Propriétés/EffetsCode ATC
L04AA37
Mécanisme d'action
Le baricitinib est un inhibiteur sélectif et réversible des Janus kinases (JAK), avec une plus forte sélectivité pour JAK 1 et JAK 2. Dans des tests d'activité d'enzymes isolées, le baricitinib a inhibé l'activité de JAK1, JAK2, de la tyrosine kinase 2 et de JAK3 avec des valeurs de CI50 de 5.9, 5.7, 53 et > 400 nM respectivement.
Les Janus kinases sont des enzymes qui sont impliquées dans la transduction des signaux intracellulaires provenant de récepteurs membranaires pour divers cytokines et facteurs de croissance impliqués dans l'hématopoïèse, l'inflammation et la fonction immunitaire. Dans la voie de signalisation intracellulaire, les JAK phosphorylent et activent des transducteurs de signaux et activateurs de transcription ((signal transducers and activators of transcription, STATs), qui activent l'expression des gènes dans la cellule. Le baricitinib module ces voies de signalisation par inhibition de l'activité enzymatique de JAK1 et de JAK2, réduisant ainsi la phosphorylation et l'activation des STATs.
Le baricitinib a été identifié comme un inhibiteur de NAK (numb-associated kinase), avec une affinité élevée pour la protéine kinase 1 associée à AP2 (AAK1) - 8.2 nM, pour BIKE - 20 nM et pour GAK - 120 nM. Les deux NAK, AAK1 et GAK, sont tout particulièrement associées à la pénétration du SARS-CoV-2 (COVID-19) dans les cellules humaines.
Pharmacodynamique
Inhibition de la phosphorylation de STAT3 induite par l'IL-6
L'administration du baricitinib a entraîné une inhibition dose-dépendante de la phosphorylation de STAT3 induite par l'IL-6 dans le sang total de sujets sains, avec une inhibition maximale qui a été observée 2 heures après l'administration de la dose et est revenue à une valeur proche de celle observée à l'inclusion dans les 24 heures. Des niveaux similaires d'inhibition ont été observés en utilisant l'IL-6 ou la thrombopoïétine (TPO) comme stimulus.
Immunoglobulines
Les valeurs moyennes d'IgG, d'IgM et d'IgA sériques ont diminué dans les 12 semaines suivant le début du traitement par Olumiant et sont restées stables pendant au moins 104 semaines. Chez la plupart des patients, les modifications des immunoglobulines sont restées dans l'intervalle normal de référence.
Des élévations des anticorps IgG dirigés contre les antigènes S1/S2 du SARS-CoV-2 ont été observées dans un échantillon limité de patients COVID-19 modérément à sévèrement atteints, hospitalisés et traités avec le baricitinib.
Lymphocytes
Le nombre absolu moyen de lymphocytes a augmenté dans la semaine suivant l'instauration du traitement par Olumiant, est revenu au même niveau qu'à l'inclusion à la semaine 24, puis est resté stable pendant au moins 104 semaines. Pour la plupart des patients, les modifications du nombre de lymphocytes sont restées dans l'intervalle normal de référence.
Thrombocytes
Sur le plan clinique, les thrombocytoses ont été plus fréquemment observées sous baricitinib que sous placebo (voir «Effets indésirables, Description d'effets indésirables sélectionnés, Thrombocytose»).
Protéine C-réactive
Chez les patients atteints de polyarthrite rhumatoïde, des diminutions de la protéine Créactive (CRP) sérique ont été observées dès la première semaine de traitement par Olumiant et se sont maintenues pendant toute la durée du traitement.
Peau
Dans un modèle de peau humaine traitée avec des cytokines pro-inflammatoires (c'est-à-dire IL-4, IL-13, IL-31), le baricitinib a réduit l'expression de pSTAT3 dans les kératinocytes épidermiques, et a augmenté l'expression de la filaggrine.
Biomarqueur pour la COVID-19
Le baricitinib abaisse les valeurs de cytokines et de biomarqueurs en rapport avec la COVID-19, notamment IL-6, IFN-γ, MCP-3, CXCL10, IL-10, MCP-2, CCL19, PTX3 et IL-27. En outre, les valeurs de marqueurs qui sont réduites chez des patients COVID-19 modérément à sévèrement atteints ont augmenté avec l'administration de baricitinib; ceci concerne CCL17, GDF2 et SCF.
Efficacité clinique
Polyarthrite rhumatoïde
L'efficacité et la sécurité d'Olumiant administré une fois par jour ont été évaluées dans 4 études de phase III multicentriques, randomisées, en doubleaveugle chez des patients ayant une polyarthrite rhumatoïde active, modérée à sévère, diagnostiquée conformément aux critères ACR/EULAR 2010 (voir Tableau 1). Les patients de plus de 18 ans pouvaient participer à ces études. La présence d'au moins 6 articulations douloureuses et 6 articulations gonflées était requise à l'inclusion. Tous les patients arrivés au terme de ces études étaient éligibles pour participer à une étude d'extension à long terme et recevoir jusqu'à 4 ans de traitement.
Tableau 1. Synthèse des études cliniques
|
Nom de l'étude (Durée)
|
Population
(Nombre total)
|
Bras de traitement
|
Synthèse des objectifs principaux des études
| |
RA-BEGIN
(52 semaines)
|
Naïfs de MTX1
(584)
|
Olumiant 4 mg 1x/j
Olumiant 4 mg 1x/j + MTX
MTX
|
Critère principal: ACR20 à la semaine 24
Capacité fonctionnelle (HAQ-DI)
Progression radiographique (mTSS)
Faible activité de la maladie et rémission (SDAI)
| |
RA-BEAM
(52 semaines)
|
MTX-RI2
(1305)
|
Olumiant 4 mg 1x/j
Adalimumab 40 mg SC 1x/2sem
Placebo
Tous les patients étaient sous MTX en traitement de fond
|
Critère principal: ACR20 à la semaine 12
Capacité fonctionnelle (HAQ-DI)
Progression radiographique (mTSS)
Faible activité de la maladie et rémission (SDAI)
Raideur articulaire matinale
| |
RA-BUILD
(24 semaines)
|
cDMARD-RI3
(684)
|
Olumiant 4 mg 1x/j
Olumiant 2 mg 1x/j
Placebo
Sous DMARDs conventionnels (cDMARDs) en traitement de fond5, si l'administration du cDMARD était stable à l'entrée dans l'étude.
|
Critère principal: ACR20 à la semaine 12
Capacité fonctionnelle (HAQ-DI)
Faible activité de la maladie et rémission (SDAI)
Progression radiographique (mTSS)
Raideur articulaire matinale
| |
RA-BEACON
(24 semaines)
|
TNF-RI4
(527)
|
Olumiant 4 mg 1x/j
Olumiant 2 mg 1x/j
Placebo
Sous cDMARDs en traitement de fond5
|
Critère principal: ACR20 à la semaine 12
Capacité fonctionnelle (HAQ-DI)
Faible activité de la maladie et rémission (SDAI)
|
1x/j = une fois par jour; 1x/2sem = toutes les 2 semaines; SC = sous-cutané
1 Patients ayant reçu moins de 3 administrations de méthotrexate (MTX); naïfs de traitement par d'autres antirhumatismaux modificateurs de la maladie conventionnels (cDMARDs) ou biologiques (bDMARDs).
2 Patients ayant eu une réponse inadéquate au MTX (+/- d'autres cDMARDs); naïfs de traitement biologique.
3 Patients ayant eu une réponse inadéquate ou étant intolérant à au moins 1 cDMARD; naïfs de traitement biologique.
4 Patients ayant eu une réponse inadéquate ou étant intolérant à au moins 1 bDMARD; incluant au moins un inhibiteur du TNF.
Réponse clinique
Dans les 3 études contrôlées contre placebo, une proportion statistiquement significativement plus élevée de patients traités par Olumiant 4 mg une fois par jour a montré une réponse ACR20, ACR50 et ACR70 à 12 et 24 semaines comparé au placebo (tableau 2). Le délai d'apparition de l'efficacité a été court pour toutes les mesures, avec des réponses significativement plus élevées dès la semaine 1. Des taux de réponse persistants et durables ont été observés, avec des réponses ACR20/50/70 qui se sont maintenus pendant au moins 2 ans, y compris pendant l'étude d'extension à long terme.
