Propriétés/EffetsCode ATC
L01FF05
Mécanisme d'action
La liaison du PD-L1 aux récepteurs PD-1 et B7.1 sur les cellules T provoque une suppression de l'activité cytotoxique des lymphocytes T par inhibition de la prolifération et de la production de cytokines par les lymphocytes T. Le PD-L1 peut être exprimé sur les cellules tumorales (TC) ainsi que les cellules immunitaires infiltrant les tumeurs (IC) et contribuer à inhiber la réponse immunitaire antitumorale dans le microenvironnement.
L'atézolizumab est un anticorps monoclonal humanisé de type immunoglobuline G1 (IgG1) à fragment Fc modifié, qui se lie directement à PD-L1 et provoque un blocage des interactions avec les récepteurs PD-1 et B7.1. Cela permet de lever l'inhibition de la réponse immunitaire médiée par la voie de signalisation PD-L1/PD-1 et entraîne une réactivation de la réponse immunitaire antitumorale. L'atézolizumab ne modifie pas l'interaction PD-L2/PD-1. Dans des modèles tumoraux syngéniques murins, le blocage de l'activité du PD-L1 a entraîné une diminution de la croissance tumorale.
La double inhibition des voies de signalisation PD-1/PD-L1 et MAPK ainsi que BRAF et MEK entrave la croissance tumorale dans les modèles de cancer chez les souris et améliore l'immunogénicité des tumeurs en augmentant la présentation de l'antigène ainsi que l'activation et l'infiltration des cellules T par rapport à la thérapie ciblée seule.
Efficacité clinique
Cancer du poumon non à petites cellules
1L en cas de NSCLC non épidermoïde métastatique
IMpower150
Une étude clinique de phase III, randomisée, en ouvert, GO29436 (IMpower150), a été menée afin d'évaluer l'efficacité́ et la sécurité́ de Tecentriq en association au paclitaxel et au carboplatine, avec ou sans bévacizumab, chez des patients atteints d'un NSCLC non épidermoïde métastatique sans chimiothérapie préalable. Au total, 1202 patients ont été inclus dans l'étude et randomisés selon un rapport 1:1:1 pour recevoir l'un des régimes thérapeutiques décrits ci-dessous. La randomisation a été stratifiée selon le sexe, la présence de métastases hépatiques et le statut d'expression de PD-L1 sur les cellules tumorales (TC) et les cellules immunitaires infiltrant les tumeurs (IC).
Les patients ont été randomisés dans un des trois groupes de traitement suivants:
·Groupe A: Tecentriq 1200 mg, paclitaxel 175 mg/m² ou 200 mg/m² et carboplatine AUC 6 mg/ml/min le jour 1 de chaque cycle de 21 jours pendant 4 ou 6 cycles au maximum.
·Groupe B: Tecentriq 1200 mg, bévacizumab 15 mg/kg, paclitaxel 175 mg/m² ou 200 mg/m² et carboplatine AUC 6 mg/ml/min le jour 1 de chaque cycle de 21 jours pendant 4 ou 6 cycles au maximum.
·Groupe C: bévacizumab 15 mg/kg, paclitaxel 175 mg/m² ou 200 mg/m² et carboplatine AUC 6 mg/ml/min le jour 1 de chaque cycle de 21 jours pendant 4 ou 6 cycles au maximum.
Le carboplatine et le paclitaxel ont été administrés jusqu'à la fin des 4 ou 6 cycles ou jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable, selon l'événement survenant le premier. Compte tenu d'un degré globalement plus élevé de toxicités hématologiques chez les patients originaires de pays asiatiques par rapport aux patients issus de pays non-asiatiques, les patients d'ethnie asiatique ont reçu une dose initiale de 175 mg/m2 de paclitaxel.
Les patients sans progression de la maladie à l'issue de la chimiothérapie à base de platine ou après son interruption ont reçu:
·Groupe A: Tecentriq 1200 mg par voie intraveineuse le jour 1 de chaque cycle de 21 jours jusqu'à la perte du bénéfice clinique selon l'évaluation du médecin investigateur.
·Groupe B: Tecentriq 1200 mg et bévacizumab 15 mg/kg par voie intraveineuse le jour 1 de chaque cycle de 21 jours. L'administration de Tecentriq s'est poursuivie jusqu'à la perte du bénéfice clinique selon l'évaluation du médecin investigateur, et l'administration de bévacizumab a été maintenue jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
·Groupe C: bévacizumab 15 mg/kg par voie intraveineuse le jour 1 de chaque cycle de 21 jours jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
Les patients ont été exclus de la participation à l'étude s'ils souffraient d'une maladie auto-immune, s'ils avaient reçu un vaccin vivant atténué dans les 28 jours précédant la randomisation, s'ils avaient reçu un traitement immunostimulant systémique dans les 4 semaines ou un traitement immunosuppresseur systémique dans les 2 semaines précédant la randomisation, s'ils présentaient des métastases du SNC actives ou non traitées, une infiltration tumorale évidente des grands vaisseaux thoraciques ou une cavitation manifeste des lésions pulmonaires à l'imagerie. Des évaluations tumorales ont été́ réalisées toutes les 6 semaines pendant les 48 premières semaines suivant le jour 1 du cycle 1, puis toutes les 9 semaines par la suite.
Les échantillons tumoraux ont été analysés à la recherche de l'expression de PD-L1 sur les cellules tumorales (TC) et les cellules immunitaires infiltrant les tumeurs (IC), et les résultats ont été utilisés pour définir les sous-groupes d'expression de PD-L1 pour les analyses décrites ci-après.
Les données démographiques et les caractéristiques de la maladie au début du traitement de la population étudiée (ITT) étaient équilibrées entre les deux groupes de traitement. Dans la présente étude, l'âge médian s'élevait à 63 ans (fourchette: de 31 à 90) et 60% des patients étaient de sexe masculin. La majorité des sujets étaient de type caucasien (82%). Près de 10% des patients présentaient des mutations de l'EGFR connues, 4% présentaient des translocations de l'ALK connues, 14% avaient des métastases hépatiques à l'inclusion dans l'étude et la plupart des patients étaient fumeurs ou anciens fumeurs (80%). Au total, 95% des patients étaient atteints d'un NSCLC non épidermoïde avec une histologie d'adénocarcinome. L'indice de performance ECOG à l'inclusion était de 0 (43%) ou 1 (57%).
Les données démographiques et les caractéristiques de la maladie au début du traitement de patients dont le statut d'expression de PD-L1 était de ≥1% sur les TC ou de ≥1% sur les IC (ci-après dénommé «expression de PD-L1 ≥1%»), sans mutations de l'EGFR ni translocations de l'ALK, étaient équilibrées entre les deux groupes de traitement. 51% des tumeurs des patients présentaient une expression de PD-L1 ≥1% et 49% une expression de PD-L1< 1% sur les TC et < 1% sur les IC.
La population en ITT-WT est définie comme l'ensemble des patients randomisés à l'exception de ceux présentant des aberrations génomiques tumorales de l'EGFR et de l'ALK.
La durée de suivi médiane était de 39,3 mois au moment de l'analyse finale de l'OS pour Tecentriq + carboplatine + paclitaxel (CP), comparé à bévacizumab + CP. Pour la population en ITT-WT regroupant des patients dont les tumeurs présentaient une expression de PD-L1 ≥1%, un allongement de l'OS a été constaté dans le groupe Tecentriq + CP, comparé au groupe bévacizumab + CP (HR non stratifié: 0,71; [IC à 95%: 0,55, 0,91] pas testé formellement en raison du concept statistique); l'OS médiane s'est élevée à 24,4 mois (8,4 mois de plus que dans le groupe bévacizumab + CP, qui a révélé une OS de 16,0 mois). Ceci correspond à une réduction du risque relatif de mortalité de 29% associée à Tecentriq + CP, par rapport à bévacizumab + CP, dans cette population. L'analyse par landmark révèle que, dans la population en ITT-WT (expression de PD-L1 ≥1%), les taux de survie après un an étaient de 70,6% dans le groupe Tecentriq + CP, contre 55,9% dans le groupe bévacizumab + CP, et de 51,5% et 36,9% respectivement après deux ans.
Au moment de l'analyse finale de la PFS, le hazard ratio de la PFS non stratifié s'élevait à 0,74 [IC à 95%: 0,58, 0,94] chez les patients sous Tecentriq + CP, comparé à bévacizumab + CP (population en ITT-WT avec une expression de PD-L1 ≥1%).
IMpower130
Un essai clinique de phase III, randomisé, en ouvert, GO29537 (IMpower130), a été́ mené́ afin d'évaluer l'efficacité́ et la sécurité́ de Tecentriq en association au nab-paclitaxel et au carboplatine, chez des patients naïfs de chimiothérapie atteints d'un NSCLC non épidermoïde métastatique. Les patients, y compris ceux avec aberrations génomiques tumorales du gène EGFR ou ALK, ont été́ inclus et randomisés selon un rapport 2:1 pour recevoir l'un des traitements décrits dans le tableau 5. La randomisation a été́ stratifiée selon le sexe, la présence de métastases hépatiques et le statut d'expression de PD-L1 sur les TC et les IC. Les patients recevant le traitement B ont pu changer de traitement et recevoir le Tecentriq en monothérapie après progression de la maladie.
