CompositionPrincipes actifs
Patiromer (sous forme de patiromer sorbitol calcium (patiromer sorbitex calcium)).
Excipients
Gomme xanthan.
Indications/Possibilités d’emploiVeltassa est indiqué dans le traitement de l'hyperkaliémie chez les adultes
Posologie/Mode d’emploiInstauration du traitement
La dose initiale recommandée de Veltassa est d'au moins 8,4 g de patiromer une fois par jour.
En général, l'intervalle entre la prise de Veltassa et celle d'un autre médicament par voie orale devrait être de 3 heures. Aucun intervalle n'est nécessaire avec certains médicaments (voir rubrique Interactions).
En raison du début d'action retardé, Veltassa ne doit pas remplacer un traitement d'urgence en cas d'hyperkaliémie menaçant la vie du patient.
Ajustement de la posologie et traitement d'entretien
La dose quotidienne de Veltassa doit être ajustée en fonction de la kaliémie et de l'intervalle cible souhaité. La dose quotidienne peut être augmentée ou diminuée avec des intervalles d'une semaine par paliers de 8,4 g selon les besoins pour atteindre l'intervalle cible souhaité, jusqu'à une dose maximale de 25,2 g par jour. Il est possible d'utiliser plusieurs sachets pour obtenir la dose désirée. Si la kaliémie diminue en dessous de l'intervalle cible souhaité, l'administration de Veltassa doit être diminuée ou arrêtée.
En cas d'oubli d'une dose de Veltassa, la dose oubliée doit être prise le plus tôt possible le même jour. La dose oubliée ne doit pas être prise avec la dose suivante.
Instructions posologiques particulières
Patients dialysés
Seules des données limitées sont disponibles concernant l'utilisation de Veltassa chez les patients sous dialyse. Dans les études cliniques, aucune directive spécifique en matière de posologie et d'administration n'a été appliquée chez ces six patients.
Patients âgés
Sur le nombre total de patients traités par Veltassa lors des études cliniques, 1 307 (61,2 %) patients étaient âgés de 65 ans et plus. Parmi ceux-ci, 500 (23,4 %) avaient 75 ans et plus. Dans ces études, aucune directive spécifique en matière de posologie n'a été appliquée chez ces patients.
Enfants et adolescents
La sécurité et l'efficacité de Veltassa n'ont pas encore été établies chez les enfants et les adolescents de moins de 18 ans. Aucune donnée n'est disponible.
Mode d'emploi
Voie orale. Veltassa devrait être mélangé avec de l'eau, d'autres liquides ou les aliments mous mentionnés ci-dessous jusqu'à l'obtention d'une consistance uniforme, conformément aux étapes suivantes:
La totalité de la dose de Veltassa devrait d'abord être mélangée à environ 40 ml d'eau (3 cuillères à soupe) dans un verre. Ajouter ensuite environ 40 ml d'eau et mélanger soigneusement la suspension. La poudre ne se dissoudra pas. En cas de besoin, il est possible d'ajouter de l'eau au mélange pour obtenir la consistance souhaitée.
Le mélange devrait être bu immédiatement. S'il reste de la poudre dans le verre après l'ingestion du mélange, ajouter de l'eau, mélanger et boire immédiatement. Recommencer si nécessaire pour garantir l'administration de la totalité de la dose.
Les boissons ou aliments mous suivants peuvent être utilisés à la place de l'eau pour effectuer le mélange conformément aux étapes décrites ci-dessus: jus de pomme, jus de canneberge, jus d'ananas, jus d'orange, jus de raisin, jus de poire, nectar d'abricot, nectar de pêche, yaourt, lait, épaississant, compote de pomme, pudding à la vanille ou au chocolat.
La teneur en potassium des boissons ou aliments mous utilisés pour le mélange devrait être prise en compte dans l'apport alimentaire en potassium recommandé individuellement au patient.
Veltassa peut être pris avec ou indépendamment des repas. Veltassa ne devrait pas être chauffé (par exemple au microondes) ou ajouté à des aliments ou liquides chauds. Veltassa ne devrait pas être pris sous sa forme sèche.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients.
Mises en garde et précautionsHypomagnésémie
Patiromer peut provoquer une hypomagnésiémie en liant le magnésium dans le colon. Dans les études cliniques, des valeurs de magnésémie < 1,4 mg/dL ont été observées chez 7,1 % des patients traités par Veltassa, 0,3 % des patients développant une magnésémie < 1,0 mg/dL. Les diminutions moyennes de magnésémie sont survenues au début du traitement par Veltassa et étaient ≤0,118 mg/dL une semaine après l'instauration du traitement par Veltassa. La magnésémie doit être surveillée pendant un mois après l'instauration du traitement par Veltassa et la surveillance doit être poursuivie en cas de diminution du taux sérique de magnésium. Une supplémentation en magnésium doit être envisagée chez les patients qui développent une hypomagnésémie pendant le traitement par Veltassa.
Troubles gastrointestinaux
Les patients ayant des antécédents d'occlusion intestinale ou de chirurgie digestive lourde, de gastroparésie diabétique, présentant des troubles gastrointestinaux sévères ou des troubles de la déglutition n'étaient pas inclus dans les études cliniques. Chez ces patients, un traitement par Veltassa devrait être évité.
Arrêt du traitement par Veltassa
Lors de l'arrêt du traitement par Veltassa, la kaliémie peut augmenter, en particulier en cas de poursuite du traitement par un inhibiteur du système rénineangiotensinealdostérone (SRAA). Les patients doivent être informés qu'ils ne doivent pas arrêter le traitement sans consulter leur médecin. Les augmentations de la kaliémie peuvent survenir dès le 2e jour suivant la dernière prise de Veltassa.