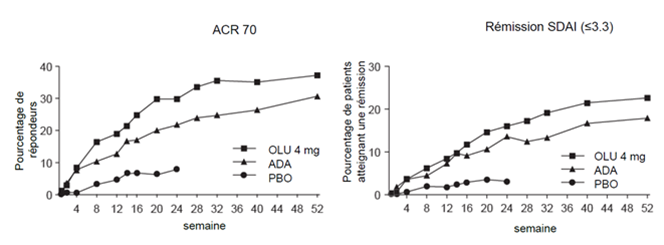
Dans l'étude avec contrôle actif RA-BEAM, une proportion statistiquement significativement plus élevée de patients traités avec Olumiant a obtenu une réponse ACR20/50/70 après 12 semaines par rapport à adalimumab et les différences ont été maintenues pendant plus de 52 semaines (Figure 1). Radiographiquement, aussi bien Olumiant qu'adalimumab ont montré des effets statistiquement significatifs sur l'inhibition des lésions structurales articulaires par rapport au placebo. Adalimumab a été numériquement un peu meilleur que Olumiant (voir ci-dessous «Réponse radiographique»).
Le traitement par Olumiant 4 mg, seul ou en association avec des cDMARDs, a entraîné une amélioration significative de toutes les composantes individuelles de la réponse ACR, incluant le nombre d'articulations douloureuses et gonflées, les évaluations globales du patient et du médecin, l'indice HAQ-DI (Health Assessment Questionnaire-Disability Index), l'évaluation de la douleur et la CRP, par rapport au placebo ou au MTX en monothérapie. Dans l'étude RA-BEAM, le traitement par Olumiant a entraîné une amélioration significative des évaluations globales du patient et du médecin, de l'indice HAQ-DI, de l'évaluation de la douleur et de la CRP aux semaines 12, 24 et 52 par rapport à l'adalimumab.
Rémission et faible niveau d'activité de la maladie
La proportion de patients atteignant la rémission, définie par un SDAI ≤3.3 et un CDAI ≤2.8, aux semaines 12 et 24, était statistiquement significativement plus élevée dans le groupe Olumiant 4 mg que dans le groupe placebo ou MTX (tableau 2). Dans l'étude RA-BEAM, Olumiant et adalimumab étaient tous deux supérieurs comparés au placebo, mesuré par un SDAI ≤3.3 à la semaine 12 et les différences ont été maintenues jusqu'à la semaine 52 (Figure 1).
Dans les 4 études, la proportion de patients atteignant un faible niveau d'activité de la maladie ou la rémission (DAS-28 lié à la vitesse de sédimentation DAS28-VS ou DAS-28 lié au dosage à ultrasensible de la CRP DAS28-CRPus ≤3.2 et DAS28-VS ou DAS28-CRPus < 2.6) aux semaines 12 et 24 était significativement plus élevée dans le groupe Olumiant 4 mg que dans le groupe placebo ou MTX.
En tenant compte de l'étude d'extension à long terme, les taux de rémission et de faible niveau d'activité de la maladie se sont maintenus pendant au moins 2 ans.
Tableau 2. Réponse, rémission et capacité fonctionnelle
|
Étude
|
RA-BEGIN
Patients naïfs de MTX
|
RA-BEAM
Patients MTX-RI
|
RA-BUILD
Patients cDMARD-RI
|
RA-BEACON
Patients TNF-RI
| |
Groupe de traitement
|
MTX
|
OLU
4 mg
|
OLU
4 mg
+ MTX
|
PBO
|
OLU
4 mg
|
ADA
40 mg 1x/2 sem.
|
PBO
|
OLU
2 mg
|
OLU 4 mg
|
PBO
|
OLU 2 mg
|
OLU
4 mg
| |
N
|
210
|
159
|
215
|
488
|
487
|
330
|
228
|
229
|
227
|
176
|
174
|
177
| |
ACR20:
| |
Semaine 12
|
59 %
|
79 %***
|
77 %***
|
40 %
|
70 %***†
|
61 %***
|
39 %
|
66 %***
|
62 %***
|
27 %
|
49 %***
|
55 %***
| |
Semaine 24
|
62 %
|
77 %**
|
78 %***
|
37 %
|
74 %***†
|
66 %***
|
42 %
|
61 %***
|
65 %***
|
27 %
|
45 %***
|
46 %***
| |
Semaine 52
|
56 %
|
73 %***
|
73 %***
|
|
71 %††
|
62 %
|
|
|
|
|
|
| |
ACR50:
| |
Semaine 12
|
33 %
|
55 %***
|
60 %***
|
17 %
|
45 %***††
|
35 %***
|
13 %
|
33 %***
|
34 %***
|
8 %
|
20 %**
|
28 %***
| |
Semaine 24
|
43 %
|
60 %**
|
63 %***
|
19 %
|
51 %***
|
45 %***
|
21 %
|
41 %***
|
44 %***
|
13 %
|
23 %*
|
29 %***
| |
Semaine 52
|
38 %
|
57 %***
|
62 %***
|
|
56 %†
|
47 %
|
|
|
|
|
|
| |
ACR70:
| |
Semaine 12
|
16 %
|
31 %***
|
34 %***
|
5 %
|
19 %***†
|
13 %***
|
3 %
|
18 %***
|
18 %***
|
2 %
|
13 %***
|
11 %**
| |
Semaine 24
|
21 %
|
42 %***
|
40 %***
|
8 %
|
30 %***†
|
22 %***
|
8 %
|
25 %***
|
24 %***
|
3 %
|
13 %***
|
17 %***
| |
Semaine 52
|
25 %
|
42 %***
|
46 %***
|
|
37 %
|
31 %
|
|
|
|
|
|
| |
DAS28-hsCRP ≤3.2:
| |
Semaine 12
|
30 %
|
47 %***
|
56 %***
|
14 %
|
44 %***††
|
35 %***
|
17 %
|
36 %***
|
39 %***
|
9 %
|
24 %***
|
32 %***
| |
Semaine 24
|
38 %
|
57 %***
|
60 %***
|
19 %
|
52 %***
|
48 %***
|
24 %
|
46 %***
|
52 %***
|
11 %
|
20 %*
|
33 %***
| |
Semaine 52
|
38 %
|
57 %***
|
63 %***
|
|
56 %†
|
48 %
|
|
|
|
|
|
| |
DAS28-VS ≤3.2:
| |
Semaine 12
|
15 %
|
21 %
|
34 %***
|
7 %
|
24 %***
|
21 %***
|
7 %
|
21 %***
|
22 %***
|
4 %
|
13 %**
|
12 %**
| |
Semaine 24
|
23 %
|
36 %**
|
39 %***
|
10 %
|
32 %***
|
34 %***
|
10 %
|
29 %***
|
32 %***
|
7 %
|
11 %
|
17 %**
| |
Semaine 52
|
27 %
|
36 %
|
45 %***
|
|
39 %
|
36 %
|
|
|
|
|
|
| |
SDAI ≤3.3:
| |
Semaine 12
|
6 %
|
14 %*
|
20 %***
|
2 %
|
8 %***
|
7 %***
|
1 %
|
9 %***
|
9 %***
|
2 %
|
2 %
|
5 %
| |
Semaine 24
|
10 %
|
22 %**
|
23 %***
|
3 %
|
16 %***
|
14 %***
|
4 %
|
17 %***
|
15 %***
|
2 %
|
5 %
|
9 %**
| |
Semaine 52
|
13 %
|
25 %**
|
30 %***
|
|
23 %
|
18 %
|
|
|
|
|
|
| |
CDAI ≤2.8:
| |
Semaine 12
|
7 %
|
14 %*
|
19 %***
|
2 %
|
8 %***
|
7 %**
|
2 %
|
10 %***
|
9 %***
|
2 %
|
3 %
|
6 %
| |
Semaine 24
|
11 %
|
21 %**
|
22 %**
|
4 %
|
16 %***
|
12 %***
|
4 %
|
15 %***
|
15 %***
|
3 %
|
5 %
|
9 %*
| |
Semaine 52
|
16 %
|
25 %*
|
28 %**
|
|
22 %
|
18 %
|
|
|
|
|
|
| |
HAQ-DI (changement depuis l'inclusion):
| |
Semaine 12
|
-0.61
|
-0.92***
|
-0.98***
|
-0.34
|
-0.66***††
|
-0.56***
|
-0.36
|
-0.57***
|
-0.56***
|
-0.17
|
-0.37***
|
-0.41***
| |
Semaine 24
|
-0.74
|
-1.04***
|
-1.03***
|
-0.35
|
-0.75***††
|
-0.63***
|
-0.38
|
-0.62***
|
-0.62***
|
-0.15
|
-0.38***
|
-0.43***
| |
Semaine 52
|
-0.71
|
-0.99***
|
-1.06***
|
|
-0.77††
|
-0.66
|
|
|
|
|
|
|
ADA = adalimumab; MTX = méthotrexate; OLU = Olumiant; PBO = Placebo
* p ≤0.05; ** p ≤0.01; *** p ≤0.001 versus placebo (versus MTX pour l'étude RA-BEGIN)
† p ≤0.05; †† p ≤0.01; ††† p ≤0.001 versus adalimumab
Figure 1. ACR70 et rémission SDAI, étude RA-BEAM
Réponse radiographique
Les effets d'Olumiant sur la progression des lésions structurales articulaires a été évalué radiographiquement dans les études RA-BEGIN (objectif principal), RA-BEAM (objectif principal) et RA-BUILD (objectif exploratoire), au moyen du Score Total de Sharp modifié (mTSS) et de ses composantes, du score d'érosion et du score de rétrécissement de l'espace articulaire (Joint Space Narrowing Score).
