins) diminue la possibilité d’une adsorption de la substance active par le cathéter. VIZAMYL est prévu pour un usage multidose. Il ne doit pas être dilué.
La dose est administrée par injection intraveineuse en bolus de 40 secondes environ. En cas d’utilisation d’un cathéter intraveineux, faire suivre l’injection d’un rinçage par injection intraveineuse de 5 à 15 ml d’une solution injectable isotonique stérile de chlorure de sodium 9 mg/ml (0,9 %) afin d’assurer l’administration complète de la dose requise. L’injection de flutémétamol (18F) doit se faire par voie intraveineuse afin d’éviter toute irradiation due à une extravasation locale, ainsi que des artefacts d’imagerie.
Acquisition des images
L’acquisition des images VIZAMYL doit débuter 90 minutes après l’injection, à l’aide d’un tomographe PET en mode 3D avec corrections appropriées des données. Positionner le patient en décubitus dorsal ; le cerveau du patient (y compris le cervelet) doit être positionné de façon à figurer sur un seul champ de vue de la caméra. La tête du patient doit être inclinée de sorte que le plan commissure antérieure-commissure postérieure (CA-CP) fasse un angle droit avec l’axe d’entrefer du tomographe PET. Pour ce faire, la tête doit être positionnée dans une têtière adéquate. Il est parfois nécessaire de limiter les mouvements de la tête à l’aide de bandes de contention adhésives ou de tout autre moyen de contention souple pour la tête.
Il est recommandé d’utiliser pour la reconstruction itérative ou par rétroprojection filtrée, une épaisseur de coupe de 2 à 4 mm, et une taille de matrice de 128 x 128 avec des tailles de pixel d’environ 2 mm. Lorsqu’un filtre de postlissage est appliqué avec une largeur totale à mi-hauteur (FWHM) de 5 mm au maximum, le filtre FWHM recommandé doit être choisi pour optimiser le rapport signal/bruit tout en préservant la netteté de l’image reconstruite. La durée de l’examen sera généralement de 20 minutes.
Exposition aux rayonnements
Le Tableau 1 ci-dessous montre la dosimétrie telle que calculée en utilisant le logiciel OLINDA/EXM (Organ Level INternal Dose Assessment/Exponential Modeling). L’estimation des doses de rayonnement absorbées pour les adultes suivant l’injection intraveineuse de VIZAMYL est indiquée au Tableau 1. Les valeurs ont été calculées en supposant une vidange de la vessie à intervalles de 3,5 heures et en utilisant les données de biodistribution chez l’homme et le logiciel OLINDA/EXM.
Tableau 1 Estimation des doses de rayonnement absorbées après administration intraveineuse de VIZAMYL (adultes)
|
Organe/Tissus
|
Dose absorbée par activité administrée [mGy/MBq]
| |
Surrénales
|
0,013
| |
Cerveau
|
0,011
| |
Seins
|
0,005
| |
Paroi de la vésicule biliaire
|
0,287
| |
Cœur
|
0,014
| |
Reins
|
0,031
| |
Foie
|
0,057
| |
Paroi du gros intestin inférieur
|
0,042
| |
Poumons
|
0,016
| |
Muscles
|
0,009
| |
Cellules ostéogéniques
|
0,011
| |
Ovaires
|
0,025
| |
Pancréas
|
0,015
| |
Moelle rouge
|
0,013
| |
Peau
|
0,005
| |
Paroi de l’intestin grêle
|
0,102
| |
Rate
|
0,015
| |
Estomac
|
0,012
| |
Testicules
|
0,008
| |
Thymus
|
0,006
| |
Thyroïde
|
0,006
| |
Paroi du gros intestin supérieur
|
0,117
| |
Vessie
|
0,145
| |
Utérus
|
0,025
| |
Autres organes
|
0,012
| |
Dose efficace (mSv/MBq)
|
0,032
|
La dose adulte efficace résultant de l’administration d’une activité maximale recommandée de 185 MBq pour un adulte de 70 kg est d’environ 5,9 mSv. Pour une activité administrée de 185 MBq, la dose type de rayonnement à l’organe cible (cerveau) est de 2,0 mGy. Si un examen TDM est effectué simultanément dans le cadre de la procédure PET, l’exposition aux rayonnements ionisants augmentera selon une quantité dépendant des réglages utilisés lors de l’acquisition TDM.
Pour une activité administrée de 185 MBq, les doses types de rayonnement délivrées aux organes critiques, comme la vésicule biliaire, la paroi de la vessie, la paroi du gros intestin supérieur, la paroi du gros intestin inférieur, l’intestin grêle et le foie sont de 53,1 mGy, 26,8 mGy, 21,8 mGy, 7,8 mGy, 18,9 mGy et 10,5 mGy, respectivement.
Contre-indicationsHypersensibilité au principe actif ou à l’un des excipients, voir la rubrique «Forme pharmaceutique et quantité de principe actif par unité».