Taux de potassium sérique
Le potassium sérique doit être surveillé lorsque la situation clinique l'exige, notamment après que des modifications aient été apportées à des médicaments ayant un effet sur la concentration sérique de potassium (par ex. inhibiteurs du système rénine-angiotensine-aldostérone ou des diurétiques) et après adaptation posologique de Veltassa.
Sorbitol
Veltassa contient du sorbitol comme composant du complexe contre-ion. La teneur en sorbitol correspond à environ 4 g (10,4 kcal) pour 8,4 g de patiromer. Les patients présentant une intolérance héréditaire au fructose (IHF) ne doivent pas prendre/recevoir ce médicament.
Fluor
Veltassa contient du fluor. Chez des patients présentant une insuffisance rénale chronique (IRC) sévère, toute autre source de fluor devrait être limitée.
Informations sur le calcium
Veltassa contient du calcium dans le cadre du complexe contre-ion. Le calcium est partiellement libéré et une partie pourrait être absorbée. Cependant, lors d'études cliniques d'une durée jusqu'à un an, aucune modification des taux sériques moyens de calcium n'a été observée. Les bénéfices et les risques de l'administration du médicament doivent être soigneusement évalués par le médecin chez les patients à risque.
InteractionsVeltassa a le potentiel de lier certains médicaments coadministrés par voie orale, ce qui pourrait diminuer leur absorption gastrointestinale. Le patiromer n'étant pas absorbé ni métabolisé par l'organisme, son effet sur le mécanisme d'action d'autres médicaments est limité.
Le tableau 1 présente les médicaments dont l'interaction avec Veltassa a été évaluée et les recommandations concernant leur administration en concomitance avec Veltassa. Pour des raisons de prudence, un intervalle d'au moins 3 heures devrait être respecté entre la prise de patiromer et celle de médicaments par voie orale non mentionnés.
Tableau 1: recommandations concernant l'administration de médicaments dont l'interaction avec Veltassa a été évaluée
|
Médicament
|
Recommandation clinique
|
Motif de la recommandation
| |
Inhibiteurs de l'enzyme de conversion de l'angiotensine (ECA)
| |
Bénazépril, captopril, énalapril, fosinopril, lisinopril, périndopril, quinapril, ramipril, trandolapril
|
Aucun intervalle nécessaire avec Veltassa
|
In vitro: aucune interaction observée
| |
Antagonistes des récepteurs de l'angiotensine II (ARA)
| |
Telmisartan
|
Intervalle d'au moins 3 heures avec Veltassa
|
In vitro: liaison observée
| |
Azilsartan, candésartan, irbésartan, losartan, olmésartan, valsartan
|
Aucun intervalle nécessaire avec Veltassa
|
In vitro: aucune interaction observée
| |
Antagonistes des récepteurs β-adrénergiques (bêta-bloquants)
| |
Bisoprolol, carvédilol, nébivolol
|
Intervalle d'au moins 3 heures avec Veltassa
|
In vitro: liaison observée
| |
Métoprolol
|
Aucun intervalle nécessaire avec Veltassa
|
In vivo: aucune interaction observée
| |
Diurétiques de l'anse
| |
Furosémide
|
Aucun intervalle nécessaire avec Veltassa
|
In vivo: aucune interaction observée
| |
Bumétanide, torasémide
|
In vitro: aucune interaction observée
| |
Antagonistes du récepteur des minéralocorticoïdes (ARM)
| |
Éplérénone, finérénone, spironolactone
|
Aucun intervalle nécessaire avec Veltassa
|
In vitro: aucune interaction observée
| |
Inhibiteurs de la néprilysine
| |
Sacubitril
|
Aucun intervalle nécessaire avec Veltassa
|
In vitro: aucune interaction observée
| |
Inhibiteurs du co-transporteur sodium-glucose de type 2 (SGLT-2)
| |
Canagliflozine, dapagliflozine, empagliflozine
|
Aucun intervalle nécessaire avec Veltassa
|
In vitro: aucune interaction observée
| |
Antibiotiques
| |
Ciprofloxacine
|
Intervalle d'au moins 3 heures avec Veltassa
|
In vivo: interaction observée, mais pas avec un intervalle de 3 heures
| |
Triméthoprime
|
Aucun intervalle nécessaire avec Veltassa
|
In vivo: aucune interaction observée
| |
Amoxicilline, céphalexine
|
In vitro: aucune interaction observée
| |
Anticoagulants
| |
Warfarine
|
Aucun intervalle nécessaire avec Veltassa
|
In vivo: aucune interaction observée
| |
Apixaban, rivaroxaban
|
In vitro: aucune interaction observée
| |
Médicaments visant à baisser le taux de PTH et hormones thyroïdiennes
| |
Lévothyroxine
|
Intervalle d'au moins 3 heures avec Veltassa
|
In vivo: interaction observée, mais pas avec un intervalle de 3 heures
| |
Cinacalcet
|
Aucun intervalle nécessaire avec Veltassa
|
In vivo: aucune interaction observée
| |
Antithrombotiques
| |
Clopidogrel
|
Aucun intervalle nécessaire avec Veltassa
|
In vivo: aucune interaction observée
| |
Acide acétylsalicylique
|
In vitro: aucune interaction observée
| |
Antidiabétiques
| |
Metformine
|
Intervalle d'au moins 3 heures avec Veltassa
|
In vivo: interaction observée, mais pas avec un intervalle de 3 heures
| |
Glipizide
|
Aucun intervalle nécessaire avec Veltassa
|
In vitro: aucune interaction observée
| |
Inhibiteurs des canaux calciques
| |
Amlodipine, vérapamil
|
Aucun intervalle nécessaire avec Veltassa
|
In vitro: aucune interaction observée
| |
Immunosuppresseurs
| |
Mycophénolate mofétil
|
Intervalle d'au moins 3 heures avec Veltassa
|
In vitro: liaison observée
| |
Tacrolimus
|
Aucun intervalle nécessaire avec Veltassa
|
In vitro: aucune interaction observée
| |
Autres
| |
Quinidine
|
Intervalle d'au moins 3 heures avec Veltassa
|
In vitro: liaison observée
| |
Lithium
|
Aucun intervalle nécessaire avec Veltassa
|
In vivo: aucune interaction observée
| |
Allopurinol, atorvastatine, digoxine, phénytoïne, riboflavine, sévélamer
|
In vitro: aucune interaction observée
|
Effet des aliments sur Veltassa
Veltassa peut être pris avec ou indépendamment des repas. Dans une étude ouverte, 114 patients souffrant d'hyperkaliémie ont été randomisés pour la prise Veltassa une fois par jour, avec ou indépendamment d'un repas. La kaliémie à la fin du traitement, la différence par rapport à la kaliémie de départ et la dose moyenne de Veltassa étaient comparables dans les deux groupes.