Dans RA-BEAM, le traitement par Olumiant 4 mg ainsi que l'adalimumab ont montré une inhibition statistiquement significative de la progression des lésions structurales articulaires par rapport au placebo (tableau 3). Les analyses des scores d'érosions et du rétrécissement de l'espace articulaire étaient cohérentes avec les scores globaux. La proportion de patients sans progression radiographique (changement mTSS ≤0) a été significativement plus élevée dans le groupe Olumiant 4 mg que dans le groupe placebo aux semaines 24 et 52.
Tableau 3. Modifications radiographiques
|
Étude
|
RA-BEGIN
Patients naïfs de MTX
|
RA-BEAM
Patients sous MTX-RI
|
RA-BUILD°
Patients sous cDMARDs-RI
| |
Groupe de traitement
|
MTX
|
OLU 4 mg
|
OLU 4 mg
+ MTX
|
PBOa
|
OLU 4 mg
|
ADA 40 mg 1x/ 2sem.
|
PBO
|
OLU 2 mg
|
OLU 4 mg
| |
N
|
210
|
159
|
215
|
488
|
487
|
330
|
228
|
229
|
227
| |
Score Total de Sharp modifié, changement moyen depuis l'inclusion:
| |
Semaine 16
|
|
|
|
0.69
|
0.35***
|
0.28***
|
|
|
| |
Semaine 24
|
0.61
|
0.39
|
0.29*
|
0.90
|
0.41***
|
0.33***
|
0.70
|
0.33*
|
0.15**
| |
Semaine 52
|
1.02
|
0.80
|
0.40**
|
1.80
|
0.71***
|
0.60***
|
|
|
| |
Score d'érosion, changement moyen depuis l'inclusion:
| |
Semaine 16
|
|
|
|
0.50
|
0.25***
|
0.21***
|
|
|
| |
Semaine 24
|
0.47
|
0.33
|
0.26*
|
0.61
|
0.29***
|
0.24***
|
0.47
|
0.30
|
0.11**
| |
Semaine 52
|
0.81
|
0.55
|
0.34**
|
1.23
|
0.51***
|
0.42***
|
|
|
| |
Score de rétrécissement de l'espace articulaire, changement moyen depuis l'inclusion:
| |
Semaine 16
|
|
|
|
0.20
|
0.11
|
0.08*
|
|
|
| |
Semaine 24
|
0.14
|
0.06
|
0.03
|
0.29
|
0.12**
|
0.10**
|
0.23
|
0.03*
|
0.04*
| |
Semaine 52
|
0.21
|
0.25
|
0.06
|
0.58
|
0.21***
|
0.19**
|
|
|
| |
Proportion de patients sans progression radiographiqueb:
| |
Semaine 16
|
|
|
|
72 %
|
81 %**
|
82 %**
|
|
|
| |
Semaine 24
|
68 %
|
76 %
|
81 %**
|
70 %
|
81 %***
|
83 %***
|
74 %
|
72 %
|
80 %
| |
Semaine 52
|
66 %
|
69 %
|
80 %**
|
70 %
|
79 %**
|
81 %**
|
|
|
|
ADA = adalimumab; MTX = méthotrexate; OLU = Olumiant; PBO = Placebo
a Données avec le placebo à la semaine 52 issues d'une extrapolation linéaire
b Aucune progression = modification mTSS ≤0.
* p ≤0.05; ** p ≤0.01; *** p ≤0.001 versus placebo (versus MTX pour l'étude RA-BEGIN)
° objectif exploratoire dans RA-BUILD
Réponse fonctionnelle et résultats liés à l'état de santé
Le traitement par Olumiant 4 mg, seul ou en association avec des cDMARDs, a entraîné une amélioration significative de la capacité fonctionnelle par rapport à tous les comparateurs (placebo, MTX, adalimumab), telle que mesurée par l'indice HAQ-DI, aux semaines 12, 24 et 52 (tableau 2). Des améliorations ont été observées dès la semaine 1 et, dans les études RA-BEGIN et RA-BEAM, ces améliorations se sont maintenues jusqu'à 52 semaines. Dans l'étude RA-BEAM, 67% des patients sous Olumiant 4 mg ont atteint au-moins une différence cliniquement importante (diminution du score HAQ-DI de ≥0.30) à la semaine 24 par rapport à 37% avec le placebo et 60% sous adalimumab (p <0.001 et p = 0.049 respectivement). L'amélioration avec Olumiant a persisté au moins 2 ans.
Le traitement par Olumiant 4 mg, seul ou en association avec des cDMARDs, a entraîné une amélioration significative de la douleur par rapport à tous les comparateurs (placebo, MTX, adalimumab), mesurée sur une échelle visuelle analogique de 0 à 100, à 12, 24 et 52 semaines. Dans l'étude RA-BEAM, la variation moyenne du score de la douleur sous Olumiant 4 mg par rapport à la valeur d'inclusion était de -33.6 à la semaine 24 comparé à -17.5 pour le placebo et -28.8 dans le groupe de traitement avec adalimumab (p < 0.001 et p = 0.004 respectivement).
Dans les études RA-BEAM et RA-BUILD, la durée et la sévérité de la raideur articulaire matinale ont été évaluées à l'aide d'un journal quotidien du patient pendant 12 semaines. Dans l'étude RA-BEAM, les patients sous Olumiant ont montré après 12 semaines une durée médiane de la raideur articulaire matinale de 27 minutes comparé à 60 minutes sous placebo et 37 minutes sous adalimumab (p < 0.001 et p = 0.024 respectivement). Dans l'étude RA-BEAM, les patients sous Olumiant ont montré après 12 semaines une sévérité moyenne de la raideur articulaire matinale de 3.0 comparé à 4.1 sous placebo et 3.5 sous adalimumab, mesurée par une échelle d'évaluation numérique de 0 à 10, avec 0 pour aucune raideur articulaire (p < 0.001 et p = 0.002 respectivement). Dans l'étude RA-BUILD, des résultats similaires versus placebo ont été observés.
Dans toutes les études, les patients traités par Olumiant ont rapporté des améliorations de la qualité de vie, mesurée par le score du statut fonctionnel du questionnaire abrégé (Short Form 36) de qualité de vie SF-36 et de la fatigue, mesurée par le score d'évaluation fonctionnelle du traitement des maladies chroniques (FACIT-F, Functional Assessment of Chronic Illness Therapy-Fatigue) et la productivité au travail mesurée par le Work Productivity and Activity Impairment Questionnaire: Rheumatoid arthritis (WPAI-RA).
Olumiant 4 mg vs. 2 mg
Dans les études cliniques incluant les doses d'Olumiant de 2 mg et 4 mg une fois par jour (RA-BUILD et RA-BEACON), l'efficacité sur les signes et les symptômes a été montrée pour les deux dosages. Un début d'action plus rapide avec une proportion numériquement plus élevée de patients atteignant un faible niveau d'activité de la maladie et une progression radiographique plus faible des lésions articulaires a été constatée avec la dose de 4 mg par rapport à 2 mg. Les différences ont été observées principalement chez les patients de la population bDMARD-RI (RA BEACON), mais très peu chez les patients qui n'avaient encore jamais été traités par un médicament biologique.
Dans l'étude d'extension à long terme, les patients des études RA-BEAM, RA-BUILD et RA-BEACON ayant atteint un état de faible niveau d'activité de la maladie prolongé ou de rémission (CDAI ≤10) après au moins 15 mois de traitement avec Olumiant 4 mg une fois par jour, ont été de nouveau randomisés selon un rapport 1:1, en double-aveugle, pour continuer à recevoir la dose de 4 mg une fois par jour ou réduire la dose à 2 mg une fois par jour.
La majorité des patients a maintenu un état de faible niveau d'activité de la maladie ou sont restés en rémission aussi après la diminution à 2 mg:
·à la semaine 12: 234/251 (93 %) ayant continué la dose de 4 mg et 207/251 (82 %) ayant reçu la dose réduite à 2 mg (p ≤0.001)
·à la semaine 24: 163/191 (85 %) ayant continué la dose de 4 mg et 144/189 (76 %) ayant reçu la dose réduite à 2 mg (p ≤0.05)
·à la semaine 48: 57/73 (78 %) ayant continué la dose de 4 mg et 51/86 (59 %) ayant reçu la dose réduite à 2 mg (p ≤0.05)
La majorité des patients qui n'a pas conservé un état de faible niveau d'activité de la maladie ou ne sont pas restés en rémission après la réduction de la dose ont pu retrouver le contrôle de la maladie après la réinstauration de la dose de 4 mg.
