CompositionPrincipes actifs
Atezolizumabum (produit par génie génétique à l'aide de cellules CHO [Chinese Hamster Ovary]).
Excipients
L-histidinum, acidum aceticum glaciale, saccharum, polysorbatum 20 (produit à partir de maïs génétiquement modifié), aqua ad iniectabile.
Indications/Possibilités d’emploiCancer du poumon non à petites cellules (CPNPC ou NSCLC) au stade précoce
Tecentriq en monothérapie est indiqué pour le traitement adjuvant des patients atteints d'un NSCLC de stade II ou IIIA (système de classification de l'UICC/AJCC, 7e édition) dont la tumeur n'a pas progressé après une chimiothérapie à base de cisplatine et présente une expression tumorale de PD-L1 ≥50% (voir «Propriétés/Effets»).
Cancer du poumon non à petites cellules (CPNPC ou NSCLC) métastatique
Tecentriq, en association au nab-paclitaxel et au carboplatine, est indiqué pour le traitement de première intention des patients atteints d'un NSCLC non épidermoïde métastatique sans aberrations génomiques tumorales du gène EGFR ou ALK.
Tecentriq, en association au paclitaxel et au carboplatine, est indiqué pour le traitement de première intention des patients atteints d'un NSCLC non épidermoïde métastatique sans aberrations génomiques tumorales du gène EGFR ou ALK et dont les tumeurs présentent une expression de PD-L1 ≥1%.
Tecentriq, en monothérapie, est indiqué pour le traitement des patients atteints d'un cancer du poumon CPNPC (ou NSCLC) localement avancé ou métastatique, après chimiothérapie préalable.
Cancer du poumon à petites cellules (CPPC ou SCLC)
Tecentriq, en association avec le carboplatine et l'étoposide, est indiqué dans le traitement de première intention des patients atteints d'un cancer du poumon à petites cellules de stade avancé (CPPC-SA ou ES-SCLC, extensive-stage small cell lung cancer).
Carcinome urothélial
Tecentriq, en monothérapie, est indiqué pour le traitement de patients adultes atteints d'un carcinome urothélial localement avancé ou métastatique, après chimiothérapie préalable à base de platine.
Cancer du sein triplement négatif
Tecentriq est indiqué en combinaison avec le nab-paclitaxel dans le traitement des patientes adultes atteintes d'un cancer du sein triplement négatif (TNBC) non résécable localement avancé ou métastatique, dont les tumeurs présentent une expression de PD-L1 ≥1% et qui n'ont pas reçu précédemment de chimiothérapie ni de traitement systémique ciblé en raison de leur maladie au stade avancé (posologie du nab-paclitaxel, voir «Posologie/Mode d'emploi»).
Tecentriq ne doit pas être utilisé en combinaison avec le paclitaxel dans le traitement de patientes adultes atteintes d'un TNBC non résécable, localement avancé ou métastatique (voir «Posologie/Mode d'emploi» et «Mises en garde et précautions»).
Mélanome
Tecentriq est indiqué en association avec du cobimétinib et du vémurafénib dans le traitement des patients adultes atteints d'un mélanome métastatique ou non résécable, testés positifs pour la mutation BRAF-V600E (voir «Efficacité clinique»).
Carcinome hépatocellulaire
Tecentriq est indiqué en association avec le bévacizumab dans le traitement des patients atteints de carcinome hépatocellulaire (CHC) inopérable ou métastatique, qui n'ont pas reçu de traitement systémique antérieur (voir rubriques «Mises en garde et précautions» et «Propriétés/Effets»).
Posologie/Mode d’emploiGénéralités
Tecentriq doit être administré sous la surveillance d'un professionnel de la santé qualifié. Il est impératif de vérifier la désignation du produit afin de s'assurer que les patients reçoivent la formulation correcte, conformément à la prescription (Tecentriq pour administration i.v. ou Tecentriq pour administration s.c.).
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Tecentriq dans sa formulation pour administration intraveineuse (Tecentriq i.v.) n'est pas destiné à être administré par voie sous-cutanée. Tecentriq doit être administré en perfusion intraveineuse (i.v.). Ne pas l'administrer en injection i.v. rapide ou bolus. Ne pas administrer d'autres médicaments simultanément par le même cathéter de perfusion. La dose initiale de Tecentriq doit être administrée pendant 60 minutes. Si la première perfusion a été bien tolérée, toutes les perfusions suivantes peuvent être administrées en 30 minutes.
Sélection des patients
Test de PD-L1 pour les patients atteints d'un TNBC qui doivent être traités par Tecentriq en combinaison avec le nab-paclitaxel, pour les patients atteints d'un NSCLC au stade précoce qui doivent être traités par Tecentriq en monothérapie, et pour les patients atteints d'un NSCLC métastatique en 1L qui doivent être traités par Tecentriq en combinaison avec le paclitaxel et le carboplatine
Pour le traitement par Tecentriq, les patients adultes doivent présenter une expression positive de PD-L1 déterminée à l'aide d'un test validé pour Tecentriq (voir rubrique «Propriétés/Effets»).
Test pour les patients atteints de mélanome
Pour être traités par Tecentriq en association avec du cobimétinib et du vémurafénib, les patients atteints de mélanome doivent présenter une mutation BRAF V600E confirmée par un test validé.
Monothérapie par Tecentriq
Tableau 1: Posologie recommandée pour la monothérapie par Tecentriq en perfusion intraveineuse
|
Indication
|
Posologie recommandée et calendrier
|
Durée du traitement
(voir «Efficacité clinique»)
| |
Carcinome urothélial en 2e ligne de traitement (2L)
|
1200 mg toutes les 3 semaines
|
Jusqu'à perte du bénéfice clinique ou survenue d'une toxicité qui ne peut pas être traitée
| |
NSCLC métastatique en 2L
| |
NSCLC au stade précoce
|
Pendant 1 an, sauf si survenue d'une récidive ou d'une toxicité inacceptable
|
Traitement combiné avec Tecentriq
Pour l'utilisation de Tecentriq en traitement combiné, il convient de tenir également compte de l'information professionnelle complète du produit associé.
Tableau 2: Posologie recommandée pour le traitement combiné avec Tecentriq en perfusion intraveineuse
|
Indication
|
Posologie recommandée et calendrier
|
Durée du traitement
(voir «Efficacité clinique»)
| |
Tecentriq
|
Médicament associé
| |
NSCLC non épidermoïde métastatique en 1L
Tecentriq avec paclitaxel et carboplatine
|
Phase d'induction:
1200 mg toutes les 3 semaines
Tecentriq doit être administré avant le traitement combiné lorsque l'administration a lieu le même jour.
Phase d'entretien (sans chimiothérapie):
1200 mg toutes les 3 semaines
|
Phase d'induction:
·Tecentriq, suivi du paclitaxel, puis du carboplatine en perfusion i.v., sont administrés toutes les 3 semaines.
|
Phase d'induction:
·pendant quatre ou six cycles
Phase d'entretien:
·jusqu'à perte du bénéfice clinique ou survenue d'une toxicité qui ne peut pas être traitée
| |
NSCLC non épidermoïde métastatique en 1L
Tecentriq avec nab-paclitaxel et carboplatine
|
Phase d'induction:
·Le nab-paclitaxel et le carboplatine sont administrés toutes les trois semaines en perfusion intraveineuse.
·Lors de chaque cycles de 21 jours, Tecentriq, le nab-paclitaxel et le carboplatine sont administrés au jour 1.
·Le nab-paclitaxel est également administré aux jours 8 et 15.
| |
ES-SCLC en 1L
Tecentriq avec carboplatine et étoposide
|
Phase d'induction:
·Le carboplatine, puis l'étoposide sont administrés toutes les trois semaines en perfusion intraveineuse.
·Tecentriq, le carboplatine, puis l'étoposide sont administrés au jour 1 de chaque cycle.
·L'étoposide est également administré aux jours 2 et 3 en perfusion intraveineuse.
|
Phase d'induction:
·pendant quatre cycles
Phase d'entretien:
·jusqu'à perte du bénéfice clinique ou survenue d'une toxicité qui ne peut pas être traitée
| |
TNBC en 1L
Tecentriq avec nab-paclitaxel
|
840 mg toutes les 2 semaines
Tecentriq doit être administré avant le traitement combiné lorsque l'administration a lieu le même jour.
|
·Le nab-paclitaxel est administré à la dose de 100 mg/m2 aux jours 1, 8 et 15 de chaque cycle de 28 jours en perfusion intrveineuse.
·Tecentriq est administré aux jours 1 et 15.
·La substitution du nab-paclitaxel par d'autres formulations de paclitaxel pour le traitement du TNBC n'est pas autorisée (voir «Indications/Possibilités d'emploi» et «Mises en garde et précautions»).
|
Jusqu'à progression de la maladie ou survenue d'une toxicité inacceptable
| |
Mélanome
Tecentriq avec cobimétinib et vémurafénib
|
840 mg toutes les 2 semaines
|
·Avant l'instauration du traitement par Tecentriq, les patients reçoivent pendant un cycle de 28 jours 60 mg de cobimétinib une fois par jour par voie orale (21 jours avec médication et 7 jours de pause) et 960 mg de vémurafénib deux fois par jour par voie orale aux jours 1-21 et 720 mg de vémurafénib deux fois par jour par voie orale aux jours 22-28.
·Pendant le traitement par Tecentriq, les patients reçoivent 60 mg de cobimétinib une fois par jour (21 jours avec médication et 7 jours de pause)et 720 mg de vémurafénib deux fois par jour par voie orale (voir «Efficacité clinique»).
|
Jusqu'à progression de la maladie ou survenue d'une toxicité inacceptable
| |
CHC en 1L
Tecentriq avec bévacizumab
|
1200 mg toutes les 3 semaines
Tecentriq doit être administré avant le traitement combiné lorsque l'administration a lieu le même jour.
|
·Tecentriq, suivi de 15 mg de bévacizumab par kg de poids corporel en perfusion intraveineuse, sont administrés toutes les 3 semaines.
|
Jusqu'à perte du bénéfice clinique ou survenue d'une toxicité qui ne peut pas être traitée
|
Ajustement de la posologie
Aucune réduction de la dose de Tecentriq n'est recommandée.
Ajustement de la posologie du fait d'effets indésirables à médiation immunitaire/d'interactions
Les recommandations relatives à certaines réactions médicamenteuses indésirables sont présentées dans le tableau 3 (voir rubriques «Mises en garde et précautions» et «Effets indésirables»).
Tableau 3: Recommandations pour l'ajustement posologique de Tecentriq
|
Réaction indésirable à médiation immunitaire
|
Degré de sévérité
|
Ajustement du traitement
| |
Infections
|
Grade 3 ou 4
|
Interrompre l'administration de la dose jusqu'à une amélioration au grade 1 ou jusqu'à résolution complète
| |
Réactions liées à la perfusion
|
Grade 1 ou 2
|
Diminuer la vitesse de perfusion ou interrompre le traitement. Le traitement peut être repris après la disparition de l'événement.
Pour les doses suivantes, une prémédication par des antipyrétiques et des antihistaminiques peut être envisagée.
| |
Grade 3 ou 4
|
Arrêter définitivement Tecentriq
| |
Lymphohistiocytose hémophagocytaire
|
Suspicion de lymphohistiocytose hémophagocytaire (quelle que soit la sévérité)
|
Arrêter définitivement Tecentriq
| |
Pneumopathie inflammatoire à médiation immunitaire
|
Grade 2
|
Interrompre la perfusion de Tecentriq
En cas d'amélioration de l'événement indésirable au grade 0 ou 1 en l'espace de 12 semaines et de réduction des doses de corticostéroïdes à ≤10 mg par jour de prednisone ou équivalent, le traitement peut être repris.
| |
Grade 3 ou 4
|
Arrêter définitivement Tecentriq
| |
Hépatite à médiation immunitaire chez les patients sans CHC
|
Grade 2:
(ALAT ou ASAT > 3 à 5 × la limite supérieure de la norme [LSN]
ou
bilirubinémie > 1,5 à 3 × LSN)
|
Interrompre la perfusion de Tecentriq
En cas d'amélioration de l'événement indésirable au grade 0 ou 1 en l'espace de 12 semaines et de réduction des doses de corticostéroïdes à ≤10 mg par jour de prednisone ou équivalent, le traitement peut être repris.
| |
Grade 3 ou 4:
(ALAT ou ASAT > 5 × LSN
ou
bilirubinémie > 3 × LSN)
|
Arrêter définitivement Tecentriq
| |
Hépatite à médiation immunitaire chez les patients atteints de CHC
|
Taux initiaux d'ASAT/ALAT dans les limites de la norme et augmentation à > 3 × à ≤ 10 × LSN
Taux initiaux d'ASAT/ALAT > 1 à ≤ 3 × LSN et augmentation à > 5 × à ≤ 10 × LSN
Taux initiaux d'ASAT/ALAT > 3 × à ≤ 5 × LSN et augmentation à > 8 × à ≤ 10 × LSN
|
Interrompre la perfusion de Tecentriq
Le traitement peut être repris lorsque l'effet indésirable s'améliore jusqu'au grade 0 ou au grade 1 en l'espace de 12 semaines et que la dose de corticostéroïdes a été réduite à ≤10 mg de prednisone ou équivalent par jour.
| |
Augmentation des taux d'ASAT/ALAT à > 10 × LSN ou augmentation de la bilirubine totale à > 3 × LSN
|
Arrêter définitivement Tecentriq
| |
Colite à médiation immunitaire
|
Diarrhée de grade 2 ou 3 (augmentation de ≥4 selles/jour par rapport au début du traitement)
ou
colite symptomatique
|
Interrompre la perfusion de Tecentriq
En cas d'amélioration de l'événement indésirable au grade 0 ou 1 en l'espace de 12 semaines et de réduction des doses de corticostéroïdes à ≤10 mg par jour de prednisone ou équivalent, le traitement peut être repris.
| |
Diarrhée ou colite de grade 4 (potentiellement fatale; indication pour une intervention d'urgence)
|
Arrêter définitivement Tecentriq
| |
Hypothyroïdie ou hyperthyroïdie à médiation immunitaire
|
Symptomatique
|
Interrompre la perfusion de Tecentriq
Hypothyroïdie:
Lorsque les symptômes sont contrôlés par un traitement substitutif de la thyroïde et que les taux de TSH redescendent, le traitement peut être repris.
Hyperthyroïdie:
Lorsque les symptômes sont contrôlés par un médicament suppresseur de la thyroïde et que la fonction thyroïdienne s'améliore, le traitement peut être repris.
| |
Insuffisance surrénalienne à médiation immunitaire
|
Symptomatique
|
Interrompre la perfusion de Tecentriq
En cas d'amélioration des symptômes au grade 0 ou 1 en l'espace de 12 semaines et de réduction des doses de corticostéroïdes à ≤10 mg par jour de prednisone ou équivalent, et pour autant que le patient soit stable sous la thérapie substitutive, le traitement peut être repris.
| |
Hypophysite à médiation immunitaire
|
Grade 2 ou 3
|
Interrompre la perfusion de Tecentriq
En cas d'amélioration des symptômes au grade 0 ou 1 en l'espace de 12 semaines et de réduction des doses de corticostéroïdes à ≤10 mg par jour de prednisone ou équivalent, et pour autant que le patient soit stable sous la thérapie substitutive, le traitement peut être repris.
| |
Grade 4
|
Arrêter définitivement Tecentriq
| |
Diabète sucré de type 1 à médiation immunitaire
|
Hyperglycémie de grade 3 ou 4 (glycémie à jeun > 250 mg/dl ou 13,9 mmol/l)
|
Interrompre la perfusion de Tecentriq
En présence d'un contrôle métabolique obtenu par l'insulinothérapie substitutive, le traitement peut être repris.
| |
Syndrome myasthénique/
myasthénie grave,
syndrome de Guillain-Barré
et méningoencéphalite, à médiation immunitaire
|
Tous les grades
|
Arrêter définitivement Tecentriq
| |
Myélite à médiation immunitaire
|
Tous les grades
|
Arrêter définitivement Tecentriq
| |
Parésie faciale à médiation immunitaire
|
Grade 1 ou 2
|
Interrompre la perfusion de Tecentriq
Un traitement par des corticostéroïdes (1–2 mg/kg/jour de prednisone ou équivalent) doit être instauré. En cas de disparition complète ou partielle de l'événement (grade 0 à 1) en l'espace de 12 semaines et après réduction des doses de corticostéroïdes à ≤10 mg/jour de prednisone orale ou équivalent, le traitement par Tecentriq peut être repris.
| |
Grade 3 ou 4
|
Arrêter définitivement Tecentriq
| |
Pancréatite à médiation immunitaire
|
Élévation de grade 3 ou 4 des taux sériques d'amylase ou des taux sériques de lipase (> 2× LSN)
ou pancréatite de grade 2 ou 3
|
Interrompre la perfusion de Tecentriq
En cas d'amélioration des taux sériques d'amylase et de lipase au grade 0 ou 1 en l'espace de 12 semaines, ou après la disparition des symptômes de la pancréatite et la réduction des doses de corticostéroïdes à ≤10 mg par jour de prednisone ou équivalent, le traitement peut être repris.
| |
Pancréatite récidivante de grade 4 ou de tout grade
|
Arrêter définitivement Tecentriq
| |
Myocardite à médiation immunitaire
|
Grade 2 ou supérieur
|
Arrêter définitivement Tecentriq
| |
Myosite à médiation immunitaire
|
Grade 2 ou 3
|
Interrompre la perfusion de Tecentriq
En cas d'amélioration des symptômes au grade 0 ou 1 en l'espace de 12 semaines et de réduction des doses de corticostéroïdes à ≤10 mg par jour de prednisone ou équivalent, le traitement peut être repris.
| |
Grade 4 ou myosite récidivante de grade 3
|
Arrêter définitivement Tecentriq
| |
Néphrite à médiation immunitaire
|
Grade 2:
(taux de créatinine > 1,5 - 3,0 × valeur initiale au début du traitement ou > 1,5 - 3,0 × LSN)
|
Interrompre la perfusion de Tecentriq
En cas d'amélioration de l'événement indésirable au grade 0 ou 1 en l'espace de 12 semaines et de réduction des doses de corticostéroïdes à ≤10 mg par jour de prednisone ou équivalent, le traitement peut être repris.
| |
Grade 3:
(taux de créatinine > 3,0 × valeur initiale au début du traitement ou > 3,0 - 6,0 × LSN)
ou grade 4:
(taux de créatinine > 6,0 × LSN)
|
Arrêter définitivement Tecentriq
| |
Affections du péricarde à médiation immunitaire
|
Péricardite de grade 1
|
Interrompre la perfusion de Tecentriq
Effectuer un bilan cardiologique détaillé afin de déterminer l'étiologie et de traiter de manière appropriée.
| |
Grade 2 ou supérieur
|
Arrêter définitivement Tecentriq
| |
Éruption cutanée/Réactions cutanées sévères à médiation immunitaire
|
Grade 3
ou suspicion de syndrome de Stevens-Johnson (SSJ) ou de nécrolyse épidermique toxique (NET)
|
Interrompre la perfusion de Tecentriq
Après la disparition de l'éruption cutanée et la réduction des doses de corticostéroïdes à ≤10 mg par jour de prednisone ou équivalent, le traitement peut être repris.
| |
Grade 4
ou confirmation d'un syndrome de Stevens-Johnson (SSJ) ou d'une nécrolyse épidermique toxique (NET)
|
Arrêter définitivement Tecentriq
| |
Autres réactions indésirables à médiation immunitaire
|
Grade 2 ou grade 3
|
Interruption jusqu'à une amélioration des réactions indésirables au grade 0-1 en l'espace de 12 semaines et une réduction des doses de corticostéroïdes à ≤10 mg par jour de prednisone ou équivalent.
| |
Grade 4 ou récidivantes de grade 3
|
Arrêter définitivement Tecentriq (excepté en cas d'endocrinopathies contrôlées par traitement hormonal substitutif)
| |
Effet indésirable persistant à médiation immunitaire de grade 2 ou 3 (à l'exception des endocrinopathies)
|
Effet indésirable à médiation immunitaire de grade 2 ou 3 qui ne s'améliore pas jusqu'au grade 0 ou 1 dans les 12 semaines qui suivent l'administration de la dernière dose de Tecentriq
|
Arrêter définitivement le traitement
| |
Impossibilité de réduire progressivement la dose de corticostéroïdes
|
Impossibilité de réduire à une dose inférieure ou égale à 10 mg par jour de prednisone ou équivalent dans les 12 semaines qui suivent l'administration de la dernière dose de Tecentriq
|
Arrêter définitivement le traitement
| |
Effet indésirable récurrent à médiation immunitaire de grade 3 ou 4
|
Effet indésirable récurrent à médiation immunitaire de grade 3 ou 4 (sévère ou engageant le pronostic vital)
|
Arrêter définitivement le traitement
|
Remarque: Les grades de toxicité correspondent aux critères généraux de terminologie pour les événements indésirables de l'Institut national du cancer (National Cancer Institute Common Terminology Criteria for Adverse Event), version 5.0 (NCI-CTCAE v.5.).
Prise retardée
Si une dose prévue de Tecentriq est omise, elle doit être administrée dès que possible, sans attendre la dose suivante prévue. Le schéma d'administration devrait être ajusté afin de maintenir un intervalle de trois semaines entre les doses.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Une analyse pharmacocinétique de population a révélé qu'aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère et modérée. Aucune donnée n'existe pour les patients présentant une insuffisance hépatique sévère (voir «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
D'après les résultats d'une analyse pharmacocinétique de population, aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance rénale légère ou modérée (voir section «Pharmacocinétique»). Les données concernant les patients présentant une insuffisance rénale sévère sont trop limitées pour permettre de tirer des conclusions sur cette population.
Patients âgés
Sur 3040 patients atteints de carcinome urothélial, de cancer du poumon, de cancer du sein triple négatif, de carcinome hépatocellulaire et de mélanome traités avec Tecentriq lors des études cliniques, 43% étaient âgés de 65 ans et plus, et 12% de 75 ans et plus. Une analyse pharmacocinétique de population a révélé qu'aucun ajustement de la dose de Tecentriq n'est nécessaire chez les patients ≥65 ans (voir «Pharmacocinétique»).