Grossesse, allaitementGrossesse
Il n'existe pas de données cliniques concernant l'utilisation de Veltassa chez la femme enceinte. Les études chez l'animal n'ont révélé aucune toxicité directe ou indirecte ayant une incidence sur la grossesse, le développement embryonnaire, le développement fœtal et/ou le développement postnatal.
À titre de précaution, il est préférable d'éviter l'utilisation de Veltassa pendant la grossesse.
Allaitement
Aucun effet sur le nouveauné/nourrisson allaité n'est attendu, puisque l'exposition systémique au patiromer chez la femme qui allaite est négligeable. Une décision doit être prise quant à l'arrêt de l'allaitement ou à l'arrêt/l'abstention d'un traitement par patiromer, en prenant en compte le bénéfice de l'allaitement pour l'enfant et le bénéfice du traitement pour la mère.
Fertilité
Il n'existe pas de données concernant l'effet de Veltassa sur la fertilité humaine. Les études chez l'animal n'ont pas révélé d'effets sur les fonctions de reproduction ou sur la fertilité (voir rubrique Données précliniques).
Effet sur l’aptitude à la conduite et l’utilisation de machinesVeltassa n'a aucune influence ou une influence négligeable sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
Le profil de sécurité actuel de Veltassa est basé sur un total de 2 135 patients ayant participé aux études cliniques, ainsi que sur l'expérience après la mise sur le marché.
La majorité des effets indésirables (EI) rapportés étaient l'hypomagnésémie et des troubles gastrointestinaux, les EI les plus fréquemment observés étant: constipation, diarrhée, douleurs abdominales, nausées, flatulences, vomissements et hypersensibilité. Les troubles gastrointestinaux étaient en général d'intensité légère à modérée, ne semblaient pas être liés à la dose, se sont généralement résolus spontanément ou avec un traitement, et aucun n'a été décrit comme grave. Les réactions d'hypersensibilité englobaient des éruptions cutanées, de l'urticaire, des gonflements de la cavité buccale et des lèvres et étaient d'intensité légère à modérée.
Remarque: la diarrhée est un terme générique englobant la diarrhée et la défécation fréquente. La douleur abdominale est un terme générique englobant les termes préférentiels gêne abdominale, douleur abdominale et douleur abdominale haute. La constipation est un terme générique englobant les termes préférentiels constipation et selles dures.
Liste des effets indésirables
Les effets indésirables sont présentés par classe de système d'organes MedDRA et par fréquence selon la convention suivante:
«très fréquents» (≥1/10),
«fréquents» (≥1/100 à <1/10),
«occasionnels» (≥1/1 000 à <1/100),
«rares» (≥1/10 000 à <1/1 000),
«très rares» (<1/10 000),
«fréquence inconnue» (ne peut être estimée sur la base des données disponibles).
Affections du système immunitaire
Fréquence inconnue: Hypersensibilité
Troubles du métabolisme et de la nutrition
Fréquents: Hypomagnésémie
Troubles gastrointestinaux
Fréquents: Constipation, diarrhée, douleurs abdominales, nausées, flatulences.
Occasionnels: Vomissements.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDes doses de Veltassa supérieures à 50,4 g de patiromer par jour n'ont pas été étudiées. Des doses excessives de Veltassa pouvant entraîner une hypokaliémie, le taux sérique de potassium doit être surveillé. S'il s'avère qu'une intervention médicale est nécessaire, des mesures appropriées pour rétablir une kaliémie normale peuvent être envisagées.
Propriétés/EffetsCode ATC
V03AE09
Classe pharmacothérapeutique: traitement de l'hyperkaliémie.
Mécanisme d'action
Veltassa est un polymère échangeur de cations non absorbé qui contient un complexe sorbitolcalcium comme contreion.
Veltassa augmente l'excrétion fécale du potassium en liant le potassium dans la lumière du tractus gastro-intestinal. La liaison du potassium diminue la concentration de potassium libre dans la lumière gastrointestinale, ce qui entraîne une diminution de la kaliémie.
Pharmacodynamique
Il a été démontré que Veltassa lie le potassium in vitro et in vivo dans des modèles animaux expérimentaux.
Dans une étude de phase I menée chez des volontaires sains adultes (6 à 8 sujets par groupe), Veltassa (de 0 g à 50,4 g de patiromer par jour) administré trois fois par jour pendant 8 jours a induit une augmentation dosedépendante de l'excrétion fécale de potassium. Une diminution dosedépendante correspondante de la kaliurèse sans modification du potassium sérique a également été observée. Par rapport au placebo, Veltassa aux doses de 25,2 g et 50,4 g par jour a diminué significativement la kaliurèse quotidienne moyenne.