Dermatite atopique
L'efficacité et la sécurité d'Olumiant en monothérapie ou en association avec des corticostéroïdes topiques (CST) ont été évaluées dans 3 études cliniques de phase III randomisées, en double aveugle, contrôlées versus placebo, d'une durée de 16 semaines (BREEZE-AD1, -AD2 et -AD7). Les études ont inclus 1568 patients atteints d'une dermatite atopique modérée à sévère définie par un score Investigator's Global Assessment (IGA) ≥3, un score Eczema Area and Severity Index (EASI) (≥16, et un Body Surface Area (surface corporelle, BSA) ≥10 %. Les patients éligibles étaient âgés de plus de 18 ans et présentaient une réponse insuffisante ou une intolérance aux médicaments topiques. Les patients étaient autorisés à recevoir un traitement de secours (comprenant un traitement topique ou systémique) mais étaient dès lors considérés comme non-répondeurs. Tous les patients ayant terminé ces études étaient éligibles pour participer à une étude d'extension à long terme (BREEZE-AD3) jusqu'à 2 ans de traitement continu.
L'étude clinique de phase III, BREEZE-AD4, randomisée, en double-aveugle, contrôlée versus placebo, a évalué l'efficacité du baricitinib en association avec des corticostéroïdes topiques chez des patients atteints de DA modérée à sévère et présentant un échec thérapeutique, une intolérance ou une contre-indication à un traitement oral à la ciclosporine.
Tableau 4. Synthèse des études cliniques
|
Nom de l'étude (Durée)
|
Nombre de patients traités (N)
|
Traitement concomitanta
|
Groupes de traitement (QD)
|
Mesures des résultats
| |
BREEZE AD-1
(16 semaines)
|
624
|
aucun
|
OLU 4 mg
OLU 2 mg
OLU 1 mg Placebo
|
·Critère principal: IGA 0 ou 1b à la semaine 16
·Amélioration de ≥50%, 75 % ou 90 % de l'Eczema Area and Severity Index par rapport à la valeur initiale (Baseline) (EASI 50, 75, 90)
·Amélioration de ≥75 % sur l'échelle SCORing Atopic Dermatitis (SCORAD)
·Amélioration du Itch Numerical Rating Scale (NRS) ≥4 points
·Effet du prurit sur le sommeil, tel que mesuré par l'Atopic Dermatitis Sleep Scale (ADSS)
·Gravité de la douleur cutanée, telle que mesurée par le Skin Pain Numerical Rating Scale (NRS)
·Patient-Oriented Eczema Measure (POEM)
·Index de qualité de vie en dermatologie (DLQI)
·Hospital Anxiety and Depression Scale (HADS)
| |
BREEZE AD-2
(16 semaines)
|
615
|
aucun
| |
BREEZE AD-7
(16 semaines)
|
329
|
CST;
ICT en cas de besoin
|
OLU 4 mg
OLU 2 mg Placebo
| |
BREEZE AD-4
(jusqu'à 200 semaines)
|
462
|
CST;
ICT en cas de besoin
|
OLU 4 mg
OLU 2 mg
OLU 1 mg
Placebo
|
OLU = Olumiant; QD = une fois par jour; ICT = inhibiteur de la calcineurin topique; CST = corticostéroïdes topiques
a Les patients ont utilisé un traitement concomitant tout au long de l'étude
b Investigators Global Assessment Score de 0 («disparition») ou 1 («quasi disparition») avec une réduction ≥2 points sur une échelle de sévérité de 5 points de 0 à 4.
Caractéristiques à l'inclusion
Dans les études en monothérapie (BREEZE-AD1 et BREEZE-AD2) la moyenne d'âge dans tous les groupes de traitement était de 35.2 ans, le poids moyen était de 73.3 kg, 37.7 % étaient des femmes, 63.5 % étaient caucasiens, 30 % étaient asiatiques et 0.2 % avaient la peau noire. Dans ces études, 54 % des patients avaient un score IGA à l'inclusion de 3 (DA modérée), 46 % avaient un score IGA de base de 4 (DA sévère) et 59.9 % des patients avaient reçu un traitement systémique préalable pour la dermatite atopique. Le score EASI moyen à l'inclusion était de 32.2, le score BSA moyen à l'inclusion était de 52.3, le score NRS du prurit moyen hebdomadaire à l'inclusion était de 6.6, le score SCORAD moyen à l'inclusion était de 67.8, le score POEM moyen à l'inclusion était de 20.6, le score DLQI moyen à l'inclusion était de 14.0, le score HADS moyen pour la dépression à l'inclusion était de 5.0 et le score HADS moyen pour l'anxiété à l'inclusion était de 6.1.
Dans l'étude en association avec les CST (BREEZE-AD7), la moyenne d'âge dans tous les groupes de traitement était de 33.8 ans, le poids moyen était de 72.9 kg, 34.3 % étaient des femmes, 45.6 % étaient caucasiens et 51.1 % étaient asiatiques. Dans cette étude, 54.9% des patients avaient un score IGA à l'inclusion de 3, 45.1% avaient un score IGA à l'inclusion de 4 et 66.4% des patients avaient reçu un traitement systémique préalable. Le score EASI moyen à l'inclusion était de 29.6, le score BSA moyen à l'inclusion était de 50.3, le score NRS du prurit moyen à l'inclusion était de 7.1, le score SCORAD moyen à l'inclusion était de 67.2, le POEM moyen à l'inclusion était de 21.1, le DLQI moyen à l'inclusion était de 14.9, le score HADS moyen pour la dépression à l'inclusion était de 5.5 et le score HADS moyen pour l'anxiété à l'inclusion était de 6.6.
Dans l'étude en association avec les CST (BREEZE-AD4) la moyenne d'âge dans tous les groupes de traitement était de 38.2 ans, le poids moyen était de 75.5 kg, 35.9 % étaient des femmes, 77.8 % étaient caucasiens et 19.2 % étaient asiatiques. Dans cette étude, 48.7 % des patients avaient un score IGA à l'inclusion de 3, 51.3 % avaient un score IGA de base de 4 et 78.8 % des patients avaient reçu un traitement systémique préalable. Le score EASI moyen à l'inclusion était de 31.8, le score BSA moyen à l'inclusion était de 51.8, le score NRS duprurit moyen hebdomadaire à l'inclusion était de 6.8, le score SCORAD moyen à l'inclusion était de 68.8, le score POEM moyen à l'inclusion était de 21.2, le score DLQI moyen à l'inclusion était de 14.0, le score HADS moyen pour la dépression à l'inclusion était de 5.0 et le score HADS moyen pour l'anxiété à l'inclusion était de 6.2.
Réponse clinique
Études cliniques en monothérapie sur 16 semaines (BREEZE-AD1 et BREEZE-AD2)
Dans les études BREEZE-AD1 et BREEZE-AD2, une proportion significativement plus élevée de patients randomisés pour recevoir baricitinib 4 mg a obtenu une réponse IGA 0 ou 1, un EASI75, ou une amélioration ≥4 points sur l'échelle du prurit (Itch NRS) par rapport au placebo à la semaine 16 (Tableau 5).
Une proportion significativement plus élevée de patients randomisés pour recevoir baricitinib 4 mg a atteint une amélioration rapide du score NRS duprurit par rapport au placebo (défini comme ≥4 points d'amélioration dès la première semaine de traitement; p < 0.001).
Les figures 2 et 3 montrent, respectivement, le pourcentage moyen de changement par rapport aux valeurs de l'EASI à l'inclusion et la proportion de patients présentant au moins 4 points d'amélioration sur l'échelle du prurit (Itch NRS). Les résultats pour la période contrôlée par placebo sont présentés jusqu'à la semaine 16, avec des résultats à long terme disponibles jusqu'aux semaines 68 et 32 pour les figures 2 et 3, respectivement.
Les effets du traitement dans les sous-groupes de patients (poids, âge, sexe, origine ethnique, sévérité de la maladie et traitement antérieur, y compris immunosuppresseurs) dans BREEZE-AD1 et BREEZE-AD2 étaient cohérents avec les résultats dans la population globale de l'étude.