Enfants et adolescents
L'utilisation de Tecentriq n'est pas autorisée chez les patients âgés de moins de 18 ans. La sécurité et l'efficacité de Tecentriq pour cette population ne sont pas établies. Dans une étude clinique, Tecentriq n'a présenté aucun bénéfice clinique chez les enfants et les adolescents (voir «Propriétés/Effets: Efficacité clinique»).
Génotype/Polymorphismes génétiques
Patients asiatiques
Il est recommandé d'administrer une dose initiale de 175 mg/m2 de paclitaxel toutes les trois semaines aux patients asiatiques, en raison d'une toxicité hématologique accrue observée dans cette population au cours de l'étude IMpower150.
Contre-indicationsTecentriq est contre-indiqué chez les patients présentant une hypersensibilité connue à l'atézolizumab ou à l'un des excipients.
Mises en garde et précautionsInfections
Tecentriq peut entraîner des infections sévères susceptibles d'être fatales (voir «Effets indésirables, Études cliniques»). Les patients doivent être surveillés pour détecter tout signe et symptôme éventuel d'infection. En présence d'infections de grade 3 ou supérieur, l'administration de Tecentriq doit être interrompue et ne peut être reprise que lorsqu'une stabilisation clinique s'est produite (voir «Posologie/Mode d'emploi, Tableau 1: Recommandations pour l'ajustement posologique de Tecentriq»).
Lors de l'étude portant sur le mélanome (CO39262), la fréquence de survenue des infections a été plus élevée chez les patients ayant reçu Tecentriq en association avec le cobimétinib et le vémurafénib (voir «Effets indésirables»).
Réactions liées à la perfusion
Des réactions liées à la perfusion ont été observées au cours d'études cliniques réalisées avec Tecentriq (voir «Effets indésirables»).
Chez les patients présentant des réactions liées à la perfusion de grade 1 ou 2, il convient de diminuer la vitesse de perfusion ou d'interrompre le traitement. Chez les patients présentant des réactions liées à la perfusion de grade 3 ou 4, il convient d'arrêter définitivement Tecentriq. Chez les patients présentant des réactions liées à la perfusion de grade 1 ou 2, le traitement par Tecentriq peut être poursuivi sous surveillance étroite; une prémédication par antipyrétiques et antihistaminiques peut être envisagée.
Les données précliniques préliminaires (voir «Données précliniques») indiquent que l'atézolizumab pourrait diminuer les réponses primaires et secondaires des IgG (immunoglobulines G) aux antigènes T-dépendants.
Pneumopathie inflammatoire à médiation immunitaire
Des cas de pneumopathies inflammatoires, dont certains d'issue fatale, ont été observés au cours d'études cliniques réalisées avec Tecentriq (voir «Effets indésirables, Études cliniques»). Les patients doivent être surveillés à la recherche de signes et de symptômes d'une pneumopathie inflammatoire.
En cas de pneumopathie inflammatoire de grade 2, il convient d'interrompre le traitement par Tecentriq et d'instaurer un traitement par 1 à 2 mg/kg de prednisone ou équivalent par jour. En cas d'amélioration des symptômes à un grade ≤1, les corticostéroïdes doivent être arrêtés progressivement sur une période de ≥1 mois. Le traitement par Tecentriq peut être repris en cas d'amélioration de l'événement à un grade ≤1 en l'espace de 12 semaines et de réduction des doses de corticostéroïdes à ≤10 mg de prednisone orale ou équivalent par jour. En cas de pneumopathie inflammatoire de grade 3 ou 4, il convient d'arrêter définitivement le traitement par Tecentriq.
Lors de l'étude portant sur le mélanome (CO39262), la fréquence de survenue d'une pneumopathie inflammatoire d'origine immunologique a été plus élevée chez les patients ayant reçu Tecentriq en association avec le cobimétinib et le vémurafénib (voir «Effets indésirables»).
Hépatite à médiation immunitaire
Des cas d'hépatites, dont certains d'issue fatale, ont été observés au cours d'études cliniques réalisées avec Tecentriq (voir «Effets indésirables, Études cliniques»). Les patients doivent être surveillés à la recherche de signes et de symptômes d'une hépatite. Les taux d'aspartate aminotransférase (ASAT), d'alanine aminotransférase (ALAT) et de bilirubine doivent être contrôlés avant le traitement et régulièrement au cours du traitement par Tecentriq. Chez les patients présentant des anomalies du bilan hépatique (BH) avant le début du traitement, des mesures appropriées doivent être envisagées.
En cas de persistance des symptômes de grade 2 (ALAT ou ASAT > 3× la limite supérieure de la norme (LSN) ou bilirubinémie > 1,5× LSN) pendant plus de 5 à 7 jours, il convient d'interrompre le traitement et d'instaurer un traitement par 1 à 2 mg/kg de prednisone ou équivalent par jour. En cas d'amélioration des anomalies du BH à un grade ≤1, les corticostéroïdes doivent être arrêtés progressivement sur une période de ≥1 mois. Le traitement par Tecentriq peut être repris en cas d'amélioration de l'événement à un grade ≤1 en l'espace de 12 semaines et de réduction des doses de corticostéroïdes à ≤10 mg de prednisone orale ou équivalent par jour. En cas d'événements de grade 3 ou 4 (ALAT ou ASAT > 5,0× LSN ou bilirubinémie > 3× LSN), il convient d'arrêter définitivement le traitement par Tecentriq.
Lors de l'étude portant sur le mélanome (CO39262), la fréquence de survenue d'une hépatite d'origine immunologique a été plus élevée chez les patients ayant reçu Tecentriq en association avec le cobimétinib et le vémurafénib (voir «Effets indésirables»).
Colite à médiation immunitaire
Des cas de diarrhées, de colite et d'autres événements gastro-intestinaux tels qu'une perforation intestinale ont été observés au cours d'études cliniques réalisées avec Tecentriq (voir «Effets indésirables, Études cliniques»). Les patients doivent être surveillés à la recherche de signes et de symptômes d'une colite.
En cas de diarrhées de grade 2 ou 3 (augmentation de ≥4 selles/jour par rapport au début du traitement) ou de colite (symptomatique), le traitement par Tecentriq doit être interrompu. Lors de diarrhées ou de colite de grade 2, un traitement par 1 à 2 mg/kg de prednisone ou équivalent par jour doit être instauré en cas de persistance des symptômes pendant plus de 5 jours ou de réapparition de ces symptômes. Une diarrhée ou une colite de grade 3 doit être traitée par des corticostéroïdes i.v. (1 à 2 mg/kg de méthylprednisolone ou équivalent par jour). Après amélioration, il faut passer à des corticostéroïdes oraux (1 à 2 mg/kg de prednisone ou équivalent par jour). En cas d'amélioration des symptômes à un grade ≤1, les corticostéroïdes doivent être arrêtés progressivement sur une période de ≥1 mois. Le traitement par Tecentriq peut être repris en cas d'amélioration de l'événement à un grade ≤1 en l'espace de 12 semaines et de réduction des doses de corticostéroïdes à ≤10 mg de prednisone orale ou équivalent par jour. En cas de diarrhées ou de colite de grade 4 (menaçant le pronostic vital: intervention d'urgence indiquée), il convient d'arrêter définitivement le traitement par Tecentriq.
Lors de l'étude portant sur le mélanome (CO39262), la fréquence de survenue de diarrhée et de colite a été plus élevée chez les patients ayant reçu Tecentriq en association avec le cobimétinib et le vémurafénib (voir «Effets indésirables»).
Endocrinopathies à médiation immunitaire
Des cas d'hypothyroïdie, d'hyperthyroïdie, d'insuffisance surrénalienne, d'hypophysite et de diabète sucré de type 1, y compris d'acidocétose diabétique, ont été observés au cours d'études cliniques réalisées avec Tecentriq (voir «Effets indésirables, Études cliniques»). Les patients doivent être surveillés à la recherche de signes et de symptômes d'endocrinopathies. La fonction thyroïdienne doit être contrôlée avant le traitement et régulièrement au cours du traitement par Tecentriq. Chez les patients présentant des anomalies du bilan thyroïdien avant le début du traitement, des mesures appropriées doivent être envisagées.
Les patients asymptomatiques présentant des résultats anormaux aux tests de la fonction thyroïdienne peuvent être traités par Tecentriq. Le traitement par Tecentriq doit être interrompu en cas d'hypothyroïdie symptomatique et une thérapie de substitution par hormones thyroïdiennes doit être instaurée en cas de besoin. Une hypothyroïdie isolée peut éventuellement être traitée par une thérapie de substitution sans corticostéroïdes. En cas d'hyperthyroïdie symptomatique, le traitement par Tecentriq doit être interrompu et un traitement par un thyréostatique, par ex. le méthimazole ou le carbimazole, doit être instauré en cas de besoin. Le traitement par Tecentriq peut être repris dès que les symptômes ont pu être contrôlés et que la fonction thyroïdienne s'est améliorée.
En cas d'insuffisance surrénalienne symptomatique, le traitement par Tecentriq doit être interrompu et un traitement par 1 à 2 mg/kg/jour de méthylprednisolone i.v. ou équivalent doit être instauré. Dès que les symptômes se sont améliorés, un traitement par 1 à 2 mg/kg/jour de prednisone orale ou équivalent doit alors suivre. En cas d'amélioration des symptômes à un grade ≤1, les corticostéroïdes doivent être arrêtés progressivement sur une période de ≥1 mois. Le traitement par Tecentriq peut être repris en cas d'amélioration de l'événement à un grade ≤1 en l'espace de 12 semaines, de réduction des doses de corticostéroïdes à ≤10 mg de prednisone orale ou équivalent par jour et pour autant que le patient soit stable sous thérapie de substitution (si nécessaire).
En cas d'hypophysite de grade 2 ou 3, le traitement par Tecentriq doit être interrompu. Un traitement par 1 à 2 mg/kg/jour de méthylprednisolone i.v. ou d'un équivalent doit être instauré, en cas de besoin, une hormonothérapie de substitution peut également s'avérer nécessaire. Une fois l'amélioration des symptômes obtenue, il convient de passer à un corticostéroïde par voie orale (1 à 2 mg/kg/jour de prednisone ou équivalent). En cas d'amélioration des symptômes à un grade ≤1, les corticostéroïdes doivent être arrêtés progressivement sur une période de ≥1 mois. Le traitement par Tecentriq peut être repris en cas d'amélioration de l'événement à un grade ≤1 en l'espace de 12 semaines, de réduction des doses de corticostéroïdes à ≤10 mg de prednisone orale ou équivalent par jour et pour autant que le patient soit stable sous thérapie de substitution (si nécessaire). En cas d'hypophysite de grade 4, le traitement par Tecentriq doit être définitivement arrêté.
En cas de diabète sucré de type 1, il convient d'instaurer une insulinothérapie. En cas d'hyperglycémie de grade ≥3 (glycémie à jeun > 250 mg/dl), il convient d'interrompre le traitement par Tecentriq. Le traitement par Tecentriq peut être repris lorsque le contrôle du métabolisme est obtenu sous l'insulinothérapie substitutive.
Lors de l'étude portant sur le mélanome (CO39262), la fréquence de survenue d'hypothyroïdie et d'hyperthyroïdie a été plus élevée chez les patients ayant reçu Tecentriq en association avec le cobimétinib et le vémurafénib (voir «Effets indésirables»).
Méningoencéphalite à médiation immunitaire
Des cas de méningoencéphalites ont été observés au cours d'études cliniques réalisées avec Tecentriq (voir «Effets indésirables, Études cliniques»). Les patients doivent être surveillés à la recherche de signes cliniques et de symptômes de méningite ou d'encéphalite.
Le traitement par Tecentriq doit être arrêté définitivement en cas de méningite ou d'encéphalite de quelque grade que ce soit. Un traitement par 1 à 2 mg/kg de méthylprednisolone i.v. ou équivalent par jour doit être administré. Après amélioration de l'état du patient, il faut remplacer ce traitement par 1 à 2 mg/kg de prednisone orale ou équivalent par jour. En cas d'amélioration des symptômes à un grade ≤1, les corticostéroïdes doivent être arrêtés progressivement sur une période de ≥1 mois.
Neuropathies à médiation immunitaire
Un syndrome myasthénique/une myasthénie grave ou un syndrome de Guillain-Barré (pouvant menacer le pronostic vital) et une parésie faciale ont été rapportés chez les patients ayant reçu Tecentriq (voir «Effets indésirables, Études cliniques»). Les patients doivent être surveillés à la recherche de symptômes d'une neuropathie motrice ou sensitive.
En cas de syndrome myasthénique/myasthénie grave ou de syndrome de Guillain-Barré de quelque grade que ce soit, il convient d'arrêter définitivement le traitement par Tecentriq. L'instauration d'un traitement par des corticostéroïdes systémiques à une dose de 1-2 mg/kg de prednisone orale ou équivalent par jour doit être envisagée.
Myélite à médiation immunitaire
Des cas de myélite ont été observés au cours d'études cliniques réalisées avec Tecentriq (voir «Effets indésirables, Études cliniques» et «Effets indésirables, Données post-commercialisation»). Les patients doivent être surveillés à la recherche de signes et de symptômes évocateurs d'une myélite. Voir «Posologie/Mode d'emploi» en ce qui concerne les ajustements posologiques recommandés.
Pancréatite à médiation immunitaire
Des cas de pancréatite, y compris d'augmentation des taux sériques d'amylase et de lipase, ont été observés au cours d'études cliniques réalisées avec Tecentriq (voir «Effets indésirables, Études cliniques»). Les patients doivent être étroitement surveillés à la recherche de signes et de symptômes évocateurs d'une pancréatite aiguë.
En cas d'augmentation des taux sériques d'amylase ou de lipase de grade ≥3 (> 2,0× LSN) ou de pancréatite de grade 2 ou 3, il convient d'interrompre le traitement par Tecentriq et d'instaurer un traitement par 1 à 2 mg/kg de méthylprednisolone i.v. ou équivalent par jour. Dès que les symptômes s'améliorent, il faut passer à un traitement par 1 à 2 mg/kg de prednisone orale ou équivalent par jour. Le traitement par Tecentriq peut être repris en cas d'amélioration des taux sériques d'amylase et de lipase à un grade ≤1 en l'espace de 12 semaines et après la disparition des symptômes de la pancréatite, ainsi qu'en cas de réduction des doses de corticostéroïdes à ≤10 mg de prednisone orale ou équivalent par jour. En cas de pancréatite de grade 4 ou de réapparition d'une pancréatite de quelque grade que ce soit, il convient d'arrêter définitivement le traitement par Tecentriq.
Affections cardiaques
Myocardite à médiation immunitaire
Une myocardite, y compris des cas d'issue fatale, a été observée au cours des essais cliniques avec Tecentriq (voir «Effets indésirables, Études cliniques»). Les patients doivent être surveillés à la recherche de signes et/ou de symptômes de myocardite. Le traitement par Tecentriq doit être définitivement arrêté dès qu'une myocardite de grade ≥2 est constatée (voir «Posologie/Mode d'emploi»). Une myocardite peut également être une manifestation clinique d'une myosite et doit être traitée en conséquence.
Affections du péricarde à médiation immunitaire
Des affections du péricarde, incluant péricardite, épanchement péricardique et tamponnade cardiaque, dont certaines d'issue fatale, ont été observées lors d'études cliniques réalisées avec Tecentriq (voir «Effets indésirables: Études cliniques et Données post-commercialisation»). Les patients doivent être surveillés à la recherche de signes et/ou de symptômes cliniques d'affections du péricarde. Les recommandations pour l'ajustement posologique figurent à la rubrique «Posologie/Mode d'emploi».
Myosite à médiation immunitaire
Au cours d'essais cliniques portant sur Tecentriq, des cas de myosite, dont certains d'issue fatale (incluant également une atteinte cardiaque), se sont produits (voir «Effets indésirables, études cliniques»). Les patients doivent faire l'objet d'une surveillance à la recherche de signes et de symptômes d'une myosite. Si un patient développe des signes et des symptômes d'une myosite, une surveillance étroite doit être mise en place et le patient doit être adressé immédiatement à un service spécialisé à des fins d'évaluation et de traitement. Les patients chez lesquels une myosite est suspectée doivent faire l'objet d'une surveillance à la recherche de signes d'une myocardite. En ce qui concerne les ajustements posologiques recommandés, se reporter à la section «Posologie/Mode d'emploi».
Néphrite à médiation immunitaire
Des néphrites ont été observées au cours d'études cliniques réalisées avec Tecentriq (voir «Effets indésirables, Études cliniques»). Les patients doivent être surveillés à la recherche d'altérations de leur fonction rénale. Voir «Posologie/Mode d'emploi» en ce qui concerne les ajustements posologiques recommandés.
Autres événements d'origine immunologique
Lymphohistiocytose hémophagocytaire (LHH)
Des cas de lymphohistiocytose hémophagocytaire (LHH), également d'issue fatale, sont survenus chez des patients traités par Tecentriq (voir «Effets indésirables, Données post-commercialisation»). Une LHH doit être envisagée en cas de tableau clinique atypique ou prolongé de syndrome de relargage des cytokines. Les patients doivent être surveillés à la recherche de signes et de symptômes cliniques d'une LHH. Voir «Posologie/Mode d'emploi» en ce qui concerne les ajustements posologiques recommandés.
La LHH est un syndrome potentiellement mortel s'accompagnant d'une activation pathologique des défenses immunitaires. En l'absence de diagnostic et de traitement précoces, la LHH a fréquemment une évolution létale. Cette maladie se caractérise par des signes et symptômes cliniques d'une inflammation systémique sévère, tels que fièvre, éruption cutanée, hépatosplénomégalie, cytopénie (surtout anémie et thrombocytopénie), lymphadénopathie, symptômes neurologiques, taux élevé de ferritine sérique, hypertriglycéridémie, ainsi que troubles de la fonction hépatique et de la coagulation. Les patients présentant de tels signes et symptômes doivent immédiatement être examinés et leur état doit être évalué en vue d'un éventuel diagnostic de LHH. L'administration de Tecentriq doit être suspendue tant qu'une autre étiologie n'a pas pu être établie.
Sous traitement par Tecentriq et/ou par d'autres inhibiteurs de point de contrôle immunitaire, des cas d'anémie hémolytique et d'anémie aplasique ont été observés. Les patients doivent être surveillés à la recherche de signes et de symptômes évocateurs de ces effets indésirables à médiation immunitaire.
Réactions cutanées sévères à médiation immunitaire
Des réactions cutanées sévères à médiation immunitaire (Severe Cutaneous Adverse Reactions – SCAR) ont été rapportées chez des patients sous traitement par Tecentriq, dont des cas de syndrome de Stevens-Johnson (SSJ) et de nécrolyse épidermique toxique (NET; y compris d'évolution fatale). Les patients doivent être surveillés à la recherche de réactions cutanées sévères. Il convient d'interrompre Tecentriq en cas de réactions cutanées de grade 3 jusqu'au retour à un grade ≤1 et de l'arrêter définitivement en cas de réactions cutanées de grade 4, et d'administrer des corticostéroïdes (voir «Posologie/Mode d'emploi»).
En cas de suspicion de SCAR, il convient d'adresser les patients à un spécialiste afin de réaliser une évaluation et un traitement adéquats. En cas de suspicion d'un SSJ ou d'une NET, le traitement par Tecentriq doit être interrompu. Si le SSJ ou la NET sont confirmés, le traitement par Tecentriq doit être arrêté définitivement.
La prudence est recommandée lorsqu'on envisage d'utiliser Tecentriq chez un patient ayant présenté une réaction cutanée indésirable sévère ou menaçant le pronostic vital lors d'un traitement anticancéreux antérieur par d'autres principes actifs immunostimulants.
Effets de classe des inhibiteurs de point de contrôle immunitaire
Pendant le traitement par d’autres inhibiteurs de point de contrôle immunitaire, des cas d’insuffisance pancréatique exocrine, qui peut aussi survenir sous traitement par atézolizumab, ont été rapportés.
Maladie cœliaque
Des cas de maladie cœliaque ont été rapportés sous traitement par Tecentriq.
Immunogénicité
Des méta-analyses exploratoires, non ajustées, portant sur l'efficacité (sans ajustement de déséquilibres des caractéristiques initiales) montrent une diminution de l'efficacité de Tecentriq concernant la survie globale chez les patients ADA-positifs par rapport aux patients ADA-négatifs (voir «Propriétés/Effets: Efficacité clinique» et «Pharmacocinétique»).
Patients atteints d’une maladie auto-immune préexistante
Des données issues d’études d’observation indiquent que les patients atteints d’une maladie auto-immune (MAI) préexistante présentent un risque accru d’effets indésirables à médiation immunitaire après un traitement par un inhibiteur de point de contrôle immunitaire par rapport aux patients sans MAI préexistante. Par ailleurs, des poussées de la MAI sous-jacente sont survenues plus fréquemment, mais elles étaient la plupart du temps légères et faciles à traiter.
Précautions spécifiques à la maladie
Utilisation de l'atézolizumab en association avec le bévacizumab en cas de carcinome hépatocellulaire
Les patients traités par le bévacizumab présentent un risque hémorragique accru. Des cas d'hémorragies gastro-intestinales sévères, y compris des événements d'issue fatale, ont été rapportés chez des patients atteints de carcinome hépatocelllulaire (CHC) traités par l'atézolizumab en association avec le bévacizumab. Avant d'instaurer le traitement par l'association d'atézolizumab et de bévacizumab, il convient de rechercher la présence de varices œsophagiennes et éventuellement d'administrer un traitement conforme aux pratiques cliniques chez les patients atteints de CHC. Le bévacizumab doit être définitivement arrêté pendant le traitement combiné chez les patients présentant des hémorragies de grade 3 ou 4. Consulter l'information professionnelle du bévacizumab.