Tableau 5: Traitement intraveineux dans IMpower130
|
Traitement
|
Induction
(quatre ou six cycles de 21 jours)
|
Entretien
(cycles de 21 jours)
| |
A
|
Tecentriq (1200 mg) a + nabpaclitaxel (100 mg/m2) b, c + carboplatine (AUC 6) c
|
Tecentriq (1200 mg) a
| |
B
|
Nab-paclitaxel (100 mg/m2) b + carboplatine (AUC 6) c
|
Meilleurs soins de soutien ou pémétrexed
|
a LeTecentriq est administré jusqu'à̀ perte du bénéfice clinique évaluée par le médecin investigateur.
b Le nab-paclitaxel est administré les jours 1, 8 et 15 de chaque cycle.
c Le nab-paclitaxel et le carboplatine sont administrés jusqu'à la fin de 4-6 cycles ou progression de la maladie ou survenue d'une toxicité inacceptable, selon l'événement survenant en premier.
Les patients ont été́ exclus s'ils présentaient un antécédent de maladie auto-immune, s'ils avaient reçu un vaccin vivant atténué́ dans les 28 jours précédant la randomisation, s'ils avaient reçu un traitement immunostimulant systémique dans les 4 semaines ou un traitement immunosuppresseur systémique dans les 2 semaines précédant la randomisation, s'ils présentaient des métastases cérébrales actives ou non traitées. Des évaluations tumorales ont été́ réalisées toutes les 6 semaines pendant les 48 premières semaines suivant le Jour 1 du cycle 1, puis toutes les 9 semaines par la suite.
Les caractéristiques démographiques et pathologiques à l'inclusion de la population étudiée (n = 723) étaient bien équilibrées entre les bras de traitement. L'âge médian s'élevait à 64 ans (entendue de 18 à 86 ans). La majorité́ des patients était de sexe masculin (57%) et de type caucasien (90%). 14,8% des patients avaient des métastases hépatiques à l'inclusion et la plupart des patients étaient fumeurs ou anciens fumeurs (88%). La majorité́ des patients présentaient un indice de performance ECOG de 1 (58,7%).
L'analyse principale a été́ réalisée chez tous les patients, en excluant ceux avec aberrations génomiques tumorales du gène EGFR ou ALK (n = 685), définie comme la population ITT-WT. Les deux critères d'évaluation principaux étaient l'OS et la PFS évaluée par le médecin investigateur. Les principaux résultats de cette étude lors d'un suivi médian de 18,6 mois de la durée de vie restante sont résumés ci-dessous.
Le traitement par atezo + carboplatine + nab-paclitaxel (atezo + CnP) été associé à un allongement statistiquement significatif de l'OS par rapport au carboplatine et nab-paclitaxel (CnP). Pour les patients de la population en ITT-WT, l'OS médiane était 4,7 mois plus longue dans le bras de traitement sous atezo + CnP, où elle était de 18,6 mois (IC à 95%: 15,8; 21,2) dans le bras de traitement sous atezo + CnP contre 13,9 mois (IC à 95%: 12,0; 18,7) dans le bras de traitement sous CnP (valeur de p stratifiée = 0,030). Le hazard ratio (HR) stratifié était de 0,79 (IC à 95%: 0,64; 0,98); cela correspond à une réduction relative du risque de mortalité de 21% sous atezo + CnP par rapport au CnP.
Par rapport au CnP, le traitement par atezo + CnP était associé à une amélioration statistiquement significative de la PFS évaluée par le médecin investigateur (selon RECIST v1.1). Pour les patients de la population en ITT-WT, la PFS médiane était numériquement plus longue dans le bras de traitement sous atezo + CnP: 7,0 mois (IC à 95%: 6,3; 7,3) dans le bras de traitement atezo + CnP contre 5,5 mois (IC à 95%: 4,4; 5,9) dans le bras sous CnP (valeur de p stratifiée < 0,0001). Le HR stratifié était de 0,64 (IC à 95%: 0,54; 0,76).
La proportion de patients de la population ITT-WT avec un taux de réponse objective évalué et confirmé par un médecin investigateur selon RECIST v1.1 était supérieur de 17,6% dans le bras de traitement sous atezo + CnP par rapport au bras sous CnP (49,3% vs 31,7%).
Dans tous les sous-groupes PD-L1, indépendamment de l'expression, un bénéfice concernant l'OS et la PFS a été obtenu hormis les patients avec des métastases hépatiques. Ainsi que les résultats d'une analyse en sous-groupes d'un nombre limité de patients (n = 101) avec des métastases hépatiques l'ont montré, l'atézolizumab, le nab-paclitaxel et le carboplatine ne semblent pas entraîner d'améliorations de l'effet thérapeutique dans ce collectif partiel par rapport au nab-paclitaxel et au carboplatine (HR: 0,96; IC à 95%: 0,61; 1,51 pour la PFS et HR: 1,21; IC à 95%: 0,73; 2,00 pour l'OS).
NSCLC au stade précoce
IMpower010
Une étude de phase III multicentrique, randomisée et en ouvert, GO29527 (IMpower010), a été réalisée afin d'évaluer l'efficacité et la sécurité de Tecentriq pour le traitement adjuvant de patients atteints de NSCLC de stade IB (tumeurs ≥4 cm) à IIIA (selon de système de classification de l'UICC/AJCC, 7e édition). Parmi les patients inclus dans l'étude ayant subi une reséction tumorale complète, un total de 1280 patients ont été éligibles pour recevoir jusqu'à 4 cycles d'une chimiothérapie à base de cisplatine. Le tableau 6 décrit les schémas de chimiothérapies à base de cisplatine.
Tableau 6: Schémas thérapeutiques pour la chimiothérapie intraveineuse de l'étude IMpower010
|
Chimiothérapie adjuvante à base de cisplatine
Cisplatine 75 mg/m2 i.v. au jour 1 de chaque cycle de 21 jours avec l'un de ces schémas thérapeutiques
|
Vinorelbine 30 mg/m2 i.v., jours 1 et 8
| |
Docétaxel 75 mg/m2 i.v., jour 1
| |
Gemcitabine 1250 mg/m2 i.v., jours 1 et 8
| |
Pémétrexed 500 mg/m2 i.v., jour 1
|
À l'issue de la chimiothérapie à base de cisplatine (jusqu'à 4 cycles), au total 1005 patients ont été randomisés selon un rapport de 1:1 pour recevoir Tecentriq (bras A) ou le meilleur traitement de soutien (bras B). Tecentriq a été administré à la dose fixe de 1200 mg en perfusion i.v. toutes les 3 semaines pendant 16 cycles, sauf en cas d'apparition d'une récidive de la maladie ou de survenue d'une toxicité inacceptable. La randomisation a été stratifiée selon le sexe, le stade de la maladie, l'histologie et le statut d'expression de PD-L1 (estimé au moyen du test VENTANA PD-L1 [SP142]).
Les patients ont été exclus de l'étude s'ils présentaient des antécédents de maladie auto-immune, s'ils avaient reçu un vaccin vivant atténué́ dans les 28 jours précédant la randomisation, ou encore s'ils avaient reçu un traitement immunostimulant systémique dans les 4 semaines ou un traitement immunosuppresseur systémique dans les 2 semaines précédant la randomisation. Des évaluations tumorales ont été réalisées au début de la phase de randomisation, au cours de la première année après le cycle 1, au jour 1 tous les 4 mois, puis tous les 6 mois jusqu'à l'année 5, puis une fois par an.
Les données démographiques et les caractéristiques pathologiques étaient bien équilibrées entre les bras de traitement. L'âge médian était de 62 ans (intervalle: de 26 à 84 ans), 67% des patients étaient de sexe masculin. La majorité des patients était de type caucasien (73%), 24% étaient d'origine asiatique. La plupart des patients étaient des fumeurs ou d'anciens fumeurs (78%); au début de l'étude, l'indice de performance ECOG était de 0 (55%) ou 1 (44%). Le maladie était de stade IB chez 12% des patients, de stade II chez 47% et de stade IIIA chez 41%. Sur la base des estimations réalisées au moyen du test VENTANA PD-L1 (SP263), la proportion de patients atteints de tumeurs présentant une expression de PD-L1 ≥1% sur les cellules tumorales (TC) était de 55% et la proportion de patients atteints de tumeurs présentant une expression de PD-L1 ≥50% sur les TC était de 26%.
Le critère d'évaluation principal de l'efficacité était la survie sans maladie (DFS) selon l'évaluation du médecin investigateur. La DFS était définie comme le temps écoulé entre la randomisation et la survenue de l'un des événements suivants: première récidive documentée de la maladie, nouveau NSCLC primaire ou décès toutes causes confondues, selon ce qui survenait en premier. L'objectif principal en lien avec l'efficacité consistait à évaluer la DFS dans la population de patients présentant une expression de PD-L1 ≥1% sur les TC (sur la base du test SP263) et une maladie de stade II à IIIA. Des critères d'évaluation secondaires en lien avec l'efficacité étaient l'évaluation de la DFS dans la population de patients présentant une expression de PD-L1 ≥50% sur les TC (sur la base du test SP263) et une maladie de stade II à IIIA, ainsi que la survie globale (OS) dans la population en intention de traiter (ITT).
