Propriétés / EffetsCode ATC
L04AB01
Benepali est un biosimilaire.
Mécanisme d’action
L'étanercept est une protéine de fusion du récepteur p75 du facteur nécrosant des tumeurs. L'étanercept est produit par la méthode des recombinants d'ADN (génie génétique) et exprimé dans des cellules ovariennes de hamster chinois (CHO). L'étanercept est un dimère d'une protéine chimère génétiquement modifiée qui a été obtenue en fusionnant le domaine de liaison extracellulaire du récepteur 2 humain au facteur nécrosant des tumeurs (TNFR2/p75) et le domaine Fc de l'IgG1 humaine. Ce fragment Fc contient les régions charnières, CH2 et CH3, mais pas la région CH1 de l'IgG1. L'étanercept est constitué de 934 acides aminés et son poids moléculaire est d'environ 150 kilodaltons. L’activité est déterminée en mesurant la capacité de l’étanercept à neutraliser l’inhibition de la croissance des cellules A375 induite par le TNFα. L'activité spécifique de l'étanercept est de 1.7 × 106 unités/mg.
Pharmacodynamique
Le facteur nécrosant des tumeurs (TNF) est une cytokine dominante dans le processus inflammatoire de la polyarthrite rhumatoïde (PR). Des taux élevés de TNF sont également retrouvés dans les membranes synoviales et les plaques de psoriasis des patients atteints de rhumatisme psoriasique, et dans le sérum et le tissu synovial des patients atteints de spondylarthrite ankylosante.
Dans les plaques de psoriasis, l'infiltration par les cellules inflammatoires, y compris les cellules T, conduit à une augmentation des taux de TNF dans les lésions psoriasiques, comparativement aux taux observés au niveau des zones non atteintes de la peau.
L'étanercept est un inhibiteur compétitif de la liaison du TNF à ses récepteurs de surface inhibant ainsi l'activité biologique du TNF.
Le TNF et la lymphotoxine sont des cytokines pro-inflammatoires qui lient deux récepteurs distincts à la surface des cellules: les récepteurs du facteur nécrosant des tumeurs (TNFR) de 55 kilodaltons (p55) et de 75 kilodaltons (p75). Ces deux TNFR existent naturellement sous des formes membranaires et solubles. On pense que les TNFR solubles régulent l'activité biologique du TNF.
Le TNF et la lymphotoxine existent principalement sous forme d'homotrimères, leur activité biologique étant dépendante de la réticulation des TNFR à la surface des cellules. Les récepteurs dimères solubles tels que l'étanercept présentent une affinité plus marquée pour le TNF que les récepteurs monomères et sont des inhibiteurs compétitifs beaucoup plus puissants de la liaison du TNF à ses récepteurs cellulaires. En outre, l'utilisation d'une région Fc d'immunoglobuline en tant qu'élément de fusion dans la construction d'un récepteur dimère confère à la molécule une demi-vie plasmatique plus longue.
L’étanercept peut également moduler les réponses biologiques contrôlées par d’autres molécules agissant en aval sur l’inflammation (p.ex. les cytokines, adhésines ou protéinases) dont l'activité est induite ou régulée par le TNF.
Efficacité clinique
Le programme de développement clinique visant à prouver la comparabilité clinique de Benepali et de l’étanercept-le produit de référence, a été mené avec des patients atteints de polyarthrite rhumatoïde (voir rubrique «Études cliniques sur l’efficacité et la sécurité de Benepali» à la fin de cette section).
Cette rubrique présente les données issues de quatre études contrôlées randomisées et de deux études supplémentaires en ouvert chez l’adulte (V et VI) atteint de polyarthrite rhumatoïde, de trois études sur l’arthrite juvénile idiopathique, d’une étude chez l’adulte atteint de rhumatisme psoriasique, de quatre études chez l’adulte atteint de spondylarthrite ankylosante, d’une étude chez le patient pédiatrique atteint de psoriasis en plaques ainsi que de quatre études en ouvert chez l’adulte atteint de psoriasis en plaques.
Patients adultes atteints de polyarthrite rhumatoïde
L’efficacité et la sécurité de l’étanercept ont été étudiées chez 2'680 patients atteints de polyarthrite rhumatoïde (PR) active.
La sécurité et l’efficacité de l’étanercept ont été étudiées dans quatre études randomisées, en double aveugle, contrôlées contre placebo et dans deux études supplémentaires (V et VI).
La première étude a évalué 234 patients atteints de PR active, âgées d’au moins 18 ans, dont le traitement par au moins et au plus quatre antirhumatismaux modificateurs de la maladie (ARMM, tels que hydroxycloroquine, or injectable ou par voie orale, méthotrexate, azathioprine, D-pénicillamine, sulfasalazine) avait échoué et qui présentaient au moins 12 articulations douloureuses, au moins 10 articulations enflées, et soit une VS ≥28 mm/h, une concentration de CRP ˃2.0 mg/dl soit une raideur matinale ≥ 45 min. Des doses de 10 mg ou 25 mg d'étanercept ou de placebo ont été administrées par voie sous-cutanée 2× par semaine pendant 6 mois consécutifs.
Les résultats de cette étude contrôlée ont été exprimés en pourcentage d'amélioration de la PR, en utilisant les critères de réponse de l'American College of Rheumatology (ACR). Le critère d’évaluation principal était l’obtention d’une réponse ACR 20 après 3 mois. Conformément à la définition, une réponse ACR 20 est obtenue lorsqu’un patient obtient une amélioration de 20% du nombre de ses articulations douloureuses et du nombre de ses articulations enflées, plus une amélioration d’au moins 20% dans au moins trois des 5 critères suivants: (1) évaluation de la douleur par les patients, (2) appréciation globale par les patients, (3) évaluation globale par le médecin, (4) auto-évaluation de la limitation fonctionnelle par les patients et (5) réaction de phase aiguë (VS ou CRP). Les réponses ACR 50 et 70 sont définies conformément aux mêmes critères comme une amélioration de 50% ou de 70%.
Les réponses ACR 20 et ACR 50 étaient supérieures chez les patients traités par l'étanercept par rapport au placebo à 3 et 6 mois (ACR 20: étanercept 62% et 59%, placebo 23% et 11% respectivement à 3 et 6 mois; ACR 50: étanercept 41% et 40%, placebo 8% et 5% respectivement à 3 et 6 mois; p< 0.01 étanercept contre placebo à tous les moments de mesure pour les réponses ACR 20 et ACR 50).
Environ 15% des patients recevant l'étanercept ont obtenu une réponse ACR 70 à 3 mois et à 6 mois, comparativement à moins de 5% des patients sous placebo.