Tableau 5. Efficacité du baricitinib en monothérapie à la semaine 16 (FASa)
|
Etude
|
BREEZE-AD1
|
BREEZE-AD2
| |
Groupe de traitement
|
PBO
|
OLU
2 mg
|
OLU
4 mg
|
PBO
|
OLU
2 mg
|
OLU
4 mg
| |
N
|
N = 249
|
N = 123
|
N = 125
|
N = 244
|
N = 123
|
N = 123
| |
IGA 0 ou 1,
% de répondeursb, c
|
4.8 %
|
11.4 %*
|
16.8 %***
|
4.5 %
|
10.6 %*
|
13.8 %***
| |
EASI-75,
% de répondeursc
|
8.8 %
|
18.7 %**
|
24.8 %***
|
6.1 %
|
17.9 %***
|
21.1 %***
| |
SCORAD75,
% de répondeursc
|
1.2 %
|
7.3 %+
|
10.4 %***
|
1.6 %
|
7.3 %**
|
11.4 %***
| |
Itch NRS (amélioration ≥4 points), % de répondeursc,d
|
7.2 %
|
12.0% +
|
21.5 %***
|
4.7 %
|
15.1 %**
|
18.7 %***
|
OLU = Olumiant; PBO = Placebo
*p ≤0.05; **p ≤0.01; ***p ≤0.001 +non significatif en partie en raison de la hiérarchie des tests par rapport au placebo.
a Full analysis set (FAS, Analyse de la population totale de l'étude), inclut tous les patients randomisés.
b Un répondeur est défini comme un patient ayant atteint un score IGA de 0 ou 1 («disparition» ou «quasi disparition») avec une réduction ≥2 points sur l'échelle IGA 0-4.
c Imputation des non-répondeurs: les patients ayant reçu un traitement de secours ont été définitivement stoppés du traitement à l'étude. Les patients avec des données manquantes ont été considérés comme non-répondeurs.
d Résultats présentés dans un sous-ensemble de patients éligibles pour une évaluation (patients avec un score NRS duprurit ≥4 à l'inclusion).
Figure 2. Variation moyenne en pourcentage du score EASI par rapport à l'inclusion dans BREEZE-AD1et BREEZE-AD2 (FAS)a avec les données à long terme de BREEZE-AD3b, c
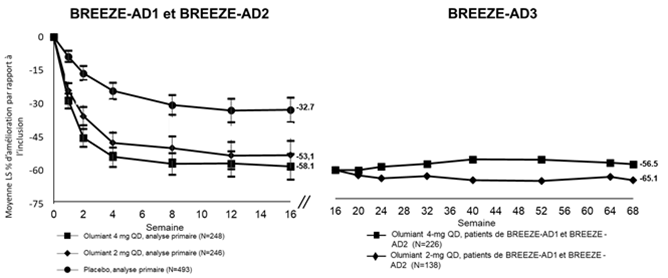
a Full analysis set (FAS) inclut tous les patients randomisés. Les données recueillies après un traitement de secours ou après un arrêt définitif du médicament à l'étude ont été considérées comme manquantes. Les moyennes LS proviennent d'analyses MMRM.
b La modified last observation carried forward (mLOCF) a été utilisée. La technique d'imputation mLOCF remplace les données manquantes par la dernière évaluation après inclusion non manquante.
c Dans BREEZE-AD3, la taille de l'échantillon de patients pour Olumiant 2 mg est réduite car les non-répondeurs à cette dose dans BREEZE-AD1 et BREEZE-AD2 ont été re-randomisés à la dose de 2 mg ou 4 mg 1:1. Les répondeurs et répondeurs partiels sont restés à la même dose.
Remarque: les barres d'erreur représentent un IC de 95%.
Figure 3. Proportion de patients présentant au moins 4 points d'amélioration du Itch NRS dans BREEZE-AD1 and BREEZE-AD2 a avec les données à long terme de BREEZE-AD3b
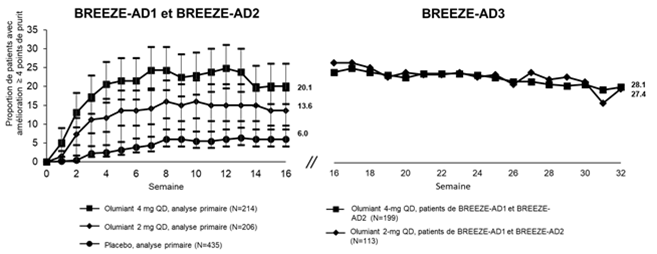
a Les résultats ont été présentés dans un sous-ensemble de patients éligibles à l'évaluation (patients atteints avec score prurit NRS≥4 à l'inclusion). Les patients qui ont reçu un traitement de secours ont été définitivement stoppés du traitement à l'étude. Les patients dont les données étaient manquantes ont été considérés comme non-répondeurs (imputation des non-répondeurs).
b Dans BREEZE-AD3, la taille de l'échantillon de patients pour Olumiant 2 mg est réduite parce que les non-répondeurs à cette dose dans BREEZE-AD1 et BREEZE-AD2 ont été re-randomisés à la dose de 2 mg ou 4 mg 1:1. Les répondeurs et les répondeurs partiels sont restés à la même dose. Les patients ayant cessé définitivement de prendre le médicament de l'étude ou pour lesquels des données manquaient ont été comptés comme non-répondeurs (Non Responder Imputation).
Remarque: les barres d'erreur représentent un IC de 95 %. L'augmentation du pourcentage observé dans BREEZE-AD3 est due au fait que les patients qui ont été secourus avec des CST dans BREEZE-AD1 et -AD2 n'ont pas été considérés comme des non-répondeurs.
Étude clinique sur 16 semaines en association avec des CST (BREEZE-AD7)
Dans BREEZE-AD7, une proportion significativement plus élevée de patients randomisés pour recevoir baricitinib 4 mg+CST a atteint une réponse IGA de 0 ou 1, unEASI75, ou une amélioration ≥4 points du score NRS duprurit par rapport au placebo à la semaine 16 (Tableau 6).
Une proportion significativement plus élevée de patients randomisés pour recevoir baricitinib 4 mg a atteint une amélioration rapide du score NRS duprurit par rapport au placebo (défini comme ≥4 points d'amélioration dès la deuxième semaine; p < 0.001).
Les figures 4 et 5 montrent, respectivement, le pourcentage moyen de changement par rapport aux valeurs de l'EASI à l'inclusion et la proportion de patients présentant au moins 4 points d'amélioration du score NRS duprurit. Les résultats pour la période contrôlée par placebo sont présentés jusqu'à la semaine 16, avec des résultats à long terme disponibles jusqu'aux semaines 68 et 32 pour les figures 4 et 5, respectivement.
Les effets du traitement dans les sous-groupes de patients (poids, âge, sexe, origine ethnique, sévérité de la maladie et traitement antérieur, y compris immunosuppresseurs) dans BREEZE-AD7 étaient cohérents avec les résultats dans la population globale de l'étude.
Tableau 6. Efficacité du baricitinib en association avec des CSTa à la semaine 16 (FAS)b
|
Etude
|
BREEZE- AD7
| |
Groupe de traitement
|
PBOa
|
OLU 2 mg a
|
OLU 4 mg a
| |
N
|
109
|
109
|
111
| |
IGA 0 ou 1,
% de répondeursc, d
|
14.7 %
|
23.9 %
|
30.6 %**
| |
EASI-75,
% de répondeursd
|
22.9 %
|
43.1 %+
|
47.7 %***
| |
SCORAD75,
% de répondeursd
|
7.3 %
|
11.0 %+
|
18.0 %+
| |
Itch NRS (amélioration ≥4 points), % de répondeursd, e
|
20.2 %
|
38.1 %+
|
44.0 %***
|
OLU = Olumiant; PBO = Placebo
*p ≤0.05; **p ≤0.01; ***p ≤0.001 vs. +non significatif en partie en raison de la hiérarchie des tests, par rapport au placebo.
a Tous les patients ont reçu une thérapie concomitante avec des corticostéroïdes topiques et ont été autorisés à utiliser des inhibiteurs topiques de la calcineurine.
b Full Analysis Set (FAS) inclut tous les patients randomisés.
c Un répondeur est défini comme un patient avec un IGA 0 ou 1 («disparition» ou «quasi disparition») avec une réduction de ≥2 points sur l'échelle IGA de 0 à 4.
d Imputation des non-répondeurs: les patients ayant bénéficié d'une thérapie de secours ont été définitivement stoppés du traitement à l'étude. Les patients dont les données étaient manquantes ont été considérés comme non-répondeurs.
eRésultats présentés dans un sous-ensemble de patients éligibles pour évaluation (patients avec un score NRS du prurit ≥4 à l'inclusion).