Chez les patients présentant une expression de PD-L1 ≥50% sur les TC et une maladie de stade II à IIIA (n = 229) une amélioration cliniquement pertinente de la DFS a été démontrée, avec un HR non stratifié de 0,43 (IC à 95%: 0,27, 0,68). La DFS médiane n'a pas été atteinte chez les patients du bras sous Tecentriq (IC à 95%: 42,3 mois, NE) et a été de 35,7 mois chez les patients du bras recevant le meileur traitement de soutien (IC à 95%: 29,7, NE). Les données d'OS n'étaient pas matures au moment de l'analyse intermédiaire de la DFS, avec une mortalité d'environ 16,2% dans les deux bras rapportée dans la population présentant une expression de PD-L1 ≥50% sur les TC et une maladie de stade II à IIIA. Une analyse exploratoire de l'OS a révélé une tendance en faveur du Tecentriq par rapport au meilleur traitement de soutien dans cette population de patients (HR non stratifé = 0,37; [IC à 95%: 0,18, 0,74]).
NSCLC prétraité par chimiothérapie
OAK
GO28915 (OAK), une étude de phase III ouverte, multicentrique, internationale et randomisée, a été réalisée pour évaluer l'efficacité et la sécurité de Tecentriq par rapport au docétaxel chez des patients atteints de NSCLC localement avancé ou métastatique, dont la maladie a progressé pendant ou après une chimiothérapie à base de platine. Au total, 1225 patients ont été inclus dans cette étude, parmi lesquels les 850 premiers patients randomisés ont été inclus dans l'analyse primaire de l'efficacité; l'analyse secondaire de l'efficacité a reposé sur 1225 patients. Les patients éligibles ont été stratifiés selon le statut d'expression du PD-L1 des IC infiltrant les tumeurs, le nombre de protocoles de chimiothérapie antérieurs et le type histologique. Les patients ont été randomisés (1:1) pour recevoir soit Tecentriq soit le docétaxel. Les patients présentant des antécédents connus de maladie auto-immune ou des métastases cérébrales évolutives ou corticodépendantes, ainsi que les patients ayant utilisé des vaccins vivants atténués dans les 28 jours précédant l'inclusion dans l'étude ou des principes actifs immunostimulants systémiques dans les 4 semaines précédant l'inclusion dans l'étude ou des médicaments immunosuppresseurs systémiques dans les 2 semaines précédant l'inclusion dans l'étude ont été exclus de l'étude. Les évaluations de la tumeur ont été réalisées toutes les 6 semaines durant les 36 premières semaines, puis toutes les 9 semaines. Une évaluation prospective de l'expression du PD-L1 sur les TC et les IC a été réalisée dans des échantillons de tissus tumoraux; les résultats ont été utilisés pour définir les sous-groupes d'expression du PD-L1 dans les analyses décrites ci-dessous.
Les données démographiques et les caractéristiques de la maladie au début du traitement des 850 premiers patients randomisés inclus dans l'analyse primaire de l'efficacité étaient bien équilibrées dans les deux bras thérapeutiques. L'âge médian était de 64 ans (intervalle: de 33 à 85); 61% des patients étaient de sexe masculin. La majorité des patients étaient des Blancs (70%). Presque les trois quarts des patients (74%) présentaient un carcinome d'histologie non épidermoïde, 10% présentaient une mutation de l'EGFR connue, 0,2% avait une translocation de l'ALK connue, 10% avaient des métastases dans le SNC au début du traitement et la plupart des patients étaient des fumeurs ou d'anciens fumeurs (82%). Le score de performance ECOG au début du traitement était de 0 (37%) ou de 1 (63%). 75% des patients n'avaient reçu auparavant qu'un seul protocole thérapeutique à base de platine. Les données démographiques et les caractéristiques de la maladie observées dans le cadre de l'analyse secondaire de l'efficacité sont en accord avec les observations réalisées lors de l'analyse primaire de l'efficacité.
Tecentriq a été administré en perfusion i.v. à la dose fixe de 1200 mg toutes les 3 semaines. Des réductions de la dose n'étaient pas autorisées. Les patients ont été traités jusqu'à la perte du bénéfice clinique selon l'évaluation du médecin investigateur. Le docétaxel a été administré en perfusion i.v. à la dose de 75 mg/m2 au jour 1 de chaque cycle de 21 jours, jusqu'à la progression de la maladie. Rapporté à l'ensemble des patients traités, la durée médiane de traitement a été de 2,1 mois dans le bras sous le docétaxel et de 3,4 mois dans le bras sous Tecentriq lors de l'analyse primaire de l'efficacité.
Le critère d'évaluation principal de l'efficacité était la survie globale (overall survival, OS). Les principaux résultats de cette étude, qui ont été rapportés dans le cadre de l'analyse primaire de l'efficacité effectuée chez les 850 premiers patients randomisés avec un suivi médian de la survie de 21 mois, sont résumés ci-dessous (date limite de collecte des données: 07.07.2016). Le traitement par Tecentriq a été associé à un allongement cliniquement et statistiquement significatif de l'OS par rapport au docétaxel. Dans le bras sous Tecentriq, l'OS médiane a été de 4,2 mois supérieure chez les 850 premiers patients randomisés de la population en ITT (intention to treat): elle était de 9,6 mois (IC à 95%: 8,6; 11,2) dans le bras sous le docétaxel contre 13,8 mois (IC à 95%: 11,8; 15,7) dans le bras sous Tecentriq (valeur de p stratifiée = 0,0003). Le hazard ratio (HR) a été de 0,73 (IC à 95%: 0,62; 0,87); cela correspond à une réduction relative du risque de mortalité de 27% sous Tecentriq par rapport au groupe sous le docétaxel (analyse stratifiée). Les courbes de Kaplan-Meier ont montré à partir de 3 mois env. une séparation claire en faveur du bras sous Tecentriq, laquelle s'est maintenue par la suite. De la même façon, dans le sous-groupe de patients avec une expression du PD-L1 ≥1% sur les TC ou les IC, le traitement par Tecentriq a également été associé à un allongement cliniquement et statistiquement significatif de l'OS par rapport au docétaxel. Dans le bras sous Tecentriq, l'OS médiane des patients a été de 5,4 mois supérieure: elle était de 10,3 mois (IC à 95%: 8,8; 12,0) dans le bras sous le docétaxel contre 15,7 mois (IC à 95%: 12,6; 18,0) dans le bras sous Tecentriq (valeur de p stratifiée = 0,0102). L'HR a été de 0,74 (IC à 95%: 0,58; 0,93); cela correspond à une réduction relative du risque de mortalité de 26% sous Tecentriq par rapport au groupe sous le docétaxel (analyse stratifiée).
L'OS dans les sous-groupes de patients avec une expression du PD-L1 ≥50% sur les TC ou ≥10% sur les IC, une expression du PD-L1 ≥5% sur les TC ou les IC, ou encore une expression du PD-L1< 1% sur les TC et les IC a montré que la durée médiane de l'OS était plus longue dans tous les sous-groupes d'expression du PD-L1 évalués. Les valeurs ont été les suivantes: expression du PD-L1 ≥50% sur les TC ou ≥10% sur les IC: OS médiane de 20,5 mois (IC à 95%: 17,5; NA) dans le bras sous Tecentriq contre 8,9 mois (IC à 95%: 5,6; 11,6) dans le bras sous le docétaxel (HR=0,41, IC à 95%: 0,27; 0,64); expression du PD-L1 ≥5% sur les TC ou les IC: OS médiane de 16,3 mois (IC à 95%: 13,3; 20,1) dans le bras sous Tecentriq contre 10,8 mois (IC à 95%: 8,8; 12,7) dans le bras sous le docétaxel (HR=0,67, IC à 95%: 0,49; 0,90); expression du PD-L1< 1% sur les TC et les IC: OS médiane de 12,6 mois (IC à 95%: 9,6; 15,2) dans le bras sous Tecentriq contre 8,9 mois (IC à 95%: 7,7; 11,5) dans le bras sous le docétaxel (HR=0,75, IC à 95%: 0,59; 0,96).
Dans l'ensemble, les résultats concernant la survie globale observés dans le cadre de l'analyse primaire de l'efficacité (850 patients) dans le groupe en ITT et dans les sous-groupes d'expression du PD-L1 ont montré sous Tecentriq un bénéfice en termes d'OS dans tous les sous-groupes, y compris dans celui avec une expression du PD-L1< 1% sur les TC et les IC.
La proportion des patients en ITT présentant une réponse confirmée évaluée par le médecin investigateur selon les critères RECIST V1.1 était comparable dans les deux bras: 13,4% (IC à 95%: 10,32; 17,02) dans le bras sous le docétaxel et 13,6% (IC à 95%: 10,53; 17,28) dans le bras sous Tecentriq. Le traitement par Tecentriq a été associé à l'obtention d'une réponse durable. Chez les répondeurs, la DOR médiane a été significativement plus longue (16,3 mois) dans le bras sous Tecentriq que dans le bras sous le docétaxel (6,2 mois). Dans le groupe en ITT, la survie sans progression (PFS) médiane a été de 2,8 mois (IC à 95%: 2,6; 3,0) dans le bras sous Tecentriq et de 4,0 mois (IC à 95%: 3,3; 4,2) dans le bras sous le docétaxel pour un HR de 0,95 (IC à 95% 0,82; 1,10).
Une amélioration de l'OS sous Tecentriq par rapport au docétaxel a été observée non seulement chez les patients atteints d'un NSCLC non épidermoïde) (HR de 0,73; IC à 95%: 0,60-0,89; OS médiane de 15,6 mois sous Tecentriq contre 11,2 mois sous le docétaxel), mais également chez les patients atteints d'un NSCLC épidermoïde (HR de 0,73; IC à 95%: 0,54-0,98; OS médiane de 8,9 mois sous Tecentriq contre 7,7 mois sous le docétaxel).