Parmi les patients recevant l'étanercept, les réponses cliniques ont généralement débuté 1 à 2 semaine(s) après l'initiation du traitement, et ont été quasiment toujours obtenues dans les 3 mois. Une réponse dose-dépendante a été observée; les résultats avec 10 mg étaient intermédiaires entre le placebo et 25 mg. L'étanercept était significativement supérieur au placebo sur tous les items des critères ACR, ainsi que sur les autres mesures d'activité de la polyarthrite rhumatoïde non compris dans ces critères de réponse ACR, comme la durée de la raideur matinale. L'échelle HAQ (Health Assessment Questionnaire), incluant le handicap, l'activité, l'état mental, l'état général, l'état des fonctions articulaires, a été évaluée tous les 3 mois pendant l'étude. Tous les domaines de l'échelle HAQ ont été améliorés chez les patients traités par l'étanercept comparés aux témoins à 3 et 6 mois.
Après l'arrêt de l'étanercept, les symptômes d'arthrite sont généralement réapparus au cours du mois suivant. Selon les résultats des études en ouvert, la reprise du traitement par l'étanercept après des arrêts allant jusqu'à 24 mois a entraîné la même amplitude de réponse que chez les patients recevant l'étanercept sans interruption de traitement. Des réponses durables ont été observées chez des patients recevant l'étanercept sans interruption jusqu'à 10 ans dans les études d’extension en ouvert.
L'efficacité de l'étanercept a été comparée avec le méthotrexate par voie orale dans une étude randomisée, contrôlée contre traitement actif avec des examens radiographiques réalisés en aveugle comme critère d'évaluation principal, chez 632 patients adultes ayant une polyarthrite rhumatoïde active (de durée <3 ans) qui n'avaient jamais reçu de traitement par méthotrexate. Les critères d’inclusion étaient plus de 12 articulations sensibles à la pression, plus de 10 articulations enflées, et une vitesse de sédimentation supérieure à 28 mm/h, une concentration sérique de la protéine C-réactive supérieure à 2.0 mg/dl ou une rigidité matinale > 45 min. Des doses de 10 mg ou de 25 mg d'étanercept ont été administrées par voie sous-cutanée 2× par semaine jusqu'à 24 mois. Les doses de méthotrexate ont été augmentées de 7.5 mg/semaine à 20 mg/semaine maximum au cours des 8 premières semaines de l'étude et maintenues jusqu'à 24 mois. Avec l'étanercept 25 mg, l'amélioration clinique, y compris le délai d'action sous deux semaines, a été similaire à celle observée lors des études précédentes, et s'est maintenue jusqu'à 24 mois. Le délai d’action était plus court avec l’étanercept 25 mg comparé au méthotrexate. À l'inclusion, les patients avaient un degré d'invalidité modéré, avec des scores moyens de HAQ de 1.4 à 1.5. Le traitement par l'étanercept à 25 mg a entraîné une amélioration importante à 12 mois, avec environ 44% de patients obtenant un score de HAQ normal (score de HAQ < 0.5). Ce bénéfice a été maintenu la deuxième année de cette étude.
Dans cette étude, les dommages structuraux articulaires ont été évalués radiographiquement et exprimés en modification du Score Total de Sharp (STS) et de ses composants: le score d'érosion et le Score de Pincement Articulaire (SPA). Les radiographies des mains/poignets et pieds ont été lues à l'inclusion puis à 6, 12 et 24 mois. La dose de 10 mg d'étanercept a eu constamment moins d'effet sur les dommages structuraux que la dose de 25 mg. Dans l’étude de 24 mois, l'étanercept à 25 mg a été significativement supérieur au méthotrexate pour le STS et les scores d'érosion. Les résultats radiographiques sont présentés dans la figure ci-dessous.
Progression radiographique: comparaison étanercept vs méthotrexate chez des patients ayant une polyarthrite rhumatoïde d'ancienneté <3 ans
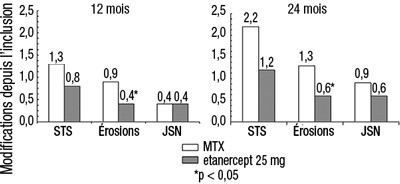
|
|
Méthotrexate
| |
|
| |
|
Étanercept 25 mg
|
*p < 0.05
Dans une autre étude contrôlée contre traitement actif, randomisée, en double aveugle, l'efficacité clinique, la tolérance, et l'évolution radiographique chez des patients atteints de polyarthrite rhumatoïde traités par étanercept seul (25 mg 2× par semaine), ou méthotrexate seul (7.5 à 20 mg par semaine, dose médiane 20 mg) ou étanercept associé au méthotrexate débutés simultanément, ont été comparées chez 682 patients adultes ayant une polyarthrite rhumatoïde active d'ancienneté de 6 mois à 20 ans (médiane 5 ans) et qui avaient eu une réponse insuffisante à au moins un ARMM autre que le méthotrexate.
Les patients traités par l'étanercept associé au méthotrexate avaient des réponses ACR 20, ACR 50 et ACR 70 ainsi qu'une amélioration des scores DAS (Disease Activity Score) et HAQ significativement plus élevées à la fois à 24 et 52 semaines, comparativement aux patients de chacun des groupes en monothérapie (résultats présentés dans le tableau ci-dessous). Des avantages significatifs avec l'étanercept associé au méthotrexate comparé à l'étanercept en monothérapie et au méthotrexate en monothérapie ont aussi été observés après 24 mois.
Résultats d'efficacité clinique à 12 mois: comparaison étanercept vs méthotrexate vs étanercept associé au méthotrexate chez des patients avec une polyarthrite rhumatoïde d'ancienneté de 6 mois à 20 ans
|
Critère
|
Méthotrexate
(n=228)
|
Étanercept
(n=223)
|
Étanercept + méthotrexate
(n=231)
| |
Réponses ACRa
| |
ACR 20
|
58.8%
|
65.5%
|
74.5%†,φ
| |
ACR 50
|
36.4%
|
43.0%
|
63.2%†,φ
| |
ACR 70
|
16.7%
|
22.0%
|
39.8%†,φ
| |
DAS
| |
Score à l’inclusionb
|
5.5
|
5.7
|
5.5
| |
Semaine 52b
|
3.0
|
3.0
|
2.3†,φ
| |
Rémissionc
|
14%
|
18%
|
37%†,φ
| |
HAQ
| |
Score à l’inclusion
|
1.7
|
1.7
|
1.8
| |
Semaine 52
|
1.1
|
1.0
|
0.8†,φ
|
a: Les patients qui n'avaient pas terminé les 12 mois de l'étude ont été considérés comme non-répondeurs.
b: Les valeurs du DAS sont des moyennes.
c: La rémission est définie par un DAS < 1.6.