Risque accru de mortalité pour les patientes atteintes de TNBC métastatique lors de l'utilisation de Tecentriq avec le paclitaxel
Dans une étude randomisée menée chez des patientes atteintes de TNBC métastatique, un risque accru de mortalité a été observé dans la population PD-L1-positive chez les patientes traitées par Tecentriq plus paclitaxel par rapport à celles traitées par un placebo et le paclitaxel. La survie globale dans la population présentant une expression du PD-L1, dans laquelle la mortalité était de 42%, a donné un HR de 1,11 (IC à 95%: 0,76; 1,64). La survie médiane a été de 22,1 mois (IC à 95%: 19,2; 30,5) chez les patientes ayant reçu Tecentriq en association avec le paclitaxel et de 28,3 mois (IC à 95%: 19,1; NE) chez les patientes ayant reçu un placebo et le paclitaxel (voir «Indications/Possibilités d'emploi» et «Posologie/Mode d'emploi»). La cause de décès la plus fréquente était une progression de la maladie et aucun déséquilibre de la mortalité liée à la toxicité n'a été observé entre les bras de l'étude.
L'efficacité de Tecentriq en association avec le paclitaxel n'a été pas démontrée chez les patientes atteintes d'un TNBC localement avancé ou métastatique. Pour traiter le TNBC métastatique dans la pratique clinique en dehors d'études contrôlées, le paclitaxel lié aux protéines ne doit pas être remplacé par le paclitaxel en association avec Tecentriq.
Groupes de patients particuliers
Patients exclus des études cliniques
Les patients présentant les affections ou les caractéristiques suivantes ont été exclus de la participation aux études cliniques:
·antécédents de pneumopathie inflammatoire,
·métastases cérébrales actives,
·infection par le VIH, l'hépatite B ou l'hépatite C,
·patients ayant reçu un vaccin vivant atténué dans les 28 jours précédant l'inclusion dans l'étude,
·patients ayant reçu un traitement immunostimulant systémique dans les 4 semaines ou un traitement immunosuppresseur systémique dans les 2 semaines précédant l'inclusion dans l'étude,
·antécédents de maladies auto-immunes,
·patients atteints de maladies cardiovasculaires importantes et patients présentant une altération de la fonction hématologique ou une insuffisance des organes cibles.
Les patients avec un score de performance ECOG ≥2 ont été exclus de la participation aux études cliniques en cas de NSCLC et de CU sous traitement de seconde intention.
Toxicité embryonnaire et fœtale
En raison de son mécanisme d'action, l'utilisation de Tecentriq peut avoir des effets délétères sur le fœtus. Les expérimentations menées chez l'animal ont montré que l'inhibition de la voie de signalisation PD-L1/PD-1 peut être associée à un risque accru de rejet à médiation immunitaire du fœtus en développement et peut par conséquent entraîner la mort du fœtus.
Les femmes enceintes doivent être informées du risque potentiel pour le fœtus. Les femmes en âge de procréer doivent être informées de la nécessité d'utiliser des méthodes contraceptives hautement efficaces pendant le traitement par Tecentriq et pendant les 5 mois suivant la dernière dose (voir «Grossesse» et «Données précliniques: Tératogénicité»).
Effets indésirables chez les patients transplantés
Chez les patients traités par des inhibiteurs du PD-1/PD-L1, un rejet de greffes d'organes solides a été observé dans le contexte post-commercialisation. Le traitement par Tecentriq est susceptible d'accroître le risque de rejet chez les receveurs de greffes d'organes solides. Chez ces patients, le bénéfice du traitement par l'atézolizumab doit être évalué par rapport au risque de rejet possible de l'organe.
InteractionsInteractions pharmacocinétiques
Aucune étude formelle d'interactions pharmacocinétiques n'a été réalisée avec l'atézolizumab. Étant donné que l'atézolizumab est éliminé de la circulation sanguine par dégradation catabolique, il ne faut s'attendre à aucune interaction médicamenteuse métabolique.
Interactions pharmacodynamiques
L'emploi de corticostéroïdes systémiques ou d'immunosuppresseurs avant le début du traitement par l'atézolizumab doit être évité en raison d'une possible altération de l'activité pharmacodynamique et de l'efficacité de l'atézolizumab. Des corticostéroïdes systémiques ou d'autres immunosuppresseurs peuvent toutefois être administrés après l'instauration du traitement par l'atézolizumab pour traiter des effets indésirables d'origine immunologique (voir «Mises en garde et précautions»).
Grossesse, allaitementGrossesse
En raison de son mécanisme d'action, Tecentriq peut avoir des effets délétères sur le fœtus lorsqu'il est administré à une femme enceinte. Il n'existe pas d'études cliniques concernant l'emploi de Tecentriq chez la femme enceinte. Les expérimentations menées chez l'animal ont montré que l'inhibition de la voie de signalisation PD-L1/PD-1 peut être associée à un risque accru de rejet immunologique du fœtus en développement, suivi de la mort du fœtus. Tecentriq ne doit pas être administré pendant la grossesse ni aux femmes en âge de procréer qui n'utilisent pas de méthode contraceptive efficace, sauf en cas de nécessité absolue. Si le médicament est utilisé pendant la grossesse ou si la patiente débute une grossesse pendant le traitement, il convient d'informer celle-ci du risque potentiel pour le fœtus.
Les patientes en âge de procréer doivent utiliser des méthodes contraceptives hautement efficaces et prendre des mesures actives pour éviter une grossesse pendant le traitement par Tecentriq et pendant au moins les 5 mois suivant la dernière dose (voir «Mises en garde et précautions: Toxicité embryonnaire et fœtale» et «Données précliniques: Tératogénicité»).
Travail et accouchement
L'utilisation de Tecentriq pendant le travail et l'accouchement n'a pas été évaluée.
Allaitement
On ignore si Tecentriq est excrété dans le lait maternel humain. Aucune étude n'a été réalisée pour évaluer les effets de Tecentriq sur la production de lait ou sur sa présence dans le lait maternel. Comme de nombreux médicaments, dont des anticorps, sont excrétés dans le lait maternel, un risque pour le nouveau-né/l'enfant en bas âge ne peut pas être exclu. En raison des éventuels effets nocifs pour le nourrisson allaité, il est recommandé de ne pas allaiter pendant le traitement par Tecentriq et pendant au moins les 5 mois suivant la dernière dose.
Fertilité
Les expérimentations menées chez l'animal indiquent que Tecentriq peut avoir un effet sur le cycle menstruel des femmes en âge de procréer (voir «Données précliniques»). Les patientes en âge de procréer doivent utiliser des méthodes contraceptives hautement efficaces et prendre des précautions actives pour éviter une grossesse pendant le traitement par Tecentriq et pendant au moins les 5 mois suivant la dernière dose.
Effet sur l’aptitude à la conduite et l’utilisation de machinesTecentriq a une légère influence sur l'aptitude à la conduite ou l'utilisation de machines. Il convient de recommander aux patients ressentant de la fatigue de ne pas conduire de véhicule ni d'utiliser des machines jusqu'à la disparition des symptômes.
Effets indésirablesÉtudes cliniques
L'évaluation de la sécurité de Tecentriq en monothérapie repose sur les données regroupées de 5464 patients atteints de différents types de tumeurs et sur des données complémentaires issues de l'exposition cumulée de > 13'000 patients inclus dans l'ensemble des études cliniques (voir tableau 4). Les effets indésirables les plus fréquemment identifiés dans les études cliniques en rapport avec Tecentriq en monothérapie (> 10%) étaient fatigue (27,9%), diminution de l'appétit (19,3%), éruption cutanée (18,8%), nausées (17,9%), diarrhée (17,1%), pyrexie (17,0%), toux (16,8%), constipation (16,2%), dyspnée (15,6%), arthralgie (15,5%), anémie (14,5%), prurit (13,0%), asthénie (12,6%), dorsalgies (12,0%), infection des voies urinaires (11,5%), hépatite [anomalies des valeurs de laboratoire] (11,2%) et vomissements (11,1%).
Le tableau 4 présente également sous une forme résumée les effets indésirables (EI) observés durant les études cliniques sous traitement combiné avec Tecentriq (n = 5196). Les effets indésirables les plus fréquents (≥10%) étaient les suivants: anémie (35,3%), neutropénie (35,0%), nausées (34,5%), fatigue (32,4%), éruption cutanée (30,4%), alopécie (29,8%), diarrhées (28,6%), neuropathie périphérique (24,8%), constipation (24,7%), thrombocytopénie (24,2%), diminution de l'appétit (23,4%), arthralgies (20,9%), pyrexie (19,2%), vomissements (18,9%), asthénie (18,6%), anomalies des valeurs de laboratoire pour l'hépatite (18,4%), toux (17,9%), dyspnée (15,0%), douleurs musculosquelettiques (15,6%), céphalées (14,5%), prurit (13,9%), leucopénie (13,9%), hypothyroïdie (13,7%), hypertension (13,2%), infection pulmonaire (12,3%), dorsalgies (11,4%), augmentation du taux d'ALAT (11,4%), augmentation du taux d'ASAT (11,0%), œdèmes périphériques (10,8%), et rhinopharyngite (10,3%).
Traitement combiné avec Tecentriq lors de mélanome
Dans l'étude CO39262 (IMspire150), les patients qui recevaient Tecentriq i.v. en association avec du cobimétinib et du vémurafénib ont eu une fréquence plus élevée d'anomalies hépatiques de laboratoire (114/230, 49,6%), de pancréatite (89/230, 38,7%), d'hypothyroïdie (60/230, 26,1%), d'hyperthyroïdie (43/230, 18,7%), de pneumopathie inflammatoire (29/230, 12,6%), de méningo-encéphalite (6/230, 2,6%), de diabète sucré (4/230, 1,7%), de myosite (3/230, 1,3%), de néphrite (3/230, 1,3%) et d'hypophysite (2/230, 0,9%). Les effets indésirables graves les plus fréquents (≥2%) étaient les suivants: pyrexie (5,7%) et élévation de l'alanine aminotransférase (2,2%).
D'autres informations concernant les éventuels effets indésirables graves figurent à la rubrique «Description de certains effets indésirables».
Les catégories de fréquence utilisées sont les suivantes: «très fréquents» (≥1/10), «fréquents» (≥1/100, < 1/10), «occasionnels» (≥1/1000, < 1/100), «rares» (≥1/10'000, < 1/1000) ou «très rares» (< 1/10'000).
Le tableau 4 résume tous les effets indésirables importants rapportés sous Tecentriq sur la base de données groupées issues de monothérapies et de traitements combinés lors de différents types de tumeurs.
Tableau 4: Résumé des effets indésirables rapportés chez les patients traités par Tecentriq i.v. ou s.c.dans le cadre d'études cliniques
|
|
Monothérapie par Tecentriq
n = 5464
|
Traitement combiné avec Tecentriq
n = 5196
| |
Infections et infestations
| |
Très fréquents
|
Infection des voies urinaires a (tous grades confondus: 11,5%, grade 3-4: 3,1%)
|
Infection pulmonaire b (tous grades confondus: 12,3%, grade 3-4: 4,7%, grade 5: 0,6%)
| |
Affections hématologiques et du système lymphatique
| |
Très fréquents
|
Anémie (tous grades confondus: 14,5%, grade 3-4: 3,8%)
|
Anémie (tous grades confondus: 35,3%, grade 3-4: 12,8%), thrombocytopénie c (tous grades confondus: 24,2%, grade 3-4: 9,4%, grade 5: < 0,1%), neutropénie d (tous grades confondus: 35,1%, grade 3-4: 23,1%, grade 5: 0,1%), leucopénie e (tous grades confondus 13,9%, grade 3-4: 5,6%)
| |
Fréquents
|
Thrombocytopénie c
|
Lymphopénie f
| |
Rares
|
Lymphohistiocytose hémophagocytaire **
|
Lymphohistiocytose hémophagocytaire **
| |
Affections du système immunitaire
| |
Fréquents
|
Hypersensibilité
|
| |
Affections endocriniennes
| |
Très fréquents
|
|
Hypothyroïdie g (tous grades confondus: 14,0%, grade 3-4: 0,2%)
| |
Fréquents
|
Hypothyroïdie g, hyperthyroïdie h
|
Hyperthyroïdie h
| |
Occasionnels
|
Insuffisance surrénalienne i, diabète sucré j, hypophysite k
|
Insuffisance surrénalienne i, hypophysite k
| |
Troubles du métabolisme et de la nutrition
| |
Très fréquents
|
Diminution de l'appétit (tous grades confondus: 19,3%, grade 3-4: 0,9%)
|
Diminution de l'appétit (tous grades confondus: 23,4%, grade 3-4: 1,5%, grade 5: < 0,1%)
| |
Fréquents
|
Hyperglycémie, hypokaliémie m, hyponatrémie n
|
Hypokaliémie m, hyponatrémie n, hypomagnésémie l
| |
Affections du système nerveux
| |
Très fréquents
|
Céphalées (tous grades confondus: 10,2%, grade 3-4: 0,3%)
|
Céphalées (tous grades confondus: 14,7%, grade 3-4: 0,3%), neuropathie périphérique o (tous grades confondus: 24,9%, grade 3-4: 3,0%)
| |
Fréquents
|
|
Vertiges, dysgueusie, syncope
| |
Occasionnels
|
Syndrome de Guillain-Barré p, méningoencéphalite q
|
| |
Rares
|
Syndrome myasthénique r, parésie faciale **, myélite **
|
Parésie faciale **, myélite **
| |
Affections cardiaques
| |
Fréquents
|
Affections du péricarde s
|
| |
Occasionnels
|
|
Affections du péricarde s
| |
Rares
|
Myocardite t
|
| |
Affections vasculaires
| |
Très fréquents
|
|
Hypertension artérielle u (tous grades confondus: 13,2%, grade 3-4: 5,8%)
| |
Fréquents
|
Hypotension artérielle
|
| |
Affections respiratoires, thoraciques et médiastinales
| |
Très fréquents
|
Toux (tous grades confondus: 16,8%, grade 3-4: 0,2%), dyspnée (tous grades confondus: 15,6%, grade 3-4: 2,4%)
|
Toux (tous grades confondus: 18,0%, grade 3-4: 0,2%), dyspnée (tous grades confondus: 15,1%, grade 3-4: 1,7%, grade 5: < 0,1%), rhinopharyngite v (tous grades confondus: 10,6%, grade 3-4: < 0,1%)
| |
Fréquents
|
Hypoxie w, rhinopharyngite v, pneumopathie inflammatoire x
|
Pneumopathie inflammatoire x, dysphonie
| |
Occasionnels
|
|
Hypoxie w
| |
Affections gastro-intestinales
| |
Très fréquents
|
Diarrhées y (tous grades confondus: 17,1%, grade 3-4: 1,0%), nausées (tous grades confondus: 17,9%, grade 3-4: 0,7%), vomissements (tous grades confondus: 11,1%, grade 3-4: 0,6%)
|
Constipation (tous grades confondus: 24,7%, grade 3-4: 0,5%), diarrhées y (tous grades confondus: 28,7%, grade 3-4: 2,6%), nausées (tous grades confondus: 34,5%, grade 3-4: 1,5%), vomissements (tous grades confondus: 19,0%, grade 3-4: 1,4%)
| |
Fréquents
|
Douleurs abdominales, colite z, dysphagie, douleurs oropharyngées aa, sécheresse buccale
|
Stomatite, pancréatite bb, sécheresse buccale
| |
Occasionnels
|
Pancréatite bb
|
| |
Rares
|
Maladie cœliaque **
|
Maladie cœliaque **
| |
Affections hépatobiliaires
| |
Très fréquents
|
Hépatite - anomalies des valeurs de laboratoire cc (tous grades confondus: 11,2%, grade 3-4: 3,2%)
|
Hépatite - anomalies des valeurs de laboratoire cc (tous grades confondus: 18,5%, grade 3-4: 5,6%, grade 5: < 0,1%), augmentation du taux d'ALAT (tous grades confondus: 11,4%, grade 3-4: 2,5%), augmentation du taux d'ASAT (tous grades confondus: 11,0%, grade 3-4: 2,3%)
| |
Fréquents
|
Augmentation du taux d'ALAT, augmentation du taux d'ASAT, hépatite dd
|
| |
Affections de la peau et du tissu sous-cutané
| |
Très fréquents
|
Prurit (tous grades confondus: 13,0%, grade 3-4: 0,2%), éruption cutanée ee (tous grades confondus: 18,8%, grade 3-4: 1,1%)
|
Prurit (tous grades confondus: 14,0%, grade 3-4: 0,2%), éruption cutanée ee (tous grades confondus: 30,6%, grade 3-4: 2,7%), alopécie ff (tous grades confondus: 29,8%, grade 3-4: < 0,1%)
| |
Fréquents
|
Sécheresse cutanée
|
| |
Occasionnels
|
Réactions cutanées indésirables sévères (nécrolyse épidermique toxique, syndrome de Stevens Johnson) gg, psoriasis hh
|
Réactions cutanées indésirables sévères (nécrolyse épidermique toxique, syndrome de Stevens Johnson) gg, psoriasis hh
| |
Affections musculosquelettiques et du tissu conjonctif
| |
Très fréquents
|
Arthralgies (tous grades confondus: 15,5%, grade 3-4: 0,8%), dorsalgies (tous grades confondus: 12,0%, grade 3-4: 1,1%)
|
Arthralgies (tous grades confondus: 21,8%, grade 3-4: 1,1%), dorsalgies (tous grades confondus: 11,5%, grade 3-4: 0,8%), douleurs musculosquelettiques ii (tous grades confondus: 15,1%, grade 3-4: 0,7%)
| |
Fréquents
|
Douleurs musculosquelettiques ii
|
| |
Occasionnels
|
Myosite jj, kk
|
| |
Affections du rein et des voies urinaires
| |
Fréquents
|
Créatinine sanguine augmentée ll
|
Protéinurie mm, créatinine sanguine augmentée ll
| |
Occasionnels
|
Néphrite nn
|
Néphrite nn
| |
Investigations
| |
Fréquents
|
|
Phosphatase alcaline sanguine augmentée
| |
Troubles généraux et anomalies au site d'administration
| |
Très fréquents
|
Asthénie (tous grades confondus: 12,6%, grade 3-4: 1,5%), fatigue (tous grades confondus: 27,9%, grade 3-4: 2,3%), pyrexie (tous grades confondus: 17,0%, grade 3-4: 0,5%)
|
Asthénie (tous grades confondus: 18,7%, grade 3-4: 2,8%), fatigue (tous grades confondus: 32,5%, grade 3-4: 3,5%), pyrexie (tous grades confondus: 19,3%, grade 3-4: 0,9%), œdèmes périphériques (tous grades confondus: 10,9%, grade 3-4: 0,2%)
| |
Fréquents
|
Frissons, affection grippale, réactions liées à la perfusion oo, réaction au site d'injection pp, œdèmes périphériques
|
|
a Inclut des cas rapportés d'infection urinaire, de cystite, de pyélonéphrite, d'infection urinaire à Escherichia, d'infection urinaire bactérienne, d'infection rénale, de pyélonéphrite aiguë, d'infection fongique des voies urinaires, d'infection urinaire à Pseudomonas, d'infection urinaire à entérocoques, de pyélonéphrite chronique, d'infection urinaire à staphylocoques, d'abcès rénal, de pyélite et d'urétrite, d'infection urinaire à streptocoques
b Inclut des cas rapportés de pneumonie, de bronchite, d'infection des voies respiratoires inférieures, d'exacerbation infectieuse d'une bronchopneumopathie chronique obstructive, d'épanchement pleural infectieux, de pneumonie atypique, d'abcès pulmonaire, d'infection pleurale, de pneumonie post-procédure, de trachéobronchite, de pneumonie paranéoplasique, de pyopneumothorax
c Inclut des cas rapportés de thrombocytopénie et de diminution du nombre de thrombocytes
d Inclut des cas rapportés de neutropénie, de diminution du nombre de neutrophiles, de neutropénie fébrile, de sepsis neutropénique, d'agranulocytose et de granulocytopénie
e Inclut des cas rapportés de diminution du nombre de globules blancs et de leucopénie
f Inclut des cas rapportés de lymphocytopénie et de diminution du nombre de lymphocytes
g Inclut des cas rapportés d'hypothyroïdie, d'augmentation et de diminution des taux sanguins de thyréostimuline, de thyroïdite auto-immune, de goitre, de thyroïdite, de diminution des taux sanguins de thyroxine libre ou de tri-iodothyronine, de troubles thyroïdiens, d'augmentation des taux sanguins de thyroxine libre ou de thyroxine totale, de diminution des taux sanguins de tri-iodothyronine totale, d'augmentation des taux sanguins de tri-iodothyronine libre, de taux sanguins anormaux de thyréostimuline, de syndrome euthyroïdien, de coma myxœdémateux, de paramètres fonctionnels thyroïdiens anormaux, de diminution de la thyroxine, de tri-iodothyronine anormale, de positivité des anticorps antithyroïdiens, de thyroïdite silencieuse, de thyroïdite chronique, de thyroïdite à médiation immunitaire, de myxœdème, de thyroïdite aiguë, de tri-iodothyronine libre anormale, d'augmentation de la tri-iodothyronine, d'hypothyroïdie auto-immune, d'hypothyroïdie primaire, de goitre hypothyroïdien, d'hypothyroïdie à médiation immunitaire
h Inclut des cas rapportés d'hyperthyroïdie, de maladie de Basedow, d'ophtalmopathie endocrinienne, d'exophtalmie et d'hyperthyroïdie primaire
i Inclut des cas rapportés d'insuffisance surrénalienne, de déficit en glucocorticoïdes, d'insuffisance surrénalienne primaire, de diminution du cortisol, d'insuffisance corticosurrénalienne aiguë, de test de stimulation à l'ACTH anormal, de maladie d'Addison, d'inflammation des glandes surrénales, de déficit en hormone adrénocorticotrope (ACTH), de taux sanguins anormaux de corticotropine, d'augmentation des taux sanguins de corticotropine, d'insuffisance corticosurrénalienne secondaire, de corticotropine sanguine diminuée
j Inclut des cas rapportés de diabète sucré, de diabète sucré de type 1, d'acidocétose diabétique et d'acidocétose
k Inclut des cas rapportés d'hypophysite, d'hypopituitarisme, de trouble de la régulation de la température, d'insuffisance surrénalienne secondaire
l Inclut des cas rapportés d'hypomagnésémie et de diminution du magnésium dans le sang
m Inclut des cas rapportés d'hypokaliémie et de diminution du potassium dans le sang
n Inclut des cas rapportés d'hyponatrémie et de diminution du sodium dans le sang
o Inclut des cas rapportés de neuropathie périphérique, de neuropathie à médiation immunitaire, de neuropathie sensitive périphérique, de polyneuropathie, de zona, de neuropathie motrice périphérique, de neuropathie auto-immune, d'amyotrophie névralgique, de neuropathie sensitivo-motrice périphérique, de neuropathie axonale, de plexopathie lombo-sacrée, d'arthropathie neuropathique, de neuropathie toxique, d'infection des nerfs périphériques, de névrite
p Inclut des cas rapportés de syndrome de Guillain-Barré, de polyneuropathie démyélinisante, de paralysie flasque ascendante
q Inclut des cas rapportés d'encéphalite, de méningite, de photophobie, d'encéphalite auto-immune, de méningite aseptique
r Inclut des cas rapportés de myasthénie grave