Dans le cadre de l'analyse secondaire de l'efficacité (date limite de collecte des données: 23 janvier 2017) avec une durée médiane de suivi de 26 mois, le traitement par Tecentriq était toujours associé à un allongement statistiquement significatif de l'OS par rapport au docétaxel. L'OS médiane de l'ensemble des 1125 patients randomisés a été supérieure de 3,5 mois dans le bras sous Tecentriq: 13,3 mois (IC à 95%: 11,3; 14,9) dans le bras sous Tecentriq et 9,8 mois (IC à 95%: 8,8; 11,3) dans le bras sous le docétaxel. Le HR stratifié a été de 0,80 (IC à 95%: 0,70; 0,92).
POPLAR
GO28753 (POPLAR), une étude de phase II multicentrique, internationale, randomisée, ouverte et contrôlée, a été réalisée chez des patients atteints de NSCLC localement avancé ou métastatique avec progression de la maladie pendant ou après une chimiothérapie incluant du platine. La variable d'efficacité principale était la survie globale. Au total, 287 patients ont été randomisés selon un rapport 1:1 pour recevoir soit Tecentriq soit le docétaxel. La randomisation a été stratifiée en fonction de l'expression du PD-L1, du nombre de protocoles de chimiothérapie antérieurs et du type histologique. Une analyse actualisée comprenant au total 200 décès recensés et un suivi médian de la survie de 22 mois a montré une OS médiane de 12,6 mois chez les patients traités par Tecentriq contre 9,7 mois chez les patients traités par le docétaxel (HR de 0,69; IC à 95%: 0,52-0,92). L'ORR a été de 15,3% sous Tecentriq contre 14,7% sous le docétaxel; la DOR médiane a été de 18,6 mois sous Tecentriq contre 7,2 mois sous le docétaxel.
Traitement de première intention du cancer du poumon à petites cellules de stade avancé (1L ES – SCLC)
IMpower133
L'étude de phase I/III GO30081 (IMpower133) contrôlée contre placebo, multicentrique, en double aveugle et randomisée a été réalisée pour évaluer l'efficacité et la sécurité de Tecentriq en association au carboplatine et à l'étoposide chez des patients atteints de cancer du poumon à petites cellules de stade avancé (ES-SCLC) sans chimiothérapie préalable et ayant un score de performance ECOG de 0 ou 1. Un total de 403 patients ont été randomisés (1:1) pour recevoir l'un des schémas thérapeutiques suivants: bras A: Tecentriq 1200 mg + carboplatine AUC 5 + étoposide 100 mg/m2 par perfusion intraveineuse toutes les 3 semaines pendant quatre cycles d'induction, suivi d'un traitement d'entretien par Tecentriq 1200 mg toutes les 3 semaines; ou bras B: placebo + carboplatine AUC 5 + étoposide 100 mg/m2 par perfusion intraveineuse toutes les 3 semaines pendant quatre cycles d'induction, suivis d'un traitement d'entretien par le placebo toutes les 3 semaines. Tecentriq et le carboplatine ont été administrés au jour 1 et l'étoposide aux jours 1, 2 et 3 de chaque cycle d'induction. Le carboplatine et l'étoposide ont été administrés jusqu'à la fin des quatre cycles ou jusqu'à la progression de la maladie ou jusqu'à l'apparition d'une toxicité inacceptable, selon ce qui survenait en premier. Tecentriq a été administré jusqu'à ce que, de l'avis du médecin investigateur, il n'y ait plus de bénéfice clinique. La randomisation a été stratifiée selon le sexe, le score de performance ECOG (0 ou 1) et la présence de métastases cérébrales.
Les patients présentant des métastases actives ou non traitées dans le SNC, des antécédents de maladie auto-immune, ayant reçu un vaccin vivant atténué dans les 4 semaines précédant la randomisation ou ayant reçu des médicaments immunosuppresseurs systémiques dans la semaine précédant la randomisation ont été exclus de l'étude. Les évaluations de la tumeur ont été réalisées toutes les 6 semaines durant les 48 premières semaines suivant le cycle 1 au jour 1, puis toutes les 9 semaines. Chez les patients qui ont poursuivi le traitement après la progression de la maladie, une évaluation de la tumeur a été réalisée toutes les 6 semaines jusqu'à l'arrêt du traitement.
Les données démographiques et les caractéristiques de la maladie dans la population de l'analyse primaire étaient bien équilibrées entre les bras thérapeutiques. Au total, 403 patients ont été randomisés dans l'étude: 201 patients dans le bras A recevant Tecentriq, le carboplatine et l'étoposide, et 202 patients dans le bras B recevant le placebo, le carboplatine et l'étoposide. L'âge médian était de 64 ans (intervalle: de 26 à 90 ans). La majorité des patients (65%) étaient des hommes et de race blanche (80%), 9% présentaient des métastases cérébrales et la plupart des patients (97%) étaient des fumeurs ou d'anciens fumeurs. La valeur initiale (ligne de base) du score de performance ECOG était de 0 (35%) ou de 1 (65%).
Les deux critères d'évaluation principaux étaient la survie globale (OS: overall survival) et la survie sans progression (PFS: progression-free survival) évaluée par le médecin investigateur. Les principaux résultats de cette étude lors d'un suivi médian de 13,9 mois de la durée de vie restante sont résumés ci-dessous.
Le traitement par Tecentriq + carboplatine et étoposide (CE) a été associé à un allongement statistiquement significatif de l'OS par rapport au placebo + CE. Pour les patients de la population en ITT (intention de traiter), l'OS médiane était 2 mois plus longue dans le bras de traitement sous Tecentriq + CE, où elle était de 12,3 mois (IC à 95%: 10,8; 15,9), contre 10,3 mois (IC à 95%: 9,3; 11,3) dans le bras de traitement sous placebo + CE (valeur de p stratifiée = 0,0069). Le hazard ratio (HR) stratifié était de 0,70 (IC à 95%: 0,54; 0,91); cela correspond à une réduction relative du risque de mortalité de 30% sous Tecentriq + CE par rapport au placebo + CE. Après 12 mois, le bras de traitement sous Tecentriq + CE comptait encore 51,7% patients en vie, contre 38,2% dans le bras de traitement sous placebo + CE. Chez les patients de moins de 65 ans, la valeur médiane de l'OS dans le bras sous Tecentriq + CE était de 12,1 mois, contre 11,5 mois dans le groupe sous placebo + CE (HR non stratifié: 0,92, IC à 95%: 0,64; 1,32). Chez les patients âgés de 65 ans et plus, la valeur médiane de l'OS dans le bras sous Tecentriq + CE était de 12,5 mois, contre 9,6 mois dans le groupe sous placebo + CE (HR non stratifié: 0,53, IC à 95%: 0,36; 0,77).
Les données des analyses exploratoires des sous-groupes d'expression du PD-L1, basées sur un nombre limité d'échantillons de patients dans cette étude (34% de la population en intention de traiter (ITT)), ont montré pour les patients PD-L1-négatifs (<1% sur les TC et les IC) un OS HR (hazard ratio pour la survie globale) de 0,51 (IC à 95%: 0,30; 0,89) avec une survie globale moyenne de 10,2 mois dans le bras Tecentriq + CE versus 8,3 mois dans le bras placebo + CE. Pour les patients PD-L1-positifs (≥1% sur les TC ou les IC), l'OS HR était de 0,87 (IC à 95%: 0,51; 1,49) avec une survie globale moyenne de 9,7 mois dans le bras Tecentriq + CE versus 10,6 mois dans le bras placebo + CE. Ces données sont cependant trop limitées pour pouvoir tirer des conclusions. L'incidence des anticorps anti-atézolizumab (ADA) survenus au cours du traitement a été de 18,6% (35 patients sur 188) dans le bras Tecentriq + CE. Dans une analyse sans ajustement l'OS médiane a été de 12,6 mois dans le groupe ADA-négatif et de 10,9 mois dans le groupe ADA-positif. Dans le groupe placebo + CE, l'OS a été de 10,3 mois. Étant donné le faible nombre de cas dans le groupe ADA-positif, aucune conclusion définitive ne peut être tirée concernant une éventuelle influence des ADA sur l'efficacité.
Le traitement par Tecentriq + CE a été associé à une amélioration statistiquement significative de la PFS évaluée par le médecin investigateur (selon RECIST v1.1) par rapport au placebo + CE. Chez les patients du collectif en ITT, la durée médiane de la PFS était de 5,2 mois (IC à 95%: 4,4; 5,6) dans le bras de traitement sous Tecentriq + CE, contre 4,3 mois (IC à 95%: 4,2; 4,5) dans le bras de traitement sous placebo + CE (valeur de p stratifiée = 0,0170). Le HR stratifié était de 0,77 (IC à 95%: 0,62; 0,96). Après 6 mois, la proportion de patients sans événements liés à la PFS (progression ou décès) était de 30,9% dans le bras de traitement sous Tecentriq + CE et de 22,4% dans le bras de traitement sous placebo + CE. Après 12 mois, la proportion de patients sans événements liés à la PFS était de 12,6% dans le bras de traitement sous Tecentriq + CE et de 5,4% dans le bras de traitement sous placebo + CE.
Dans le collectif en ITT, la proportion de patients dont la réponse objective a été confirmée par l'évaluation du médecin investigateur selon RECIST v1.1 était de 60,2% dans le bras de traitement sous Tecentriq + CE, contre 64,4% dans le groupe de traitement sous placebo + CE.