Valeur du p lors des comparaisons deux à deux:
†: p< 0.05 pour les comparaisons des groupes étanercept + méthotrexate vs méthotrexate.
φ: p< 0.05 pour les comparaisons des groupes étanercept + méthotrexate vs étanercept.
L'évolution radiographique à 12 mois était significativement moins importante dans le groupe étanercept que dans le groupe méthotrexate, alors que l'association était significativement meilleure que chacune des monothérapies pour ralentir l'évolution radiographique (voir figure ci-dessous).
Évolution radiographique: comparaison étanercept vs méthotrexate vs étanercept associé au méthotrexate chez des patients ayant une polyarthrite rhumatoïde d'ancienneté de 6 mois à 20 ans (résultats à 12 mois).
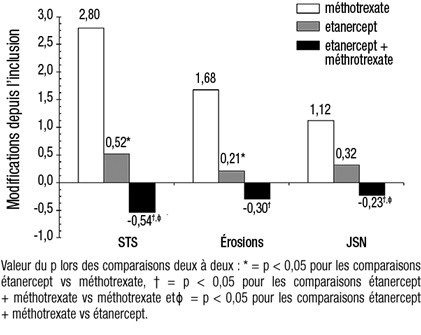
Des avantages significatifs avec l'étanercept associé au méthotrexate comparativement à l'étanercept en monothérapie et au méthotrexate en monothérapie ont aussi été observés après 24 mois. De même, des avantages significatifs avec l'étanercept en monothérapie comparativement au méthotrexate en monothérapie ont aussi été observés après 24 mois.
Dans une analyse où tous les patients sortis prématurément de l'étude quelle qu'en soit la raison étaient considérés comme s'étant aggravés, le pourcentage de patients sans aggravation (variation du STS ≤ 0.5) à 24 mois était plus élevé dans le groupe étanercept associé au méthotrexate, comparativement à l'étanercept seul et au méthotrexate seul (respectivement 62%, 50% et 36%; p < 0.05). La différence entre étanercept seul et méthotrexate seul a aussi été significative (p < 0.05). Parmi les patients ayant terminé la totalité des 24 mois de traitement dans l'étude, les taux de patients sans aggravation étaient respectivement de 78%, 70% et 61%.
La tolérance et l'efficacité de l'étanercept à la dose de 50 mg (deux injections de 25 mg en sous-cutanée) ont été évaluées dans une étude en double aveugle, contrôlée contre placebo chez 420 patients atteints de polyarthrite rhumatoïde active. Dans cette étude, 53 patients ont reçu du placebo, 214 patients ont reçu 50 mg d'étanercept 1× par semaine et 153 patients ont reçu 25 mg d'étanercept 2× par semaine. Les profils d'efficacité et de tolérance des deux schémas posologiques de l'étanercept ont été similaires à la 8ème semaine en ce qui concerne les signes et symptômes de la PR.
À la 16ème semaine de traitement, les deux schémas posologiques n’étaient plus équivalents, avec une tendance non significative en faveur de l’administration plus fréquente de l’étanercept.
Une injection unique d’étanercept 50 mg/ml était bioéquivalente à deux injections simultanées de 25 mg/ml.
L’étude V a évalué 89 patients remplissant les mêmes critères d’inclusion que dans la première étude menée, sauf que les patients de l’étude V avaient reçu en outre pendant au moins 6 mois du méthotrexate à une dose stable (12.5 à 25 mg/semaine) pendant au moins 4 semaines et présentaient au moins 6 articulations douloureuses. Les patients ont reçu, outre leur dose stable de méthotrexate pendant 6 mois consécutifs, une dose de 25 mg d’étanercept ou de placebo 2× par semaine par voie sous-cutanée.
L’étude finale VI a évalué 559 patients qui remplissaient des critères d’inclusion similaires à ceux des études précédentes. Les patients furent traités pendant un maximum de 6 mois par quatre schémas posologiques d’étanercept (10 mg 1× par semaine, 10 mg 2× par semaine, 25 mg 1× par semaine ou 25 mg 2× par semaine).
Les résultats des études contrôlées V et VI ont été indiqués en pourcentage d’amélioration de la PR conformément aux critères de réponse de l’American College of Rheumatology (ACR). Dans l’étude V, le critère d’évaluation principal était l’obtention d’une réponse ACR 20 après 6 mois. Conformément à la définition, une réponse ACR 20 est obtenue lorsqu’un patient obtient une amélioration de 20% du nombre de ses articulations douloureuses et du nombre de ses articulations enflées, plus 20% d’amélioration au minimum dans au moins trois des cinq critères suivants: (1) évaluation de la douleur par les patients, (2) évaluation globale par les patients, (3) évaluation globale par le médecin, (4) auto-évaluation de la limitation fonctionnelle par les patients et (5) réaction de phase aiguë (VS ou CRP). Les réponses ACR 50 et 70 sont définies conformément aux mêmes critères comme une amélioration de 50% ou de 70%. Dans l’étude VI, le critère d’évaluation principal était le pourcentage d’amélioration des articulations douloureuses et enflées après 3 mois.
Après 3 et 6 mois dans les études V et VI, la réponse était meilleure chez les patients traités par l’étanercept.
Dans l’étude V, environ 15% des patients ayant reçu l’étanercept ont obtenu une réponse ACR 70 à 3 et à 6 mois, comparativement à moins de 5% des patients sous placebo. Parmi les patients recevant l'étanercept, les réponses cliniques ont généralement débuté 1 à 2 semaine(s) après l'initiation du traitement, et ont été quasiment toujours obtenues dans les 3 mois.
Enfants et adolescents atteints d’arthrite juvénile idiopathique
La sécurité et l’efficacité de l’étanercept ont été évaluées dans une étude en 2 phases, chez 69 enfants atteints d’arthrite juvénile idiopathique polyarticulaire ayant présenté différentes formes de début de la maladie (polyarthrite, oligoarthrite, origine systémique). Les patients inclus dans l’étude étaient âgés de 4 à 17 ans, présentaient une arthrite juvénile idiopathique polyarticulaire active modérée à sévère et étaient réfractaires ou intolérants au méthotrexate. Une dose stable d’un seul anti-inflammatoire non stéroïdien et/ou de prednisolone (< 0.2 mg/kg/jour ou 10 mg maximum) a été maintenue chez les patients. Dans la première phase de l’étude, tous les patients ont reçu 0.4 mg/kg (maximum 25 mg par injection) d’étanercept administré en sous-cutané 2× par semaine. Dans la deuxième phase, les patients présentant une réponse clinique au 90e jour ont été randomisés soit pour rester sous étanercept, soit pour recevoir le placebo pendant 4 mois et ont été surveillés pour détecter toute flambée de la maladie. La réponse a été mesurée en utilisant le score ACR Pedi 30 (score pédiatrique de l’American College of Rheumatology), à savoir une amélioration ≥ 30% d’au moins 3 des 6 critères-clés de l’ACJ et une aggravation ≥ 30% d’au plus 1 des 6 critères. Ces critères incluent le nombre d’articulations atteintes, la limitation des mouvements, l’évaluation globale de la maladie par le médecin et le patient/parent, le handicap fonctionnel et la vitesse de sédimentation (VS). Une flambée de la maladie était définie comme une aggravation ≥ 30% de 3 des 6 critères-clés de l’ACJ et une amélioration ≥ 30% d’au plus 1 des 6 critères, ainsi qu’un minimum de 2 articulations atteintes.