Dans une étude ouverte, croisée, de phase I à doses répétées menée chez 12 volontaires sains, la dose de 25,2 g de patiromer par jour a été administrée par voie orale selon un schéma d'une fois par jour, de deux fois par jour ou de trois fois par jour pendant 6 jours selon une séquence attribuée par randomisation. Il a été observé une augmentation significative de l'excrétion fécale quotidienne moyenne de potassium et une diminution concomitante de la kaliurèse quotidienne moyenne avec les trois schémas posologiques. L'augmentation moyenne de l'excrétion fécale de potassium était de 1 283 à 1 550 mg/jour et la diminution moyenne de la kaliurèse de 1 438 à 1 534 mg/jour sur l'éventail des trois schémas posologiques. Aucune différence significative n'a été observée entre les schémas posologiques en termes d'excrétion fécale et urinaire quotidienne moyenne de potassium, tant dans la comparaison globale que dans les comparaisons par paires. L'excrétion urinaire quotidienne de calcium a augmenté de 53 mg/jour, 66 mg/jour et 73 mg/jour par rapport à la valeur initiale avec les schémas une prise par jour, deux prises par jour et trois prises par jour respectivement.
Dans une étude ouverte non contrôlée, 25 patients présentant une hyperkaliémie (kaliémie moyenne à l'inclusion de 5,9 mEq/L) et une insuffisance rénale chronique ont suivi un régime contrôlé en potassium pendant 3 jours, puis ont reçu 16,8 g de patiromer par jour (en deux doses fractionnées) pendant 2 jours tout en poursuivant le régime contrôlé. Une réduction statistiquement significative de la kaliémie (-0,2 mEq/L) a été observée 7 heures après la première dose. Les valeurs de la kaliémie ont continué à diminuer pendant la période de traitement de 48 heures (-0,8 mEq/L 48 heures après la première dose). Les taux de potassium sont restés stables pendant 24 heures après la dernière dose, puis ont augmenté pendant la période d'observation de 4 jours suivant l'arrêt de Veltassa.
Figure 1: Début d'action – Kaliémie moyenne (IC à 95 %) (mEq/L) au cours du temps
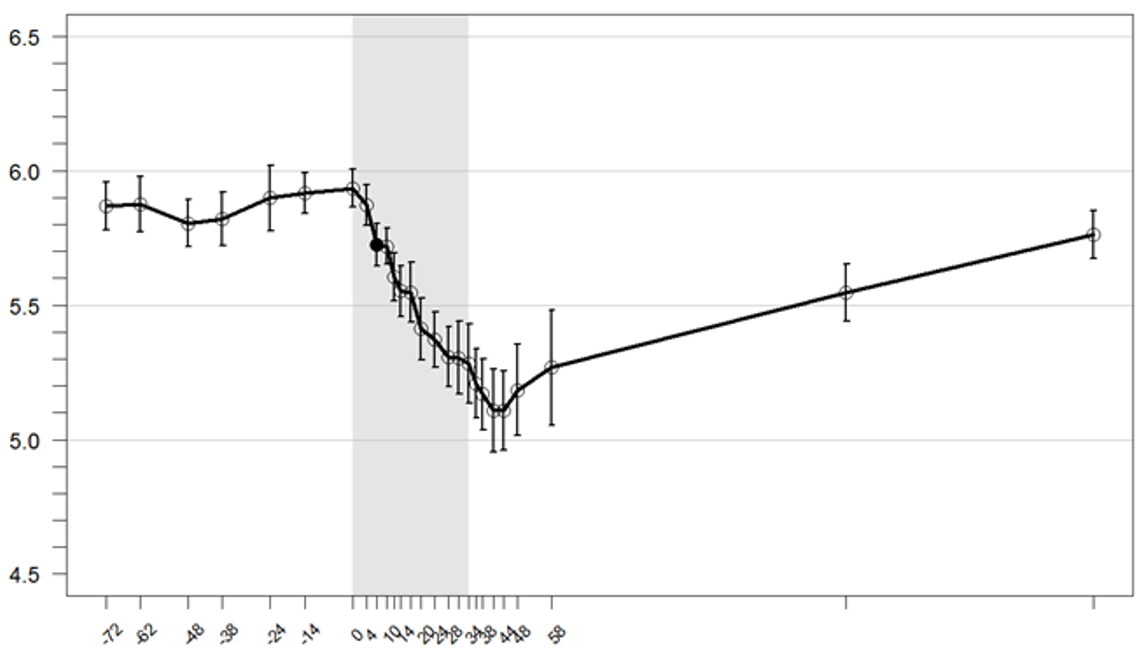
* Le cercle plein indique l'heure à laquelle la première réduction statistiquement significative a été identifiée. Une réduction moyenne de 0,8 mEq/L a été observée à l'heure 48 (p < 0,001).
Remarques: heures -72 à 0 = période de préinclusion avec régime contrôlé en potassium; heures 0 à 58 = période de traitement par Veltassa 16,8 g par jour en doses fractionnées en milieu hospitalier; heure 58 à jour 6 = période de suivi en ambulatoire.
Efficacité clinique
L'effet hypokaliémiant du patiromer a été démontré dans 5 études cliniques au cours desquelles des patients hyperkaliémiques ou normokaliémiques recevaient des inhibiteurs du SRAA et présentaient ainsi un risque accru d'hyperkaliémie. L'effet du patiromer chez les patients hyperkaliémiques atteints de maladie rénale chronique et recevant des inhibiteurs du SRAA a été évalué dans une étude en simple aveugle suivie d'une partie de sevrage randomisée. Une étude ouverte de 52 semaines a été menée avec des patients hyperkaliémiques atteints de maladie rénale chronique, d'hypertension non contrôlée et de diabète sucré de type 2, traités par inhibiteurs du SRAA. Dans deux études randomisées, en double aveugle, contrôlées par placebo, le patiromer a été administré à des patients normokaliémiques atteints d'insuffisance cardiaque ou de maladie rénale chronique et d'hypertension réfractaire ou sous instauration concomitante d'un traitement par inhibiteur du SRAA (spironolactone). Une étude en double aveugle, contrôlée par placebo a inclus des patients atteints d'insuffisance cardiaque et présentant une fraction d'éjection réduite hyperkaliémiques ou chez qui une hyperkaliémie était récemment apparue qui ont pu recevoir et continuer à prendre les doses autorisées d'inhibiteurs du SRAA et atteindre ou maintenir un taux de potassium normal pendant la prise de patiromer. Ensuite, l'effet du patiromer sur le taux de potassium sérique a été étudié lors d'une procédure de sevrage randomisée.