Traitement de première ligne du cancer du sein triplement négatif (1L TNBC)
IMpassion130
L'étude de phase III randomisée, à 2 bras, menée en double aveugle et contrôlée contre placebo WO29522 (IMpassion130) a été réalisée pour évaluer l'efficacité et la sécurité de Tecentriq en association avec le nab-paclitaxel chez des patients atteints de cancer du sein triplement négatif (TNBC) non résécable, localement avancé ou métastatique, n'ayant reçu au préalable aucune chimiothérapie ni aucun traitement systémique ciblé en raison d'un TNBC inopérable, localement avancé ou métastatique. Au total, 902 patients ont été inclus dans l'étude et le collectif a été stratifié selon la présence de métastases hépatiques, un traitement préalable à base de taxane et le statut de l'expression du PD-L1 dans les cellules immunitaires (CI) infiltrant la tumeur (cellules immunitaires [CI] infiltrant la tumeur avec coloration de PD-L1< 1% de la surface de la tumeur contre ≥1% de la surface de la tumeur), évalué à l'aide du test VENTANA PD-L1 (SP142).
Les patients ont été randomisés pour recevoir soit Tecentriq (840 mg), soit un placebo en perfusions i.v. aux jours 1 et 15 plus nab-paclitaxel (100 mg/m2) sous forme de perfusion i.v. aux jours 1, 8 et 15 de chaque cycle de 28 jours. Les patients ont reçu le traitement jusqu'à l'apparition d'une progression tumorale radiologique, selon RECIST version 1.1, ou jusqu'à la survenue d'une toxicité intolérable. Le traitement par Tecentriq a été poursuivi en cas d'interruption du nab-paclitaxel pour cause de toxicité intolérable.
Étaient notamment exclus de l'étude les patients présentant des antécédents de maladie auto-immune; les patients ayant reçu un vaccin vivant atténué au cours des 4 semaines précédant la randomisation; les patients ayant reçu des substances actives immunostimulatrices systémiques dans les 4 semaines ou des médicaments immunosuppresseurs systémiques dans les 2 semaines précédant la randomisation; les patients présentant des métastases cérébrales non traitées ou dépendantes des corticostéroïdes; les patients ayant des antécédents de transplantation de cellules souches allogéniques ou d'organe solide; les patients atteints de fibrose pulmonaire idiopathique active ou anamnestique et de pneumopathie inflammatoire (à l'exception d'une pneumopathie inflammatoire actinique dans le champ d'irradiation) et de pneumonie organisante; ou encore les patients atteints d'infections actives, telles que VIH, hépatite B et C ainsi que tuberculose. Une évaluation de la tumeur a été effectuée toutes les 8 semaines durant les 12 premiers mois, puis toutes les 12 semaines.
Au début de l'étude, la population de l'étude était comparable en termes de données démographiques et de caractéristiques de la maladie dans les deux bras de l'étude. La grande majorité des patients (99,6%) était de sexe féminin. 67,5% des patients étaient de race caucasienne, 17,8% étaient d'origine asiatique, 6,5% étaient noirs ou afroaméricains et 4,4% étaient des indiens ou des autochtones de l'Alaska. L'âge médian était de 55 ans (intervalle: de 20 à 86 ans). La valeur initiale du statut de performance ECOG était 0 (58,4%) ou 1 (41,3%). Dans l'ensemble, lors de l'inclusion dans l'étude, 41% des patients inclus présentaient une expression de PD-L1 ≥1%, 27% présentaient des métastases hépatiques et 7% avaient des métastases cérébrales. Près de la moitié des patients était sous taxane (51%) ou anthracycline (54%) en guise de traitement (néo-)adjuvant. Les données démographiques des patients et la maladie tumorale lors de l'inclusion dans l'étude de la population avec expression du PD-L1 ≥1% étaient en général représentatives de la population totale de l'étude.
Les résultats de la PFS (survie sans progression), de l'ORR (taux de réponse objective) et de la DOR (durée de réponse) issus de cette étude sont résumés dans le tableau 7 et dans le texte ci-dessous et ils concernent les patients présentant une expression du PD-L1 ≥1% avec une durée médiane de suivi des patients survivants de 13 mois.
Une analyse finale de l'OS avec une expression de PD-L1 ≥1% avec une période d'observation médiane de 19,12 mois a été réalisée. Les résultats de l'OS sont présentés dans le tableau 5.
Aucun bénéfice en termes d'OS n'a été observé sous traitement combiné par l'atézolizumab + nab-paclitaxel par rapport au placebo + nab-paclitaxel chez les patients pour lesquels ≥24 mois avaient passé entre la dernière opération et le diagnostic de maladie tumorale métastatique ou localement avancée inopérable.
Tableau 7: Résumé des données de l'efficacité chez les patients présentant une expression du PD-L1 ≥1% (IMpassion130)
|
Critères d'évaluation de l'efficacité importants
|
Tecentriq + nab-paclitaxel
|
Placebo + nab-paclitaxel
| |
Critères co-primaires d'évaluation
| |
Survie sans progression (PFS) selon l'appréciation du médecin investigateur (RECIST vers. 1.1 – Analyse primaire3)
|
n = 185
|
n = 184
| |
Nombre d'événements (%)
|
138 (74,6%)
|
57 (85,3%)
| |
Durée médiane de la survie sans progression (en mois)
|
7,5
|
5,0
| |
IC à 95%
|
(6,7; 9,2)
|
(3,8; 5,6)
| |
Hazard-Ratio stratifié ‡ (IC à 95%)
|
0,62 (0,49; 0,78)
| |
Valeur de p1
|
< 0,0001
| |
Survie sans progression de 12 mois (%)
|
29,1
|
16,4
| |
Survie sans progression (PFS) selon l'appréciation du médecin investigateur (RECIST vers. 1.1) – Analyse exploratoire actualisée 4
|
|
| |
Nombre d'événements (%)
|
149 (80,5%)
|
163 (88,6%)
| |
Durée médiane de la survie sans progression (en mois)
|
7,5
|
5,3
| |
IC à 95%
|
(6,7; 9,2)
|
(3,8; 5,6)
| |
Hazard-Ratio stratifié ‡ (IC à 95%)
|
0,63 (0,50-0,80)
| |
Survie sans progression de 12 mois (%)
|
30,3
|
17,3
| |
Analyse de la survie globale1, 2, 5
|
n = 185
|
n = 184
| |
Nombre de décès (%)
|
120 (64,9%)
|
139 (75,5%)
| |
Délai moyen jusqu'à l'événement (en mois)
|
25,4
|
17,9
| |
IC à 95%
|
(19,6; 30,7)
|
(13,6; 20,3)
| |
Hazard-Ratio ‡ stratifié (IC à 95%)
|
0,67 (0,53; 0,86)
|
1 Sur la base du test de log-rank stratifié
2 La comparaison des survies globales entre les groupes de traitement de patients présentant une expression du PD-L1 ≥1% n'a pas été testée formellement selon la hiérarchie des analyses spécifiée à l'avance
3 D'après l'analyse finale de la PFS, de l'ORR et de la DOR et une première analyse intermédiaire de l'OS au moment du cut-off clinique du 17 avril 2018
4 D'après une analyse exploratoire de la PFS au moment du cut-off clinique du 2 janvier 2019
5 D'après l'analyse finale de l'OS au moment du cut-off clinique du 14 avril 2020
‡ Stratifié en fonction de la présence de métastases hépatiques et d'un traitement antérieur par taxane
PFS = survie sans progression (progression-free survival); RECIST = Response Evaluation Criteria in Solid Tumors v1.1. (critères d'évaluation de la réponse pour les tumeurs solides, version 1.1); IC = intervalle de confiance; OS = survie globale (overall survival), NE = non appréciable (not estimable)
Chez les patients présentant une expression positive du PD-L1, un taux de réponse globale (Overall Response Rate, ORR) numériquement plus élevé a été observé dans le groupe sous Tecentriq + nab-paclitaxel (58,9%) par rapport au groupe sous placebo + nab-paclitaxel (42,6%). Parmi les répondeurs, le nombre de réponses persistantes dans le bras sous Tecentriq + nab-paclitaxel était supérieur (35,8%) à celui observé dans le bras sous placebo + nab-paclitaxel (24,4%). La durée de réponse (Duration of Response, DOR) médiane estimée pour le groupe sous Tecentriq + nab-paclitaxel était supérieure de 3 mois (8,5 mois contre 5,5 mois dans le groupe sous placebo + nab-paclitaxel, HR non stratifié: 0,60; IC à 95%; 0,43; 0,86).
Mélanome
IMspire150
CO39262 (IMspire150), une étude de phase III randomisée, en double aveugle, à deux bras, contrôlée contre placebo, a été menée afin d'évaluer l'efficacité et la sécurité de Tecentriq en association avec du cobimétinib et du vémurafénib chez des patients atteints d'un mélanome localement avancé, métastatique ou non résécable, porteur de la mutation BRAF-V600. La mutation BRAF V600 du mélanome a été détectée à l'aide d'un test disponible localement et a été confirmée de manière centralisée par le test FoundationOneTM. Les patients n'avaient pas reçu auparavant de traitement systémique pour leur mélanome. Au total, 514 patients ont été randomisés pour recevoir l'un des régimes de traitement décrits ci-dessous. La randomisation a été stratifiée en fonction de la localisation géographique et du taux initial de lactate déshydrogénase (LDH).