Mises en garde et précautionsPossibilité de réactions d’hypersensibilité ou anaphylactiques
En cas de réaction d’hypersensibilité ou anaphylactique, l’administration du produit doit être immédiatement interrompue et un traitement par voie intraveineuse doit être débuté, si nécessaire. Afin de permettre une prise en charge rapide en cas d’urgence, il convient d’avoir à disposition immédiate les médicaments et le matériel nécessaires, notamment une sonde d’intubation trachéale et du matériel de ventilation.
Bénéfice individuel/justification du risque
Pour chaque patient, l’exposition aux rayonnements ionisants doit être justifiée par le bénéfice attendu. L’activité administrée doit correspondre à la plus faible dose de rayonnements possible compatible avec l’obtention de l’information diagnostique recherchée.
Troubles de la fonction rénale/hépatique
Il convient d’évaluer attentivement le rapport bénéfice-risque chez ces patients, car une exposition accrue aux rayonnements est possible. Le flutémétamol(18F) est excrété essentiellement par le système hépatobiliaire et les patients présentant une insuffisance hépatique sont potentiellement plus exposés aux rayonnements. Voir la rubrique «Posologie/Mode d’emploi».
Enfants et adolescents
Pour plus d’informations sur l’utilisation chez les enfants et les adolescents, voir les rubriques «Posologie/Mode d’emploi» ou «Propriétés/Effets».
Interprétation des images obtenues avec VIZAMYL
Les images obtenues avec VIZAMYL ne doivent être interprétées que par des évaluateurs formés à l’interprétation d’images PET avec flutémétamol (18F). Un examen négatif indique une densité très faible à nulle de plaques ß-amyloïdes neuritiques corticales. Un examen positif indique une densité modérée à forte. Des erreurs d’interprétation des images de la densité des plaques ß--amyloïdes neuritiques du cerveau, incluant des faux négatifs et des faux positifs, ont été observées. L’interprétation des images doit être contrôlée indépendamment des données cliniques du patient. L’utilisation de données cliniques pour l’interprétation des images PET VIZAMYL n’a pas été évaluée.
Les images PET doivent être lues à l’aide d’une échelle de couleurs Sokoloff, Rainbow ou Spectrum. L’évaluateur doit comparer l’intensité du signal de la substance grise corticale par rapport à l’intensité maximale du signal de la substance blanche. Les images doivent être visualisées de manière systématique (Figure 1) en commençant au niveau du pont (p) et en défilant vers le haut:
·Lobes frontaux et cortex cingulaire antérieur (f, ac, analyse axiale)
·Cortex cingulaire postérieur et précunéus (pc, analyse sagittale)
·Aspects des zones temporo-pariétales incluant l’insula (in, analyse axiale et tp-in, analyse coronale)
·Lobes temporo- latéraux (lt, analyse axiale)
·Région striatale (s, analyse axiale)
L’interprétation des images se fait visuellement, en comparant l’activité dans la substance grise corticale et l’activité dans la substance blanche adjacente.
·Une région est considérée comme ayant une fixation négative (normale) si le signal du traceur est faible dans les régions corticales (intensité du signal nettement inférieure comparée à la substance blanche adjacente et similaire en intensité aux régions riches en substance grise du cervelet). Le signal ne sera pas complètement absent dans les régions de la substance grise, car la fixation de la substance blanche déborde sur les régions de substance grise du fait de l’effet de volume partiel lié à la résolution des caméras PET.
·Une région est considérée comme positive (anormale) si le signal du traceur dans les régions corticales semble élevé (d’une intensité environ égale ou supérieure à celle de la substance blanche adjacente et supérieure aux régions riches en substance grise du cervelet).
·Si l’une de ces régions est clairement positive (anormale), l’image doit alors être classée positive (anormale). Sinon, elle doit être classée négative (normale).
Une atrophie peut être présente dans de nombreuses zones du cerveau et peut compliquer l’interprétation des images car la perte de substance grise résultera en une fixation réduite du traceur rendant un examen positif plus difficile à détecter. Il est fortement recommandé de passer en revue les images IRM ou TDM si elles sont disponibles afin d’aider à l’interprétation de l’image VIZAMYL, en particulier si une atrophie est suspectée.
Figure 1
Cas de PET avec VIZAMYL montrant des exemples d’examens PET au flutémétamol (18F) négatifs (gauche) et positifs (droite). Les vues axiales (première ligne), sagittales (seconde ligne) et coronales (troisième ligne) sont représentées.