Dans la première phase de l’étude, 51 des 69 patients (74%) ont bénéficié d’une réponse clinique et ont été inclus dans la deuxième phase de l’étude. Dans la deuxième phase de l’étude, 6 patients sur 25 (24%) maintenus sous étanercept ont eu une flambée de la maladie, en comparaison à 20 patients sur 26 (77%) sous placebo (p = 0.007). À partir du début de la deuxième phase de l’étude, le délai moyen d’apparition de la flambée a été supérieur ou égal à 116 jours pour les patients ayant reçu de l’étanercept et de 26 jours pour les patients sous placebo. Chaque item des critères de l’ACJ s’est aggravé dans le groupe placebo et est resté stable ou s’est même amélioré dans le groupe étanercept. Les données suggèrent la possibilité d’un taux plus élevé de flambée chez les patients présentant une VS plus accélérée à l’inclusion. Parmi les patients qui ont bénéficié d’une réponse clinique à 90 jours et qui ont été inclus dans la deuxième phase de l’étude, certains des patients maintenus sous étanercept ont continué à obtenir des améliorations entre le troisième mois et le septième mois alors que ceux sous placebo n’ont montré aucune amélioration.
Chez 58 patients pédiatriques de l’étude mentionnée ci-dessus (âgés d’au moins 4 ans au moment de l’inclusion), le traitement par étanercept a été poursuivi sur une période allant jusqu’à 10 ans dans une étude à long terme ouverte destinée à examiner la sécurité. La fréquence des effets indésirables graves et des infections graves n’a pas augmenté avec l’utilisation à long terme.
La sécurité à long terme de l’étanercept en monothérapie (n = 103), de l’étanercept en association avec le méthotrexate (n = 294) ou du méthotrexate en monothérapie (n = 197) a été évaluée dans une étude de registre réalisée pendant 3 ans chez 594 enfants âgés de 2 à 18 ans atteints d’arthrite juvénile idiopathique. 39 de ces enfants étaient âgés de 2 à 3 ans. Globalement, des infections ont été rapportées plus fréquemment chez les patients traités par l’étanercept que chez les patients traités par le méthotrexate seul (3.8% vs 2%) et les infections associées à l’utilisation d’étanercept ont été plus graves.
Dans une autre étude ouverte à un bras (n=127), 60 patients atteints d’une oligoarthrite étendue (extended oligoarthritis, EO) (15 patients âgés de 2 à 4 ans, 23 patients âgés de 5 à 11 ans et 22 patients âgés de 12 à 17 ans), 38 patients atteints d’arthrite associée à une enthésite (âgés de 12 à 17 ans) et 29 patients atteints d’arthrite psoriasique (âgés de 12 à 17 ans) ont reçu une dose hebdomadaire de 0.8 mg/kg de PC d’étanercept (et au maximum 50 mg par injection) pendant 12 semaines. Dans chaque sous-type d’AJI, la majorité des patients a rempli les critères ACR Pedi 30 et a montré des améliorations cliniques dans les critères secondaires d’évaluation tels que le nombre d’articulations douloureuses et l’évaluation globale de la maladie par le médecin. Le profil de sécurité a été cohérent avec celui observé dans les autres études menées sur l’AJI.
Parmi les 127 patients de l’étude principale, 109 ont participé à l’étude d’extension ouverte et ont été suivis pendant 8 ans supplémentaires, soit jusqu’à 10 ans au total. À la fin de l’étude d’extension, 84/109 (77%) des patients avaient complété l’étude; 27 (25%) prenaient encore activement de l’étanercept, 7 (6%) avaient interrompu le traitement en raison d’une maladie faible/inactive; 5 (5%) avaient recommencé l’étanercept après un arrêt antérieur du traitement; et 45 (41%) avaient arrêté l’étanercept (mais étaient restés sous surveillance); 25/109 (23%) patients avaient définitivement arrêté l’étude (dont 1 patient en raison d’une réponse insuffisante et 1 patient en raison d’effets indésirables (maladie de Hodgkin)). Les améliorations de l’état clinique obtenues dans l’étude principale se sont généralement maintenues pour tous les critères d’efficacité tout au long de la période de suivi. Les patients qui prenaient de l’étanercept ont pu participer à une phase d’arrêt optionnelle une fois pendant l’étude de prolongation, selon l’évaluation de la réponse clinique par l’investigateur. 30 patients ont participé à la phase d’arrêt. 17 (57%) patients ont présenté une poussée de la maladie (définie comme une aggravation de ≥30% dans au moins 3 des 6 composants ACR Pedi avec une amélioration de ≥30% dans l’un au maximum des 6 composants restants et au moins 2 articulations atteintes); le délai médian de survenue d’une poussée de la maladie après l’arrêt de l’étanercept était de 190 jours (EO 190 jours, arthrite associée à l’enthésite 533 jours, arthrite psoriasique 110 jours).
Une affection maligne, la maladie de Hodgkin, a été rapportée chez un patient EO-JIA âgé de 18 ans au cours de la première année de l’étude d’extension. Le nombre (taux d’événements rapporté à l’exposition pour 100 années-patients) d’événements indésirables graves, d’affections malignes et d’infections graves était respectivement de 40 (5.85 EP100PY), 1 et 14 (2.05 EP100PY).
Aucune étude n’a été menée chez les patients atteints d’arthrite juvénile idiopathique dans le but d’évaluer l’influence de la poursuite d’un traitement par étanercept chez les patients qui n’avaient pas répondu 3 mois après le début du traitement par étanercept. En outre, aucune étude n’a été menée chez les patients atteints d’AJI afin d’examiner les effets de la diminution de la dose recommandée d’étanercept après une utilisation à long terme.