Le traitement a été effectué par cycles de 28 jours. Au cours du cycle 1, tous les patients ont reçu 960 mg de vémurafénib par voie orale deux fois par jour et 60 mg de cobimétinib par voie orale une fois par jour pendant 21 jours, puis soit 720 mg de vémurafénib + placebo deux fois par jour pendant 7 jours dans le groupe Tecentriq, soit 960 mg dans le groupe témoin. À partir du cycle 2, les patients du groupe Tecentriq ont reçu 840 mg de Tecentriq par voie intraveineuse aux jours 1 et 15 ainsi que 720 mg de vémurafénib + placebo deux fois par jour (pendant 28 jours) et 60 mg de cobimétinib une fois par jour (prise de médicament pendant 21 jours, suivi d'une pause de 7 jours). Les patients du groupe témoin ont reçu un placebo par voie intraveineuse aux jours 1 et 15 ainsi que 960 mg de vémurafénib deux fois par jour et du cobimétinib une fois par jour pendant 21 jours, suivi d'une pause de 7 jours. Des réductions de dose et des interruptions de traitement ont été autorisées pour le vémurafénib et le cobimétinib, tandis que, pour Tecentriq, seules les interruptions de traitement pour gérer des événements indésirables ont été autorisées. Le traitement a été poursuivi jusqu'à ce que la progression de la maladie, la mort ou une toxicité inacceptable aient été établies par le médecin investigateur. Le croisement n'était pas autorisé en cas de progression. Les patients n'ont pas été inclus dans l'étude s'ils présentaient des antécédents de maladie auto-immune, s'ils avaient reçu un vaccin vivant atténué dans les 28 jours précédant la randomisation, s'ils avaient reçu un traitement immunostimulant systémique dans les 4 semaines ou un traitement immunosuppresseur systémique dans les 2 semaines précédant la randomisation. Les évaluations tumorales ont été réalisées toutes les 8 semaines (± 1 semaine) pendant les 24 premiers mois et toutes les 12 semaines (± 1 semaine) par la suite.
Les données démographiques et les caractéristiques de la maladie à l'inclusion de l'étude de la population étudiée étaient équilibrées entre les deux bras de traitement. L'âge médian s'élevait à 54 ans (fourchette: de 22 à 88 ans), 58% des patients étaient de sexe masculin. La majorité des patients étaient de type caucasien (95%). 2,5% des patients avaient déjà reçu un traitement pour des métastases cérébrales, 30% des patients avaient des métastases hépatiques au moment de leur inclusion dans l'étude et 14% des patients avaient reçu un traitement adjuvant systémique. Le mélanome a été classifié, selon la classification de l'American Joint Committee on Cancer (AJCC), septième édition, comme étant de stade IIIC chez 5,8% des patients et de stade IV chez 93,8% des patients (M1A: 14,8; M1B: 19,1%; M1C: 59,9%).
Au début de l'étude, le statut de performance ECOG était de 0 (76,5%) ou de 1 (22,8%). La proportion de patients présentant une LDH élevée était de 32,8% dans le groupe de traitement avec Tecentriq et de 32,9% dans le groupe de traitement sans Tecentriq.
D'après les tests effectués de manière centralisée, 74% des patients présentaient la mutation V600E, 11% la mutation V600K et 1% la mutation V600D ou V600R.
Le critère d'évaluation principal de l'efficacité était la survie sans progression (PFS), évaluée par le médecin investigateur. À la fin de la période de collecte des données (analyse primaire), la période d'observation médiane était de 18,9 mois.
Dans le sous-groupe Tecentriq V600E, la PFS médiane était de 16,2 mois selon l'évaluation du médecin investigateur contre 10,0 mois dans le sous-groupe témoin V600E (Hazard Ratio non stratifié: 0,68; IC à 95%: 0,53; 0,88). Les critères secondaires comprenaient la PFS médiane, évaluée par une commission d'examen indépendante (IRC); elle était de 17,6 mois dans le sous-groupe Tecentriq V600E et de 11,4 mois dans le sous-groupe témoin V600E (Hazard Ratio non stratifié: 0,71; IC à 95%: 0,55; 0,93). Une analyse intermédiaire de la survie globale (OS) a montré un rapport de risque non stratifié de 0,69 (IC à 95%: 0,50; 0,95). Les taux de réponse objective (évalués par le médecin investigateur) étaient similaires dans les deux groupes; ils étaient de 68,9% dans le sous-groupe Tecentriq V600E et de 63,2% dans le sous-groupe témoin V600E. La durée médiane de la réponse (évaluée par le médecin investigateur) était de 20,4 mois dans le sous-groupe Tecentriq V600E contre 12,6 mois dans le sous-groupe témoin V600E.
Tecentriq en association avec du vémurafénib et du cobimétinib n'a pas été étudié chez les patients atteints d'un mélanome exprimant un BRAF de type sauvage.
CHC
IMbrave150
Une étude internationale de phase III, randomisée, multicentrique, en ouvert, YO40245 (IMbrave150), a été réalisée pour évaluer l'efficacité et la sécurité de Tecentriq en association avec le bévacizumab chez des patients atteints de CHC localement avancé ou métastatique et/ou inopérable n'ayant pas reçu de traitement systémique antérieur. Au total, 501 patients ont été randomisés (rapport 2:1) pour recevoir 1200 mg de Tecentriq et 15 mg/kg de bévacizumab toutes les trois semaines en perfusion intraveineuse ou 400 mg de sorafénib deux fois par jour par voie orale. La randomisation a été stratifiée en fonction de la région géographique (Asie, à l'exclusion du Japon vs reste du monde), de l'invasion macrovasculaire et/ou de l'extension extrahépatique (présente vs absente), du taux initial d'AFP (< 400 vs ≥400 ng/ml) et de l'indice de performance ECOG (0 vs 1). Dans les deux bras, les patients ont reçu le traitement jusqu'à la perte du bénéfice clinique ou la survenue d'une toxicité inacceptable. Les patients pouvaient arrêter Tecentriq ou le bévacizumab (p.ex. en raison d'effets indésirables) et poursuivre par une monothérapie jusqu'à la perte du bénéfice clinique ou la survenue d'une toxicité inacceptable associée à la monothérapie.
Les patients inclus dans l'étude étaient des adultes (Child-Pugh A, ECOG 0/1) n'ayant pas reçu de traitement systémique antérieur. Les hémorragies (dont des événements d'issue fatale) sont un effet indésirable connu du bévacizumab et les hémorragies gastro-intestinales hautes sont une complication fréquente engageant le pronostic vital chez les patients atteints d'un CHC. Par conséquent, la présence éventuelle de varices devait avoir été évaluée chez les patients dans les 6 mois précédant le traitement. Les critères d'exclusion de l'étude étaient un saignement variqueux dans les 6 mois précédant le traitement, des varices non traitées ou partiellement traitées avec saignement ou risque élevé de saignement. Les critères d'exclusion étaient un saignement variqueux dans les 6 mois précédant le traitement, des varices non traitées, une ascite modérée à sévère, des antécédents d'encéphalopathie hépatique, une anamnèse connue de maladie auto-immune, l'administration d'un vaccin vivant atténué ou d'immunostimulants systémiques dans les 4 semaines précédant la randomisation ou l'administration d'immunosuppresseurs systémiques dans les 2 semaines précédant la randomisation ainsi que des métastases cérébrales non traitées ou corticodépendantes. Les évaluations tumorales ont été effectuées toutes les 6 semaines durant les 54 premières semaines, puis toutes les 9 semaines.
Les caractéristiques démographiques et pathologiques à l'inclusion dans la population de l'étude étaient bien équilibrées entre les deux bras de traitement. L'âge médian était de 65 ans (intervalle: de 26 à 88 ans) et 83% des patients étaient de sexe masculin. La plupart des patients étaient Asiatiques (57%) et Blancs (35%). 40% des patients étaient originaires d'Asie (à l'exclusion du Japon) et 60% provenaient du reste du monde. Au départ, environ 75% des patients présentaient une invasion macrovasculaire et/ou une extension extrahépatique, et 37% avaient un taux initial d'AFP ≥400 ng/ml. L'indice de performance ECOG au début de l'étude était de 0 (62%) ou de 1 (38%). Les facteurs de risque primaires de développement d'un CHC étaient une infection par le virus de l'hépatite B chez 48% des patients, une infection par le virus de l'hépatite C chez 22% des patients et une affection non virale chez 31% des patients. 82% des patients avaient un CHC de stade C, 16% un CHC de stade B et 3% un CHC de stade A selon la classification BCLC (Barcelona Clinic Liver Cancer).
Les deux critères co-primaires d'évaluation de l'efficacité étaient la survie globale (OS) et la survie sans progression (PFS) évaluée par un centre d'examen indépendant (IRF) selon les critères RECIST v1.1. Au moment de l'analyse primaire, la durée médiane de suivi était de 8,6 mois. Dans le groupe sous Tecentriq + bévacizumab, une amélioration statistiquement significative des deux critères co-primaires d'évaluation de l'efficacité, a été obtenue dans la population en ITT par rapport au sorafénib. L'OS médiane n'a pas été atteinte (NR) dans le bras traité par l'association Tecentriq + bévacizumab (IC à 95%: NR; NR) et était de 13,2 mois dans le bras sous sorafénib (IC à 95%: 10,4; NR). Le HR stratifié était de 0,58 (IC à 95%: 0,42; 0,79; valeur de p stratifiée = 0,0006), ce qui correspond à une diminution de 42% du risque de mortalité lié à l'association Tecentriq + bévacizumab par rapport au sorafénib.