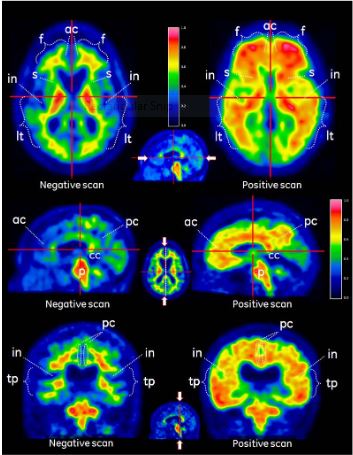
Figure 1. Vues axiales (a), sagittales (b) et coronales (c) d’images négatives et positives avec le flutémétamol (18F) (gauche et droite respectivement). Les images négatives montrent le motif sulcogyral de la substance blanche alors qu’il n’est pas discernable sur les images positives à droite. Noter l’intensité plus élevée (>60 % du maximum) dans la substance grise des images positives comparativement aux images négatives et l’extension radiale de l’intensité vers le bord convexe nettement défini des faces latérales. Les images négatives présentent une intensité dégressive vers la périphérie des tissus. Il faut noter également dans les régions médiales un plus haut niveau d’intensité dans la substance grise sur les images positives à droite.
Légende: Substance grise – f frontal et ac cingulaire antérieur, pc cingulaire postérieur et précunéus, lt temporo-latéral, tp temporo-pariétal, in insula et s striatum. Substance blanche – p pont et cc corps calleux.
L’évaluation quantitative de l’intensité du signal radioactif cortical par un logiciel validé et certifié CE peut être utilisée pour aider à l’interprétation visuelle de la distribution du signal radioactif. Un tel logiciel permet le calcul de la quantité totale d’amyloïde dans le cerveau en divisant l’intensité moyenne de l’image dans les régions corticales associées à des dépôts d’amyloïde (augmentée chez des patients atteints de MA), par l’intensité moyenne de l’image dans une région de référence telle que le pont. Cette valeur est appelée rapport de valeurs de fixation normalisée (Standard Uptake Value Ratio [SUVR]). Des évaluations visuelles dichotomiques d’examens réalisés avec le flutémétamol (18F) ont été validées vis-à-vis de la limite entre une densité faible et modérée de plaques neuritiques. Il a été déterminé que des valeurs seuils de SUVR de 0,59 à 0,61, obtenues avec un logiciel certifié CE utilisant le pont comme région de référence, présentaient une très grande concordance avec l’interprétation visuelle (voir la rubrique «Propriétés/Effets») et étaient utilisées en complément à l’interprétation visuelle des images.
Les utilisateurs doivent être formés à l’utilisation du logiciel certifié CE par le fabricant et doivent avoir suivi une formation sur l’évaluation de l’interprétation visuelle des images avec Vizamyl.
En cas de discordance entre les résultats de la quantification et ceux de l’interprétation visuelle, il convient d’envisager de suivre les étapes suivantes pour obtenir une interprétation finale.
L’image doit tout d’abord être interprétée visuellement.
Puis la quantification doit être réalisée selon les instructions du fabricant, en incluant le contrôle de qualité du processus de quantification. Le résultat de la quantification doit être comparé avec l’interprétation visuelle, en prenant garde aux plages de variation attendues pour un examen négatif ou positif. Si les taux de quantification ne concordent pas avec ceux de l’interprétation visuelle, l’évaluateur doit:
1. Vérifier la délimitation des régions d’intérêt (Regions of interest / ROIs) sur l’image. Les ROIs doivent se situer dans la substance grise du cerveau, de manière à ne pas inclure le liquide cérébrospinal ou des régions significatives de substance blanche.
2. Vérifier le positionnement de la/des ROIs de référence pour s’assurer que celles-ci sont toutes ajustées à la région de référence. En outre, il convient d’examiner l’aspect de la région de référence à la recherche d’anomalies structurelles ou de zones de moindre vascularisation.
3. Spécificités en cas de différence entre les résultats visuels et quantitatifs:
i) En cas d’évaluation visuelle positive et de résultat de quantification négatif ou limite, les régions positives à l’examen visuel doivent être comparées avec les régions équivalentes étudiées par une ROI. En cas de fixation du traceur très focalisée, il se peut que la ROI échantillonne une plus grande surface et que la moyenne de la ROI donne ainsi un résultat négatif pour l’amyloïde. De plus, des zones d’atrophie pourraient être exclues lors d’une interprétation visuelle, mais être incluses lors d’une quantification.
ii) En cas d’évaluation visuelle négative pour l’amyloïde et de résultat de quantification positif, il convient d’inspecter la région de référence. En cas de doute sur l’exactitude de la délimitation des ROIs ou en cas de réduction manifeste de la fixation, il convient d’utiliser une région alternative (le logiciel peut accepter un certain nombre de régions de référence). De plus, le positionnement des ROIs corticales doit être vérifié; si elles contiennent de la substance blanche, cela pourrait augmenter les valeurs de quantification.
4. Faire une interprétation finale de l’examen sur la base de l’interprétation visuelle après avoir réalisé les étapes 1 à 3 ci-dessus.