Figure 4. Variation moyenne en pourcentage du score EASI par rapport à l'inclusion dans BREEZE-AD7 (FAS) a avec les données à long terme de BREEZE-AD3b
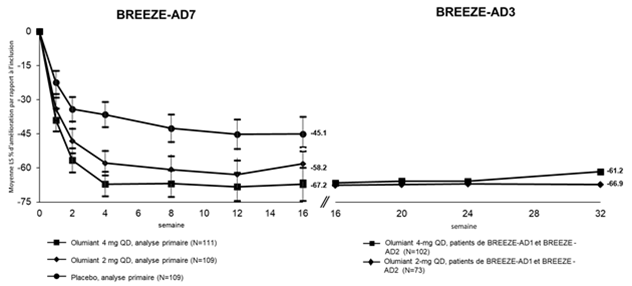
a Full analysis set (FAS) inclut tous les patients randomisés. Les données recueillies après un traitement de secours ou après un arrêt définitif du médicament à l'étude ont été considérées comme manquantes. Les moyennes LS proviennent d'analyses MMRM.
b La modified last observation carried forward (mLOCF) a été utilisée. La technique d'imputation mLOCF remplace les données manquantes par la dernière évaluation après inclusion non manquante.
Remarque: les barres d'erreur représentent un IC de 95%.
Figure 5. Proportion de patients avec au moins 4 points d'amélioration du score Itch NRS dans BREEZE-AD7 a avec les données à long terme de BREEZE-AD3 b
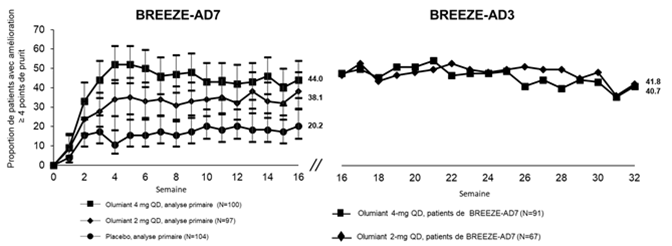
a Les résultats ont été présentés dans un sous-ensemble de patients éligibles à l'évaluation (patients avec un score prurit NRS≥4 à l'inclusion). Les patients ayant suivis un traitement de secours ont été définitivement stoppés du traitement à l'étude. Les patients dont les données étaient manquantes ont été notés comme non-répondeurs (imputation des non-répondeurs).
b Dans BREEZE-AD3, la taille de l'échantillon de patients pour Olumiant 2 mg est réduite parce que les non-répondeurs à cette dose dans BREEZE-AD7 ont été re-randomisés à la dose de 2 mg ou 4 mg 1:1. Les répondeurs et les répondeurs partiels sont restés à la même dose. Les patients ayant cessé définitivement de prendre le médicament à l'étude ou pour lesquels des données manquaient ont été comptés comme non-répondeurs (Non Responder Imputation).
Remarque: les barres d'erreur représentent un IC de 95 %.
Maintien de la réponse
Afin d'évaluer le maintien de la réponse, 1373 sujets traités par baricitinib pendant 16 semaines dans les études cliniques BREEZE-AD1 (N=541), BREEZE-AD2 (N =540) et BREEZE-AD7 (N=292) étaient éligibles pour participer à une étude d'extension à long terme BREEZE-AD3. Les données sont disponibles sur une durée cumulée maximale de 68 semaines de traitement pour les patients des études BREEZE-AD1 et BREEZE-AD2, et sur une durée cumulée maximale de 32 semaines de traitement pour les patients de l'étude BREEZE-AD7. Une différence soutenue par rapport au placebo a été observée dans les bras baricitinib. Des différences numériques constantes sont apparues entre la dose plus élevée de 4 mg et la dose de 2 mg, au détriment de la dose plus élevée de 4 mg (voir figure 6).
Figure 6. Persistance d'IGA 0 ou 1 pour baricitinib 4 mg et 2 mg dans BREEZE-AD3 jusqu'à la semaine 68a

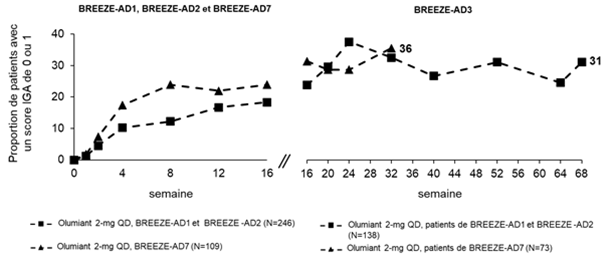
a L'imputation des non-répondeurs a été utilisée. Les patients qui ont cessé définitivement de prendre le médicament à l'étude ou pour lesquels il manquait des données ont été considérés comme non-répondeurs.
Réponse clinique chez les patients en situation d'échec au traitement à la ciclosporine, d'intolérance à celle-ci ou de contre-indication au traitement à la ciclosporine (étude BREEZE-AD4)
Au total, 462 patients ont été inclus, soit en situation d'échec au traitement (n=173), soit en cas d'intolérance (n=75) ou de contre-indication (n=126) à la ciclosporine orale.
Le critère principal d'évaluation était la proportion de patients ayant atteint un EASI-75 à la semaine 16 et a été atteint dans le groupe de traitement par le barictinib à 4 mg. Les résultats à la semaine 24 sont résumés dans le tableau 7.
Tableau 7. Efficacité du baricitinib en association avec des CST à la semaine 24 dans BREEZE-AD4 (FAS)b
|
Etude
|
BREEZE- AD4
| |
Groupe de traitement
|
PBOa
|
OLU 2 mga
|
OLU 4 mga
| |
N
|
93
|
185
|
92
| |
EASI-75,
% de répondeursc
|
15.1 %
|
27.0 %
|
28.3 %*
| |
EASI, moyenne LS % variation par rapport la valeur initiale (SE) d
|
-45.10
(4.24 )
|
-57.24 **
(2.80 )
|
-58.46 ***
(4.02 )
| |
IGA 0 ou 1,
% de répondeursc, e
|
12.9 %
|
18.9 %
|
13.0 %*
| |
Itch NRS, moyenne LS % de variation par rapport à l'inclusion (SE) d,f
|
-15.35
(5.35 )
|
-30.11 **
(3.50)
|
-33.16 **
(4.97)
| |
variation du score DLQI, moyenne (SE)d
|
-4.90 (0.74 )
|
-6.81
(0.49 )
|
-7.65 **
(0.71 )
|
SE = standard error (erreur standard). *p ≤0.05; **p ≤0.01; ***p ≤0.001, +non significant en partie en raison de la hiérarchie des tests par rapport au placebo.
Note: IGA (0,1) et EASI75 sont des paramètres gated. Tous les autres paramètres ne sont pas gated.
a Tous les patients ont reçu une thérapie concomitante avec des corticostéroïdes topiques et ont été autorisés à utiliser des inhibiteurs topiques de la calcineurine.
b Full analysis set (FAS, Analyse de la population totale de l'étude), inclut tous les patients randomisés.
c Imputation des non-répondeurs: les patients ayant bénéficié d'une thérapie de secours ont été définitivement stoppés du traitement à l'étude. Les patients dont les données étaient manquantes étaient considérés comme non-répondeurs.
d Les données recueillies après une thérapie de secours ou l'arrêt définitif du médicament à l'étude ont été considérées comme manquantes. Les valeurs moyennes LS proviennent d'analyses du MMRM qui comprennent des imputations multiples pour les données manquantes.
e Un répondeur est défini comme un patient avec un IGA 0 ou 1 («disparition» ou «quasi disparition») avec une réduction de ≥2 points sur l'échelle IGA de 0 à 4.
f L'ensemble des analyses évaluables comprend les patients ayant un NRS non nul à l'inclusion.
Réduction de dose
Dans l'étude d'extension à long terme BREEZE-AD3, les patients ayant obtenu une peau sans symptômes ou une maladie persistante minimale ou légère (IGA 0, 1 ou 2) avec Olumiant 4 mg une fois par jour ont été ré randomisés en double aveugle pour poursuivre le traitement à 4 mg une fois par jour ou pour réduire la dose à 2 mg une fois par jour. La majorité des patients re-randomisés à Olumiant 2 mg ont maintenu leur réponse 16 semaines après la re-randomisation. La majorité des patients qui ont perdu l'activité faible de la maladie ou un statut de peau sans symptômes après la réduction de la dose ont retrouvé le contrôle de la maladie après être revenus à Olumiant 4 mg.
Qualité de vie/résultats rapportés par les patients
Pendant la période de 16 semaines contrôlée par placebo, les patients traités par 2 mg et 4 mg de baricitinib ont montré une amélioration du prurit, du sommeil (scores ADSS), des douleurs cutanées et de la qualité de vie (scores DLQI) par rapport aux patients randomisés pour le placebo.