Patients adultes atteints de rhumatisme psoriasique
L'efficacité de l'étanercept a été évaluée au cours d'une étude randomisée, en double aveugle, contrôlée contre placebo, chez 205 patients atteints de rhumatisme psoriasique. Les patients étaient âgés de 18 à 70 ans et souffraient d'un rhumatisme psoriasique actif (au moins 3 articulations enflées et au moins 3 articulations douloureuses) dans au moins l'une de ces formes: (1) atteinte interphalangienne distale (AID); (2) polyarthrite (absence de nodules rhumatoïdes et présence de psoriasis); (3) arthropathie destructrice; (4) rhumatisme psoriasique asymétrique; ou (5) ankylose vertébrale de type inflammatoire. Les patients avaient également des plaques de psoriasis constituant une lésion dont le diamètre devait être d’au moins 2 cm.
Les patients étaient préalablement traités avec des AINS (86%), des traitements de fond (80%), et des corticoïdes (24%). Les patients habituellement traités par méthotrexate (stable depuis au moins 2 mois) pouvaient continuer le méthotrexate à une dose constante de 25 mg/semaine maximum. Des doses de 25 mg d'étanercept (basé sur les études de recherche de dose chez les patients atteints de polyarthrite rhumatoïde) ou de placebo étaient administrées par voie sous-cutanée 2× par semaine pendant 6 mois. À la fin de l'étude en double aveugle, les patients pouvaient entrer dans une étude d'extension en ouvert à long terme pour une durée totale allant jusqu'à 2 ans.
Les réponses cliniques ont été exprimées en pourcentages de patients atteignant une réponse ACR 20, 50 et 70 et en pourcentages de patients avec une amélioration du Critère de Réponse du Rhumatisme Psoriasique (PsARC). Les résultats sont résumés dans le tableau ci-après.
Réponses des patients atteints de rhumatisme psoriasique dans les études contrôlées contre placebo
|
|
Pourcentage de patients
| |
|
Placebo
|
Étanercepta
| |
Réponse du rhumatisme psoriasique
|
n=104
|
n=101
| |
ACR 20
|
|
| |
Mois 3
|
15
|
59b
| |
Mois 6
|
13
|
50b
| |
ACR 50
|
|
| |
Mois 3
|
4
|
38b
| |
Mois 6
|
4
|
37b
| |
ACR 70
|
|
| |
Mois 3
|
0
|
11b
| |
Mois 6
|
1
|
9c
| |
PsARC
|
|
| |
Mois 3
|
31
|
72b
| |
Mois 6
|
23
|
70b
|
a: 25 mg d'étanercept sous-cutané 2× par semaine
b : p<0.001, comparé au placebo.
c : p<0.01, comparé au placebo.
Parmi les patients atteints de rhumatisme psoriasique traités par l'étanercept, les réponses cliniques étaient visibles dès la première visite (à 4 semaines) et se maintenaient pendant les 6 mois de traitement. L'étanercept a été significativement meilleur que le placebo sur tous les paramètres évaluant l'activité de la maladie (p < 0.001), et les réponses étaient similaires avec et sans traitement concomitant par le méthotrexate. La qualité de vie des patients atteints de rhumatisme psoriasique a été évaluée à chaque moment d'évaluation à l'aide de l'indice de handicap du questionnaire HAQ. L'indice de handicap était significativement amélioré à tous les moments d'évaluation chez les patients atteints de rhumatisme psoriasique traités par l'étanercept par rapport au groupe placebo (p< 0.001).
Les modifications radiographiques ont été évaluées dans l'étude sur le rhumatisme psoriasique. Des radiographies des mains et des poignets ont été réalisées à l'inclusion et à 6, 12 et 24 mois. Le score total de Sharp (STS) modifié à 12 mois est présenté dans le tableau ci-dessous:
Évolution moyenne annualisée (ES) du score total de Sharp depuis l'inclusion
|
|
Placebo
(n=104)
|
Étanercept
(n=101)
| |
Mois 12
|
1.00 (0.29)
|
-0.03 (0.09)a
|
a: p=0.0001, comparé au placebo.
Les capacités fonctionnelles ont été améliorées avec le traitement par l'étanercept pendant la phase en double aveugle, et ce bénéfice a été maintenu au cours de l'exposition à long terme jusqu'à 2 ans.
Dans le rhumatisme psoriasique, proche de la spondylarthrite ankylosante, les preuves d'efficacité de l'étanercept sont insuffisantes en raison du nombre trop faible de patients étudiés.
Aucune étude n'a été effectuée chez des patients atteints de rhumatisme psoriasique avec le schéma posologique de 50 mg 1× par semaine. Les preuves de l'efficacité du schéma posologique d'une fois par semaine chez les patients atteints de rhumatisme psoriasique reposent sur des résultats provenant d'une étude chez des patients atteints de spondylarthrite ankylosante.
Patients adultes atteints de spondylarthrite ankylosante
L'efficacité de l'étanercept dans la spondylarthrite ankylosante a été évaluée dans quatre études randomisées, en double aveugle, chez 401 patients atteints de spondylarthrite ankylosante, qui ont comparé l'administration 2× par semaine d'étanercept 25 mg avec le placebo. Un total de 401 patients a été inclus dont 203 étaient traités par l'étanercept. La plus importante de ces études (n = 277) a inclus des patients âgés de 18 à 70 ans et qui avaient une spondylarthrite ankylosante active définie par des scores d'échelle visuelle analogique (EVA) ≥30 pour la durée et l'intensité moyennes de la raideur matinale, associée à des scores EVA ≥30 pour au moins 2 des 3 paramètres suivants: évaluation globale par le patient, moyenne des valeurs EVA pour la douleur dorsale nocturne et la douleur dorsale totale, moyenne des 10 questions de l'Indice Fonctionnel de la Spondylarthrite Ankylosante de Bath (BASFI). Les patients recevant des traitements de fond, des AINS ou des corticoïdes pouvaient continuer ces traitements à des doses constantes. Les patients présentant une ankylose complète de la colonne vertébrale n'ont pas été inclus dans l'étude. Des doses de 25 mg d'étanercept (déterminées lors des études de recherche de dose chez les patients atteints de polyarthrite rhumatoïde) ou de placebo ont été administrées par voie sous-cutanée 2× par semaine pendant 6 mois.
Le critère principal d'efficacité consistait en une amélioration de 20% conformément aux critères de réponse de l’évaluation de la spondylarthrite ankylosante (ASAS 20). Comparé au placebo, le traitement avec l'étanercept a montré des améliorations significatives des réponses ASAS 20, ASAS 50 et ASAS 70 dès la deuxième semaine après l'initiation du traitement (voir tableau ci-dessous).