Limites d’utilisation
Un examen positif ne permet pas d’établir isolément un diagnostic de MA ou d’autres déficiences cognitives puisque des plaques neuritiques peuvent être présentes dans la substance grise de patients âgés asymptomatiques et de patients atteints d’autres démences neurodégénératives (Maladie d’Alzheimer, mais aussi démence à corps de Lewy et maladie de Parkinson). L’efficacité du flutémétamol (18F) pour évaluer la réponse à un traitement n’a pas été établie (voir la rubrique «Propriétés/Effets»).
Certains examens peuvent être difficiles à interpréter en raison du bruit de fond sur l’image, d’une atrophie avec amincissement cortical, ou d’une image floue pouvant entraîner des erreurs d’interprétation. Dans les cas pour lesquels il y a incertitude quant à l’emplacement de la substance grise et de la limite substance grise/blanche sur l’examen PET, si une image TDM ou IRM couplée récente est disponible, l’évaluateur devra examiner l’image fusionnée PET-TDM ou PET-IRM pour clarifier la relation entre l’activité PET et l’anatomie de la substance grise.
Après l’examen
Tout contact étroit avec les jeunes enfants et les femmes enceintes doit être restreint dans les 24 heures suivant l’administration.
Mises en garde spécifiques
Ce médicament contient de l’éthanol (alcool) 7 % vol, autrement dit jusqu’à 552 mg (environ 0,7 ml) par dose, ce qui équivaut à 14 ml de bière ou 6 ml de vin par dose. La faible quantité d’alcool contenue dans ce médicament n’a aucun effet perceptible.
Ce médicament contient 41 mg de sodium par dose, ce qui équivaut à 2 % de l’apport alimentaire quotidien maximal recommandé par l’OMS de 2 g de sodium par adulte.
Pour les précautions liées aux risques environnementaux, voir la rubrique «Remarques particulières».
InteractionsIl n’a pas été effectué d’études d’interactions pharmacodynamiques chez des patients pour établir jusqu’à quel point, le cas échéant, des médications concomitantes peuvent altérer les résultats des images avec VIZAMYL. Aucune étude d’interactions in vivo n’a été réalisée.
Les études de fixation in vitro n’ont pas montré d’interférence sur la liaison du flutémétamol (18F) aux plaques β-amyloïdes en présence d’autres médicaments communément pris par des patients atteints de la MA.
Grossesse, AllaitementFemmes en âge de procréer
Il convient d’exclure toute grossesse avant l’administration du produit. Lorsqu’il est nécessaire d’administrer des produits radiopharmaceutiques chez la femme en âge de procréer, toute éventualité de grossesse doit être écartée, par exemple en effectuant un test de grossesse. Toute femme n’ayant pas eu ses règles doit être considérée comme enceinte jusqu’à preuve du contraire. Dans l’incertitude quant à une éventuelle grossesse (absence de règles, règles irrégulières, etc.), d’autres techniques n’utilisant pas les rayonnements ionisants (le cas échéant) doivent être envisagées.
Grossesse
Aucune étude n’a été menée chez des femmes enceintes. Aucune étude des effets du flutémétamol (18F) sur la reproduction n’a été menée chez l’animal (voir la rubrique «Données précliniques»). Chez la femme enceinte, le traitement par des radionucléides peut exposer le fœtus à des rayonnements. Par conséquent, seuls les examens essentiels doivent être réalisés, si le bénéfice attendu excède largement le risque encouru par la mère et le fœtus.
Allaitement
On ne sait pas si le flutémétamol (18F) est excrété dans le lait maternel pendant l’allaitement. Avant toute administration d’un produit radiopharmaceutique pendant l’allaitement, il convient d’envisager de repousser le traitement par des radionucléide après l’arrêt de l’allaitement, car la radioactivité est éliminée dans le lait maternel.
Si un traitement est nécessaire, l’allaitement doit être interrompu pendant 24 heures et le lait tiré doit être jeté. Tout contact étroit avec les jeunes enfants doit être restreint dans les 24 heures suivant l’administration.
Fertilité
Aucune étude sur la fertilité n’a été effectuée.
Effet sur l’aptitude à la conduite et l’utilisation de machinesVIZAMYL n’a aucune influence ou une influence négligeable sur l’aptitude à la conduite ou l’utilisation de machines.
·Toutefois, VIZAMYL peut entraîner des étourdissements et des vertiges transitoires. Par conséquent, après l’administration de VIZAMYL, il est conseillé aux patients de ne pas conduire, de ne pas utiliser des machines complexes ou de ne pas participer à des activités potentiellement dangereuses jusqu’à disparition complète de ces effets.
Effets indésirablesRésumé du profil de sécurité
Le profil de sécurité global de VIZAMYL est basé sur des données provenant de son administration à 831 sujets.