Pelade (Alopecia areata)
L'efficacité et la sécurité du baricitinib une fois par jour ont été examinées dans une étude adaptative de phase II/III (BRAVE-AA1) et une étude de phase III (BRAVE-AA2). Les deux études étaient des études randomisées, en double aveugle, contrôlées contre placebo, sur une durée de 36 semaines avec des phases de prolongation allant jusqu'à 200 semaines. Dans les deux études, les patients ont été randomisés dans un rapport de 2:2:3 pour recevoir un placebo, 2 mg ou 4 mg de baricitinib. Les patients éligibles étaient des adultes âgés de 18 à 60 ans pour les hommes et de 18 à 70 ans pour les femmes, avec un épisode actuel de plus de 6 mois de pelade sévère (perte de cheveux touchant ≥50 % du cuir chevelu). Les patients avec un épisode actuel de plus de 8 ans n'étaient pas éligibles sauf si des épisodes de repousse avaient été observés sur les zones atteintes du cuir chevelu au cours des 8 dernières années. Les patients souffrant d'hépatite virale chronique et/ou d'infections actives ont été exclus des études.
Les seuls traitement concomitants de la pelade autorisés étaient le finastéride (ou d'autres inhibiteurs de la 5-alpha-réductase), le minoxidil oral ou topique et la solution ophtalmique de bimatoprost en pour les cils, s'il y avait une dose stable à l'entrée dans l'étude.
Le critère principal d'efficacité des deux études était la proportion de sujets qui atteignaient un score SALT ≤20 (au moins 80 % du cuir chevelu couvert de cheveux) à la semaine 36. En outre, les deux études ont évalué la chute des cheveux estimée par les patients au niveau du cuir chevelu sur une échelle en 5 points (Scalp Hair Assessment PRO™) et la chute des cils et des sourcils évaluée par le médecin de l'étude à l'aide d'une échelle en 4 points (ClinRO Measure for Eyebrow Hair Loss™, ClinRO Measure for Eyelash Hair Loss™).
Caractéristiques à l'inclusion
La partie de phase III de l'étude BRAVE-AA1 et l'étude de phase III BRAVE-AA2 ont englobé 1200 patients adultes. Sur l'ensemble des groupes de traitement l'âge moyen était de 37.5 ans, 61 % des patients étaient des femmes.
La durée moyenne de la pelade depuis son apparition et la durée moyenne de l'épisode actuel de perte de cheveux étaient respectivement de 12.2 ans et 3.9 ans. Le score SALT médian sur l'ensemble des études était de 96 et environ 44 % des patients présentaient une pelade universelle. Dans l'ensemble des études, à l'inclusion, 69 % des patients présentaient une perte significative ou totale des sourcils et 58 % présentaient une perte significative ou totale des cils, mesurées par des scores de 2 ou de 3 dans les échelles ClinRO pour les sourcils et les cils. Environ 90 % des patients avaient reçu au moins un traitement contre la pelade avant de participer aux études, et 50 % au moins un immunosuppresseur systémique. L'utilisation de traitements concomitants autorisés dans la pelade n'a été rapportée que par 4.3 % des patients au cours des études.
Réponse clinique
Dans les deux études, une proportion significativement plus importante de patients randomisés sous baricitinib 4 mg une fois par jour a atteint un score SALT ≤20 à la semaine 36 comparativement à ceux sous placebo, et ce dès la semaine 8 dans l'étude BRAVE-AA1 et dès la semaine 12 dans l'étude BRAVE-AA2. Une efficacité similaire a été observée pour la plupart des critères d'évaluation secondaires (Tableau 8). La figure 7 montre la proportion de patients ayant atteint un SALT ≤20 par rapport à la valeur initiale jusqu'à la semaine 36. Les effets du traitement dans les sous-groupes (sexe, âge, poids, débit de filtration glomérulaire estimé, origine ethnique, région géographique, gravité de la maladie, durée de l'épisode actuel de pelade) étaient en accord à la semaine 36 avec les résultats obtenus pour l'ensemble de la population de l'étude.
Tableau 8. Efficacité du baricitinib jusqu'à la semaine 36 (FASa)
|
Étude
|
BRAVE-AA1
|
BRAVE-AA2
| |
Groupe de traitement
|
PBO
N=189
|
BARI 2 mg
N=184
|
BARI 4 mg
N=281
|
PBO
N=156
|
BARI 2 mg
N=156
|
BARI 4 mg
N=234
| |
SALT ≤20 à la semaine 36
|
5.3%
|
21.7%**
|
35.2%**
|
2.6%
|
17.3%**
|
32.5%**
| |
SALT ≤20 à la semaine 24
|
4.8%
|
11.4%**
|
26.7%**
|
1.3%
|
10.9%**
|
28.2%**
| |
Scalp Hair Assessment PRO de 0 ou 1 à la semaine 36 avec ≥2 points d'amélioration par rapport à la valeur initiale b
|
5.0%
|
16.0%**
|
33.1%**
|
4.0%
|
16.1%**
|
34.4%**
| |
ClinRO Measure for Eyebrow Hair Loss de 0 ou 1 à la semaine 36 avec ≥2 points d'amélioration par rapport à la valeur initiale c
|
3.2%
|
19.1%**
|
31.4%**
|
4.5%
|
11.5%*
|
34.8%**
| |
ClinRO Measure for Eyelash Hair Loss de 0 ou 1 à la semaine 36 avec ≥2 points d'amélioration par rapport à la valeur initiale d
|
3.1%
|
13.5%*
|
33.5%**
|
5.6%
|
10.1%
|
34.3%**
|
BARI = baricitinib; PBO = placebo
* statistiquement significatif versus placebo sans ajustement pour la multiplicité; ** statistiquement significatif versus placebo avec ajustement pour la multiplicité.
a Full Analysis Set (FAS, population totale), englobe tous les patients randomisés.
b 0 = pas de chute de cheveux, 1 = chute de cheveux dans une zone limitée du cuir chevelu (de 1% à 20%). Chez les patients qui ont un score Scalp Hair PRO ≥3 au début de l'étude (n=181, 175 respectivement 275 pour BRAVE-AA1 et n=151, 149 respectivement 215 pour BRAVE-AA2)
c 0 = sourcils couvrant en totalité, sans zones de perte de poils, 1 = lacunes minimes dans les poils des sourcils, avec une distribution régulière. Chez les patients qui ont un score ClinRO Measure for Eyebrow Hair loss ≥2 au début de l'étude (n=124, 136 respectivement 188 pour BRAVE-AA1 et n = 112, 104 respectivement 161 pour BRAVE-AA2)
d 0 = les cils forment une ligne continue le long des paupières des deux yeux, 1 = il y a des lacunes minimes, les cils sont implantés à intervalles réguliers le long des paupières des deux yeux. Chez les patients qui ont un score ClinRO Measure for Eyelash Hair Loss ≥2 au début de l'étude (n=96, 111 respectivement 167 pour BRAVE-AA1 et n=90, 89 respectivement 140 pour BRAVE-AA2)
Figure 7. Proportion de patients avec un SALT ≤20
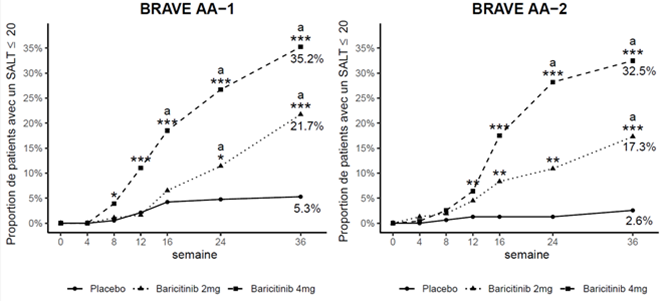
a Statistiquement significatif après ajustement pour la multiplicité.
* Valeur de p pour baricitinib versus placebo ≤0.05; ** Valeur de p pour baricitinib versus placebo ≤0.01; *** Valeur de p pour baricitinib versus placebo ≤0.001.
Efficacité jusqu'à la semaine 52
La proportion de patients traités par le baricitinib atteignant un SALT ≤20 a continué d'augmenter après la semaine 36, atteignant 39.0 % des patients sous baricitinib 4 mg à la semaine 52. Les résultats dans les sous-groupes pour la sévérité de la maladie à l'inclusion et la durée de l'épisode de pelade à la semaine 52 correspondaient à ceux observés à la semaine 36 et aux résultats de la population générale de l'étude.
Réduction de la dose
Dans l'étude BRAVE-AA2, les patients ayant reçu du baricitinib 4 mg une fois par jour depuis la randomisation initiale et ayant atteint un SALT ≤20 à la semaine 52 ont été de nouveau randomisés en double aveugle pour continuer à prendre le traitement de 4 mg une fois par jour ou pour réduire la dose à 2 mg une fois par jour. Les résultats montrent que 96 % des patients qui sont restés sous baricitinib 4 mg et 74 % des patients qui ont été rerandomisés pour prendre du baricitinib 2 mg ont maintenu leur réponse à la semaine 76.