Au total, 1 918 patients ont participé à 5 études et ont reçu au moins une dose de patiromer. Parmi eux, 64,6 % souffraient de maladie rénale chronique avec un DFGe < 60 ml/min/1,73 m2, 54,3 % de diabète sucré et 77,9 % d'insuffisance cardiaque.
Étude 1 (OPAL-HK)
Étude de sevrage randomisée en simple aveugle, en deux parties, chez des patients présentant une insuffisance rénale chronique (IRC) et une hyperkaliémie, recevant des doses stables d'au moins un inhibiteur du SRAA.
La sécurité et l'efficacité de Veltassa ont été démontrées dans une étude de sevrage randomisée en simple aveugle en deux parties ayant évalué Veltassa chez des patients présentant une IRC et une hyperkaliémie recevant des doses stables d'au moins un inhibiteur du SRAA (c'estàdire, inhibiteur de l'enzyme de conversion de l'angiotensine [IECA], antagoniste des récepteurs de l'angiotensine II [ARA] ou antagoniste du récepteur des minéralocorticoïdes [ARM]).
Dans la partie A, 243 patients ont été traités par Veltassa pendant 4 semaines. La dose initiale de Veltassa était de 8,4 g de patiromer par jour (en doses fractionnées) chez les patients ayant une kaliémie de 5,1 mEq/L à < 5,5 mEq/L lors de l'inclusion et de 16,8 g de patiromer par jour (en doses fractionnées) chez ceux qui avaient une kaliémie de 5,5 mEq/L à < 6,5 mEq/L lors de l'inclusion. La dose de Veltassa était ajustée, au besoin, sur la base de la valeur de la kaliémie, déterminée à partir du jour 3 puis lors des visites hebdomadaires (semaines 1, 2 et 3) jusqu'à la fin de la période de traitement de 4 semaines, avec pour objectif de maintenir la kaliémie dans l'intervalle cible (3,8 mEq/L à < 5,1 mEq/L).
L'âge moyen des patients était de 64 ans, 58 % des patients étaient des hommes et 98 % étaient d'origine caucasienne. Environ 97 % des patients présentaient une hypertension, 57 % un diabète de type 2 et 42 % une insuffisance cardiaque.
Les doses quotidiennes moyennes de Veltassa étaient de 13 g et 21 g chez les patients ayant une kaliémie de 5,1 à < 5,5 mEq/L et de 5,5 à < 6,5 mEq/L respectivement.
La valeur moyenne de la kaliémie était de 5,58 mEq/L lors de l'inclusion et la variation moyenne (ET) de la kaliémie entre la visite d'inclusion et la semaine 4 de la partie A était de -1,01 (0,031) mEq/L, 95%CI: [-1.07; -0.95] (voir figure 2); cette réduction moyenne de la kaliémie était statistiquement significative. Pour le critère d'évaluation secondaire de la partie A, 76 % des patients (IC à 95 %: 70 %; 81 %) avaient une kaliémie dans l'intervalle cible de 3,8 mEq/L à < 5,1 mEq/L à la semaine 4 de la partie A.
Figure 2: Moyenne estimée (IC à 95 %) de la kaliémie (mEq/L) déterminée par le laboratoire central au cours du temps
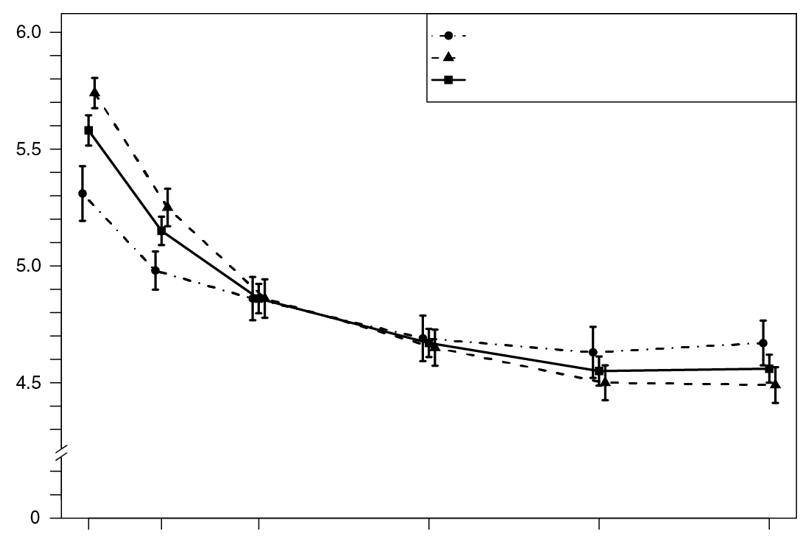
Dans la partie B, 107 patients ayant eu une kaliémie de 5,5 mEq/L à < 6,5 mEq/L lors de la visite d'inclusion dans la partie A, et dont la kaliémie était dans l'intervalle cible (3,8 mEq/L à < 5,1 mEq/L) à la semaine 4 de la partie A et recevant toujours un ou plusieurs inhibiteurs du SRAA, ont été randomisés pour continuer le traitement par Veltassa ou pour recevoir le placebo pendant 8 semaines afin d'évaluer l'effet de l'arrêt de Veltassa sur la kaliémie. Chez les patients du groupe Veltassa, la dose quotidienne moyenne était de 21 g au début et pendant la partie B.