s Inclut des cas rapportés de péricardite, d'épanchement péricardique, de tamponnade cardiaque et de péricardite constrictive
t Inclut des cas rapportés de myocardite, de myocardite auto-immune, de myocardite à médiation immunitaire (des cas de myocardite ont été rapportés dans des études en dehors des données combinées)
u Inclut des cas rapportés d'hypertension, d'augmentation de la pression artérielle, de crise hypertensive, d'augmentation de la pression artérielle systolique, d'hypertension diastolique, de pression artérielle insuffisamment contrôlée et de rétinopathie hypertensive, d'hypertension essentielle, de néphropathie hypertensive, d'hypertension orthostatique
v Inclut des cas rapportés de rhinopharyngite, de congestion nasale et de rhinorrhée
w Inclut des cas rapportés d'hypoxie, de diminution de la saturation en oxygène et de diminution de la PO2
x Inclut des cas rapportés de pneumopathie inflammatoire, d'alvéolite, de toxicité pulmonaire, d'infiltration pulmonaire, de bronchiolite, de pneumopathie interstitielle, de pneumopathie inflammatoire radique, d'opacité pulmonaire, de pneumopathie alvéolaire, de pneumonie à éosinophiles, de pneumopathie inflammatoire à médiation immunitaire, de fibrose pulmonaire, de lésions pulmonaires radiques, de maladie pulmonaire à médiation immunitaire
y Inclut des cas rapportés de diarrhées, de selles fréquentes, d'hypermotilité gastro-intestinale, de selles impérieuses, de diarrhées hémorragiques
z Inclut des cas rapportés de colite, de colite de diversion, de colite auto-immune, de colite ischémique, de colite microscopique, de colite ulcéreuse, d'entérocolite à médiation immunitaire, de colite éosinophilique
aa Inclut des cas rapportés de douleurs oropharyngées, de troubles oropharyngés et d'irritation de la gorge
bb Inclut des cas rapportés de pancréatite, de pancréatite auto-immune, de pancréatite aiguë, d'augmentation de la lipase, d'augmentation de l'amylase, d'augmentation des enzymes pancréatiques, d'amylase anormale
cc Inclut des cas rapportés d'augmentation des taux d'ALAT, d'augmentation des taux d'ASAT, d'augmentation de la bilirubine dans le sang, d'augmentation de la gamma-glutamyltransférase, d'ascite, d'augmentation des transaminases, de douleurs hépatiques, d'hyperbilirubinémie, d'augmentation des paramètres fonctionnels hépatiques, d'augmentation des enzymes hépatiques, de trouble de la fonction hépatique, d'hypertransaminasémie, d'augmentation des taux sanguins de bilirubine non conjuguée, d'hyperammoniémie, d'augmentation des taux d'acides biliaires totaux, d'augmentation des taux d'ammoniaque, d'hépatopathie congestive, d'augmentation des taux urinaires de bilirubine, d'augmentation des taux sanguins de bilirubine conjuguée, d'hépatomégalie, d'enzymes hépatiques anormaux, de paramètres fonctionnels hépatiques anormaux, de bilirubine sanguine anormale
dd Inclut des cas rapportés d'ascite, d'hépatite auto-immune, de lésion hépatocellulaire, d'hépatite, d'hépatite aiguë, d'hépatotoxicité, de maladie hépatique, de lésion hépatique d'origine médicamenteuse, de défaillance hépatique, de stéatose hépatique, de lésion hépatique, d'hémorragie de varices œsophagiennes, de varices œsophagiennes, d'hépatomégalie et d'hépatite toxique, de cytolyse hépatique, d'hépatite à médiation immunitaire, de lésion hépatique, de péritonite bactérienne spontanée
ee Inclut des cas rapportés d'éruption cutanée, d'éruption cutanée maculopapuleuse, d'érythème, d'éruption cutanée prurigineuse, de dermatite acnéiforme, d'eczéma, de dermatite, d'éruption cutanée érythémateuse, d'ulcération cutanée, d'éruption cutanée papuleuse, de folliculite, d'éruption cutanée maculeuse, de desquamation cutanée, de dermatite des mains, d'eczéma infecté, de dermatite scrotale, d'éruption cutanée nodulaire, d'éruption cutanée folliculaire, d'éruption cutanée morbilliforme, d'éruption cutanée pustuleuse, de furoncle, d'acné, d'exanthème médicamenteux, de syndrome d'érythrodysesthésie palmo-plantaire, de dermatite séborrhéique, de dermatite allergique, d'érythème de la paupière, de toxicité cutanée, d'éruption cutanée de la paupière, d'exanthème fixe, d'éruption cutanée papulo-squameuse, d'éruption cutanée vésiculaire, de bulle cutanée, de vésicules labiales, de pemphigoïde, de bulle hémorragique buccale, d'exanthème de l'accès vasculaire, de dermatite à médiation immunitaire
ff Inclut des cas rapportés d'alopécie, de madarose, d'alopécie en plaques, d'alopécie totale et d'hypotrichose
gg Inclut des cas rapportés de dermatite bulleuse, d'exanthème exfoliatif, d'érythème polymorphe, de dermatite exfoliative généralisée, d'exanthème toxique, de syndrome de Stevens-Johnson (SSJ), de dermatite exfoliative, de réaction médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), de nécrolyse épidermique toxique (NET) et de vascularite cutanée (des cas de SSJ et de DRESS ont été rapportés dans des études en dehors des données combinées)
hh Inclut des cas rapportés de dermatite psoriasiforme, de psoriasis en gouttes et de psoriasis
ii Inclut des cas rapportés de douleurs de l'appareil locomoteur, de myalgies et de douleurs osseuses
jj Inclut des cas rapportés de myosite, de rhabdomyolyse, de pseudopolyarthrite rhizomélique, de dermatomyosite, d'abcès musculaire, de présence de myoglobine dans les urines, de myopathie, de polymyosite
kk Des cas mortels ont été rapportés dans des études en dehors des données combinées
ll Inclut des cas rapportés d'augmentation de la créatinine dans le sang et d'hypercréatininémie
mm Inclut des cas rapportés de protéinurie, de microalbuminurie, de présence de protéines dans les urines, d'hémoglobinurie, de syndrome néphrotique, d'anomalie urinaire et d'albuminurie
nn Inclut des cas rapportés de néphrite, de glomérulonéphrite paranéoplasique, de glomérulonéphrite chronique, de néphropathie du purpura rhumatoïde (purpura d'Henoch-Schönlein), de néphrite tubulo-interstitielle, de néphrite d'origine auto-immune, de néphrite allergique, de glomérulonéphrite, de syndrome néphrotique et de glomérulonéphrite mésangioproliférative
oo Inclut des cas rapportés de réaction liée à la perfusion et de syndrome de relargage des cytokines, d'hypersensibilité, de réaction anaphylactique
pp Rapportée dans l'étude IMscin001 (se référant à l'administration par voie sous-cutanée). L'indication de fréquence repose sur l'exposition à Tecentriq s.c.dans l'étude IMscin001 et inclut des rapports de réactions au site d'injection, de douleurs au site d'injection, d'érythème au site d'injection et d'éruption cutanée au site d'injection
** Rapportées après la commercialisation en dehors de l'ensemble des données regroupées. L'indication de fréquence repose sur l'exposition dans la totalité du programme
Données post-commercialisation
Les réactions médicamenteuses indésirables suivantes ont été identifiées dans le cadre de la surveillance post-commercialisation de Tecentriq. Les réactions médicamenteuses indésirables observées dans le cadre de la surveillance post-commercialisation sont rangées par classe de système d'organes de la classification MedDRA.
Affections hématologiques et du système lymphatique
Rares: lymphohistiocytose hémophagocytaire a.
Affections du système nerveux
Rares: parésie faciale a.
Rares: myélite a.
Affections oculaires
Occasionnels: uvéite a.
a Rapportées dans le cadre de l'expérience postcommercialisation en dehors de l'ensemble des données combinées. La fréquence repose sur l'exposition dans la totalité du programme.
Description de certains effets indésirables
Les données ci-dessous contiennent des indications relatives aux réactions indésirables substantielles survenues sous Tecentriq en monothérapie. Les détails concernant les principales réactions indésirables de Tecentriq utilisé en traitement combiné sont mentionnés lorsque des différences cliniques pertinentes ont été constatées par rapport à Tecentriq en monothérapie. Pour la conduite à tenir lors des événements suivants, voir «Mises en garde et précautions, Généralités»:
Infections
Lors des études cliniques menées chez 5464 patients atteints de différents types de cancers, qui ont reçu Tecentriq, 41,1% des patients ont développé des infections, notamment des infections de grade 3 (8,1%), de grade 4 (1,6%) et de grade 5 (1,0%). Les infections de grade 3 ou supérieur les plus fréquentes étaient les infections urinaires et les pneumonies, survenues chez respectivement 2,5 et 2,4% des patients.
Dans l'étude sur le mélanome (CO39262), les infections étaient plus fréquentes chez les patients ayant reçu Tecentriq i.v. en association avec du cobimétinib et du vémurafénib. Des infections sont survenues chez 60,4% (139/230) des patients, dont des infections de grade 3-4 chez 8,7% (20/230) et des infections de grade 5 chez 1,7% (4/230) des patients. L'infection urinaire était l'infection la plus fréquente et elle est survenue chez 7,8% (18/230) des patients.
Lymphohistiocytose hémophagocytaire
Une lymphohistiocytose hémophagocytaire (LLH) est survenue chez < 0,1% (2/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai médian d'apparition a été de 2,7 mois (intervalle: de 1,6 à 3,8 mois). La durée a été de 1,1 mois (intervalle: de 0,9 à 1,4 mois). Chez 1 patient (< 0,1%), la LLH a entraîné l'arrêt de Tecentriq. Aucun des deux patients n'a eu besoin de corticostéroïdes.
Pneumopathie inflammatoire à médiation immunitaire
Une pneumopathie inflammatoire est survenue chez 2,9% (156/5464) des patients ayant reçu Tecentriq en monothérapie. Cet événement a eu une issue fatale chez trois des 156 patients. Le délai médian d'apparition a été de 4,0 mois (intervalle: de 0,1 à 29,8 mois). La durée médiane a été de 1,8 mois (intervalle: de 0 à 27,8+ mois; le signe + signale une valeur censurée). Chez 36 patients (0,7%), la pneumopathie inflammatoire a entraîné une interruption du traitement par Tecentriq. Une pneumopathie inflammatoire ayant nécessité l'utilisation de corticostéroïdes est survenue chez 1,6% (88/5464) des patients traités par Tecentriq.
Dans l'étude IMscin001, une pneumopathie inflammatoire est survenue chez 2,0% (5/247) des patients traités par Tecentriq s.c.
Dans l'étude sur le mélanome (CO39262), les pneumopathies inflammatoires d'origine immunologique étaient plus fréquentes chez les patients ayant reçu Tecentriq i.v. en association avec du cobimétinib et du vémurafénib. Une pneumopathie inflammatoire d'origine immunologique est survenue chez 12,6% (29/230) des patients et était de grade 3 à 4 chez 1,3% (3/230) des patients.
Hépatite à médiation immunitaire
Une hépatite est survenue chez 1,7% (90/5464) des patients ayant reçu Tecentriq en monothérapie. Cet événement a eu une issue fatale chez trois des 90 patients. Le délai médian d'apparition a été de 1,9 mois (intervalle: de 0,2 à 18,8 mois). La durée médiane a été de 1,1 mois (intervalle: de 0 à 32,4+ mois; le signe + signale une valeur censurée). Une hépatite a entraîné une interruption du traitement par Tecentriq chez 18 patients (0,3%). Une hépatite a nécessité l'administration de corticostéroïdes chez 0,6% (32/5464) des patients traités par Tecentriq.
Dans l'étude IMscin001, une hépatite à médiation immunitaire est survenue chez 1,2% (3/247) des patients traités par Tecentriq s.c.. Dans l'étude sur le mélanome (CO39262), les hépatites d'origine immunologique étaient plus fréquentes chez les patients ayant reçu Tecentriq i.v. en association avec du cobimétinib et du vémurafénib. Une hépatite d'origine immunologique est survenue chez 53,0% (122/230) des patients et était de grade 3-4 chez 22,2% (51/230) des patients et de grade 5 chez 0,9% (2/230) des patients. La plupart des événements en lien avec une hépatite d'origine immunologique signalée chez les patients traités par Tecentriq en association avec du cobimétinib et du vémurafénib étaient des valeurs de laboratoire anormales (114 des 122 patients [93,4%]).
Colite à médiation immunitaire
Une colite est survenue chez 1,1% (62/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai médian d'apparition a été de 4,9 mois (intervalle: de 0,5 à 17,2 mois). La durée médiane a été de 1,4 mois (intervalle: de 0,1 à 50,2+ mois; le signe + signale une valeur censurée). Chez 23 patients (0,4%), la colite a entraîné une interruption du traitement par Tecentriq. Chez 0,5% (29/5464) des patients sous traitement par Tecentriq, la survenue d'une colite a nécessité l'utilisation de corticostéroïdes.
Dans l'étude sur le mélanome (CO39262), les diarrhées ou les colites étaient plus fréquentes chez les patients ayant reçu Tecentriq i.v. en association avec du cobimétinib et du vémurafénib. Des diarrhées ou une colite sont survenues chez 50,4% (116/230) des patients et étaient de grade 3-4 chez 3,0% (7/230) des patients.
Endocrinopathies à médiation immunitaire
Affections de la thyroïde
Une hypothyroïdie est survenue chez 8,6% (471/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai médian d'apparition a été de 4,2 mois (intervalle: de 0 à 34,5 mois). L'hypothyroïdie a entraîné une interruption du traitement par Tecentriq chez 10 patients (0,2%). La durée médiane a été de 51,1 mois (intervalle: de 0,1+ à 56,2+ mois; le signe + signale une valeur censurée).
Une hyperthyroïdie est survenue chez 2,5% (138/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai médian d'apparition a été de 2,7 mois (intervalle: de 0 à 24,3 mois). La durée médiane a été de 2,6 mois (intervalle: de 0+ à 47,7+ mois; le signe + signale une valeur censurée). L'hyperthyroïdie a entraîné une interruption du traitement par Tecentriq chez 5 patients (< 0,1%).
Une hyperthyroïdie est survenue chez 4,9% (23/473) des patients ayant reçu Tecentriq en association avec du carboplatine et du nab-paclitaxel. Un patient (0,2%) a arrêté le traitement à cause d'une hyperthyroïdie.
Dans l'étude IMscin001, une hypothyroïdie est survenue chez 10,5% (26/247) des patients traités par Tecentriq s.c. et une hyperthyroïdie est survenue chez 2,0% (5/247) des patients traités par Tecentriq s.c.
Dans l'étude sur le mélanome (CO39262), l'hypothyroïdie et l'hyperthyroïdie étaient plus fréquentes chez les patients ayant reçu Tecentriq i.v. en association avec du cobimétinib et du vémurafénib. Une hypothyroïdie est survenue chez 26,1% (60/230) des patients; aucune n'était de grade 3-4. Une hyperthyroïdie est survenue chez 18,7% (43/230) des patients et était de grade 3-4 chez 0,9% (2/230) des patients.
Insuffisance surrénalienne
Une insuffisance surrénalienne est survenue chez 0,5% (26/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai médian d'apparition a été de 6,7 mois (intervalle: de 0,1 à 21,4 mois). L'insuffisance surrénalienne a entraîné l'interruption du traitement chez 5 patients (< 0,1%). Chez 0,4% (22/5464) des patients sous traitement par Tecentriq, la survenue d'une insuffisance surrénalienne a nécessité l'utilisation de corticostéroïdes.
Dans l'étude IMscin001, une insuffisance surrénalienne est survenue chez 0,4% (1/247) des patients traités par Tecentriq s.c.
Une insuffisance surrénalienne est survenue chez 1,5% (7/473) des patients ayant reçu Tecentriq en association avec du carboplatine et du nab-paclitaxel. Une insuffisance surrénalienne nécessitant l'utilisation de corticoïdes est survenue chez 0,8% (4/473) des patients ayant reçu Tecentriq en association avec du carboplatine et du nab-paclitaxel.
Hypophysite
Une hypophysite est survenue chez 0,1% (6/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai médian d'apparition a été de 6,1 mois chez ces patients (intervalle: de 0,8 à 13,7 mois). L'hypophysite a entraîné l'interruption de Tecentriq chez 1 patient (< 0,1%) et nécessité le recours à des corticostéroïdes chez < 0,1% (5/5464) des patients sous traitement par Tecentriq. Dans une étude en cours, une hypophysite est survenue chez 0,8% (3/393) des patients ayant reçu Tecentriq i.v. en association au bévacizumab, au paclitaxel et au carboplatine. Le délai médian d'apparition a été de 7,7 mois (intervalle: de 5,0 à 8,8 mois). Les trois patients ont nécessité un traitement par des corticostéroïdes.
Diabète sucré
Un diabète sucré est survenu chez 0,5% (26/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai médian d'apparition a été de 5,4 mois (intervalle: de 0,1 à 29,0 mois). La durée médiane a été de 9,2 mois (intervalle: de 0,1 à 51,2+ mois; le signe + signale une valeur censurée). Chez 3 patients (< 0,1%), le diabète sucré a entraîné une interruption du traitement par Tecentriq et quatre patients ont nécessité un traitement par des corticostéroïdes.
Un diabète sucré est survenu chez 2,0% (10/493) des patients atteints de CHC ayant reçu Tecentriq i.v. en association avec le bévacizumab. Le délai médian d'apparition a été de 4,4 mois (intervalle: de 1,2 à 8,3 mois). Aucun des cas de diabète sucré n'a nécessité l'arrêt de Tecentriq.
Méningoencéphalite à médiation immunitaire
Une méningoencéphalite est survenue chez 0,5% (24/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai d'apparition a été de 0,5 mois (intervalle: de 0 à 12,5 mois). La durée médiane a été de 0,7 mois (intervalle: de 0,1+ à 14,5+ mois; le signe + signale une valeur censurée). Une méningoencéphalite a entraîné l'interruption de Tecentriq chez 9 patients (0,2%) et nécessité le recours à des corticostéroïdes chez 0,3% (14/5464) des patients sous traitement par Tecentriq.
Neuropathies à médiation immunitaire
Syndrome de Guillain-Barré et polyneuropathie démyélinisante
Un syndrome de Guillain-Barré et une polyneuropathie démyélinisante sont survenus chez 0,1% (6/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai médian d'apparition de cet événement a été de 4,1 mois (intervalle: de 0,6 à 8,1 mois). La durée médiane a été de 8,0 mois (intervalle: de 0,6 à 24,5+ mois; le signe + signale une valeur censurée). Un syndrome de Guillain-Barré a entraîné l'interruption de Tecentriq chez 3 patients (< 0,1%). Chez < 0,1% (3/5464) des patients, un syndrome de Guillain-Barré a nécessité le recours à des corticostéroïdes.
Parésie faciale à médiation immunitaire
Une parésie faciale est survenue chez < 0,1% (1/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai d'apparition a été de 1,0 mois. La durée a été de 1,1 mois. Cet événement n'a pas nécessité le recours à des corticostéroïdes et n'a pas entraîné l'arrêt de Tecentriq.
Myélite à médiation immunitaire
Une myélite à médiation immunitaire est survenue chez < 0,1% (1/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai d'apparition a été de 0,76 mois. Cet événement a nécessité le recours à des corticostéroïdes, mais n'a pas entraîné l'arrêt de Tecentriq.
Pancréatite à médiation immunitaire
Une pancréatite, y compris une augmentation des taux sériques d'amylase et de lipase, est survenue chez 0,8% (43/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai médian d'apparition a été de 6,2 mois (intervalle: de 0 à 40,4 mois). La durée médiane a été de 0,9 mois (intervalle: de 0 à 40,4+ mois; le signe + signale une valeur censurée). Chez 3 patients (< 0,1%), la pancréatite a entraîné l'interruption du traitement par Tecentriq. Chez < 0,1% (7/5464) des patients ayant reçu Tecentriq, une pancréatite a nécessité le recours à des corticostéroïdes.
Myosite à médiation immunitaire
Une myosite est survenue chez 0,6% (33/5464) des patients qui avaient reçu Tecentriq en monothérapie. Le délai médian d'apparition a été de 5,0 mois (intervalle: de 0,4 à 11,5 mois). La durée médiane a été de 3,2 mois (intervalle: de 0,0 à 51,1+ mois; le signe + signale une valeur censurée). La myosite a entraîné l'interruption de Tecentriq chez 2 patients (0,1%). Chez 0,2% (11/5464) des patients sous traitement par Tecentriq, la myosite a nécessité l'administration de corticostéroïdes.
Néphrite à médiation immunitaire
Une néphrite est survenue chez 0,2% (12/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai médian d'apparition a été de 5,1 mois (intervalle: de 0,1 mois à 20,4 mois). Une néphrite a entraîné l'interruption de Tecentriq chez 6 patients (0,1%). Six patients (0,1%) ont dû suivre un traitement par des corticostéroïdes.
Réactions cutanées sévères à médiation immunitaire
Des réactions cutanées indésirables sévères (SCAR) sont survenues chez 0,6% (33/5464) des patients qui avaient reçu Tecentriq en monothérapie. Chez deux des 33 patients, cet événement a eu une issue fatale. Le délai médian d'apparition a été de 4,9 mois (intervalle: de 0,1 à 15,5 mois). La durée médiane a été de 2,8 mois (intervalle: de 0 à 37,5+ mois; le signe + signale une valeur censurée). Chez 3 patients (< 0,1%), les SCAR ont entraîné une interruption du traitement par Tecentriq. Des SCAR ayant nécessité l'utilisation systémique de corticostéroïdes sont survenues chez 0,2% (11/5464) des patients ayant reçu Tecentriq en monothérapie.