La PFS médiane était de 6,8 mois dans le bras traité par Tecentriq + bévacizumab (IC à 95%: 5,8; 8,3) et de 4,3 mois dans le bras sous sorafénib (IC à 95%: 4,0; 5,6). L'HR stratifié était de 0,59 (IC à 95%: 0,47; 0,76; valeur de p stratifiée < 0,0001), ce qui correspond à une diminution de 41% du risque de progression de la maladie ou de mortalité lié à l'association Tecentriq + bévacizumab par rapport au sorafénib.
Une amélioration statistiquement significative de l'ORR évaluée par l'IRF selon les critères RECIST v1.1, de 27,3% (IC à 95%: 22,5%, 32,5%), a été observée dans le bras traité par Tecentriq + bévacizumab contre 11,9% (IC à 95%: 7,4%, 18,0%) dans le bras sous sorafénib. Le taux de patients avec une réponse complète était de 5,5% sous Tecentriq + bévacizumab contre 0% dans le bras sous sorafénib.
Une analyse descriptive et actualisée de l'efficacité a été réalisée sur une période médiane de suivi de la survie de 15,6 mois. L'OS médiane était de 19,2 mois (IC à 95%: 17,0; 23,7) dans le bras traité par l'association Tecentriq + bévacizumab et de 13,4 mois (IC à 95%: 11,4; 16,9) dans le bras sous sorafénib. L'HR stratifié était de 0,66 (IC à 95%: 0,52; 0,85), ce qui correspond à une diminution de 34% du risque de mortalité lié à l'association Tecentriq + bévacizumab par rapport au sorafénib.
GO30140
Une étude internationale de phase Ib, en ouvert, multicentrique, à plusieurs bras (GO30140) a également été réalisée chez des patients atteints d'une tumeur solide. Un protocole randomisé a été utilisé dans le bras F de l'étude pour évaluer la sécurité et l'efficacité de l'administration de Tecentriq en association avec le bévacizumab par rapport à Tecentriq en monothérapie chez des patients atteints de CHC avancé ou métastatique et/ou inopérable qui n'avaient pas reçu de traitement systémique antérieur. Le principal critère d'évaluation de l'efficacité était la PFS évaluée par un IRF selon les critères RECIST v1.1. Au total, 119 patients ont été randomisés (rapport 1:1) pour recevoir Tecentriq (1200 mg) et le bévacizumab (15 mg/kg) toutes les 3 semaines en perfusion intraveineuse ou seulement Tecentriq (1200 mg) toutes les 3 semaines. Au moment de l'analyse primaire, la durée médiane de suivi de la survie était de 6,6 mois. L'association Tecentriq + bévacizumab a montré un bénéfice statistiquement significatif en termes de PFS par rapport à Tecentriq en monothérapie (HR: 0,55, IC à 80%: 0,40; 0,74, valeur de p = 0,0108), avec une PFS médiane de 5,6 mois chez les patients traités par Tecentriq et bévacizumab, contre 3,4 mois chez les patients traités par Tecentriq en monothérapie.
Carcinome urothélial
Utilisation de Tecentriq chez des patients atteints d'un carcinome urothélial et présentant des facteurs pronostiques défavorables après un traitement préalable par une chimiothérapie à base de platine
Avant d'instaurer un traitement par Tecentriq chez des patients présentant des facteurs pronostiques défavorables ou une maladie agressive, le médecin doit tenir compte de l'effet différé du traitement par Tecentriq. Dans le cadre de l'étude IMvigor211, une incidence de décès plus élevée a été constatée chez les patients atteints d'un carcinome urothélial pendant les 4 premiers mois du traitement par Tecentriq, par rapport à une chimiothérapie. Les facteurs associés à des décès précoces sont les suivants: ≥2 facteurs de risque de Bellmunt (composé d'un score ECOG > 0, de métastases hépatiques et d'un taux d'hémoglobine < 10 g/dl), taux d'ALP élevé et/ou métastases hépatiques à l'entrée dans l'étude.
IMvigor211
GO29294 (IMvigor211), une étude de phase III ouverte, multicentrique, internationale et randomisée, a été réalisée pour évaluer l'efficacité et la sécurité de Tecentriq par rapport à une chimiothérapie (vinflunine, docétaxel ou paclitaxel selon le choix du médecin investigateur) chez des patients atteints d'un carcinome urothélial inopérable, localement avancé ou métastatique, avec progression de la maladie pendant ou après une chimiothérapie à base de platine. Les patients présentant les antécédents suivants ont été exclus de l'étude: patients atteints d'une maladie auto-immune ou de métastases cérébrales évolutives ou corticodépendantes, patients ayant reçu un vaccin vivant atténué durant les 28 jours précédant l'inclusion dans l'étude et ayant reçu des immunostimulants systémiques durant les 4 semaines précédant l'inclusion ou des médicaments immunosuppresseurs systémiques durant les 2 semaines avant l'inclusion dans l'étude. Les évaluations de la tumeur ont été réalisées toutes les 9 semaines pendant les 54 premières semaines, puis toutes les 12 semaines. Des échantillons de tumeur ont été évalués prospectivement à la recherche de l'expression du PD-L1 sur des IC infiltrant les tumeurs, et, sur la base des résultats obtenus, des sous-groupes de l'expression du PD-L1 ont été définis pour les analyses décrites ci-après.
Au total, 931 patients ont été inclus. Les patients ont été randomisés (1:1) pour recevoir soit Tecentriq, soit une chimiothérapie. La randomisation a été stratifiée selon la chimiothérapie (vinflunine contre taxane), le statut d'expression du PD-L1 sur les IC (< 5% contre ≥5%), le nombre de facteurs de risque pronostiques (0 contre 1 à 3) et les métastases hépatiques (oui contre non). Les facteurs de risque pronostiques ont inclus une période < 3 mois depuis la dernière chimiothérapie, un score de performance ECOG > 0 et un taux d'hémoglobine < 10 g/dl.
Tecentriq a été administré en perfusion i.v. à la dose fixe de 1200 mg toutes les 3 semaines. Une réduction de la dose de Tecentriq n'était pas autorisée. Les patients ont été traités jusqu'à la perte du bénéfice clinique d'après l'évaluation du médecin investigateur ou jusqu'à l'apparition d'une toxicité inacceptable. La vinflunine a été administrée en perfusion i.v. à la dose de 320 mg/m2 au jour 1 de chaque cycle de 3 semaines jusqu'à la progression de la maladie ou jusqu'à l'apparition d'une toxicité inacceptable. Le paclitaxel a été administré en perfusion i.v. pendant 3 heures à la dose de 175 mg/m2 au jour 1 de chaque cycle de 3 semaines jusqu'à la progression de la maladie ou jusqu'à l'apparition d'une toxicité inacceptable. Le docétaxel a été administré en perfusion i.v. à la dose de 75 mg/m2 au jour 1 de chaque cycle de 3 semaines jusqu'à la progression de la maladie ou jusqu'à l'apparition d'une toxicité inacceptable. Chez les patients traités, la durée médiane de traitement a été de 2,8 mois dans le bras sous Tecentriq, de 2,1 mois dans le bras sous la vinflunine et dans le bras sous le paclitaxel, et de 1,6 mois dans le bras sous le docétaxel.
Les données démographiques et les caractéristiques de la maladie présentes au début de l'étude dans le collectif de l'analyse primaire étaient équilibrées en comparant les bras de traitement. L'âge médian était de 67 ans (intervalle: 31 à 88) et 77,1% des patients étaient de sexe masculin. 53,9% des patients dans le bras sous chimiothérapie ont reçu de la vinflunine, 71,4% des patients présentaient au moins un facteur de risque pronostique défavorable et 28,8% présentaient des métastases hépatiques au début de l'étude. Le score de performance ECOG au début de l'étude était de 0 (45,6%) ou de 1 (54,4%). Chez 71,1% des patients, la tumeur primaire se situait dans la vessie et 25,4% des patients présentaient un carcinome urothélial dans les voies urinaires supérieures. 24,2% des patients n'avaient reçu qu'une seule thérapie adjuvante ou néoadjuvante à base de platine auparavant et avaient connu une progression de la maladie dans un intervalle de 12 mois.
Le critère d'évaluation principal de l'efficacité dans l'étude IMvigor211 était la survie globale (OS). Les critères d'évaluation secondaires de l'efficacité sont le taux de réponse objective (ORR), la survie sans progression (PFS) et la durée de la réponse (DOR) évalués par le médecin investigateur selon les critères RECIST (Response Evaluation Criteria in Solid Tumors) v1.1. Les comparaisons des OS entre le bras de traitement et le bras témoin ont été testées, comme décrit ci-après, en utilisant un procédé hiérarchique avec un ordre déterminé sur la base d'un test log-rank stratifié avec un niveau bilatéral de 5%: étape 1) Sous-groupe avec une expression du PD-L1 ≥5%, étape 2) Sous-groupe avec une expression du PD-L1 ≥1%, étape 3) Tous les patients satisfaisant aux critères d'inclusion («all comers»). Les résultats en termes d'OS pour les étapes 2 et 3 ne pouvaient à chaque fois être testés formellement que lorsque le résultat était statistiquement significatif lors de l'étape précédente.
Le suivi médian sur le plan de la survie a été de 17 mois. L'étude IMvigor211 n'a pas atteint le critère d'évaluation principal pour l'OS. Dans le sous-groupe de patients présentant des tumeurs avec une expression du PD-L1 ≥5%, Tecentriq n'a pas apporté de bénéfice statistiquement significatif en termes de survie par rapport à la chimiothérapie, avec un HR pour l'OS de 0,87 (IC à 95%: 0,63; 1,21; OS médiane de 11,1 contre 10,6 mois pour Tecentriq et la chimiothérapie). La valeur de p lors du test log-rank stratifié était de 0,41. Par conséquent, aucun test statistique formel portant sur l'OS n'a été réalisé dans le sous-groupe avec une expression du PD-L1 ≥1% ou dans la population en ITT, et les résultats de ces analyses sont considérés comme exploratoires.