Le critère d'évaluation principal de la partie B était la variation de la kaliémie depuis l'inclusion dans la partie B jusqu'à la première visite au cours de laquelle la kaliémie du patient était pour la première fois en dehors de l'intervalle de 3,8 à < 5,5 mEq/L ou jusqu'à la semaine 4, si la kaliémie du patient restait dans l'intervalle. Dans la partie B, la kaliémie a augmenté de 0,72 mEq/L, 95%CI [0.46, 0.99] chez les patients recevant le placebo, tandis qu'elle n'a pas varié chez les patients qui poursuivaient le traitement par Veltassa.
Le pourcentage de patients ayant présenté une kaliémie ≥5,1 mEq/L à tout moment pendant la partie B était plus élevé dans le groupe recevant le placebo (91 % [IC à 95 %: 83 %; 99 %]) que dans le groupe traité par Veltassa (43 % [IC à 95 %: 30 %; 56 %]). Le pourcentage de patients ayant présenté une kaliémie ≥5,5 mEq/L à tout moment pendant la partie B était plus élevé dans le groupe recevant le placebo (60 % [IC à 95 %: 47 %; 74 %]) que dans le groupe traité par Veltassa (15 % [IC à 95 %: 6 %; 24 %]).
Cinquantedeux pour cent (52 %) des patients recevant le placebo ont dû arrêter le traitement par inhibiteur du SRAA en raison de la réapparition de l'hyperkaliémie versus 5 % des patients traités par Veltassa.
Étude 2 (AMETHYST-DN)
L'effet du traitement par Veltassa pendant une durée allant jusqu'à 52 semaines a été évalué dans une étude ouverte menée chez 304 patients diabétiques de type 2 présentant une IRC et une hyperkaliémie recevant des doses stables d'un inhibiteur du SRAA. Les diminutions de la kaliémie observées avec le traitement par Veltassa ont été maintenues pendant un an de traitement régulier. Chez les patients ayant une kaliémie de > 5,0 à 5,5 mEq/L lors de l'inclusion et ayant préalablement reçu une dose initiale de 8,4 g de patiromer par jour (en doses fractionnées), la dose quotidienne moyenne était de 14 g; chez ceux ayant une kaliémie de > 5,5 à < 6,0 mEq/L lors de l'inclusion et ayant préalablement reçu une dose initiale de 16,8 g de patiromer par jour (en doses fractionnées), la dose quotidienne moyenne était de 20 g pendant toute l'étude.
Figure 3: Kaliémie moyenne (IC à 95 %) au cours du temps
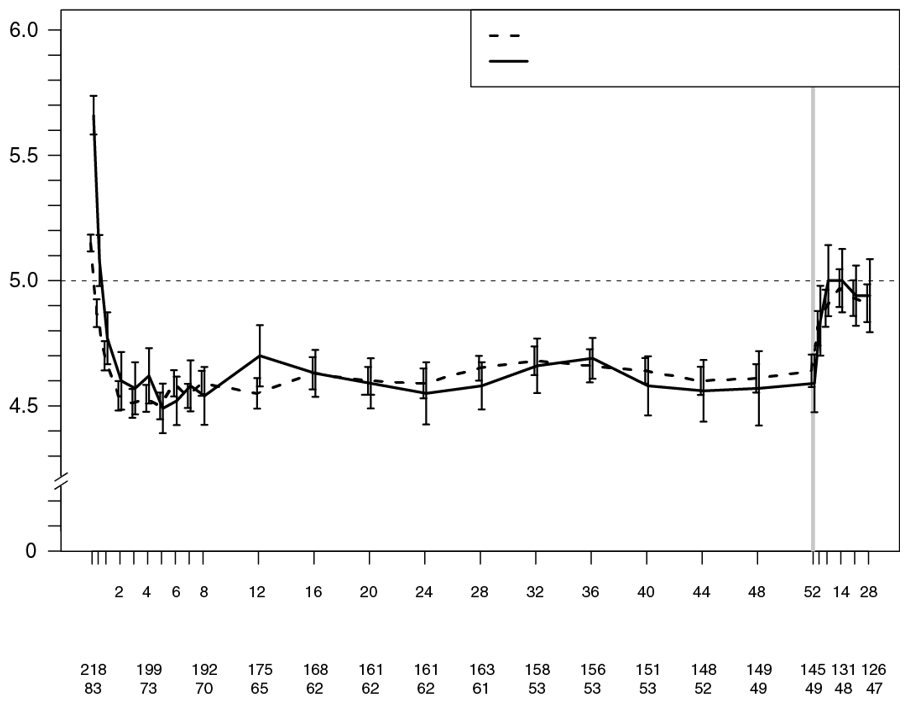
Étude 3 (PEARL-HF)
La capacité de Veltassa à fournir un traitement concomitant à la spironolactone a été évaluée dans le cadre d'une étude randomisée, en double aveugle, contrôlée par placebo et menée chez des patients atteints d'insuffisance cardiaque dont la prise d'ARM était indiquée sur le plan clinique. Les patients ont commencé à prendre de la spironolactone à raison de 25 mg/jour concomitant à leur traitement randomisé (Veltassa 12,6 g deux fois par jour ou placebo) et ont augmenté leur dose à 50 mg/jour après 14 jours si la valeur de la kaliémie était > 3,5 mEq/L ou ≤5,1 mEq/L. Sur les 105 patients randomisés recevant le médicament de l'étude (Veltassa 56; placebo 49), l'âge moyen était de 68,3 ans, 60,6 % étaient des hommes, 97,1 % étaient de race blanche et le DFGe moyen s'élevait à 81,3 ml/min. La kaliémie de base moyenne était de de 4,71 mEq/L pour Veltassa et de de 4,68 mEq/L pour le placebo.