Dans l'étude IMscin001, des SCAR sont survenues chez 0,8% (2/247) des patients traités par Tecentriq s.c.
Affections du péricarde à médiation immunitaire
Des affections du péricarde sont survenues chez 0,9% (48/5464) des patients ayant reçu Tecentriq en monothérapie. Le délai médian de survenue a été de 1,4 mois (intervalle: 0,2 à 17,5 mois). La durée médiane était de 1,4 mois (intervalle: 0 à 51,5+ mois; + signale une valeur censurée). Les affections du péricarde ont conduit à l'arrêt du traitement par Tecentriq chez 3 patients (< 0,1%). Des affections du péricarde ayant nécessité l'utilisation de corticoïdes sont survenues chez 0,1% (7/5464) des patients.
Immunogénicité
Comme toutes les protéines thérapeutiques, Tecentriq peut également potentiellement provoquer une réponse immunitaire. Dans plusieurs études de phase III, 13,1 à 38,5% des patients ont présenté des anticorps dirigés contre le médicament (ADA: anti-drug antibodies) en lien avec le traitement et 4,3% à 19,9% des patients ont présenté des anticorps neutralisants (NAb) (voir «Mises en garde et précautions», «Propriétés/Effets», «Pharmacocinétique»).
Dans les données regroupées des patients ayant reçu de l'atézolizumab en monothérapie (n = 3460) ou en traitement combiné (n = 2285), les taux suivants d'événements indésirables ont été observés pour la population ADA-positive, par rapport à la population ADA-négative: événements indésirables de grade 3-4: 45,9% contre 39,1%; événements indésirables graves: 39,4% contre 33,0%; événements indésirables ayant nécessité l'interruption du traitement: 8,4% contre 7,7% (en monothérapie); événements indésirables de grade 3-4: 63,9% contre 60,9%, événements indésirables graves: 43,9% contre 35,6%, événements indésirables ayant nécessité l'interruption du traitement: 22,8% contre 18,4% (en traitement combiné).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucun cas de surdosage n'a été rapporté.
Propriétés/EffetsCode ATC
L01FF05
Mécanisme d'action
La liaison du PD-L1 aux récepteurs PD-1 et B7.1 sur les cellules T provoque une suppression de l'activité cytotoxique des lymphocytes T par inhibition de la prolifération et de la production de cytokines par les lymphocytes T. Le PD-L1 peut être exprimé sur les cellules tumorales (TC) ainsi que les cellules immunitaires infiltrant les tumeurs (IC) et contribuer à inhiber la réponse immunitaire antitumorale dans le microenvironnement.
L'atézolizumab est un anticorps monoclonal humanisé de type immunoglobuline G1 (IgG1) à fragment Fc modifié, qui se lie directement à PD-L1 et provoque un blocage des interactions avec les récepteurs PD-1 et B7.1. Cela permet de lever l'inhibition de la réponse immunitaire médiée par la voie de signalisation PD-L1/PD-1 et entraîne une réactivation de la réponse immunitaire antitumorale. L'atézolizumab ne modifie pas l'interaction PD-L2/PD-1. Dans des modèles tumoraux syngéniques murins, le blocage de l'activité du PD-L1 a entraîné une diminution de la croissance tumorale.
La double inhibition des voies de signalisation PD-1/PD-L1 et MAPK ainsi que BRAF et MEK entrave la croissance tumorale dans les modèles de cancer chez les souris et améliore l'immunogénicité des tumeurs en augmentant la présentation de l'antigène ainsi que l'activation et l'infiltration des cellules T par rapport à la thérapie ciblée seule.
Efficacité clinique
Cancer du poumon non à petites cellules
1L en cas de NSCLC non épidermoïde métastatique
IMpower150
Une étude clinique de phase III, randomisée, en ouvert, GO29436 (IMpower150), a été menée afin d'évaluer l'efficacité́ et la sécurité́ de Tecentriq en association au paclitaxel et au carboplatine, avec ou sans bévacizumab, chez des patients atteints d'un NSCLC non épidermoïde métastatique sans chimiothérapie préalable. Au total, 1202 patients ont été inclus dans l'étude et randomisés selon un rapport 1:1:1 pour recevoir l'un des régimes thérapeutiques décrits ci-dessous. La randomisation a été stratifiée selon le sexe, la présence de métastases hépatiques et le statut d'expression de PD-L1 sur les cellules tumorales (TC) et les cellules immunitaires infiltrant les tumeurs (IC).
Les patients ont été randomisés dans un des trois groupes de traitement suivants:
·Groupe A: Tecentriq 1200 mg, paclitaxel 175 mg/m² ou 200 mg/m² et carboplatine AUC 6 mg/ml/min le jour 1 de chaque cycle de 21 jours pendant 4 ou 6 cycles au maximum.
·Groupe B: Tecentriq 1200 mg, bévacizumab 15 mg/kg, paclitaxel 175 mg/m² ou 200 mg/m² et carboplatine AUC 6 mg/ml/min le jour 1 de chaque cycle de 21 jours pendant 4 ou 6 cycles au maximum.
·Groupe C: bévacizumab 15 mg/kg, paclitaxel 175 mg/m² ou 200 mg/m² et carboplatine AUC 6 mg/ml/min le jour 1 de chaque cycle de 21 jours pendant 4 ou 6 cycles au maximum.
Le carboplatine et le paclitaxel ont été administrés jusqu'à la fin des 4 ou 6 cycles ou jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable, selon l'événement survenant le premier. Compte tenu d'un degré globalement plus élevé de toxicités hématologiques chez les patients originaires de pays asiatiques par rapport aux patients issus de pays non-asiatiques, les patients d'ethnie asiatique ont reçu une dose initiale de 175 mg/m2 de paclitaxel.
Les patients sans progression de la maladie à l'issue de la chimiothérapie à base de platine ou après son interruption ont reçu:
·Groupe A: Tecentriq 1200 mg par voie intraveineuse le jour 1 de chaque cycle de 21 jours jusqu'à la perte du bénéfice clinique selon l'évaluation du médecin investigateur.
·Groupe B: Tecentriq 1200 mg et bévacizumab 15 mg/kg par voie intraveineuse le jour 1 de chaque cycle de 21 jours. L'administration de Tecentriq s'est poursuivie jusqu'à la perte du bénéfice clinique selon l'évaluation du médecin investigateur, et l'administration de bévacizumab a été maintenue jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
·Groupe C: bévacizumab 15 mg/kg par voie intraveineuse le jour 1 de chaque cycle de 21 jours jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
Les patients ont été exclus de la participation à l'étude s'ils souffraient d'une maladie auto-immune, s'ils avaient reçu un vaccin vivant atténué dans les 28 jours précédant la randomisation, s'ils avaient reçu un traitement immunostimulant systémique dans les 4 semaines ou un traitement immunosuppresseur systémique dans les 2 semaines précédant la randomisation, s'ils présentaient des métastases du SNC actives ou non traitées, une infiltration tumorale évidente des grands vaisseaux thoraciques ou une cavitation manifeste des lésions pulmonaires à l'imagerie. Des évaluations tumorales ont été́ réalisées toutes les 6 semaines pendant les 48 premières semaines suivant le jour 1 du cycle 1, puis toutes les 9 semaines par la suite.
Les échantillons tumoraux ont été analysés à la recherche de l'expression de PD-L1 sur les cellules tumorales (TC) et les cellules immunitaires infiltrant les tumeurs (IC), et les résultats ont été utilisés pour définir les sous-groupes d'expression de PD-L1 pour les analyses décrites ci-après.
Les données démographiques et les caractéristiques de la maladie au début du traitement de la population étudiée (ITT) étaient équilibrées entre les deux groupes de traitement. Dans la présente étude, l'âge médian s'élevait à 63 ans (fourchette: de 31 à 90) et 60% des patients étaient de sexe masculin. La majorité des sujets étaient de type caucasien (82%). Près de 10% des patients présentaient des mutations de l'EGFR connues, 4% présentaient des translocations de l'ALK connues, 14% avaient des métastases hépatiques à l'inclusion dans l'étude et la plupart des patients étaient fumeurs ou anciens fumeurs (80%). Au total, 95% des patients étaient atteints d'un NSCLC non épidermoïde avec une histologie d'adénocarcinome. L'indice de performance ECOG à l'inclusion était de 0 (43%) ou 1 (57%).
Les données démographiques et les caractéristiques de la maladie au début du traitement de patients dont le statut d'expression de PD-L1 était de ≥1% sur les TC ou de ≥1% sur les IC (ci-après dénommé «expression de PD-L1 ≥1%»), sans mutations de l'EGFR ni translocations de l'ALK, étaient équilibrées entre les deux groupes de traitement. 51% des tumeurs des patients présentaient une expression de PD-L1 ≥1% et 49% une expression de PD-L1< 1% sur les TC et < 1% sur les IC.
La population en ITT-WT est définie comme l'ensemble des patients randomisés à l'exception de ceux présentant des aberrations génomiques tumorales de l'EGFR et de l'ALK.
La durée de suivi médiane était de 39,3 mois au moment de l'analyse finale de l'OS pour Tecentriq + carboplatine + paclitaxel (CP), comparé à bévacizumab + CP. Pour la population en ITT-WT regroupant des patients dont les tumeurs présentaient une expression de PD-L1 ≥1%, un allongement de l'OS a été constaté dans le groupe Tecentriq + CP, comparé au groupe bévacizumab + CP (HR non stratifié: 0,71; [IC à 95%: 0,55, 0,91] pas testé formellement en raison du concept statistique); l'OS médiane s'est élevée à 24,4 mois (8,4 mois de plus que dans le groupe bévacizumab + CP, qui a révélé une OS de 16,0 mois). Ceci correspond à une réduction du risque relatif de mortalité de 29% associée à Tecentriq + CP, par rapport à bévacizumab + CP, dans cette population. L'analyse par landmark révèle que, dans la population en ITT-WT (expression de PD-L1 ≥1%), les taux de survie après un an étaient de 70,6% dans le groupe Tecentriq + CP, contre 55,9% dans le groupe bévacizumab + CP, et de 51,5% et 36,9% respectivement après deux ans.
Au moment de l'analyse finale de la PFS, le hazard ratio de la PFS non stratifié s'élevait à 0,74 [IC à 95%: 0,58, 0,94] chez les patients sous Tecentriq + CP, comparé à bévacizumab + CP (population en ITT-WT avec une expression de PD-L1 ≥1%).
IMpower130
Un essai clinique de phase III, randomisé, en ouvert, GO29537 (IMpower130), a été́ mené́ afin d'évaluer l'efficacité́ et la sécurité́ de Tecentriq en association au nab-paclitaxel et au carboplatine, chez des patients naïfs de chimiothérapie atteints d'un NSCLC non épidermoïde métastatique. Les patients, y compris ceux avec aberrations génomiques tumorales du gène EGFR ou ALK, ont été́ inclus et randomisés selon un rapport 2:1 pour recevoir l'un des traitements décrits dans le tableau 5. La randomisation a été́ stratifiée selon le sexe, la présence de métastases hépatiques et le statut d'expression de PD-L1 sur les TC et les IC. Les patients recevant le traitement B ont pu changer de traitement et recevoir le Tecentriq en monothérapie après progression de la maladie.
Tableau 5: Traitement intraveineux dans IMpower130
|
Traitement
|
Induction
(quatre ou six cycles de 21 jours)
|
Entretien
(cycles de 21 jours)
| |
A
|
Tecentriq (1200 mg) a + nabpaclitaxel (100 mg/m2) b, c + carboplatine (AUC 6) c
|
Tecentriq (1200 mg) a
| |
B
|
Nab-paclitaxel (100 mg/m2) b + carboplatine (AUC 6) c
|
Meilleurs soins de soutien ou pémétrexed
|
a LeTecentriq est administré jusqu'à̀ perte du bénéfice clinique évaluée par le médecin investigateur.
b Le nab-paclitaxel est administré les jours 1, 8 et 15 de chaque cycle.
c Le nab-paclitaxel et le carboplatine sont administrés jusqu'à la fin de 4-6 cycles ou progression de la maladie ou survenue d'une toxicité inacceptable, selon l'événement survenant en premier.
Les patients ont été́ exclus s'ils présentaient un antécédent de maladie auto-immune, s'ils avaient reçu un vaccin vivant atténué́ dans les 28 jours précédant la randomisation, s'ils avaient reçu un traitement immunostimulant systémique dans les 4 semaines ou un traitement immunosuppresseur systémique dans les 2 semaines précédant la randomisation, s'ils présentaient des métastases cérébrales actives ou non traitées. Des évaluations tumorales ont été́ réalisées toutes les 6 semaines pendant les 48 premières semaines suivant le Jour 1 du cycle 1, puis toutes les 9 semaines par la suite.
Les caractéristiques démographiques et pathologiques à l'inclusion de la population étudiée (n = 723) étaient bien équilibrées entre les bras de traitement. L'âge médian s'élevait à 64 ans (entendue de 18 à 86 ans). La majorité́ des patients était de sexe masculin (57%) et de type caucasien (90%). 14,8% des patients avaient des métastases hépatiques à l'inclusion et la plupart des patients étaient fumeurs ou anciens fumeurs (88%). La majorité́ des patients présentaient un indice de performance ECOG de 1 (58,7%).
L'analyse principale a été́ réalisée chez tous les patients, en excluant ceux avec aberrations génomiques tumorales du gène EGFR ou ALK (n = 685), définie comme la population ITT-WT. Les deux critères d'évaluation principaux étaient l'OS et la PFS évaluée par le médecin investigateur. Les principaux résultats de cette étude lors d'un suivi médian de 18,6 mois de la durée de vie restante sont résumés ci-dessous.
Le traitement par atezo + carboplatine + nab-paclitaxel (atezo + CnP) été associé à un allongement statistiquement significatif de l'OS par rapport au carboplatine et nab-paclitaxel (CnP). Pour les patients de la population en ITT-WT, l'OS médiane était 4,7 mois plus longue dans le bras de traitement sous atezo + CnP, où elle était de 18,6 mois (IC à 95%: 15,8; 21,2) dans le bras de traitement sous atezo + CnP contre 13,9 mois (IC à 95%: 12,0; 18,7) dans le bras de traitement sous CnP (valeur de p stratifiée = 0,030). Le hazard ratio (HR) stratifié était de 0,79 (IC à 95%: 0,64; 0,98); cela correspond à une réduction relative du risque de mortalité de 21% sous atezo + CnP par rapport au CnP.
Par rapport au CnP, le traitement par atezo + CnP était associé à une amélioration statistiquement significative de la PFS évaluée par le médecin investigateur (selon RECIST v1.1). Pour les patients de la population en ITT-WT, la PFS médiane était numériquement plus longue dans le bras de traitement sous atezo + CnP: 7,0 mois (IC à 95%: 6,3; 7,3) dans le bras de traitement atezo + CnP contre 5,5 mois (IC à 95%: 4,4; 5,9) dans le bras sous CnP (valeur de p stratifiée < 0,0001). Le HR stratifié était de 0,64 (IC à 95%: 0,54; 0,76).
La proportion de patients de la population ITT-WT avec un taux de réponse objective évalué et confirmé par un médecin investigateur selon RECIST v1.1 était supérieur de 17,6% dans le bras de traitement sous atezo + CnP par rapport au bras sous CnP (49,3% vs 31,7%).
Dans tous les sous-groupes PD-L1, indépendamment de l'expression, un bénéfice concernant l'OS et la PFS a été obtenu hormis les patients avec des métastases hépatiques. Ainsi que les résultats d'une analyse en sous-groupes d'un nombre limité de patients (n = 101) avec des métastases hépatiques l'ont montré, l'atézolizumab, le nab-paclitaxel et le carboplatine ne semblent pas entraîner d'améliorations de l'effet thérapeutique dans ce collectif partiel par rapport au nab-paclitaxel et au carboplatine (HR: 0,96; IC à 95%: 0,61; 1,51 pour la PFS et HR: 1,21; IC à 95%: 0,73; 2,00 pour l'OS).
NSCLC au stade précoce
IMpower010
Une étude de phase III multicentrique, randomisée et en ouvert, GO29527 (IMpower010), a été réalisée afin d'évaluer l'efficacité et la sécurité de Tecentriq pour le traitement adjuvant de patients atteints de NSCLC de stade IB (tumeurs ≥4 cm) à IIIA (selon de système de classification de l'UICC/AJCC, 7e édition). Parmi les patients inclus dans l'étude ayant subi une reséction tumorale complète, un total de 1280 patients ont été éligibles pour recevoir jusqu'à 4 cycles d'une chimiothérapie à base de cisplatine. Le tableau 6 décrit les schémas de chimiothérapies à base de cisplatine.
Tableau 6: Schémas thérapeutiques pour la chimiothérapie intraveineuse de l'étude IMpower010
|
Chimiothérapie adjuvante à base de cisplatine
Cisplatine 75 mg/m2 i.v. au jour 1 de chaque cycle de 21 jours avec l'un de ces schémas thérapeutiques
|
Vinorelbine 30 mg/m2 i.v., jours 1 et 8
| |
Docétaxel 75 mg/m2 i.v., jour 1
| |
Gemcitabine 1250 mg/m2 i.v., jours 1 et 8
| |
Pémétrexed 500 mg/m2 i.v., jour 1
|
À l'issue de la chimiothérapie à base de cisplatine (jusqu'à 4 cycles), au total 1005 patients ont été randomisés selon un rapport de 1:1 pour recevoir Tecentriq (bras A) ou le meilleur traitement de soutien (bras B). Tecentriq a été administré à la dose fixe de 1200 mg en perfusion i.v. toutes les 3 semaines pendant 16 cycles, sauf en cas d'apparition d'une récidive de la maladie ou de survenue d'une toxicité inacceptable. La randomisation a été stratifiée selon le sexe, le stade de la maladie, l'histologie et le statut d'expression de PD-L1 (estimé au moyen du test VENTANA PD-L1 [SP142]).
Les patients ont été exclus de l'étude s'ils présentaient des antécédents de maladie auto-immune, s'ils avaient reçu un vaccin vivant atténué́ dans les 28 jours précédant la randomisation, ou encore s'ils avaient reçu un traitement immunostimulant systémique dans les 4 semaines ou un traitement immunosuppresseur systémique dans les 2 semaines précédant la randomisation. Des évaluations tumorales ont été réalisées au début de la phase de randomisation, au cours de la première année après le cycle 1, au jour 1 tous les 4 mois, puis tous les 6 mois jusqu'à l'année 5, puis une fois par an.
Les données démographiques et les caractéristiques pathologiques étaient bien équilibrées entre les bras de traitement. L'âge médian était de 62 ans (intervalle: de 26 à 84 ans), 67% des patients étaient de sexe masculin. La majorité des patients était de type caucasien (73%), 24% étaient d'origine asiatique. La plupart des patients étaient des fumeurs ou d'anciens fumeurs (78%); au début de l'étude, l'indice de performance ECOG était de 0 (55%) ou 1 (44%). Le maladie était de stade IB chez 12% des patients, de stade II chez 47% et de stade IIIA chez 41%. Sur la base des estimations réalisées au moyen du test VENTANA PD-L1 (SP263), la proportion de patients atteints de tumeurs présentant une expression de PD-L1 ≥1% sur les cellules tumorales (TC) était de 55% et la proportion de patients atteints de tumeurs présentant une expression de PD-L1 ≥50% sur les TC était de 26%.
Le critère d'évaluation principal de l'efficacité était la survie sans maladie (DFS) selon l'évaluation du médecin investigateur. La DFS était définie comme le temps écoulé entre la randomisation et la survenue de l'un des événements suivants: première récidive documentée de la maladie, nouveau NSCLC primaire ou décès toutes causes confondues, selon ce qui survenait en premier. L'objectif principal en lien avec l'efficacité consistait à évaluer la DFS dans la population de patients présentant une expression de PD-L1 ≥1% sur les TC (sur la base du test SP263) et une maladie de stade II à IIIA. Des critères d'évaluation secondaires en lien avec l'efficacité étaient l'évaluation de la DFS dans la population de patients présentant une expression de PD-L1 ≥50% sur les TC (sur la base du test SP263) et une maladie de stade II à IIIA, ainsi que la survie globale (OS) dans la population en intention de traiter (ITT).
Chez les patients présentant une expression de PD-L1 ≥50% sur les TC et une maladie de stade II à IIIA (n = 229) une amélioration cliniquement pertinente de la DFS a été démontrée, avec un HR non stratifié de 0,43 (IC à 95%: 0,27, 0,68). La DFS médiane n'a pas été atteinte chez les patients du bras sous Tecentriq (IC à 95%: 42,3 mois, NE) et a été de 35,7 mois chez les patients du bras recevant le meileur traitement de soutien (IC à 95%: 29,7, NE). Les données d'OS n'étaient pas matures au moment de l'analyse intermédiaire de la DFS, avec une mortalité d'environ 16,2% dans les deux bras rapportée dans la population présentant une expression de PD-L1 ≥50% sur les TC et une maladie de stade II à IIIA. Une analyse exploratoire de l'OS a révélé une tendance en faveur du Tecentriq par rapport au meilleur traitement de soutien dans cette population de patients (HR non stratifé = 0,37; [IC à 95%: 0,18, 0,74]).