Des réactions anaphylactoïdes sont survenues occasionnellement dans le cadre des études cliniques. Diverses réactions indésirables, comme des rougeurs, une hypertension, une gêne dans la poitrine, des nausées, des céphalées ou encore des étourdissements, se sont produites à une fréquence de 2 % maximum, avec un degré de gravité allant de léger à modéré.)
Liste des effets indésirables
Les fréquences de réactions indésirables sont définies comme suit:
Très fréquents (≥1/10), fréquents (≥1/100 à <1/10), occasionnels (≥1/1000 à <1/100), rares (≥1/10 000 à <1/1000), très rares (<1/10 000). À l’intérieur de chaque groupe de fréquences, les réactions indésirables sont présentées par ordre de gravité décroissant.
Affections du système immunitaire
Occasionnels: réaction anaphylactoïde
Affections psychiatriques
Occasionnels: anxiété
Affections du système nerveux
Occasionnels: étourdissement, céphalée, hypoesthésie, hypotonie, désordre du goût, tremblement
Affections oculaires
Occasionnels: Gonflement oculaire
Affections de l’oreille et du labyrinthe
Occasionnels: vertige
Affections cardiaques
Occasionnels: palpitations
Affections vasculaires
Fréquents: rougeur
Occasionnels: pâleur
Affections respiratoires, thoraciques et médiastinales
Occasionnels: dyspnée, hyperventilation, irritation de la gorge
Affections gastro-intestinales
Occasionnels: nausée, dyspepsie, gêne abdominale, troubles dans la cavité buccale, vomissement
Affections de la peau et du tissu sous-cutané
Occasionnels: hypoesthésie de la face, prurit, rash cutanée, tiraillement de la peau, œdème du visage
Affections musculosquelettiques et du tissu conjonctif
Occasionnels: dorsalgie, tension musculaire, douleurs musculosquelettiques
Affections des organes de reproduction et du sein
Occasionnels: dysfonction érectile
Troubles généraux et anomalies au site d’administration
Occasionnels: gêne dans la poitrine, sensation de chaleur, asthénie, fatigue, sensation de malaise, sensation de froid, douleur au site d’injection, œdème, fièvre
Investigations
Fréquents: pression artérielle élevée
Occasionnels: baisse de la glycémie, augmentation de la lactate déshydrogénase sanguine, augmentation de la numération des neutrophiles, augmentation de la fréquence respiratoire
L’exposition aux rayonnements ionisants peut entraîner le développement de cancers et d’anomalies héréditaires. Comme la dose efficace est d’environ 5,9 mSv lorsque l’activité maximale recommandée de 185 MBq de flutémétamol (18F) est administrée, la probabilité de survenue de ces évènements indésirables est faible.
Description de certains effets indésirables
Les réactions indésirables suivantes peuvent se produire sous forme de symptômes et de signes d’une réaction d’hypersensibilité à VIZAMYL ou à l’un de ses excipients (voir la rubrique «Composition»): œdème oculaire/du visage, pâleur, dyspnée, irritation de la gorge, vomissements, rash cutanée, prurit, tiraillement de la peau, gêne respiratoire (voir la rubrique «Mises en garde et précautions»).
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDu fait de la faible quantité de flutémétamol (18F) présente dans chaque dose, un surdosage ne devrait pas entraîner d’effets pharmacologiques mais devrait conduire à une exposition accrue au rayonnement. L’élimination du produit radiopharmaceutique est favorisée par une augmentation de la fréquence des mictions et de l’excrétion fécale. Il peut être utile d’estimer la dose effective qui avait été appliquée.
Propriétés/EffetsCode ATC
V09AX04
Propriétés physiques
Le fluor (18F) se désintègre en oxygène stable (18O) avec une demi-vie d’environ 110 minutes en émettant un rayonnement de positrons de 634 keV, suivi d’un rayonnement photonique d’annihilation de 511 keV.
Mécanisme d’action
Le flutémétamol (18F) se lie aux plaques β-amyloïdes neuritiques dans le cerveau.
In vitro, le flutémétamol (18F) se lie aux plaques β-amyloïdes neuritiques cérébrales et, de manière négligeable, aux enchevêtrements neurofibrillaires. Les données suggèrent que le flutémétamol peut marquer les dépôts β-amyloïdes constitués et diffus et les plaques neuritiques. Il n’existe aucune preuve suggérant que le flutémétamol (18F) se lie aux formes solubles Abeta.
Lors d’études d’imagerie PET in-vivo, une corrélation quantitative a été établie chez des patients en fin de vie entre l’activité du flutémétamol (18F) fixé par la substance grise corticale et la totalité de la charge β-amyloïde mise en évidence sur des échantillons d’autopsie par marquage par anticorps anti β-amyloïdes 4G8 au niveau des plaques neuritiques comme des plaques diffuses. In-vivo, le flutémétamol (18F) peut détecter les plaques β-amyloïdes diffuses lorsqu’elles sont fréquentes. La fixation in-vivo du flutémétamol (18F) aux autres structures β-amyloïdes ou à d’autres structures ou récepteurs cérébraux n’est pas connue.