Réponses des patients atteints de spondylarthrite ankylosante dans une étude contrôlée contre placebo
|
|
Pourcentage de patients
| |
Réponse de la spondylarthrite ankylosante
|
Placebo
n=139
|
Étanercepta
n=138
| |
ASAS 20
| |
2 semaines
|
22
|
46a
| |
3 mois
|
27
|
60a
| |
6 mois
|
23
|
58a
| |
ASAS 50
| |
2 semaines
|
7
|
24a
| |
3 mois
|
13
|
45a
| |
6 mois
|
10
|
42a
| |
ASAS 70
| |
2 semaines
|
2
|
12b
| |
3 mois
|
7
|
29b
| |
6 mois
|
5
|
28b
|
a : p< 0.001, comparé au placebo.
b : p=0.002, comparé au placebo.
Par ailleurs, une amélioration significativement plus importante d’autres paramètres d’évaluation de l’activité de la maladie a été observée chez les patients atteints de spondylarthrite ankylosante. Comparé au placebo, l’étanercept a démontré une amélioration significativement plus importante de tous les paramètres des critères de classification ASAS (évaluation globale par les patients, douleurs dorsales globales et nocturnes, BASFI et inflammation), protéine C réactive et vitesse de sédimentation, évaluation globale par le médecin ainsi que mobilisation de la colonne vertébrale (test de Schober, ampliation thoracique, distance nuque-mur).
Parmi les patients atteints de spondylarthrite ankylosante ayant reçu l'étanercept, les réponses cliniques sont apparues dès la première visite (2 semaines) et se sont maintenues au cours des 6 mois de traitement. Les réponses étaient similaires chez les patients qui initialement recevaient ou non des traitements concomitants.
Des résultats similaires ont été obtenus au cours d’études cliniques d'effectifs moins importants réalisés dans la spondylarthrite ankylosante.
Dans une quatrième étude, la tolérance et l'efficacité de l'étanercept 50 mg (deux injections sous-cutanées de 25 mg) administré 1× par semaine versus l'étanercept 25 mg administré 2× par semaine ont été évaluées dans une étude en double aveugle, contrôlée contre placebo chez 356 patients atteints de spondylarthrite ankylosante active. Les profils de tolérance et d'efficacité des deux schémas posologiques (50 mg 1× par semaine et 25 mg 2× par semaine) ont été similaires.
Patients adultes atteints de psoriasis en plaques
La sécurité et l’efficacité de l’étanercept ont été déterminées dans quatre études randomisées, en double aveugle, contrôlées contre placebo.
Les personnes atteintes de psoriasis «en échec» dans la population, sont définis comme présentant une réponse insuffisante (PASI < 50 ou PGA, Patient Global Assessment (évaluation globale par le patient) insatisfaisante), ou une aggravation de la maladie au cours du traitement avec au moins chacun des trois traitements systémiques majeurs disponibles utilisés à une posologie adéquate pendant une durée suffisamment longue pour évaluer la réponse au traitement.
L'efficacité de l'étanercept par rapport aux autres traitements systémiques chez les patients avec un psoriasis modéré à sévère (répondeurs aux autres traitements systémiques) n'a pas été évaluée dans des études comparant directement l'étanercept aux autres traitements systémiques. À la place, l'efficacité et la tolérance de l'étanercept ont été évaluées dans quatre études randomisées, en double aveugle, contrôlées contre placebo. Le critère d'efficacité principal dans les quatre études était la proportion de patients dans chaque groupe de traitement qui atteignait le PASI 75 (c'est-à-dire une amélioration par rapport à l'inclusion d'au moins 75% du score PASI) à 12 semaines.
L'étude 1 était une étude de phase II chez des patients âgés d'au moins 18 ans et présentant un psoriasis en plaques actif mais cliniquement stable atteignant au moins 10% de la surface corporelle. 112 patients ont été randomisés pour recevoir une dose de 25 mg d'étanercept (n = 57) ou du placebo (n = 55) 2× par semaine pendant 24 semaines consécutives.
L'étude 2 a évalué 652 patients atteints de psoriasis en plaques chronique avec les mêmes critères d'inclusion que dans l'étude 1 et un PASI (Area and Severity Index) d’au moins 10 au moment de la sélection. L'étanercept a été administré à des doses de 25 mg 1× par semaine, 25 mg 2× par semaine ou 50 mg 2× par semaine pendant 6 mois consécutifs. Au cours des 12 premières semaines de la période de traitement en double aveugle, les patients ont reçu du placebo ou l'une des trois doses d'étanercept décrites ci-dessus. Après 12 semaines de traitement, les patients du groupe sous placebo ont commencé le traitement en aveugle par l'étanercept (25 mg 2× par semaine); les patients dans les groupes de traitement actif ont continué le traitement jusqu'à la semaine 24, à la dose à laquelle ils avaient été initialement randomisés.
L'étude 3 a évalué 583 patients. Les critères d'inclusion étaient les mêmes que dans l'étude 2. Dans cette étude, les patients ont reçu une dose sous-cutanée de 25 mg ou 50 mg d'étanercept, ou du placebo, 2× par semaine pendant 12 semaines. Puis tous les patients ont reçu 25 mg d'étanercept 2× par semaine en ouvert pendant 24 semaines supplémentaires.
L'étude 4 a évalué 142 patients et les critères d'inclusion étaient similaires à ceux des études 1 et 2. Dans cette étude, les patients ont reçu une dose de 50 mg d'étanercept ou du placebo 1× par semaine pendant 12 semaines consécutives; puis tous les patients ont reçu 50 mg d'étanercept 1× par semaine en ouvert pendant 12 semaines supplémentaires.
Dans l'étude 1, le groupe traité par l'étanercept avait une proportion significativement plus élevée de patients présentant une réponse PASI 75 à la semaine 12 (30%) comparativement au groupe traité par placebo (2%) (p < 0.0001). À 24 semaines, 56% des patients dans le groupe traité par l'étanercept avaient atteint le PASI 75 comparativement à 5% des patients traités par placebo. Les principaux résultats des études 2, 3 et 4 sont présentés ci-dessous.