NSCLC prétraité par chimiothérapie
OAK
GO28915 (OAK), une étude de phase III ouverte, multicentrique, internationale et randomisée, a été réalisée pour évaluer l'efficacité et la sécurité de Tecentriq par rapport au docétaxel chez des patients atteints de NSCLC localement avancé ou métastatique, dont la maladie a progressé pendant ou après une chimiothérapie à base de platine. Au total, 1225 patients ont été inclus dans cette étude, parmi lesquels les 850 premiers patients randomisés ont été inclus dans l'analyse primaire de l'efficacité; l'analyse secondaire de l'efficacité a reposé sur 1225 patients. Les patients éligibles ont été stratifiés selon le statut d'expression du PD-L1 des IC infiltrant les tumeurs, le nombre de protocoles de chimiothérapie antérieurs et le type histologique. Les patients ont été randomisés (1:1) pour recevoir soit Tecentriq soit le docétaxel. Les patients présentant des antécédents connus de maladie auto-immune ou des métastases cérébrales évolutives ou corticodépendantes, ainsi que les patients ayant utilisé des vaccins vivants atténués dans les 28 jours précédant l'inclusion dans l'étude ou des principes actifs immunostimulants systémiques dans les 4 semaines précédant l'inclusion dans l'étude ou des médicaments immunosuppresseurs systémiques dans les 2 semaines précédant l'inclusion dans l'étude ont été exclus de l'étude. Les évaluations de la tumeur ont été réalisées toutes les 6 semaines durant les 36 premières semaines, puis toutes les 9 semaines. Une évaluation prospective de l'expression du PD-L1 sur les TC et les IC a été réalisée dans des échantillons de tissus tumoraux; les résultats ont été utilisés pour définir les sous-groupes d'expression du PD-L1 dans les analyses décrites ci-dessous.
Les données démographiques et les caractéristiques de la maladie au début du traitement des 850 premiers patients randomisés inclus dans l'analyse primaire de l'efficacité étaient bien équilibrées dans les deux bras thérapeutiques. L'âge médian était de 64 ans (intervalle: de 33 à 85); 61% des patients étaient de sexe masculin. La majorité des patients étaient des Blancs (70%). Presque les trois quarts des patients (74%) présentaient un carcinome d'histologie non épidermoïde, 10% présentaient une mutation de l'EGFR connue, 0,2% avait une translocation de l'ALK connue, 10% avaient des métastases dans le SNC au début du traitement et la plupart des patients étaient des fumeurs ou d'anciens fumeurs (82%). Le score de performance ECOG au début du traitement était de 0 (37%) ou de 1 (63%). 75% des patients n'avaient reçu auparavant qu'un seul protocole thérapeutique à base de platine. Les données démographiques et les caractéristiques de la maladie observées dans le cadre de l'analyse secondaire de l'efficacité sont en accord avec les observations réalisées lors de l'analyse primaire de l'efficacité.
Tecentriq a été administré en perfusion i.v. à la dose fixe de 1200 mg toutes les 3 semaines. Des réductions de la dose n'étaient pas autorisées. Les patients ont été traités jusqu'à la perte du bénéfice clinique selon l'évaluation du médecin investigateur. Le docétaxel a été administré en perfusion i.v. à la dose de 75 mg/m2 au jour 1 de chaque cycle de 21 jours, jusqu'à la progression de la maladie. Rapporté à l'ensemble des patients traités, la durée médiane de traitement a été de 2,1 mois dans le bras sous le docétaxel et de 3,4 mois dans le bras sous Tecentriq lors de l'analyse primaire de l'efficacité.
Le critère d'évaluation principal de l'efficacité était la survie globale (overall survival, OS). Les principaux résultats de cette étude, qui ont été rapportés dans le cadre de l'analyse primaire de l'efficacité effectuée chez les 850 premiers patients randomisés avec un suivi médian de la survie de 21 mois, sont résumés ci-dessous (date limite de collecte des données: 07.07.2016). Le traitement par Tecentriq a été associé à un allongement cliniquement et statistiquement significatif de l'OS par rapport au docétaxel. Dans le bras sous Tecentriq, l'OS médiane a été de 4,2 mois supérieure chez les 850 premiers patients randomisés de la population en ITT (intention to treat): elle était de 9,6 mois (IC à 95%: 8,6; 11,2) dans le bras sous le docétaxel contre 13,8 mois (IC à 95%: 11,8; 15,7) dans le bras sous Tecentriq (valeur de p stratifiée = 0,0003). Le hazard ratio (HR) a été de 0,73 (IC à 95%: 0,62; 0,87); cela correspond à une réduction relative du risque de mortalité de 27% sous Tecentriq par rapport au groupe sous le docétaxel (analyse stratifiée). Les courbes de Kaplan-Meier ont montré à partir de 3 mois env. une séparation claire en faveur du bras sous Tecentriq, laquelle s'est maintenue par la suite. De la même façon, dans le sous-groupe de patients avec une expression du PD-L1 ≥1% sur les TC ou les IC, le traitement par Tecentriq a également été associé à un allongement cliniquement et statistiquement significatif de l'OS par rapport au docétaxel. Dans le bras sous Tecentriq, l'OS médiane des patients a été de 5,4 mois supérieure: elle était de 10,3 mois (IC à 95%: 8,8; 12,0) dans le bras sous le docétaxel contre 15,7 mois (IC à 95%: 12,6; 18,0) dans le bras sous Tecentriq (valeur de p stratifiée = 0,0102). L'HR a été de 0,74 (IC à 95%: 0,58; 0,93); cela correspond à une réduction relative du risque de mortalité de 26% sous Tecentriq par rapport au groupe sous le docétaxel (analyse stratifiée).
L'OS dans les sous-groupes de patients avec une expression du PD-L1 ≥50% sur les TC ou ≥10% sur les IC, une expression du PD-L1 ≥5% sur les TC ou les IC, ou encore une expression du PD-L1< 1% sur les TC et les IC a montré que la durée médiane de l'OS était plus longue dans tous les sous-groupes d'expression du PD-L1 évalués. Les valeurs ont été les suivantes: expression du PD-L1 ≥50% sur les TC ou ≥10% sur les IC: OS médiane de 20,5 mois (IC à 95%: 17,5; NA) dans le bras sous Tecentriq contre 8,9 mois (IC à 95%: 5,6; 11,6) dans le bras sous le docétaxel (HR=0,41, IC à 95%: 0,27; 0,64); expression du PD-L1 ≥5% sur les TC ou les IC: OS médiane de 16,3 mois (IC à 95%: 13,3; 20,1) dans le bras sous Tecentriq contre 10,8 mois (IC à 95%: 8,8; 12,7) dans le bras sous le docétaxel (HR=0,67, IC à 95%: 0,49; 0,90); expression du PD-L1< 1% sur les TC et les IC: OS médiane de 12,6 mois (IC à 95%: 9,6; 15,2) dans le bras sous Tecentriq contre 8,9 mois (IC à 95%: 7,7; 11,5) dans le bras sous le docétaxel (HR=0,75, IC à 95%: 0,59; 0,96).
Dans l'ensemble, les résultats concernant la survie globale observés dans le cadre de l'analyse primaire de l'efficacité (850 patients) dans le groupe en ITT et dans les sous-groupes d'expression du PD-L1 ont montré sous Tecentriq un bénéfice en termes d'OS dans tous les sous-groupes, y compris dans celui avec une expression du PD-L1< 1% sur les TC et les IC.
La proportion des patients en ITT présentant une réponse confirmée évaluée par le médecin investigateur selon les critères RECIST V1.1 était comparable dans les deux bras: 13,4% (IC à 95%: 10,32; 17,02) dans le bras sous le docétaxel et 13,6% (IC à 95%: 10,53; 17,28) dans le bras sous Tecentriq. Le traitement par Tecentriq a été associé à l'obtention d'une réponse durable. Chez les répondeurs, la DOR médiane a été significativement plus longue (16,3 mois) dans le bras sous Tecentriq que dans le bras sous le docétaxel (6,2 mois). Dans le groupe en ITT, la survie sans progression (PFS) médiane a été de 2,8 mois (IC à 95%: 2,6; 3,0) dans le bras sous Tecentriq et de 4,0 mois (IC à 95%: 3,3; 4,2) dans le bras sous le docétaxel pour un HR de 0,95 (IC à 95% 0,82; 1,10).
Une amélioration de l'OS sous Tecentriq par rapport au docétaxel a été observée non seulement chez les patients atteints d'un NSCLC non épidermoïde) (HR de 0,73; IC à 95%: 0,60-0,89; OS médiane de 15,6 mois sous Tecentriq contre 11,2 mois sous le docétaxel), mais également chez les patients atteints d'un NSCLC épidermoïde (HR de 0,73; IC à 95%: 0,54-0,98; OS médiane de 8,9 mois sous Tecentriq contre 7,7 mois sous le docétaxel).
Dans le cadre de l'analyse secondaire de l'efficacité (date limite de collecte des données: 23 janvier 2017) avec une durée médiane de suivi de 26 mois, le traitement par Tecentriq était toujours associé à un allongement statistiquement significatif de l'OS par rapport au docétaxel. L'OS médiane de l'ensemble des 1125 patients randomisés a été supérieure de 3,5 mois dans le bras sous Tecentriq: 13,3 mois (IC à 95%: 11,3; 14,9) dans le bras sous Tecentriq et 9,8 mois (IC à 95%: 8,8; 11,3) dans le bras sous le docétaxel. Le HR stratifié a été de 0,80 (IC à 95%: 0,70; 0,92).
POPLAR
GO28753 (POPLAR), une étude de phase II multicentrique, internationale, randomisée, ouverte et contrôlée, a été réalisée chez des patients atteints de NSCLC localement avancé ou métastatique avec progression de la maladie pendant ou après une chimiothérapie incluant du platine. La variable d'efficacité principale était la survie globale. Au total, 287 patients ont été randomisés selon un rapport 1:1 pour recevoir soit Tecentriq soit le docétaxel. La randomisation a été stratifiée en fonction de l'expression du PD-L1, du nombre de protocoles de chimiothérapie antérieurs et du type histologique. Une analyse actualisée comprenant au total 200 décès recensés et un suivi médian de la survie de 22 mois a montré une OS médiane de 12,6 mois chez les patients traités par Tecentriq contre 9,7 mois chez les patients traités par le docétaxel (HR de 0,69; IC à 95%: 0,52-0,92). L'ORR a été de 15,3% sous Tecentriq contre 14,7% sous le docétaxel; la DOR médiane a été de 18,6 mois sous Tecentriq contre 7,2 mois sous le docétaxel.
Traitement de première intention du cancer du poumon à petites cellules de stade avancé (1L ES – SCLC)
IMpower133
L'étude de phase I/III GO30081 (IMpower133) contrôlée contre placebo, multicentrique, en double aveugle et randomisée a été réalisée pour évaluer l'efficacité et la sécurité de Tecentriq en association au carboplatine et à l'étoposide chez des patients atteints de cancer du poumon à petites cellules de stade avancé (ES-SCLC) sans chimiothérapie préalable et ayant un score de performance ECOG de 0 ou 1. Un total de 403 patients ont été randomisés (1:1) pour recevoir l'un des schémas thérapeutiques suivants: bras A: Tecentriq 1200 mg + carboplatine AUC 5 + étoposide 100 mg/m2 par perfusion intraveineuse toutes les 3 semaines pendant quatre cycles d'induction, suivi d'un traitement d'entretien par Tecentriq 1200 mg toutes les 3 semaines; ou bras B: placebo + carboplatine AUC 5 + étoposide 100 mg/m2 par perfusion intraveineuse toutes les 3 semaines pendant quatre cycles d'induction, suivis d'un traitement d'entretien par le placebo toutes les 3 semaines. Tecentriq et le carboplatine ont été administrés au jour 1 et l'étoposide aux jours 1, 2 et 3 de chaque cycle d'induction. Le carboplatine et l'étoposide ont été administrés jusqu'à la fin des quatre cycles ou jusqu'à la progression de la maladie ou jusqu'à l'apparition d'une toxicité inacceptable, selon ce qui survenait en premier. Tecentriq a été administré jusqu'à ce que, de l'avis du médecin investigateur, il n'y ait plus de bénéfice clinique. La randomisation a été stratifiée selon le sexe, le score de performance ECOG (0 ou 1) et la présence de métastases cérébrales.
Les patients présentant des métastases actives ou non traitées dans le SNC, des antécédents de maladie auto-immune, ayant reçu un vaccin vivant atténué dans les 4 semaines précédant la randomisation ou ayant reçu des médicaments immunosuppresseurs systémiques dans la semaine précédant la randomisation ont été exclus de l'étude. Les évaluations de la tumeur ont été réalisées toutes les 6 semaines durant les 48 premières semaines suivant le cycle 1 au jour 1, puis toutes les 9 semaines. Chez les patients qui ont poursuivi le traitement après la progression de la maladie, une évaluation de la tumeur a été réalisée toutes les 6 semaines jusqu'à l'arrêt du traitement.
Les données démographiques et les caractéristiques de la maladie dans la population de l'analyse primaire étaient bien équilibrées entre les bras thérapeutiques. Au total, 403 patients ont été randomisés dans l'étude: 201 patients dans le bras A recevant Tecentriq, le carboplatine et l'étoposide, et 202 patients dans le bras B recevant le placebo, le carboplatine et l'étoposide. L'âge médian était de 64 ans (intervalle: de 26 à 90 ans). La majorité des patients (65%) étaient des hommes et de race blanche (80%), 9% présentaient des métastases cérébrales et la plupart des patients (97%) étaient des fumeurs ou d'anciens fumeurs. La valeur initiale (ligne de base) du score de performance ECOG était de 0 (35%) ou de 1 (65%).
Les deux critères d'évaluation principaux étaient la survie globale (OS: overall survival) et la survie sans progression (PFS: progression-free survival) évaluée par le médecin investigateur. Les principaux résultats de cette étude lors d'un suivi médian de 13,9 mois de la durée de vie restante sont résumés ci-dessous.
Le traitement par Tecentriq + carboplatine et étoposide (CE) a été associé à un allongement statistiquement significatif de l'OS par rapport au placebo + CE. Pour les patients de la population en ITT (intention de traiter), l'OS médiane était 2 mois plus longue dans le bras de traitement sous Tecentriq + CE, où elle était de 12,3 mois (IC à 95%: 10,8; 15,9), contre 10,3 mois (IC à 95%: 9,3; 11,3) dans le bras de traitement sous placebo + CE (valeur de p stratifiée = 0,0069). Le hazard ratio (HR) stratifié était de 0,70 (IC à 95%: 0,54; 0,91); cela correspond à une réduction relative du risque de mortalité de 30% sous Tecentriq + CE par rapport au placebo + CE. Après 12 mois, le bras de traitement sous Tecentriq + CE comptait encore 51,7% patients en vie, contre 38,2% dans le bras de traitement sous placebo + CE. Chez les patients de moins de 65 ans, la valeur médiane de l'OS dans le bras sous Tecentriq + CE était de 12,1 mois, contre 11,5 mois dans le groupe sous placebo + CE (HR non stratifié: 0,92, IC à 95%: 0,64; 1,32). Chez les patients âgés de 65 ans et plus, la valeur médiane de l'OS dans le bras sous Tecentriq + CE était de 12,5 mois, contre 9,6 mois dans le groupe sous placebo + CE (HR non stratifié: 0,53, IC à 95%: 0,36; 0,77).
Les données des analyses exploratoires des sous-groupes d'expression du PD-L1, basées sur un nombre limité d'échantillons de patients dans cette étude (34% de la population en intention de traiter (ITT)), ont montré pour les patients PD-L1-négatifs (<1% sur les TC et les IC) un OS HR (hazard ratio pour la survie globale) de 0,51 (IC à 95%: 0,30; 0,89) avec une survie globale moyenne de 10,2 mois dans le bras Tecentriq + CE versus 8,3 mois dans le bras placebo + CE. Pour les patients PD-L1-positifs (≥1% sur les TC ou les IC), l'OS HR était de 0,87 (IC à 95%: 0,51; 1,49) avec une survie globale moyenne de 9,7 mois dans le bras Tecentriq + CE versus 10,6 mois dans le bras placebo + CE. Ces données sont cependant trop limitées pour pouvoir tirer des conclusions. L'incidence des anticorps anti-atézolizumab (ADA) survenus au cours du traitement a été de 18,6% (35 patients sur 188) dans le bras Tecentriq + CE. Dans une analyse sans ajustement l'OS médiane a été de 12,6 mois dans le groupe ADA-négatif et de 10,9 mois dans le groupe ADA-positif. Dans le groupe placebo + CE, l'OS a été de 10,3 mois. Étant donné le faible nombre de cas dans le groupe ADA-positif, aucune conclusion définitive ne peut être tirée concernant une éventuelle influence des ADA sur l'efficacité.
Le traitement par Tecentriq + CE a été associé à une amélioration statistiquement significative de la PFS évaluée par le médecin investigateur (selon RECIST v1.1) par rapport au placebo + CE. Chez les patients du collectif en ITT, la durée médiane de la PFS était de 5,2 mois (IC à 95%: 4,4; 5,6) dans le bras de traitement sous Tecentriq + CE, contre 4,3 mois (IC à 95%: 4,2; 4,5) dans le bras de traitement sous placebo + CE (valeur de p stratifiée = 0,0170). Le HR stratifié était de 0,77 (IC à 95%: 0,62; 0,96). Après 6 mois, la proportion de patients sans événements liés à la PFS (progression ou décès) était de 30,9% dans le bras de traitement sous Tecentriq + CE et de 22,4% dans le bras de traitement sous placebo + CE. Après 12 mois, la proportion de patients sans événements liés à la PFS était de 12,6% dans le bras de traitement sous Tecentriq + CE et de 5,4% dans le bras de traitement sous placebo + CE.
Dans le collectif en ITT, la proportion de patients dont la réponse objective a été confirmée par l'évaluation du médecin investigateur selon RECIST v1.1 était de 60,2% dans le bras de traitement sous Tecentriq + CE, contre 64,4% dans le groupe de traitement sous placebo + CE.
Traitement de première ligne du cancer du sein triplement négatif (1L TNBC)
IMpassion130
L'étude de phase III randomisée, à 2 bras, menée en double aveugle et contrôlée contre placebo WO29522 (IMpassion130) a été réalisée pour évaluer l'efficacité et la sécurité de Tecentriq en association avec le nab-paclitaxel chez des patients atteints de cancer du sein triplement négatif (TNBC) non résécable, localement avancé ou métastatique, n'ayant reçu au préalable aucune chimiothérapie ni aucun traitement systémique ciblé en raison d'un TNBC inopérable, localement avancé ou métastatique. Au total, 902 patients ont été inclus dans l'étude et le collectif a été stratifié selon la présence de métastases hépatiques, un traitement préalable à base de taxane et le statut de l'expression du PD-L1 dans les cellules immunitaires (CI) infiltrant la tumeur (cellules immunitaires [CI] infiltrant la tumeur avec coloration de PD-L1< 1% de la surface de la tumeur contre ≥1% de la surface de la tumeur), évalué à l'aide du test VENTANA PD-L1 (SP142).
Les patients ont été randomisés pour recevoir soit Tecentriq (840 mg), soit un placebo en perfusions i.v. aux jours 1 et 15 plus nab-paclitaxel (100 mg/m2) sous forme de perfusion i.v. aux jours 1, 8 et 15 de chaque cycle de 28 jours. Les patients ont reçu le traitement jusqu'à l'apparition d'une progression tumorale radiologique, selon RECIST version 1.1, ou jusqu'à la survenue d'une toxicité intolérable. Le traitement par Tecentriq a été poursuivi en cas d'interruption du nab-paclitaxel pour cause de toxicité intolérable.
Étaient notamment exclus de l'étude les patients présentant des antécédents de maladie auto-immune; les patients ayant reçu un vaccin vivant atténué au cours des 4 semaines précédant la randomisation; les patients ayant reçu des substances actives immunostimulatrices systémiques dans les 4 semaines ou des médicaments immunosuppresseurs systémiques dans les 2 semaines précédant la randomisation; les patients présentant des métastases cérébrales non traitées ou dépendantes des corticostéroïdes; les patients ayant des antécédents de transplantation de cellules souches allogéniques ou d'organe solide; les patients atteints de fibrose pulmonaire idiopathique active ou anamnestique et de pneumopathie inflammatoire (à l'exception d'une pneumopathie inflammatoire actinique dans le champ d'irradiation) et de pneumonie organisante; ou encore les patients atteints d'infections actives, telles que VIH, hépatite B et C ainsi que tuberculose. Une évaluation de la tumeur a été effectuée toutes les 8 semaines durant les 12 premiers mois, puis toutes les 12 semaines.
Au début de l'étude, la population de l'étude était comparable en termes de données démographiques et de caractéristiques de la maladie dans les deux bras de l'étude. La grande majorité des patients (99,6%) était de sexe féminin. 67,5% des patients étaient de race caucasienne, 17,8% étaient d'origine asiatique, 6,5% étaient noirs ou afroaméricains et 4,4% étaient des indiens ou des autochtones de l'Alaska. L'âge médian était de 55 ans (intervalle: de 20 à 86 ans). La valeur initiale du statut de performance ECOG était 0 (58,4%) ou 1 (41,3%). Dans l'ensemble, lors de l'inclusion dans l'étude, 41% des patients inclus présentaient une expression de PD-L1 ≥1%, 27% présentaient des métastases hépatiques et 7% avaient des métastases cérébrales. Près de la moitié des patients était sous taxane (51%) ou anthracycline (54%) en guise de traitement (néo-)adjuvant. Les données démographiques des patients et la maladie tumorale lors de l'inclusion dans l'étude de la population avec expression du PD-L1 ≥1% étaient en général représentatives de la population totale de l'étude.
Les résultats de la PFS (survie sans progression), de l'ORR (taux de réponse objective) et de la DOR (durée de réponse) issus de cette étude sont résumés dans le tableau 7 et dans le texte ci-dessous et ils concernent les patients présentant une expression du PD-L1 ≥1% avec une durée médiane de suivi des patients survivants de 13 mois.
Une analyse finale de l'OS avec une expression de PD-L1 ≥1% avec une période d'observation médiane de 19,12 mois a été réalisée. Les résultats de l'OS sont présentés dans le tableau 5.
Aucun bénéfice en termes d'OS n'a été observé sous traitement combiné par l'atézolizumab + nab-paclitaxel par rapport au placebo + nab-paclitaxel chez les patients pour lesquels ≥24 mois avaient passé entre la dernière opération et le diagnostic de maladie tumorale métastatique ou localement avancée inopérable.