Pharmacodynamique
Aux faibles niveaux de concentration présents dans VIZAMYL, le flutémétamol (18F) n’a pas d’activité pharmacodynamique détectable.
La fixation dans le cerveau et la distribution du flutémétamol (18F) n’ont pas été évaluées dans une étude spécifique visant à évaluer ses caractéristiques pharmacodynamiques. Dans deux études similaires de biodistribution et une étude clinique de phase II, les valeurs quantitatives moyennes de fixation issues des images PET différaient entre les sujets présentant une MA cliniquement probable et les volontaires sains dans la plupart des zones étudiées du cerveau.
Après l’injection intraveineuse, le flutémétamol (18F) se diffuse à travers la barrière hémato-encéphalique humaine et produit dans le cerveau un signal radioactif pouvant être détecté. Ensuite, sous l’effet de l’irrigation sanguine du cerveau, la concentration en flutémétamol (18F) diminue, avec une différence de concentration du traceur entre les régions corticales qui présentent des dépôts β-amyloïdes et les zones exemptes de dépôts. Les courbes temps-activité du flutémétamol (18F) dans le cerveau des participants de l’étude dont les examens sont positifs montrent une augmentation continue du signal de 0 à 30 minutes suivant l’injection. Ensuite, les valeurs restent stables pendant au moins 120 minutes après l’injection. L’interprétation des images repose sur les variations d’intensité du signal entre les zones du cerveau présentant une concentration spécifique de flutémétamol (18F) et celles présentant une concentration non spécifique du traceur.
La répartition «examen – reprise d’examen» du flutémétamol (18F) a été évaluée chez 5 participants de l’étude présentant un diagnostic clinique de MA probable. Dans un intervalle d’1 à 4 semaines, ces patients ont reçu 2 injections de flutémétamol (18F), suivies d’un examen PET. L’évaluation semi-quantitative effectuée au moyen d’une estimation de l’unité de fixation normalisée dans les zones corticales du cerveau détectées au préalable a permis de démontrer la reproductibilité des images.
Efficacité clinique
Une étude pivot chez 68 patients en fin de vie avait pour objectif d’établir les performances diagnostiques du flutémétamol (18F) pour la détection de la densité des plaques neuritiques corticales. Les résultats de l’examen PET ont été comparés à la densité des plaques neuritiques mesurée sur des coupes de huit régions cérébrales prédéfinies des patients à l’autopsie. Les régions histopathologiques incluaient, mais n’étaient pas limitées aux régions définies par le CERAD. L’état cognitif des patients n’avait pas été déterminé. Chez les 68 patients, l’interprétation visuelle en aveugle des examens PET effectuée par 5 évaluateurs sur la base de la majorité des interprétations des images a présenté une sensibilité de 86 % (IC à 95 %: 72 à 95 %) et une spécificité de 92 % (IC à 95 %: 74 à 99 %).
Une autre étude a permis d’étudier également la sensibilité et la spécificité du flutémétamol (18F) pour évaluer les dépôts β-amyloïdes en faisant appel à 5 autres lecteurs différents qui avaient suivi un programme de formation électronique et qui ont évalué en aveugle les images des 68 patients de l’étude pivot suivis jusqu’à autopsie. Les résultats d’histopathologie de l’étude pivot ont été utilisés. Sur la base de la majorité des interprétations de ces images, la sensibilité et la spécificité étaient respectivement de 93 % (IC à 95 %: 81 à 99 %) et de 84 % (IC à 95 %: 64 à 96 %).
Dans une étude de relecture qui incluait 38 patients supplémentaires ayant fait l’objet d’une autopsie du cerveau, permettant ainsi d’augmenter la population de l’étude pivot (à 106 patients), la sensibilité et la spécificité de détection de la densité des plaques β-amyloïdes neuritiques modérées à fréquentes a été en première analyse respectivement de 91 % (IC à 95 %: 82 à 96 %) et de 90 % (IC à 95 %: 74 à 98 %) sur la base de la majorité des interprétations (c.a.d. : concordance d’interprétation pour au moins 3 évaluateurs sur 5 ayant suivi le programme de formation électronique). Dans une analyse secondaire, utilisant comme standard de preuve le score de densité maximum des plaques neuritiques dans trois régions du néocortex recommandées initialement par le CERAD, la sensibilité était de 92 % (IC à 95 %: 83 à 97 %) et la spécificité de 88 % (IC à 95 %: 71,0 à 97 %).