La survie moyenne sans progression a été de 2,1 mois (intervalle de confiance à 95%: 2,1; 2,2) dans le bras sous Tecentriq et de 4,0 mois (IC à 95%: 3,4; 4,2) dans le bras sous chimiothérapie, avec un hazard ratio de 1,10 (IC à 95%: 0,95; 1,26). La proportion de patients en ITT présentant une réponse positive selon les critères RECIST version 1.1 confirmée par le médecin investigateur s'est révélée similaire dans les deux bras de l'étude: 13,4% (IC à 95%: 10,45; 16,87) dans le bras sous Tecentriq et 13,4% (IC à 95%: 10,47; 16,91) dans le bras sous chimiothérapie. Chez les patients répondeurs, la durée médiane de la réponse au traitement a été significativement plus longue dans le bras sous Tecentriq (21,7 mois) que dans le bras sous chimiothérapie (7,4 mois).
Dans l'analyse exploratoire finale (date de clôture du recueil des données: 8 novembre 2018) de la survie globale avec une durée médiane de suivi de 34 mois dans la population en ITT, la survie médiane était de 8,6 mois dans le bras Tecentriq (IC à 95%: 7,8; 9,6) et de 8,0 mois dans le bras chimiothérapie (IC à 95%: 7,2; 8,6) avec un hazard ratio de 0,82 (IC à 95%: 0,71; 0,94). Dans la population en ITT, des taux numériquement plus élevés d'OS ont été constatés à 12, 24 et 30 mois chez les patients du bras Tecentriq par rapport à ceux du bras chimiothérapie. Le pourcentage de patients en vie à 12 mois (estimation de KM) était de 32,5% dans le bras chimiothérapie et de 39,2% dans le bras Tecentriq. À 24 mois, 12,7% de patients étaient en vie dans le bras chimiothérapie et 22,5% dans le bras Tecentriq (estimation de KM). À 30 mois (estimations de KM), ils étaient 9,8% dans le bras chimiothérapie et 18,1% dans le bras Tecentriq.
Étude d'appoint chez des patients présentant un carcinome urothélial localement avancé ou métastatique, ayant reçu au préalable une chimiothérapie à base de platine: cohorte 2 de IMvigor210
GO29293 (IMvigor210), une étude clinique de phase II multicentrique, internationale, à un bras, avec deux cohortes, a été réalisée chez des patients présentant un carcinome urothélial localement avancé ou métastatique. Au total, 438 patients ont été inclus dans l'étude, qui était composée de deux cohortes. La cohorte 2 était constituée de patients ayant reçu au moins une chimiothérapie à base de platine pour traiter un carcinome urothélial localement avancé ou métastatique ou chez lesquels une progression de la maladie avait été constatée dans un délai de 12 mois après un traitement par une chimiothérapie adjuvante ou néoadjuvante à base de platine.
Les critères d'évaluation co-principaux dans la cohorte 2 étaient l'ORR confirmé par un centre d'examen indépendant (IRF) selon les critères RECIST v1.1, ainsi que l'ORR évalué par le médecin investigateur selon les critères RECIST modifiés (mRECIST). 310 patients ont été traités par Tecentriq en perfusion i.v. à la dose de 1200 mg toutes les 3 semaines jusqu'à la perte du bénéfice clinique. L'analyse primaire de la cohorte 2 a été effectuée après la réalisation d'un suivi de 24 semaines au minimum chez tous les patients. Dans la cohorte 2, l'étude a atteint les deux critères d'évaluation principaux, à savoir des ORR statistiquement significatifs dans le cadre de l'évaluation de l'IRF selon les critères RECIST v1.1 et dans le cadre de l'évaluation par le médecin investigateur selon les critères mRECIST, en comparaison avec un taux de rémission témoin historique de 10% préalablement déterminé.
Par ailleurs, une analyse a également été réalisée dans la cohorte 2 après une durée de suivi médiane de la survie de 21,1 mois. Les ORR confirmés par l'IRF selon les critères RECIST v1.1 ont été de 15,8% (IC à 95%: 11,9-20,4) au sein de la population en ITT, l'ORR confirmé par l'évaluation du médecin investigateur selon les critères mRECIST a été de 19,7% (IC à 95%: 15,4-24,6) et le taux d'OS après 12 mois a été de 37%. La DOR médiane n'a pas encore été atteinte dans la population en ITT.
Enfants et adolescents
Une étude multicentrique en ouvert d'une phase précoce été réalisée chez des patients pédiatriques (< 18 ans, n = 69) et chez des patients adultes jeunes (de 18 à 30 ans, n = 18) atteints d'une tumeur solide récurrente ou progressive ou d'un lymphome de Hodgkin ou non-hodgkinien afin d'évaluer la sécurité et la pharmacocinétique de Tecentriq. Les patients ont reçu 15 mg/kg d'atézolizumab par voie i.v. toutes les 3 semaines. Les principaux indicateurs d'efficacité étaient le taux de réponse objective (objective response rate, ORR), la survie sans progression (progression-free survival, PFS) et le taux de réponse avec bénéfice clinique (clinical benefit response rate, CBRR; seulement chez les patients atteints d'ostéosarcome).
Au moment de l'analyse primaire, l'ORR était de 4,6% (IC à 95%: 1,59; 10,89) et la durée moyenne de la PFS était de 1,3 mois (IC à 95%: 1,2; 1,4). Comme aucun patient de la cohorte atteinte d'ostéosarcome n'a obtenu de rémission complète ou partielle ou de stabilisation de la maladie pendant au moins 6 mois, le CBRR était de 0. Le bénéfice clinique de l'atézolizumab n'a pas été démontré chez les patients pédiatriques atteints de tumeurs solides récurrentes/réfractaires.
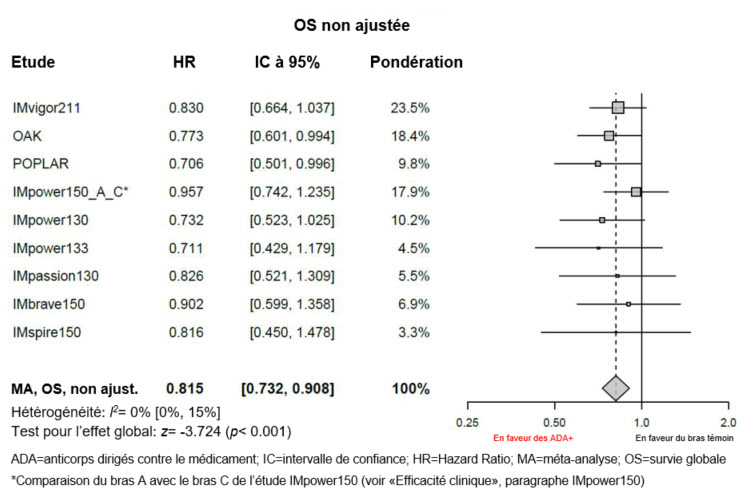
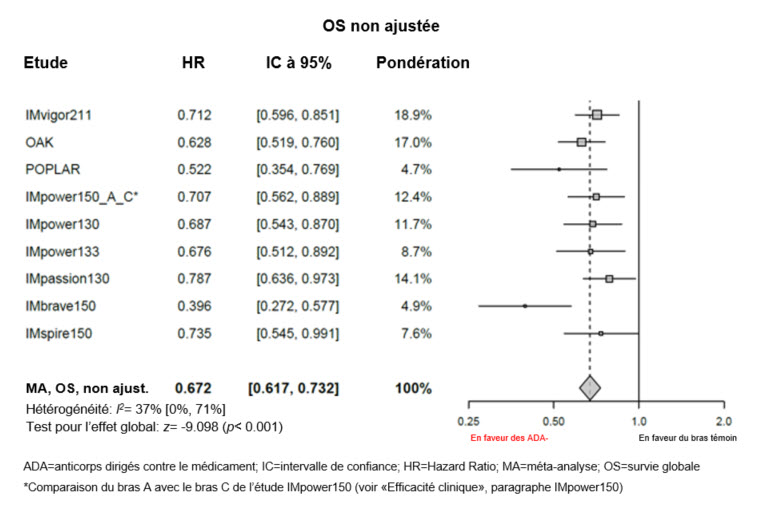
Efficacité de l'atézolizumab en présence d'anticorps dirigés contre le médicament
Des méta-analyses exploratoires, non ajustées, portant sur l'efficacité (sans ajustements de déséquilibres des caractéristiques initiales) ont été réalisées. Selon ces méta-analyses, la valeur estimée du HR pour la survie globale (OS) était de 0,815 entre le sous-groupe ADA-positif et le bras témoin (IC à 95%: 0,732; 0,908) et la valeur estimée du HR pour l'OS était de 0,672 entre le sous-groupe ADA-négatif et le bras témoin (IC à 95%: 0,617; 0,732), voir graphiques 1 et 2.
Graphique 1: Méta-analyse sur l'OS (non ajustée) de patients ADA-positifs par rapport au bras témoin
Graphique 2: Méta-analyse sur l'OS (non ajustée) de patients ADA-négatifs par rapport au bras témoin
|