Tableau 7: Résumé des données de l'efficacité chez les patients présentant une expression du PD-L1 ≥1% (IMpassion130)
|
Critères d'évaluation de l'efficacité importants
|
Tecentriq + nab-paclitaxel
|
Placebo + nab-paclitaxel
| |
Critères co-primaires d'évaluation
| |
Survie sans progression (PFS) selon l'appréciation du médecin investigateur (RECIST vers. 1.1 – Analyse primaire3)
|
n = 185
|
n = 184
| |
Nombre d'événements (%)
|
138 (74,6%)
|
57 (85,3%)
| |
Durée médiane de la survie sans progression (en mois)
|
7,5
|
5,0
| |
IC à 95%
|
(6,7; 9,2)
|
(3,8; 5,6)
| |
Hazard-Ratio stratifié ‡ (IC à 95%)
|
0,62 (0,49; 0,78)
| |
Valeur de p1
|
< 0,0001
| |
Survie sans progression de 12 mois (%)
|
29,1
|
16,4
| |
Survie sans progression (PFS) selon l'appréciation du médecin investigateur (RECIST vers. 1.1) – Analyse exploratoire actualisée 4
|
|
| |
Nombre d'événements (%)
|
149 (80,5%)
|
163 (88,6%)
| |
Durée médiane de la survie sans progression (en mois)
|
7,5
|
5,3
| |
IC à 95%
|
(6,7; 9,2)
|
(3,8; 5,6)
| |
Hazard-Ratio stratifié ‡ (IC à 95%)
|
0,63 (0,50-0,80)
| |
Survie sans progression de 12 mois (%)
|
30,3
|
17,3
| |
Analyse de la survie globale1, 2, 5
|
n = 185
|
n = 184
| |
Nombre de décès (%)
|
120 (64,9%)
|
139 (75,5%)
| |
Délai moyen jusqu'à l'événement (en mois)
|
25,4
|
17,9
| |
IC à 95%
|
(19,6; 30,7)
|
(13,6; 20,3)
| |
Hazard-Ratio ‡ stratifié (IC à 95%)
|
0,67 (0,53; 0,86)
|
1 Sur la base du test de log-rank stratifié
2 La comparaison des survies globales entre les groupes de traitement de patients présentant une expression du PD-L1 ≥1% n'a pas été testée formellement selon la hiérarchie des analyses spécifiée à l'avance
3 D'après l'analyse finale de la PFS, de l'ORR et de la DOR et une première analyse intermédiaire de l'OS au moment du cut-off clinique du 17 avril 2018
4 D'après une analyse exploratoire de la PFS au moment du cut-off clinique du 2 janvier 2019
5 D'après l'analyse finale de l'OS au moment du cut-off clinique du 14 avril 2020
‡ Stratifié en fonction de la présence de métastases hépatiques et d'un traitement antérieur par taxane
PFS = survie sans progression (progression-free survival); RECIST = Response Evaluation Criteria in Solid Tumors v1.1. (critères d'évaluation de la réponse pour les tumeurs solides, version 1.1); IC = intervalle de confiance; OS = survie globale (overall survival), NE = non appréciable (not estimable)
Chez les patients présentant une expression positive du PD-L1, un taux de réponse globale (Overall Response Rate, ORR) numériquement plus élevé a été observé dans le groupe sous Tecentriq + nab-paclitaxel (58,9%) par rapport au groupe sous placebo + nab-paclitaxel (42,6%). Parmi les répondeurs, le nombre de réponses persistantes dans le bras sous Tecentriq + nab-paclitaxel était supérieur (35,8%) à celui observé dans le bras sous placebo + nab-paclitaxel (24,4%). La durée de réponse (Duration of Response, DOR) médiane estimée pour le groupe sous Tecentriq + nab-paclitaxel était supérieure de 3 mois (8,5 mois contre 5,5 mois dans le groupe sous placebo + nab-paclitaxel, HR non stratifié: 0,60; IC à 95%; 0,43; 0,86).
Mélanome
IMspire150
CO39262 (IMspire150), une étude de phase III randomisée, en double aveugle, à deux bras, contrôlée contre placebo, a été menée afin d'évaluer l'efficacité et la sécurité de Tecentriq en association avec du cobimétinib et du vémurafénib chez des patients atteints d'un mélanome localement avancé, métastatique ou non résécable, porteur de la mutation BRAF-V600. La mutation BRAF V600 du mélanome a été détectée à l'aide d'un test disponible localement et a été confirmée de manière centralisée par le test FoundationOneTM. Les patients n'avaient pas reçu auparavant de traitement systémique pour leur mélanome. Au total, 514 patients ont été randomisés pour recevoir l'un des régimes de traitement décrits ci-dessous. La randomisation a été stratifiée en fonction de la localisation géographique et du taux initial de lactate déshydrogénase (LDH).
Le traitement a été effectué par cycles de 28 jours. Au cours du cycle 1, tous les patients ont reçu 960 mg de vémurafénib par voie orale deux fois par jour et 60 mg de cobimétinib par voie orale une fois par jour pendant 21 jours, puis soit 720 mg de vémurafénib + placebo deux fois par jour pendant 7 jours dans le groupe Tecentriq, soit 960 mg dans le groupe témoin. À partir du cycle 2, les patients du groupe Tecentriq ont reçu 840 mg de Tecentriq par voie intraveineuse aux jours 1 et 15 ainsi que 720 mg de vémurafénib + placebo deux fois par jour (pendant 28 jours) et 60 mg de cobimétinib une fois par jour (prise de médicament pendant 21 jours, suivi d'une pause de 7 jours). Les patients du groupe témoin ont reçu un placebo par voie intraveineuse aux jours 1 et 15 ainsi que 960 mg de vémurafénib deux fois par jour et du cobimétinib une fois par jour pendant 21 jours, suivi d'une pause de 7 jours. Des réductions de dose et des interruptions de traitement ont été autorisées pour le vémurafénib et le cobimétinib, tandis que, pour Tecentriq, seules les interruptions de traitement pour gérer des événements indésirables ont été autorisées. Le traitement a été poursuivi jusqu'à ce que la progression de la maladie, la mort ou une toxicité inacceptable aient été établies par le médecin investigateur. Le croisement n'était pas autorisé en cas de progression. Les patients n'ont pas été inclus dans l'étude s'ils présentaient des antécédents de maladie auto-immune, s'ils avaient reçu un vaccin vivant atténué dans les 28 jours précédant la randomisation, s'ils avaient reçu un traitement immunostimulant systémique dans les 4 semaines ou un traitement immunosuppresseur systémique dans les 2 semaines précédant la randomisation. Les évaluations tumorales ont été réalisées toutes les 8 semaines (± 1 semaine) pendant les 24 premiers mois et toutes les 12 semaines (± 1 semaine) par la suite.
Les données démographiques et les caractéristiques de la maladie à l'inclusion de l'étude de la population étudiée étaient équilibrées entre les deux bras de traitement. L'âge médian s'élevait à 54 ans (fourchette: de 22 à 88 ans), 58% des patients étaient de sexe masculin. La majorité des patients étaient de type caucasien (95%). 2,5% des patients avaient déjà reçu un traitement pour des métastases cérébrales, 30% des patients avaient des métastases hépatiques au moment de leur inclusion dans l'étude et 14% des patients avaient reçu un traitement adjuvant systémique. Le mélanome a été classifié, selon la classification de l'American Joint Committee on Cancer (AJCC), septième édition, comme étant de stade IIIC chez 5,8% des patients et de stade IV chez 93,8% des patients (M1A: 14,8; M1B: 19,1%; M1C: 59,9%).
Au début de l'étude, le statut de performance ECOG était de 0 (76,5%) ou de 1 (22,8%). La proportion de patients présentant une LDH élevée était de 32,8% dans le groupe de traitement avec Tecentriq et de 32,9% dans le groupe de traitement sans Tecentriq.
D'après les tests effectués de manière centralisée, 74% des patients présentaient la mutation V600E, 11% la mutation V600K et 1% la mutation V600D ou V600R.
Le critère d'évaluation principal de l'efficacité était la survie sans progression (PFS), évaluée par le médecin investigateur. À la fin de la période de collecte des données (analyse primaire), la période d'observation médiane était de 18,9 mois.
Dans le sous-groupe Tecentriq V600E, la PFS médiane était de 16,2 mois selon l'évaluation du médecin investigateur contre 10,0 mois dans le sous-groupe témoin V600E (Hazard Ratio non stratifié: 0,68; IC à 95%: 0,53; 0,88). Les critères secondaires comprenaient la PFS médiane, évaluée par une commission d'examen indépendante (IRC); elle était de 17,6 mois dans le sous-groupe Tecentriq V600E et de 11,4 mois dans le sous-groupe témoin V600E (Hazard Ratio non stratifié: 0,71; IC à 95%: 0,55; 0,93). Une analyse intermédiaire de la survie globale (OS) a montré un rapport de risque non stratifié de 0,69 (IC à 95%: 0,50; 0,95). Les taux de réponse objective (évalués par le médecin investigateur) étaient similaires dans les deux groupes; ils étaient de 68,9% dans le sous-groupe Tecentriq V600E et de 63,2% dans le sous-groupe témoin V600E. La durée médiane de la réponse (évaluée par le médecin investigateur) était de 20,4 mois dans le sous-groupe Tecentriq V600E contre 12,6 mois dans le sous-groupe témoin V600E.
Tecentriq en association avec du vémurafénib et du cobimétinib n'a pas été étudié chez les patients atteints d'un mélanome exprimant un BRAF de type sauvage.
CHC
IMbrave150
Une étude internationale de phase III, randomisée, multicentrique, en ouvert, YO40245 (IMbrave150), a été réalisée pour évaluer l'efficacité et la sécurité de Tecentriq en association avec le bévacizumab chez des patients atteints de CHC localement avancé ou métastatique et/ou inopérable n'ayant pas reçu de traitement systémique antérieur. Au total, 501 patients ont été randomisés (rapport 2:1) pour recevoir 1200 mg de Tecentriq et 15 mg/kg de bévacizumab toutes les trois semaines en perfusion intraveineuse ou 400 mg de sorafénib deux fois par jour par voie orale. La randomisation a été stratifiée en fonction de la région géographique (Asie, à l'exclusion du Japon vs reste du monde), de l'invasion macrovasculaire et/ou de l'extension extrahépatique (présente vs absente), du taux initial d'AFP (< 400 vs ≥400 ng/ml) et de l'indice de performance ECOG (0 vs 1). Dans les deux bras, les patients ont reçu le traitement jusqu'à la perte du bénéfice clinique ou la survenue d'une toxicité inacceptable. Les patients pouvaient arrêter Tecentriq ou le bévacizumab (p.ex. en raison d'effets indésirables) et poursuivre par une monothérapie jusqu'à la perte du bénéfice clinique ou la survenue d'une toxicité inacceptable associée à la monothérapie.
Les patients inclus dans l'étude étaient des adultes (Child-Pugh A, ECOG 0/1) n'ayant pas reçu de traitement systémique antérieur. Les hémorragies (dont des événements d'issue fatale) sont un effet indésirable connu du bévacizumab et les hémorragies gastro-intestinales hautes sont une complication fréquente engageant le pronostic vital chez les patients atteints d'un CHC. Par conséquent, la présence éventuelle de varices devait avoir été évaluée chez les patients dans les 6 mois précédant le traitement. Les critères d'exclusion de l'étude étaient un saignement variqueux dans les 6 mois précédant le traitement, des varices non traitées ou partiellement traitées avec saignement ou risque élevé de saignement. Les critères d'exclusion étaient un saignement variqueux dans les 6 mois précédant le traitement, des varices non traitées, une ascite modérée à sévère, des antécédents d'encéphalopathie hépatique, une anamnèse connue de maladie auto-immune, l'administration d'un vaccin vivant atténué ou d'immunostimulants systémiques dans les 4 semaines précédant la randomisation ou l'administration d'immunosuppresseurs systémiques dans les 2 semaines précédant la randomisation ainsi que des métastases cérébrales non traitées ou corticodépendantes. Les évaluations tumorales ont été effectuées toutes les 6 semaines durant les 54 premières semaines, puis toutes les 9 semaines.
Les caractéristiques démographiques et pathologiques à l'inclusion dans la population de l'étude étaient bien équilibrées entre les deux bras de traitement. L'âge médian était de 65 ans (intervalle: de 26 à 88 ans) et 83% des patients étaient de sexe masculin. La plupart des patients étaient Asiatiques (57%) et Blancs (35%). 40% des patients étaient originaires d'Asie (à l'exclusion du Japon) et 60% provenaient du reste du monde. Au départ, environ 75% des patients présentaient une invasion macrovasculaire et/ou une extension extrahépatique, et 37% avaient un taux initial d'AFP ≥400 ng/ml. L'indice de performance ECOG au début de l'étude était de 0 (62%) ou de 1 (38%). Les facteurs de risque primaires de développement d'un CHC étaient une infection par le virus de l'hépatite B chez 48% des patients, une infection par le virus de l'hépatite C chez 22% des patients et une affection non virale chez 31% des patients. 82% des patients avaient un CHC de stade C, 16% un CHC de stade B et 3% un CHC de stade A selon la classification BCLC (Barcelona Clinic Liver Cancer).
Les deux critères co-primaires d'évaluation de l'efficacité étaient la survie globale (OS) et la survie sans progression (PFS) évaluée par un centre d'examen indépendant (IRF) selon les critères RECIST v1.1. Au moment de l'analyse primaire, la durée médiane de suivi était de 8,6 mois. Dans le groupe sous Tecentriq + bévacizumab, une amélioration statistiquement significative des deux critères co-primaires d'évaluation de l'efficacité, a été obtenue dans la population en ITT par rapport au sorafénib. L'OS médiane n'a pas été atteinte (NR) dans le bras traité par l'association Tecentriq + bévacizumab (IC à 95%: NR; NR) et était de 13,2 mois dans le bras sous sorafénib (IC à 95%: 10,4; NR). Le HR stratifié était de 0,58 (IC à 95%: 0,42; 0,79; valeur de p stratifiée = 0,0006), ce qui correspond à une diminution de 42% du risque de mortalité lié à l'association Tecentriq + bévacizumab par rapport au sorafénib.
La PFS médiane était de 6,8 mois dans le bras traité par Tecentriq + bévacizumab (IC à 95%: 5,8; 8,3) et de 4,3 mois dans le bras sous sorafénib (IC à 95%: 4,0; 5,6). L'HR stratifié était de 0,59 (IC à 95%: 0,47; 0,76; valeur de p stratifiée < 0,0001), ce qui correspond à une diminution de 41% du risque de progression de la maladie ou de mortalité lié à l'association Tecentriq + bévacizumab par rapport au sorafénib.
Une amélioration statistiquement significative de l'ORR évaluée par l'IRF selon les critères RECIST v1.1, de 27,3% (IC à 95%: 22,5%, 32,5%), a été observée dans le bras traité par Tecentriq + bévacizumab contre 11,9% (IC à 95%: 7,4%, 18,0%) dans le bras sous sorafénib. Le taux de patients avec une réponse complète était de 5,5% sous Tecentriq + bévacizumab contre 0% dans le bras sous sorafénib.
Une analyse descriptive et actualisée de l'efficacité a été réalisée sur une période médiane de suivi de la survie de 15,6 mois. L'OS médiane était de 19,2 mois (IC à 95%: 17,0; 23,7) dans le bras traité par l'association Tecentriq + bévacizumab et de 13,4 mois (IC à 95%: 11,4; 16,9) dans le bras sous sorafénib. L'HR stratifié était de 0,66 (IC à 95%: 0,52; 0,85), ce qui correspond à une diminution de 34% du risque de mortalité lié à l'association Tecentriq + bévacizumab par rapport au sorafénib.
GO30140
Une étude internationale de phase Ib, en ouvert, multicentrique, à plusieurs bras (GO30140) a également été réalisée chez des patients atteints d'une tumeur solide. Un protocole randomisé a été utilisé dans le bras F de l'étude pour évaluer la sécurité et l'efficacité de l'administration de Tecentriq en association avec le bévacizumab par rapport à Tecentriq en monothérapie chez des patients atteints de CHC avancé ou métastatique et/ou inopérable qui n'avaient pas reçu de traitement systémique antérieur. Le principal critère d'évaluation de l'efficacité était la PFS évaluée par un IRF selon les critères RECIST v1.1. Au total, 119 patients ont été randomisés (rapport 1:1) pour recevoir Tecentriq (1200 mg) et le bévacizumab (15 mg/kg) toutes les 3 semaines en perfusion intraveineuse ou seulement Tecentriq (1200 mg) toutes les 3 semaines. Au moment de l'analyse primaire, la durée médiane de suivi de la survie était de 6,6 mois. L'association Tecentriq + bévacizumab a montré un bénéfice statistiquement significatif en termes de PFS par rapport à Tecentriq en monothérapie (HR: 0,55, IC à 80%: 0,40; 0,74, valeur de p = 0,0108), avec une PFS médiane de 5,6 mois chez les patients traités par Tecentriq et bévacizumab, contre 3,4 mois chez les patients traités par Tecentriq en monothérapie.
Carcinome urothélial
Utilisation de Tecentriq chez des patients atteints d'un carcinome urothélial et présentant des facteurs pronostiques défavorables après un traitement préalable par une chimiothérapie à base de platine
Avant d'instaurer un traitement par Tecentriq chez des patients présentant des facteurs pronostiques défavorables ou une maladie agressive, le médecin doit tenir compte de l'effet différé du traitement par Tecentriq. Dans le cadre de l'étude IMvigor211, une incidence de décès plus élevée a été constatée chez les patients atteints d'un carcinome urothélial pendant les 4 premiers mois du traitement par Tecentriq, par rapport à une chimiothérapie. Les facteurs associés à des décès précoces sont les suivants: ≥2 facteurs de risque de Bellmunt (composé d'un score ECOG > 0, de métastases hépatiques et d'un taux d'hémoglobine < 10 g/dl), taux d'ALP élevé et/ou métastases hépatiques à l'entrée dans l'étude.
IMvigor211
GO29294 (IMvigor211), une étude de phase III ouverte, multicentrique, internationale et randomisée, a été réalisée pour évaluer l'efficacité et la sécurité de Tecentriq par rapport à une chimiothérapie (vinflunine, docétaxel ou paclitaxel selon le choix du médecin investigateur) chez des patients atteints d'un carcinome urothélial inopérable, localement avancé ou métastatique, avec progression de la maladie pendant ou après une chimiothérapie à base de platine. Les patients présentant les antécédents suivants ont été exclus de l'étude: patients atteints d'une maladie auto-immune ou de métastases cérébrales évolutives ou corticodépendantes, patients ayant reçu un vaccin vivant atténué durant les 28 jours précédant l'inclusion dans l'étude et ayant reçu des immunostimulants systémiques durant les 4 semaines précédant l'inclusion ou des médicaments immunosuppresseurs systémiques durant les 2 semaines avant l'inclusion dans l'étude. Les évaluations de la tumeur ont été réalisées toutes les 9 semaines pendant les 54 premières semaines, puis toutes les 12 semaines. Des échantillons de tumeur ont été évalués prospectivement à la recherche de l'expression du PD-L1 sur des IC infiltrant les tumeurs, et, sur la base des résultats obtenus, des sous-groupes de l'expression du PD-L1 ont été définis pour les analyses décrites ci-après.
Au total, 931 patients ont été inclus. Les patients ont été randomisés (1:1) pour recevoir soit Tecentriq, soit une chimiothérapie. La randomisation a été stratifiée selon la chimiothérapie (vinflunine contre taxane), le statut d'expression du PD-L1 sur les IC (< 5% contre ≥5%), le nombre de facteurs de risque pronostiques (0 contre 1 à 3) et les métastases hépatiques (oui contre non). Les facteurs de risque pronostiques ont inclus une période < 3 mois depuis la dernière chimiothérapie, un score de performance ECOG > 0 et un taux d'hémoglobine < 10 g/dl.
Tecentriq a été administré en perfusion i.v. à la dose fixe de 1200 mg toutes les 3 semaines. Une réduction de la dose de Tecentriq n'était pas autorisée. Les patients ont été traités jusqu'à la perte du bénéfice clinique d'après l'évaluation du médecin investigateur ou jusqu'à l'apparition d'une toxicité inacceptable. La vinflunine a été administrée en perfusion i.v. à la dose de 320 mg/m2 au jour 1 de chaque cycle de 3 semaines jusqu'à la progression de la maladie ou jusqu'à l'apparition d'une toxicité inacceptable. Le paclitaxel a été administré en perfusion i.v. pendant 3 heures à la dose de 175 mg/m2 au jour 1 de chaque cycle de 3 semaines jusqu'à la progression de la maladie ou jusqu'à l'apparition d'une toxicité inacceptable. Le docétaxel a été administré en perfusion i.v. à la dose de 75 mg/m2 au jour 1 de chaque cycle de 3 semaines jusqu'à la progression de la maladie ou jusqu'à l'apparition d'une toxicité inacceptable. Chez les patients traités, la durée médiane de traitement a été de 2,8 mois dans le bras sous Tecentriq, de 2,1 mois dans le bras sous la vinflunine et dans le bras sous le paclitaxel, et de 1,6 mois dans le bras sous le docétaxel.
Les données démographiques et les caractéristiques de la maladie présentes au début de l'étude dans le collectif de l'analyse primaire étaient équilibrées en comparant les bras de traitement. L'âge médian était de 67 ans (intervalle: 31 à 88) et 77,1% des patients étaient de sexe masculin. 53,9% des patients dans le bras sous chimiothérapie ont reçu de la vinflunine, 71,4% des patients présentaient au moins un facteur de risque pronostique défavorable et 28,8% présentaient des métastases hépatiques au début de l'étude. Le score de performance ECOG au début de l'étude était de 0 (45,6%) ou de 1 (54,4%). Chez 71,1% des patients, la tumeur primaire se situait dans la vessie et 25,4% des patients présentaient un carcinome urothélial dans les voies urinaires supérieures. 24,2% des patients n'avaient reçu qu'une seule thérapie adjuvante ou néoadjuvante à base de platine auparavant et avaient connu une progression de la maladie dans un intervalle de 12 mois.
Le critère d'évaluation principal de l'efficacité dans l'étude IMvigor211 était la survie globale (OS). Les critères d'évaluation secondaires de l'efficacité sont le taux de réponse objective (ORR), la survie sans progression (PFS) et la durée de la réponse (DOR) évalués par le médecin investigateur selon les critères RECIST (Response Evaluation Criteria in Solid Tumors) v1.1. Les comparaisons des OS entre le bras de traitement et le bras témoin ont été testées, comme décrit ci-après, en utilisant un procédé hiérarchique avec un ordre déterminé sur la base d'un test log-rank stratifié avec un niveau bilatéral de 5%: étape 1) Sous-groupe avec une expression du PD-L1 ≥5%, étape 2) Sous-groupe avec une expression du PD-L1 ≥1%, étape 3) Tous les patients satisfaisant aux critères d'inclusion («all comers»). Les résultats en termes d'OS pour les étapes 2 et 3 ne pouvaient à chaque fois être testés formellement que lorsque le résultat était statistiquement significatif lors de l'étape précédente.