Dans une étude longitudinale, 232 patients cliniquement diagnostiqués avec un trouble cognitif léger de type amnésique (aMCI) ont subi un examen PET initial avec le flutémétamol (18F) et ont été suivis pendant 36 mois afin de déterminer la relation entre l’imagerie au flutémétamol (18F) et les changements de statut diagnostique. 98 (42 %) des 232 patients avaient un examen anormal (positif) au flutémétamol (18F). Sur les 232 patients inclus, 224 ont eu au moins une évaluation supplémentaire post-examen PET par un comité indépendant et ont été inclus dans l’analyse. Après un suivi de 36 mois, la conversion vers une MA a été cliniquement diagnostiquée chez 81 (35 %) patients. Sur les 97 patients aMCI qui avaient un examen PET positif et au moins une évaluation par le comité indépendant, pour 52 (54 %) la conversion vers une MA a été cliniquement diagnostiquée après 36 mois comparé aux 29 (23 %) patients sur 127 qui présentaient un examen négatif et au moins une évaluation par le comité indépendant. À 36 mois, la sensibilité de l’examen au flutémétamol (18F) pour la prédiction de la conversion d’un trouble cognitif léger de type amnésique (aMCI) vers une MA chez les 81 patients ayant converti est de 64 % (IC à 95 %: 54 à 75 %) et la spécificité chez les de 143 patients n’ayant pas converti est de 69 % (IC à 95 %: 60 à 76 %). Sur la base de la majorité des interprétations des images, les rapports de probabilité positif et négatif sont respectivement de 2,04 et de 0,52.
Études cliniques sur l’utilisation supplémentaire d’informations quantitatives pour l’interprétation des images
La fiabilité de l’utilisation d’informations quantitatives pour aider à l’interprétation visuelle a été évaluée dans deux études cliniques ayant comparé la concordance entre les deux méthodes d’interprétation. Un logiciel certifié CE de quantification des dépôts amyloïdes a été utilisé dans les deux études (n(total) = 379)et le pourcentage de concordance entre l’interprétation visuelle et la quantification était de 98,8% à 99%.
Dans la première étude, les valeurs seuils pour la quantification des dépôts amyloïdes ont été calculées à partir du statut amyloïde post-mortem confirmé du cerveau comme valeur étalon (à partir de l’étude de cohorte pivot sur réalisation d’autopsies n = 68) et la plage de référence permettant de définir les valeurs quantitatives normales a été obtenue chez des volontaires sains (n = 105). Les valeurs seuils en découlant ont été utilisées pour catégoriser une cohorte de 172 images (33 MA probables, 80 MCI de type amnésique et 59 volontaires sains) comme négatives ou positives et comparer le résultat avec le classement obtenu avec l’examen visuel. La concordance était de 98,8% (170/172 examens).
La deuxième étude a évalué l’impact du PET amyloïde au flutémétamol (18F) sur le diagnostic et le traitement de patients consultant dans un centre tertiaire de la mémoire. Les images de 207 patients ont été évaluées par un examen visuel ou avec le logiciel certifié CE et la concordance des deux méthodes était de 99% (205/207 images).
Sécurité et efficacité en pédiatrie
La maladie ou le trouble pour lequel le médicament spécifique est prévu affecte uniquement les adultes (voir la rubrique «Posologie/Mode d’emploi; Enfants et adolescents»).
PharmacocinétiqueAbsorption
La fixation maximale du flutémétamol (18F) dans le cerveau d’environ 7 % de la dose injectée se produit dans les deux minutes qui suivent l’administration. Ceci est suivi par une clairance cérébrale rapide au bout des 90 premières minutes (temps recommandé pour commencer l’examen), suivi d’une clairance plus graduelle. Les cinq organes/tissus présentant les activités cumulées les plus élevées sont la paroi de l’intestin grêle, le foie, la paroi de la vessie, la paroi du gros intestin supérieur et la paroi de la vésicule biliaire.
Des témoins sains montrent de faibles niveaux de rétention du flutémétamol (18F) dans le cortex cérébral. Le niveau de fixation le plus élevé se situe dans le pont et d’autres régions de la substance blanche. Chez les patients atteints de la MA, les régions corticales et striatales montrent une fixation significativement supérieure par rapport aux régions corticales des contrôles. Chez les patients atteints de la MA, comme chez les contrôles, on retrouve une rétention élevée dans le pont et d’autres zones de substance blanche.
Les bases biophysiques de la rétention du flutémétamol (18F) dans la substance blanche du cerveau humain vivant ne sont pas totalement expliquées. L’hypothèse est que la solubilité du radiopharmaceutique dans les lipides contenus dans les tissus du cerveau pourrait contribuer à sa rétention dans la substance blanche.
Distribution
Le flutémétamol (18F) est distribué dans tout l’organisme quelques minutes après l’injection.
Après 20 minutes, environ 20 % du principe actif flutémétamol (18F) reste dans la circulation, ce taux tombe à 10 % après 180 minutes.