Le critère d'efficacité primaire, soit la variation de la kaliémie entre le début et la fin de la période de traitement de 28 jours, était significativement plus faible dans le groupe Veltassa (p < 0,001) (LSQ moyenne [SEM]: -0,21 [0,07] mEq/l) que dans le groupe placebo (LSQ moyenne [SEM]: +0,23 [0,07] mEq/l). Dans le groupe Veltassa, le nombre de patients présentant une kaliémie > 5,5 mEq/l (7,3 % contre 24,5 %; p = 0,027) est resté plus faible et le nombre de patients recevant une dose de spironolactone de 50 mg/jour s'est avéré significativement plus élevé (90,9 % contre 73,5 %, p = 0,022).
Étude 4 (AMBER)
Une autre étude randomisée, en double aveugle et contrôlée par placebo, a analysé l'effet du patiromer sur la tolérance du traitement par la spironolactone chez les patients présentant une hypertension résistante au traitement et une IRC. Dans cette étude, les patients ont été randomisés dans le groupe patiromer ou le groupe placebo en plus de l'administration de spironolactone (25 mg/jour; augmentation à 50 mg/jour à partir de la troisième semaine chez les patients présentant une tension artérielle systolique ≥120 mmHg et un taux de potassium sérique ≤5,1 mEq/l). Pour maintenir un taux de potassium sérique compris entre ≥4,0 mEq/l et ≤5,1 mEq/l, la dose de patiromer/placebo pouvait être augmentée chaque semaine de la dose initiale de 8,4 g/jour jusqu'à une dose maximale de 25,2 g/jour. Au total, 295 patients (patiromer 147; placebo 148) dont l'âge moyen était de 68,1 ans et le DFGe moyen de 35,73 ml/min/1,73 m² ont été randomisés, stratifiés en fonction du taux de potassium sérique de référence (taux moyen de 4,71 mEq/l dans le bras patiromer et 4,68 mEq/l dans le bras de contrôle sous placebo) et des antécédents de diabète sucré (de type 1 ou de type 2). Le critère d'évaluation principal était la proportion de patients qui ont poursuivi le traitement par la spironolactone jusqu'à la fin de l'étude (12 semaines). Cette proportion a été significativement plus élevée chez les patients du groupe sous patiromer que chez les patients du groupe sous placebo (85,7 % vs 66,2 %; p < 0,0001). Les patients du bras patiromer ont reçu une dose de spironolactone de 50 mg/jour plus souvent (69,4 % versus 51,4 % dans le groupe de contrôle sous placebo) et ont plus rarement présenté des taux de potassium sérique ≥5,5 mEq/l que ceux du groupe de contrôle sous placebo (35,4 % vs 64,2 %; p < 0,001).
Étude 5 (DIAMOND)
L'effet du patiromer sur la kaliémie a été évalué dans une étude de sevrage randomisé à groupes parallèles en double aveugle, contrôlée par placebo chez des patients ayant reçu les doses autorisées d'inhibiteurs du SRAA dans le traitement de l'insuffisance cardiaque et ayant atteint ou maintenu un taux de potassium normal pendant la prise de patiromer [39]. Avant la randomisation, les patients étaient hyperkaliémiques pendant le traitement par inhibiteurs du SRAA ou normokaliémiques avec des antécédents d'hyperkaliémie pendant la prise d'inhibiteurs du SRAA au cours des 12 derniers mois, ayant entraîné une diminution de la dose ou un arrêt du traitement par inhibiteurs du SRAA. Le patiromer a été instauré chez tous les patients à une dose de 8,4 g par jour, avec une augmentation progressive hebdomadaire jusqu'à une dose maximale de 25,2 g par jour, afin de maintenir un taux de potassium sérique ≥4,0 mEq/l et ≤5,0 mEq/l. Les patients hyperkaliémiques ont reçu le patiromer directement au début; les patients normokaliémiques ont reçu le patiromer à la semaine 1 ou plus tard. En parallèle, la médication de l'inhibiteur du SRAA a été optimisée, avec instauration, escalade de dose et entretien d'au moins 50 % des doses cibles d'iECA/ARA/ARNI prédéfinies et 100 % des doses cibles d'ARM prédéfinies. Dès que ces doses cibles d'inhibiteurs du SRAA ont été atteintes et que les patients ont présenté une kaliémie stable ≥4,0 mEq/l et ≤5,0 mEq/l depuis au moins une semaine, ils ont été randomisés pour recevoir le patiromer ou le placebo.
Sur les 1 168 patients chez qui un traitement par patiromer a été instauré, 130 n'ont pas achevé la partie de pré-inclusion. Sur les 1 038 patients qui ont achevé la pré-inclusion, 878 (85 %) ont été randomisés (patiromer: 439, placebo: 439). L'âge moyen des patients randomisés était de 66,9 ans, 72,9 % des patients étaient des hommes, 97,9 % étaient de race blanche et le DFGe moyen était de 63,0 ml/min/1,73 m2. Au moment de la randomisation, la kaliémie moyenne à l'inclusion était de 4,72 mEq/l.
Tous les participants avaient des antécédents d'hyperkaliémie. Au total, 40,3 % des participants étaient hyperkaliémiques au début de l'étude et 59,7 % étaient normokaliémiques, mais présentaient des antécédents d'hyperkaliémie au cours des 12 mois précédents.