Réponses des patients atteints de psoriasis dans les études 2, 3 et 4
|
|
Étude 2
|
Étude 3
|
Étude 4
| |
|
Placebo
|
Étanercept
|
Placebo
|
Étanercept
|
Placebo
|
Étanercept
| |
|
|
25 mg
2× par semaine
|
50 mg
2× par semaine
|
|
25 mg
2× par semaine
|
50 mg
2× par semaine
|
|
50 mg 1× par semaine
|
50 mg
1× par semaine
| |
|
n=166
|
n=162
|
n=162
|
n=164
|
n=164
|
n=193
|
n=196
|
n=196
|
n=46
|
n=96
|
n=90
| |
Réponse
|
Semaine
12
|
Semaine
12
|
Semaine
24a
|
Semaine
12
|
Semaine
24a
|
Semaine
12
|
Semaine 12
|
Semaine
12
|
Semaine
12
|
Semaine
12
|
Semaine
24a
| |
PASI 50 [%]
|
14
|
58*
|
70
|
74*
|
77
|
9
|
64*
|
77*
|
9
|
69*
|
83
| |
PASI 75 [%]
|
4
|
34*
|
44
|
49*
|
59
|
3
|
34*
|
49*
|
2
|
38*
|
71
| |
DSGAb, pas ou presque pas de lésions apparentes [%]
|
5
|
34*
|
39
|
49*
|
55
|
4
|
39*
|
57*
|
4
|
39*
|
64
|
*: p≤ 0.0001, comparé au placebo.
a: Aucune comparaison statistique versus placebo n'a été faite à la semaine 24 dans les études 2 et 4 étant donné que le groupe initialement sous placebo a commencé à recevoir l'étanercept 25 mg 2× par semaine ou 50 mg 1× par semaine à partir de la semaine 13 jusqu'à la semaine 24.
b: Dermatologist Static Global Assessment. Pas de lésions apparentes ou presque pas de lésions apparentes, défini par 0 ou 1 sur une échelle de 0 à 5.
Parmi les patients atteints de psoriasis en plaques qui recevaient l'étanercept, des réponses significatives comparativement au placebo sont apparues dès la première visite (2 semaines) et se sont maintenues durant les 24 semaines de traitement.
L'étude 2 comprenait également une période d'arrêt du traitement au cours de laquelle les patients qui avaient atteint une amélioration du PASI d'au moins 50% à la semaine 24 arrêtaient le traitement. L'apparition d'un rebond (PASI ≥150% de la valeur à l'inclusion) et le délai de rechute (définie par la perte d'au moins la moitié de l'amélioration obtenue entre l'inclusion et la semaine 24) ont été observés chez les patients qui n'étaient plus sous traitement. Au cours de la période sans traitement, les symptômes du psoriasis sont progressivement réapparus avec un délai médian de rechute de 3 mois. Aucun effet rebond de la maladie et aucun événement indésirable grave lié au psoriasis n'ont été observés. Il existe des données montrant le bénéfice de la reprise du traitement par l'étanercept chez les patients qui répondaient initialement au traitement.
Dans l'étude 3, la majorité des patients (77%) qui étaient initialement randomisés à la dose de 50 mg 2× par semaine et dont la dose d'étanercept avait été réduite à 25 mg 2× par semaine à la semaine 12 ont maintenu une réponse PASI 75 jusqu'à la semaine 36. Pour les patients qui recevaient 25 mg 2× par semaine tout au long de l'étude, la réponse PASI 75 continuait de s'améliorer entre les semaines 12 et 36.
Dans l'étude 4, le groupe traité par l'étanercept avait une proportion plus élevée de patients avec une réponse PASI 75 à la semaine 12 (38%) comparativement au groupe traité par placebo (2%) (p < 0.0001). Pour les patients qui recevaient 50 mg 1× par semaine tout au long de l'étude, les réponses d'efficacité ont continué à s'améliorer avec un PASI 75 à la semaine 24 atteignant 71%.
Dans les études à long terme (jusqu'à 34 mois) et en ouvert au cours desquelles l'étanercept avait été administré sans interruption, les réponses cliniques étaient maintenues et la sécurité était comparable aux études à court terme.
Une analyse des données cliniques n'a révélé aucune caractéristique de la maladie à l'instauration du traitement qui pourrait conduire les médecins à sélectionner le type de posologie le plus approprié (intermittent ou continu). En conséquence, le choix d'un traitement intermittent ou continu doit être basé sur l’expérience thérapeutique individuelle du patient.
Enfants et adolescents atteints de psoriasis en plaques
L’efficacité de l’étanercept a été évaluée dans une étude randomisée, en double aveugle, contrôlée contre placebo, chez 211 enfants et adolescents âgés de 4 à 17 ans, atteints de psoriasis en plaques modéré à sévère (défini par un score sPGA ≥ 3, une surface cutanée atteinte ≥ 10% (BSA) et un PASI ≥ 12). Les patients inclus avaient déjà reçu une photothérapie ou un traitement systémique ou avaient présenté une réponse insuffisante à un traitement topique.
Pendant 12 semaines, les patients ont reçu soit 0.8 mg d’étanercept/kg de PC 1× par semaine (jusqu’à 50 mg) soit un placebo. À la semaine 12, davantage de patients étaient répondeurs en ce qui concerne l’efficacité (p.ex. PASI 75) dans le groupe étanercept que dans le groupe placebo.
|
Résultats à 12 semaines chez des enfants et adolescents atteints de psoriasis en plaques
| |
|
Étanercept
0.8 mg/kg 1× par semaine
|
Placebo
| |
|
(n = 106)
|
(n = 105)
| |
PASI 75, n (%)
|
60 (57%)a
|
12 (11%)
| |
PASI 50, n (%)
|
79 (75%)a
|
24 (23%)
| |
sPGA pas de lésions apparentes ou minimal, n (%)
|
56 (53%)a
|
14 (13%)
| |
Abréviation: sPGA-static Physician Global Assessment.
a: p< 0.0001, par rapport au placebo.
|
Après la phase de traitement en double aveugle de 12 semaines, tous les patients ont reçu 0.8 mg d’étanercept/kg de PC (jusqu’à 50 mg) 1× par semaine pendant 24 semaines supplémentaires. Les taux de réponse observés pendant la phase ouverte de l’étude ont été comparables à ceux observés pendant la phase en double aveugle.
Par rapport aux patients à nouveau randomisés dans le groupe étanercept, un nombre significativement plus élevé de patients à nouveau randomisés dans le groupe placebo ont présenté une rechute de la maladie (perte de la réponse au PASI 75) pendant la phase randomisée d’arrêt. En cas de traitement continu, les taux de réponse se sont maintenus pendant une période allant jusqu’à 48 semaines.
La sécurité et l’efficacité continues de l’étanercept 0.8 mg/kg de PC (jusqu’à une dose maximale de 50 mg) 1× par semaine, observées au cours de l’étude de 48 semaines mentionnée ci-dessus, ont été évaluées au cours d’une étude d’extension ouverte allant jusqu’à 2 ans, qui incluait 181 patients pédiatriques atteints de psoriasis en plaques. L’utilisation de l’étanercept sur une longue durée n’a révélé aucun nouvel aspect concernant la sécurité. Le rapport global bénéfice-risque est resté positif et comparable aux résultats de l’étude initiale de 48 semaines.