COVID-19
Dans une étude clinique randomisée, en double aveugle, contrôlée contre placebo (ACTT-2), le baricitinib 4 mg une fois par jour + remdésivir a été comparé au placebo + remdésivir chez 1033 patients COVID-19 adultes hospitalisés.
L'échelle de classification en 8 points (Ordinal Score, OS) du NIAID (National Institute of Allergy and Infectious Diseases) suivante a été utilisée pour classer la maladie de base en fonction de son degré de gravité:
8. Mort;
7. Hospitalisé, avec ventilation mécanique invasive ou ECMO (oxygénation par membrane extracorporelle);
6. Hospitalisé, avec ventilation non invasive ou oxygénothérapie à haut débit;
5. Hospitalisé, avec supplémentation d'oxygène à bas débit;
4. Hospitalisé, sans supplémentation d'oxygène à bas débit, mais avec traitement médical (en lien avec la COVID-19 ou autre);
3. Hospitalisé, sans supplémentation d'oxygène à bas débit - ne nécessitant plus de traitement médical;
2. Non hospitalisé, avec limitations des activités et/ou supplémentation d'oxygène à domicile;
1. Non hospitalisé, sans limitations des activités
L'étude incluait 14% de patients avec OS 4, 55% de patients avec OS 5, 21% de patients avec OS 6 et 11 % de patients avec OS 7.
Le critère clinique principal a été le temps écoulé jusqu'à la guérison dans les 29 jours qui ont suivi la randomisation, la guérison étant définie comme l'obtention d'un OS de la catégorie 1, 2 ou 3.
Les patients ont été randomisés dans un rapport de 1:1, stratifiés selon le degré de gravité de la maladie au moment de l'inclusion dans l'étude, pour recevoir un traitement de baricitinib + remdésivir (n=515) ou de placebo + remdésivir (n=518). Les patients ont été traités selon le schéma suivant:
·Baricitinib 4 mg ou placebo une fois par jour (par voie orale) sur 14 jours ou jusqu'à la sortie de l'hôpital
·Remdésivir 200 mg le jour 1, suivi de 100 mg une fois par jour (par perfusion intraveineuse) les jours suivants, sur une période de traitement totale de 10 jours ou jusqu'à la sortie de l'hôpital.
L'âge moyen des patients au début de l'étude était de 55 ans, 30% des patients ayant 65 ans ou plus. 63 % des patients étaient des hommes, 48 % étaient caucasiens, 15 % avaient la peau noire et 10% étaient asiatiques. 83% des patients présentaient des comorbidités, l'obésité (56 %), l'hypertension (52 %) et le diabète de type 2 (37 %) étant les comorbidités les plus fréquentes. Les caractéristiques démographiques et les caractéristiques liées à la maladie étaient équilibrées dans le groupe de l'association et dans le groupe du placebo.
En ce qui concerne la population totale (ITT), le temps moyen écoulé jusqu'à la guérison a été de 7 jours sous baricitinib + remdésivir, par rapport à 8 jours sous placebo + remdésivir [Hazard Ratio: 1.15 (IC 95 % 1.00, 1.31); p=0.047]. Le bénéfice clinique sous baricitibib + remdésivir a été le plus prononcé chez les patients qui nécessitaient une supplémentation d'oxygène à bas débit (OS 5), une ventilation non invasive ou une oxygénothérapie à haut débit (OS 6) (voir Tableau 9). Chez les patients qui nécessitaient une ventilation mécanique invasive ou une ECMO (OS 7), la durée moyenne jusqu'à la guérison n'a pas pu être estimée. Chez les patients qui ne nécessitaient pas d'oxygène (OS 4), il n'y a pas eu d'avantage manifeste avec le baricitinib + remdésivir au niveau du temps médian écoulé jusqu'à la guérison (5 jours) par rapport au remdésivir (4 jours) (Recovery Rate Ratio 0.88 [IC 95 % 0.62–1.23]).
Tableau 9. Résultats de guérison en fonction du score ordinal au début de l'étude pour les patients nécessitant de l'oxygène - Étude ACTT-2a
|
|
Score ordinal au début de l'étude - Population ITT
| |
|
5
|
6
|
7
| |
|
Oxygène à
bas débit
|
Ventilation non invasive ou oxygène à haut débit
|
Ventilation mécanique invasive/ECMO
| |
|
BARI
+ RDV
(n=288)
|
PBO
+ RDV
(n=276)
|
BARI
+ RDV
(n=103)
|
PBO
+ RDV
(n=113)
|
BARI
+ RDV
(n=54)
|
PBO
+ RDV
(n=57)
| |
Nombre de guérisons
|
262
|
243
|
82
|
73
|
22
|
21
| |
Temps médian écoulé jusqu'à la guérison
Jours (IC à 95 %)
|
5
(5, 6)
|
6
(5, 6)
|
10
(9, 13)
|
18
(13, 21)
|
NE
(25, NE)
|
NE
(26, NE)
| |
Recovery Rate Ratio
Joursa (IC 95 %)
|
1.17
(0.98, 1.39)
|
1.51
(1.10, 2.08)
|
1.08
(0.59, 1.97)
|
ECMO = oxygénation extracorporelle par membrane, ITT = Intention to Treat, RDV = remdésivir, PBO = placebo, BARI= baricitinib
a Recovery Rate Ratio calculé selon le modèle stratifié de Cox. Un Recovery Rate Ratio >1 indique un avantage pour le baricitinib + remdésivir. NE = non évaluable.
La mortalité au jour 29 dans la population totale s'est élevée à 4.7 % (n=24/515) dans le groupe baricitinib/remdésivir, par rapport à 7.1 % (n=37/518) dans le groupe placebo/remdésivir (Hazard Ratio: 0.65; [IC 95 % de 0.39 à 1.09]; p=0.102). Le bénéfice clinique du baricitinib était le plus prononcé chez les patients qui nécessitaient une supplémentation d'oxygène à bas débit, une ventilation non invasive ou une oxygénothérapie à haut débit (voir Tableau 10):
Tableau 10. Résultats de mortalité au jour 29 en fonction du score ordinal au début de l'étude pour les patients nécessitant de l'oxygène - Étude ACTT-2
|
|
Score ordinal au début de l'étude – Population ITTc
| |
|
5
|
6
|
7
| |
|
Oxygène à bas débit
|
Ventilation non invasive ou oxygène à haut débit
|
Ventilation mécanique invasive/ECMO
| |
|
BARI
+ RDV
(n=288)
|
PBO
+ RDV
(n=276)
|
BARI
+ RDV
(n= 103)
|
PBO
+ RDV
(n=113)
|
BARI
+ RDV
(n=54)
|
PBO
+ RDV
(n=57)
| |
Mortalité au jour 29 – N (%)a
|
5
(1.9%)
|
12
(4.7%)
|
7
(7.5%)
|
13
(13.0%)
|
12 (23.1%)
|
12
(22.6%)
| |
Hazard Ratiob
(IC 95 %)
|
0.4
(0.14, 1.14)
|
0.55
(0.22, 1.38)
|
1.00 (0.45, 2.22)
|
a Les indications de pourcentages se basent sur la méthode de Kaplan-Meier.
b Les Hazard Ratios pour les sous-groupes de l'échelle ordinale au début de l'étude proviennent de modèles non stratifiés de Proportional-Hazard de Cox.
c ITT = Intention-To-Treat Population = tous les patients randomisés .
Des analyses par sous-groupes ont montré pour les patients âgés de moins de 40 ans et pour les patients sans comorbidités une durée plus longue du temps écoulé jusqu'à la guérison lorsqu'ils étaient traités avec le baricitinib+remdésivir plutôt qu'avec le remdésivir seul, mais leurs chances de guérison (Recovery Rate Ratio) se situaient un peu au-dessus de celles du traitement avec remdésivir seul:
·Patients de moins de 40 ans (n=162/1033): 6 versus 5 jours; Recovery Rate Ratio (IC 95 %): 1.01 (0.74-1.38)
·Patients sans comorbidités (n=138/1033): 7 versus 6 jours; Recovery Rate Ratio (IC 95 %): 1.11 (0.79-1.55)
Des analyses de sous-groupes ont montré pour les femmes et pour les patients non obèses aucune différence dans le temps écoulé jusqu'à la guérison lorsqu'ils étaient traités avec le baricitinib+remdésivir plutôt qu'avec le remdésivir seul mais leurs chances de guérison (Recovery Rate Ratio) se situaient un peu au-dessus de celles du traitement avec remdésivir seul:
·Femmes (n=316/1033): 7 versus 7 jours; Recovery Rate Ratio (IC 95 %): 1.06 (0.85-1.32)
·Patients non obèses (n=362/1033): 9 versus 9 jours; Recovery Rate Ratio (IC 95 %): 1.07 (0.87-1.32)
|