Le critère d'évaluation principal de l'efficacité était la différence de variation moyenne ajustée (erreur-type [ET]) du taux de potassium sérique par rapport à l'inclusion, analysée à l'aide d'un modèle mixte à mesures répétées (MMRM). La différence des variations moyennes ajustées par rapport à l'inclusion était de -0,097 mEq/l (IC à 95 %: -0,128; -0,067). La variation moyenne de la kaliémie par rapport à l'inclusion dans le groupe patiromer (0,029 [0,019] mEq/l) était plus faible, de manière statistiquement significative, (p < 0,001) que dans le groupe placebo (0,127 [0,019] mEq/l).
Le délai d'apparition du premier événement d'hyperkaliémie (défini comme une kaliémie > 5,5 mEq/l) était significativement plus court dans le groupe patiromer que dans le groupe placebo (p = 0,006), avec un hazard ratio de 0,63 (IC à 95 %: 0,45; 0,87).
La proportion de patients dont le médicament ARM (spironolactone ou éplérénone) a été réduit sous la dose cible était de 13,9 % dans le groupe patiromer vs 18,9 % dans le groupe placebo. Le hazard ratio pour le délai de la première réduction de dose de l'ARM dans le groupe patiromer vs placebo était de 0,62 (IC à 95 %: 0,45; 0,87) et la différence entre les traitements était statistiquement significative (p = 0,006).
Dans les sous-groupes prédéfinis DFGe ml/min/1,73 m2, ml/min/1,73 m2 avec analyse post-hoc du DFGe ml/min/1,73 m2, le patiromer a permis l'utilisation de doses d'inhibiteurs du SRAA efficaces, a contrôlé la kaliémie et a réduit le risque d'hyperkaliémie chez les patients atteints de HFrEF et de maladie rénale chronique légère à sévère.
Au total, 31,2 % des patients du groupe patiromer et 45,1 % des patients du groupe placebo ont présenté au moins un événement d'hyperkaliémie signalé par le médecin de l'étude avec une kaliémie de > 5,5 mEq/l. Le rapport du taux d'événement annualisé pour le patiromer vs placebo était de 0,658 (IC à 95 %: 0,534; 0,810), ce qui était statistiquement significatif (p < 0,001).
Pour deux critères d'évaluation secondaires testés hiérarchiquement, une approche «win-ratio» décalée a été utilisée: une approche a été adoptée pour les critères d'évaluations fermes groupés liés à l'hyperkaliémie, notamment le décès de cause cardiovasculaire, les hospitalisations pour cause de maladie cardiovasculaire et les événements d'hyperkaliémie de trois grades, présentant un win-ratio de 1,526 (IC à 95 %:1,231; 1,906; p < 0,001), ce qui est favorable à un traitement par patiromer, et une autre approche a employé un nouveau score pour l'utilisation d'inhibiteurs du SRAA (0–8 points), qui indique un taux favorable d'obtention d'un score plus élevé d'utilisation d'inhibiteurs du SRAA dans le groupe patiromer vs groupe placebo, avec un win-ratio de 1,248 (IC à 95 %: 1,003; 1,564; p = 0,048).
PharmacocinétiqueAbsorption
Veltassa agit en liant le potassium dans le tractus gastro-intestinal; par conséquent, la concentration sérique n'est pas pertinente pour son efficacité. Du fait de l'insolubilité et des caractéristiques de nonabsorption de Veltassa, de nombreuses études pharmacocinétiques classiques n'ont pas pu être réalisées.
Dans les études ADME (absorption, distribution, métabolisme, élimination) avec radiomarquage effectuées chez le rat et le chien, le patiromer n'était pas absorbé au niveau systémique.
Distribution
Les analyses par autoradiographie quantitative du corps entier chez le rat ont démontré que la radioactivité était limitée au tractus gastro-intestinal, sans niveau détectable de radioactivité dans d'autres tissus ou organes.
Métabolisme
Sans objet, en raison de l'insolubilité et des caractéristiques de nonabsorption de Veltassa.
Élimination
Dans les études ADME avec radiomarquage chez le rat et le chien, le patiromer était éliminé dans les fèces.
Données précliniquesLes données non cliniques issues des études conventionnelles sur la pharmacologie de sécurité, la toxicité en administration répétée, la génotoxicité et la toxicité pour la reproduction et le développement n'ont pas révélé de risque particulier pour l'homme.
Le patiromer n'a pas été génotoxique dans les essais de mutation réverse (test d'Ames), d'aberrations chromosomiques ou des micronoyaux chez le rat.
Aucune étude de carcinogénicité n'a été réalisée.
Le patiromer n'a pas altéré la fertilité chez le rat mâle ou femelle à des doses allant jusqu'à 10 fois la dose quotidienne maximale recommandée chez l'homme (DMRH). Le patiromer n'a pas non plus eu d'effets délétères sur le développement embryonnaire et fœtal lorsqu'il était administré à des rates gestantes à des doses allant jusqu'à 6 g/kg, fournissant ainsi une marge d'exposition 12 fois supérieure à la DMRH et administré à des lapines gestantes à des doses allant jusqu'à 3 g/kg, fournissant ainsi une marge d'exposition 6 fois supérieure à la DMRH.
Remarques particulièresIncompatibilités
Non pertinent.
Stabilité
Veltassa ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Transporter et conserver au réfrigérateur (2-8°C).
Ne pas exposer à des températures supérieures à 40°C.
Une fois délivré au patient, Veltassa peut être conservé à température ambiante (en dessous de 25°C) pendant 6 mois au maximum.
Ne pas utiliser Veltassa après la date de péremption imprimée sur le sachet.
Conserver hors de portée des enfants.
Numéro d’autorisation66411(Swissmedic)
PrésentationVeltassa est disponible en sachets contenant 8,4 g ou 16,8 g de patiromer (sous forme de patiromer sorbitol calcium (patiromer sorbitex calcium)).
Boîtes contenant 30 sachets. [B]
Titulaire de l’autorisationVifor Fresenius Medical Care Renal Pharma Ltd.
Saint-Gall.
Mise à jour de l’informationMai 2024
|