Études cliniques sur l’efficacité et la sécurité de Benepali
Polyarthrite rhumatoïde
L’efficacité, la sécurité/tolérance, la PK et l’immunogénicité de Benepali comparé à l’étanercept-la préparation de référence, ont été évaluées dans une étude d’équivalence de phase III (étude SB4-G31-RA) randomisée contrôlée en double aveugle. Des patients atteints de PR active qui, avant la randomisation, n’avaient pas répondu suffisamment à un traitement par le MTX d’au moins 6 mois ont été recrutés dans l’étude.
Dans l’étude SB4-G31-RA de phase III, 596 patients âgés de 18 à 75 ans avec une maladie active moyenne à sévère malgré un traitement par MTX ont été évalués. Les patients avaient été randomisés dans un rapport de 1:1 dans le bras de traitement par Benepali 50 mg (n = 299) ou par l’étanercept-la préparation de référence 50 mg (n = 297). Les médicaments ont été administrés en injection sous-cutanée 1× par semaine. En outre, chaque patient a reçu le MTX en une dose stable de 10-25 mg/semaine à compter de la Semaine 4 avant la sélection jusqu’à la visite de fin de traitement (Semaine 52). Pendant l’administration de MTX dans l’étude, les patients devaient prendre 5-10 mg d’acide folique par semaine. Les patients ont participé à l’étude pendant 56 semaines après le recrutement et la randomisation, phase de suivi incluse, soit 52 semaines de traitement actif et 4 semaines de suivi pour la sécurité.
L’objectif principal visait à prouver que Benepali et l’étanerceptle produit de référence, sont équivalents à la Semaine 24 en termes de ACR 20, le critère de réponse à 20% conformément à la définition de l’American College of Rheumatology. Pour pouvoir établir une équivalence entre les bras de traitement, l’intervalle de confiance bilatéral à 95% pour la différence des taux de réponse ACR 20 entre les bras de traitement devait se situer dans des limites d’équivalence prédéfinies [–15%, 15%]. Le deuxième objectif était l’évaluation de l’efficacité à l’aide des critères d’évaluation de l’efficacité à part l’ACR 20 ainsi que l’évaluation de la sécurité/tolérance, la PK et l’immunogénicité de Benepali comparativement à la préparation de référence.
Les taux de réponse ACR 20 pour Benepali et l’étanercept-la préparation de référence étaient équivalents à la Semaine 24 dans la population per protocole 1 (PPS1). La proportion de patients qui avaient obtenu une réponse ACR 20 dans la PPS1 s’élevait à 78.1% (193/247) ou 80.5% (190/236) dans le bras de traitement sous Benepali et celui sous l’étanercept-la préparation de référence (différence entre les traitements ajustée -2.37% [IC à 95%: -9.54%, -4.80%]). Dans l’analyse en intention de traiter (ITT) à 24 semaines, qui incluait tous les patients admis dans l’étude SB4-G31-RA de phase III, ce résultat a été confirmé avec 73,6% (220/299) pour Benepali contre 71,7% (213/297) pour l’étanercept-la préparation de référence (différence entre les traitements ajustée à l’analyse des non-répondeurs 1.66% [IC à 95% -5.50% - 8.82%]).
Analyse du taux de réponse ACR 20 à la semaine 24 (étude SB4-G31-RA de phase III)
|
Bras de traitement
|
Répondeur
n/n’ (%)
|
Pourcentage de différence ajusté
|
IC à 95% pour la différence entre les traitementsc
| |
Population per protocole 1a
| |
Benepali 50 mg (N = 247)
|
193/247 (78.1)
|
–2.37%
|
–9.54%, 4.80%
| |
Étanercept-préparation de référence 50 mg (N = 236)
|
190/236 (80.5)
| |
Échantillon complet d’analyseb
| |
Benepali 50 mg (N = 299)
|
220/299 (73.6)
|
1.66%
|
–5.50%, 8.82%
| |
Étanercept-préparation de référence 50 mg (N = 297)
|
213/297 (71.7)
|
ACR 20 = Critères pour une réponse à 20% selon la définition de l’American College of Rheumatology
IC = intervalle de confiance; N = nombre de patients dans la population
n’ = nombre de patients avec une évaluation; n = nombre de répondeurs
a: Population per protocole 1 (PPS1) = la PPS1 comprenait tous les patients de l’échantillon complet d’analyse, qui avaient terminé la Semaine 24 et dont l’observance du traitement (début de l’étude jusqu’à la Semaine 24) se situait entre 80-120%, et qui ne compromettaient pas l’évaluation de l’efficacité tant en termes du nombre attendu d’injections de Benepali/d’étanerceptpréparation de référence qu’en termes de la quantité attendue de doses de MTX sans déviations importantes au protocole (PD: protocol deviations). La PPS1 était le groupe d’analyse principal.
b: Échantillon complet d’analyse (Full-Analysis-Set (FAS)) = le FAS comprenait tous les patients randomisés à la visite de randomisation. Conformément au principe de l’intention de traiter, les patients ont été analysés selon le traitement qui leur avait été attribué lors de la randomisation.
Les pourcentages s’appuyent sur le nombre de participants à l’étude dans la population per protocole 1 ou dans l’échantillon complet d’analyse.
c: L’équivalence thérapeutique des deux bras de traitement était établie si l’intervalle de confiance (IC) bilatéral à 95% de la différence des 2 proportions se situait entièrement dans des marges d’équivalence prédéfinies de [−15%, 15%].
À la suite de la phase en double aveugle randomisée de 52 semaines (étude SB4-G31-RA), l’efficacité à long terme, la sécurité/tolérance et l’immunogénicité de Benepali chez les patients atteints de PR qui avaient été précédemment traités par Benepali ou par l’étanercept-la préparation de référence ont été évaluées. Dans la phase d’extension en ouvert (Semaine 52 à Semaine 104), 126 patients au total du bras de traitement par Benepali ont continué à recevoir Benepali (Benepali/Benepali) et 119 patients du bras de traitement sous la préparation de référence sont passés à Benepali (étanerceptpréparation de référence/Benepali).
La période comprenait 48 semaines de traitement actif (Benepali 50 mg 1× par semaine en injection s.c.) et 4 semaines de suivi pour la sécurité.
Les taux de réponse conformément aux critères ACR entre les bras de traitement Benepali/Benepali et étanercept-préparation de référence/Benepali étaient comparables de la Semaine 52 à la Semaine 100. Lors de la comparaison entre la phase randomisée en double aveugle et la phase d’extension en ouvert, aucune modification pertinente d’autres paramètres d’évaluation de l’efficacité n’a été observée.
|