Métabolisme
Voir la rubrique «Absorption»
Élimination
Le flutémétamol (18F) est rapidement éliminé de la circulation (par les voies intestinale et urinaire). Vingt minutes après l’injection, 75 % de la radioactivité dans le plasma est présente sous forme de métabolites polaires. À 180 minutes, 90 % de la radioactivité dans le plasma est présente sous forme de métabolites polaires. L’élimination du flutémétamol (18F) est approximativement rénale à 37 % et hépatobiliaire à 52 %. La demi-vie d’élimination apparente est de 4,5 heures alors que la période radioactive du flutémétamol (18F) est de 110 minutes.
Cinétique pour certains groupes de patients
Troubles de la fonction rénale/hépatique
La pharmacocinétique chez des patients atteints d’insuffisance rénale ou hépatique n’a pas été caractérisée.
Données précliniquesLes données non cliniques issues des études conventionnelles sur la pharmacologie de sécurité et la toxicité lors d’une administration répétée n’ont mis en évidence aucun risque particulier pour l’être humain.
Le flutémétamol (18F) est positif dans des tests de génotoxicité in vitro sur les bactéries et les cellules de mammifères mais négatif dans trois études in-vivo différentes à des doses suffisamment élevées. Tout potentiel mutagène pertinent du point de vue clinique est par conséquent considéré comme fortement improbable.
Aucune étude de carcinogénicité et de toxicité sur la reproduction n’a été effectuée avec le flutémétamol (18F).
Remarques particulièresIncompatibilités
Aucune étude de tolérance n’ayant été effectuée, ce médicament ne doit pas être mélangé à d’autres médicaments.
Stabilité
Dix heures à compter de la fin de la synthèse.
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l’emballage.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 30°C. Tenir hors de portée des enfants.
Les médicaments radiopharmaceutiques doivent être stockés conformément à la réglementation nationale relative aux substances radioactives.
Remarques concernant la manipulation
Les prélèvements doivent être effectués dans des conditions d’asepsie. VIZAMYL ne doit pas être dilué. Les flacons ne doivent pas être ouverts avant désinfection du bouchon. La solution doit ensuite être prélevée à travers le bouchon à l’aide soit d’une seringue à usage unique munie d’une protection blindée appropriée et d’une aiguille stérile à usage unique, soit à l’aide d’un système automatisé autorisé. Si l’intégrité physique du flacon est compromise, le médicament ne doit pas être utilisé.
Contrôle de la qualité
Un contrôle visuel de la solution doit être effectué avant utilisation. Seules des solutions limpides et exemptes de particules visibles doivent être utilisées.
VIZAMYL est un médicament radioactif qui émet des positrons qui s’annihilent par capture électronique pour produire des rayons gamma ; des mesures de sécurité doivent être prises pour la manipulation du produit afin de limiter l’exposition du personnel soignant et des patients au rayonnement. Afin de réduire l’irradiation de la vessie, il faut recommander au patient de boire abondamment avant et après l’administration de VIZAMYL et de vider sa vessie aussi souvent que possible. Encouragez le patient à vider fréquemment sa vessie avant et après l’examen avec VIZAMYL, et pendant les 24 heures qui suivent.
Si, à un moment quelconque lors de la préparation de ce produit, l’intégrité du flacon est compromise, le produit ne doit pas être utilisé. Les procédures d’administration doivent être effectuées de manière à limiter le risque de contamination du médicament et d’irradiation des opérateurs. Un blindage adéquat est obligatoire. L’administration de produits radiopharmaceutiques présente des risques pour l’entourage du patient en raison de l’irradiation externe ou de la contamination par l’urine, les vomissements, etc.
Dispositions légales
En Suisse, l’utilisation de substances radioactives chez l’homme est réglementée par la dernière version en vigueur de l’ordonnance sur la radioprotection. Selon cette ordonnance, seules les personnes agréées par l’Office fédéral de la santé publique sont autorisées à utiliser des produits radiopharmaceutiques. Lors de la manipulation de substances radioactives et de l’élimination de déchets radioactifs, les mesures de protection stipulées dans l’ordonnance susmentionnée doivent être respectées, afin d’éviter toute irradiation inutile des patients et du personnel. Il est recommandé de retirer les étiquettes de désignation du produit avant l’élimination.
Numéro d’autorisation66110 (Swissmedic)
Présentation10 ml et 15 ml (A)
VIZAMYL est présenté en flacon de 10 ml et 15 ml en verre de Type I avec bouchon en caoutchouc halobutyle et scellé par une capsule en aluminium.
Un flacon multidose contient de 1 à 10 ml de solution, correspondant à 150 à 1500 MBq à la date et à l’heure de la calibration.
Tous les conditionnements peuvent ne pas être commercialisés. Du fait du processus de fabrication, certains flacons sont distribués avec des bouchons en caoutchouc perforés.
Titulaire de l’autorisationGE Healthcare AG, Opfikon
Mise à jour de l’informationOctobre 2020
|