Le suivi médian sur le plan de la survie a été de 17 mois. L'étude IMvigor211 n'a pas atteint le critère d'évaluation principal pour l'OS. Dans le sous-groupe de patients présentant des tumeurs avec une expression du PD-L1 ≥5%, Tecentriq n'a pas apporté de bénéfice statistiquement significatif en termes de survie par rapport à la chimiothérapie, avec un HR pour l'OS de 0,87 (IC à 95%: 0,63; 1,21; OS médiane de 11,1 contre 10,6 mois pour Tecentriq et la chimiothérapie). La valeur de p lors du test log-rank stratifié était de 0,41. Par conséquent, aucun test statistique formel portant sur l'OS n'a été réalisé dans le sous-groupe avec une expression du PD-L1 ≥1% ou dans la population en ITT, et les résultats de ces analyses sont considérés comme exploratoires.
La survie moyenne sans progression a été de 2,1 mois (intervalle de confiance à 95%: 2,1; 2,2) dans le bras sous Tecentriq et de 4,0 mois (IC à 95%: 3,4; 4,2) dans le bras sous chimiothérapie, avec un hazard ratio de 1,10 (IC à 95%: 0,95; 1,26). La proportion de patients en ITT présentant une réponse positive selon les critères RECIST version 1.1 confirmée par le médecin investigateur s'est révélée similaire dans les deux bras de l'étude: 13,4% (IC à 95%: 10,45; 16,87) dans le bras sous Tecentriq et 13,4% (IC à 95%: 10,47; 16,91) dans le bras sous chimiothérapie. Chez les patients répondeurs, la durée médiane de la réponse au traitement a été significativement plus longue dans le bras sous Tecentriq (21,7 mois) que dans le bras sous chimiothérapie (7,4 mois).
Dans l'analyse exploratoire finale (date de clôture du recueil des données: 8 novembre 2018) de la survie globale avec une durée médiane de suivi de 34 mois dans la population en ITT, la survie médiane était de 8,6 mois dans le bras Tecentriq (IC à 95%: 7,8; 9,6) et de 8,0 mois dans le bras chimiothérapie (IC à 95%: 7,2; 8,6) avec un hazard ratio de 0,82 (IC à 95%: 0,71; 0,94). Dans la population en ITT, des taux numériquement plus élevés d'OS ont été constatés à 12, 24 et 30 mois chez les patients du bras Tecentriq par rapport à ceux du bras chimiothérapie. Le pourcentage de patients en vie à 12 mois (estimation de KM) était de 32,5% dans le bras chimiothérapie et de 39,2% dans le bras Tecentriq. À 24 mois, 12,7% de patients étaient en vie dans le bras chimiothérapie et 22,5% dans le bras Tecentriq (estimation de KM). À 30 mois (estimations de KM), ils étaient 9,8% dans le bras chimiothérapie et 18,1% dans le bras Tecentriq.
Étude d'appoint chez des patients présentant un carcinome urothélial localement avancé ou métastatique, ayant reçu au préalable une chimiothérapie à base de platine: cohorte 2 de IMvigor210
GO29293 (IMvigor210), une étude clinique de phase II multicentrique, internationale, à un bras, avec deux cohortes, a été réalisée chez des patients présentant un carcinome urothélial localement avancé ou métastatique. Au total, 438 patients ont été inclus dans l'étude, qui était composée de deux cohortes. La cohorte 2 était constituée de patients ayant reçu au moins une chimiothérapie à base de platine pour traiter un carcinome urothélial localement avancé ou métastatique ou chez lesquels une progression de la maladie avait été constatée dans un délai de 12 mois après un traitement par une chimiothérapie adjuvante ou néoadjuvante à base de platine.
Les critères d'évaluation co-principaux dans la cohorte 2 étaient l'ORR confirmé par un centre d'examen indépendant (IRF) selon les critères RECIST v1.1, ainsi que l'ORR évalué par le médecin investigateur selon les critères RECIST modifiés (mRECIST). 310 patients ont été traités par Tecentriq en perfusion i.v. à la dose de 1200 mg toutes les 3 semaines jusqu'à la perte du bénéfice clinique. L'analyse primaire de la cohorte 2 a été effectuée après la réalisation d'un suivi de 24 semaines au minimum chez tous les patients. Dans la cohorte 2, l'étude a atteint les deux critères d'évaluation principaux, à savoir des ORR statistiquement significatifs dans le cadre de l'évaluation de l'IRF selon les critères RECIST v1.1 et dans le cadre de l'évaluation par le médecin investigateur selon les critères mRECIST, en comparaison avec un taux de rémission témoin historique de 10% préalablement déterminé.
Par ailleurs, une analyse a également été réalisée dans la cohorte 2 après une durée de suivi médiane de la survie de 21,1 mois. Les ORR confirmés par l'IRF selon les critères RECIST v1.1 ont été de 15,8% (IC à 95%: 11,9-20,4) au sein de la population en ITT, l'ORR confirmé par l'évaluation du médecin investigateur selon les critères mRECIST a été de 19,7% (IC à 95%: 15,4-24,6) et le taux d'OS après 12 mois a été de 37%. La DOR médiane n'a pas encore été atteinte dans la population en ITT.
Enfants et adolescents
Une étude multicentrique en ouvert d'une phase précoce été réalisée chez des patients pédiatriques (< 18 ans, n = 69) et chez des patients adultes jeunes (de 18 à 30 ans, n = 18) atteints d'une tumeur solide récurrente ou progressive ou d'un lymphome de Hodgkin ou non-hodgkinien afin d'évaluer la sécurité et la pharmacocinétique de Tecentriq. Les patients ont reçu 15 mg/kg d'atézolizumab par voie i.v. toutes les 3 semaines. Les principaux indicateurs d'efficacité étaient le taux de réponse objective (objective response rate, ORR), la survie sans progression (progression-free survival, PFS) et le taux de réponse avec bénéfice clinique (clinical benefit response rate, CBRR; seulement chez les patients atteints d'ostéosarcome).
Au moment de l'analyse primaire, l'ORR était de 4,6% (IC à 95%: 1,59; 10,89) et la durée moyenne de la PFS était de 1,3 mois (IC à 95%: 1,2; 1,4). Comme aucun patient de la cohorte atteinte d'ostéosarcome n'a obtenu de rémission complète ou partielle ou de stabilisation de la maladie pendant au moins 6 mois, le CBRR était de 0. Le bénéfice clinique de l'atézolizumab n'a pas été démontré chez les patients pédiatriques atteints de tumeurs solides récurrentes/réfractaires.
Efficacité de l'atézolizumab en présence d'anticorps dirigés contre le médicament
Des méta-analyses exploratoires, non ajustées, portant sur l'efficacité (sans ajustements de déséquilibres des caractéristiques initiales) ont été réalisées. Selon ces méta-analyses, la valeur estimée du HR pour la survie globale (OS) était de 0,815 entre le sous-groupe ADA-positif et le bras témoin (IC à 95%: 0,732; 0,908) et la valeur estimée du HR pour l'OS était de 0,672 entre le sous-groupe ADA-négatif et le bras témoin (IC à 95%: 0,617; 0,732), voir graphiques 1 et 2.
Graphique 1: Méta-analyse sur l'OS (non ajustée) de patients ADA-positifs par rapport au bras témoin
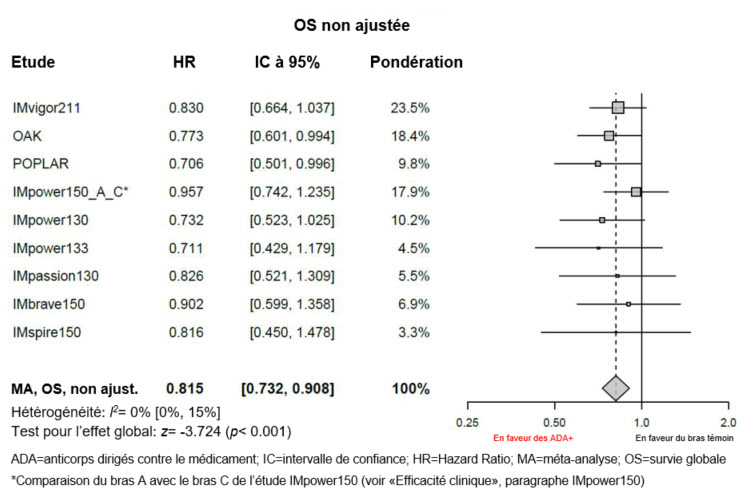
Graphique 2: Méta-analyse sur l'OS (non ajustée) de patients ADA-négatifs par rapport au bras témoin
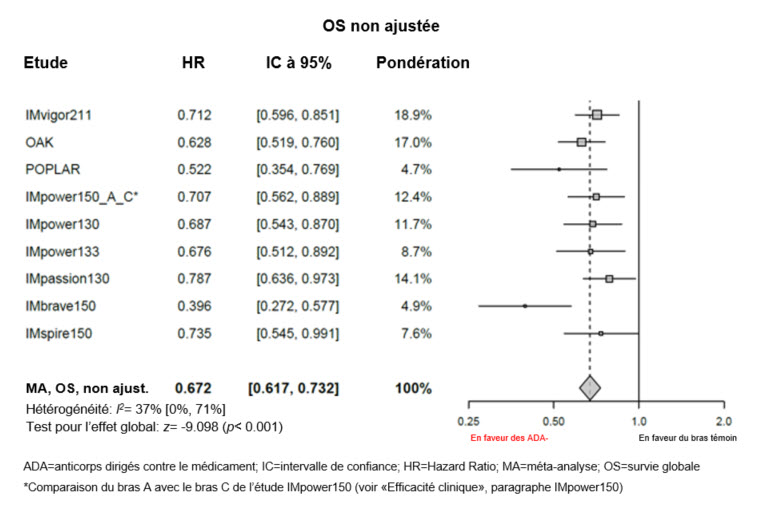
PharmacocinétiqueLa pharmacocinétique de Tecentriq a été caractérisée chez des patients dans plusieurs études cliniques, à des doses allant de 0,01 mg/kg à 20 mg/kg toutes les 3 semaines, ainsi qu'à la dose fixe de 1200 mg. L'exposition à Tecentriq a augmenté proportionnellement à la dose dans l'intervalle posologique de 1 mg/kg à 20 mg/kg. Une analyse de population portant sur 472 patients a décrit la pharmacocinétique de Tecentriq dans l'intervalle posologique compris entre 1 et 20 mg/kg avec un modèle d'ordonnancement linéaire bicompartimental et une cinétique d'élimination de premier ordre. Les propriétés pharmacocinétiques de Tecentriq à la posologie de 840 mg toutes les 2 semaines et celles à la posologie de 1200 mg toutes les 3 semaines sont comparables. Une analyse pharmacocinétique de population indique que l'état d'équilibre est atteint après 6 à 9 semaines (deux à trois cycles) après administration de doses répétée. Le taux d'accumulation systémique maximal de 3,3 pour tous les schémas posologiques.
L'analyse des données portant sur l'exposition, la sécurité et l'efficacité n'a pas révélé d'effet cliniquement significatif des facteurs suivants: âge (21-89 ans), poids corporel, sexe, taux d'albumine, charge tumorale, région ou appartenance ethnique, insuffisance rénale, insuffisance hépatique légère, grade de l'expression du PD-L1 et statut ECOG. Concernant les effets du statut ADA sur l'efficacité, voir «Mises en garde et précautions» et «Propriétés/Effets», paragraphe «Efficacité clinique».
Absorption
Tecentriq est administré en perfusion i.v.
Distribution
Une analyse pharmacocinétique de population a montré que chez un patient typique, le volume de distribution du compartiment central (V1) est de 3,28 l et le volume de distribution à l'état d'équilibre (Vss) de 6,91 l.
Métabolisme
Le métabolisme de Tecentriq n'a pas été directement étudié. Les anticorps sont en principe éliminés par catabolisme.
Élimination
Une analyse pharmacocinétique de population a montré que la clairance de Tecentriq est de 0,200 l/jour et la demi-vie d'élimination typique (t1/2) de 27 jours. Dans les études cliniques menées avec Tecentriq administré par voie intraveineuse, la clairance médiane de l'atézolizumab était augmentée de 22% (intervalle de 18% à 49%) chez les patients présentant des ADA en lien avec le traitement par rapport aux patients sans ADA liés au traitement.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Aucune étude spécifique n'a été réalisée avec Tecentriq chez des patients insuffisants hépatiques. L'analyse pharmacocinétique de population n'a montré aucune différence cliniquement significative concernant la clairance de Tecentriq, administré par voie intraveineuse ou sous-cutanée, entre les patients présentant une insuffisance hépatique légère (bilirubine ≤ LSN et ASAT > LSN ou bilirubine < 1,0 jusqu'à 1,5 × LSN et quel que soit le taux d'ASAT) ou modérée (bilirubine > 1,5 jusqu'à 3 × LSN et quel que soit le taux d'ASAT) et ceux présentant une fonction hépatique normale (bilirubine et ASAT ≤ LSN). Parmi les patients traités par Tecentriq présentant une insuffisance hépatique modérée, un seul a reçu Tecentriq par voie sous-cutanée. Il n'existe aucune donnée chez les patients présentant une insuffisance hépatique sévère (bilirubine > 3,0 × LSN, quel que soit le taux d'ASAT). Les troubles de la fonction hépatique ont été définis selon les critères de l'insuffisance hépatique établis par le National Cancer Institute (NCI) (voir «Posologie/Mode d'emploi: Instructions posologiques particulières»).
Troubles de la fonction rénale
Aucune étude spécifique n'a été réalisée avec Tecentriq chez des patients insuffisants rénaux. Chez les patients ayant reçu Tecentriq soit par voie sous-cutanée soit par voie intraveineuse, l'analyse pharmacocinétique de population n'a montré aucune différence cliniquement significative en termes de clairance en cas d'insuffisance rénale légère (DFGe de 60 à 89 ml/min/1,73 m²; n = 111 par traitement s.c. ou n = 208 par traitement i.v.) ou modérée (DFGe de 30 à 59 ml/min/1,73 m²; n = 32 par traitement s.c. ou n = 116 par traitement i.v.), par rapport aux patients présentant une fonction rénale normale (DFGe supérieur ou égal à 90 ml/min/1,73 m²; n = 103 par traitement s.c. ou n = 140 par traitement i.v.). Seuls quelques patients traités par Tecentriq par voie intraveineuse présentaient une insuffisance rénale sévère (DFGe de 15 à 29 ml/min/1,73 m²; n = 8) (voir «Posologie/Mode d'emploi: Instructions posologiques particulières»).
Patients âgés
Aucune étude spécifique n'a été réalisée avec Tecentriq chez des patients âgés. L'influence de l'âge sur la pharmacocinétique de Tecentriq a été évaluée dans une analyse pharmacocinétique de population. Chez les patients âgés de 21 à 89 ans (n = 472), avec un âge médian de 62 ans, l'âge n'a pas été identifié comme une covariable significative affectant la pharmacocinétique de Tecentriq administré par voie intraveineuse. Aucune différence cliniquement significative concernant la pharmacocinétique de Tecentriq, administré par voie sous-cutanée ou par voie intraveineuse, n'a été observée entre les patients < 65 ans (n = 138 par traitement s.c. ou n = 274 par traitement i.v.), les patients âgés de 65 à 75 ans (n = 89 par traitement s.c. ou n = 152 par traitement i.v.) et les patients > 75 ans (n = 19 par traitement s.c. ou n = 46 par traitement i.v.) (voir «Posologie/Mode d'emploi: Instructions posologiques particulières»).
Données précliniquesPharmacologie de sécurité
Les critères d'évaluation pharmacologiques de sécurité de Tecentriq administré par voie intraveineuse ont été évalués dans le cadre de deux études portant sur la toxicité chronique, l'une de 8 semaines et l'autre de 26 semaines, menées chez le singe cynomolgus. Elles n'ont révélé aucun risque particulier pour l'être humain.
Propriétés toxicologiques chez l'animal
Dans une souche spécifique de souris, des neuropathies minimes du nerf sciatique ont été observées à des doses ≥10 mg/kg administrées par voie intraveineuse. Des artérites/périartérites minimes à légères touchant de multiples organes sont survenues chez le singe cynomolgus après administration sous-cutanée de doses ≥15 mg/kg et après administration intraveineuse de doses de 50 mg/kg. Chez les singes femelles, des cycles sexuels irréguliers ont été observés à des doses de 50 mg/kg administrées par voie intraveineuse. Une augmentation des inflammations sous-cutanées (irritations légères) au niveau du site d'injection a été observée chez le singe.
Une augmentation de la réponse immunitaire ayant entraîné une mortalité accrue a été observée dans le modèle murin d'infection virale par le LCMV CL-13, mais pas par le LCMV Armstrong.
Dans une étude sur des singes cynomolgus exposés à l'hémocyanine de patelle (Keyhole Limpet Hemocyanin, KLH) utilisée comme antigène T-dépendant, l'administration de 50 mg d'atézolizumab par kg en injection i.v. pendant une période allant jusqu'à 26 semaines a été bien tolérée. Chez les singes cynomolgus ayant reçu la KLH, aucune toxicité inattendue ou nouvelle n'est survenue. L'administration au long cours d'atézolizumab a entraîné une diminution non significative des réponses primaires des IgM et des réponses primaires et secondaires des IgG à la KLH. Même si la signification toxicologique est incertaine en raison du faible nombre d'animaux utilisés dans cette étude et de la grande variabilité entre les animaux, on peut en conclure que l'atézolizumab pourrait diminuer les réponses primaires et secondaires des IgG aux antigènes T-dépendants.
Mutagénicité
Aucune étude de mutagénicité n'a été réalisée avec Tecentriq.
Carcinogénicité
Aucune étude de carcinogénicité n'a été réalisée avec Tecentriq.
Toxicité sur la reproduction
Aucune étude de reproduction ou de tératogénicité n'a été réalisée chez l'animal avec Tecentriq. L'importance capitale de la voie de signalisation du PD-L1/PD-1 pour la tolérance maternelle et fœtale ainsi que pour la survie embryonnaire et fœtale pendant la gestation est bien démontrée. On peut s'attendre à ce que l'administration de Tecentriq ait des effets néfastes sur la grossesse et expose le fœtus humain à un risque, notamment de mort de l'embryon.
Fertilité
Aucune étude de fertilité n'a été réalisée avec Tecentriq. L'étude portant sur la toxicité chronique a cependant comporté une évaluation des organes reproducteurs de singes cynomolgus mâles et femelles. Chez tous les singes femelles du groupe ayant reçu la dose de 50 mg/kg, Tecentriq administré par voie intraveineuse a eu des effets sur le cycle menstruel, caractérisés par des cycles irréguliers pendant la phase d'administration et par l'absence de corps jaunes nouvellement formés dans les ovaires à l'autopsie. Ces effets sont survenus à une AUC estimée environ 6 fois supérieure à l'AUC observée chez les patients après administration de la dose recommandée; ils ont été réversibles au cours de la phase de récupération sans administration. Aucun effet sur les organes reproducteurs mâles n'a été constaté.
Remarques particulièresIncompatibilités
Aucune incompatibilité n'a été observée entre Tecentriq et les poches de perfusion dont les surfaces en contact avec le produit sont en polychlorure de vinyle (PVC), polyoléfine, polyéthylène (PE) ou polypropylène (PP). En outre, aucune incompatibilité n'a été constatée avec des membranes filtrantes pour perfusion en polyéthersulfone ou polysulfone ainsi qu'avec des sets de perfusion et autres accessoires de perfusion en PVC, PE, polybutadiène ou polyétheruréthane.
Utiliser uniquement une solution de chlorure de sodium pour préparations injectables à 0,9% pour réaliser la dilution.
Ce médicament ne doit pas être mélangé avec d'autres médicaments.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Stabilité après ouverture
La stabilité chimique et physique de la solution pour perfusion préparée a été démontrée pendant 30 jours à 2-8 °C et pendant 24 heures à ≤25 °C.
Pour des raisons microbiologiques, la solution diluée doit être utilisée immédiatement.
Si la solution n'est pas utilisée immédiatement, les conditions de stockage et la durée du stockage relèvent de la responsabilité de l'utilisateur et ne doivent normalement pas dépasser 24 heures à une température comprise entre 2 et 8 °C, sauf si la dilution se déroule dans des conditions aseptiques contrôlées et validées.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Ne pas agiter. Ne pas congeler.
Conserver le récipient dans son carton pour le protéger de la lumière.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Instructions de dilution
Tecentriq doit être préparé par un professionnel de la santé, dans des conditions d'asepsie. Utiliser une aiguille et une seringue stériles pour préparer Tecentriq. Prélevez le volume nécessaire de solution à diluer Tecentriq du flacon et diluez-le dans 250 ml d'une solution de chlorure de sodium à 0,9%. Pour la dilution, utilisez exclusivement une solution de chlorure de sodium à 0,9% pour préparations injectables. Après la dilution, la concentration finale de la solution préparée doit être comprise entre 3,2 et 16,8 mg/ml.
Tecentriq ne contient pas de conservateur. Chaque flacon est donc à usage unique. Tout médicament non utilisé doit être éliminé.
Élimination des médicaments non utilisés/périmés
Le rejet de médicaments dans l'environnement doit être réduit au minimum. Les médicaments ne doivent pas être éliminés avec les eaux usées. Éviter toute élimination avec des déchets ménagers. Utilisez les systèmes collecteurs établis disponibles sur place.
Numéro d’autorisation66152 (Swissmedic).
PrésentationFlacon de 20 ml de solution à diluer pour perfusion (60 mg/ml) [A]
Flacon de 14 ml de solution à diluer pour perfusion (60 mg/ml) [A]
Titulaire de l’autorisationRoche Pharma (Suisse) SA, Bâle.
Mise à jour de l’informationSeptembre 2024.
|