Propriétés/EffetsCode ATC
C10AA07
Mécanisme d'action
La rosuvastatine est un inhibiteur sélectif et compétitif de l'HMG-CoA réductase, l'enzyme limitant la vitesse de transformation de la 3-hydroxy-3-méthylglutaryl coenzyme A en mévalonate, précurseur du cholestérol.
L'effet de la rosuvastatine sur le profil lipidique a lieu de deux manières: d'une part, la rosuvastatine augmente le nombre des récepteurs des LDL à la surface des hépatocytes, augmentant ainsi la captation et le catabolisme des LDL, et d'autre part elle inhibe la synthèse hépatique des VLDL, réduisant ainsi le nombre total de particules de VLDL et de LDL.
Pharmacodynamique
Voir aussi sous «Mécanisme d'action».
Efficacité clinique
Rosuvastatine réduit les taux élevés de LDL-cholestérol, de cholestérol total et de triglycérides, et augmente le taux de HDL-cholestérol. Il réduit également les taux des ApoB, du non HDL-C, du VLDL-C, du VLDL-TG et augmente le taux d'ApoA-I (voir tableaux 2 et 3).
Rosuvastatine réduit également les ratios LDL-C/HDL-C, cholestérol total/HDL-C, non HDL-C/HDL-C et ApoB/ApoA-I.
Un effet thérapeutique de rosuvastatine est obtenu durant la première semaine de traitement et 90% de la réponse maximale sont normalement observés après 2 semaines. La réponse maximale est habituellement atteinte en 4 semaines et persiste par la suite.
Tableau 2: Effets dose-dépendants chez les patients avec hypercholestérolémie primaire, types IIa et IIb (moyenne ajustée en % par rapport à la valeur initiale)
|
Dose
|
N
|
LDL-C
|
C total
|
HDL-C
|
TG
|
Non HDL-C
|
ApoB
|
ApoA-I
| |
Placebo
|
13
|
-7
|
-5
|
3
|
-3
|
-7
|
-3
|
0
| |
5 mg
|
17
|
-45
|
-33
|
13
|
-35
|
-44
|
-38
|
4
| |
10 mg
|
17
|
-52
|
-36
|
14
|
-10
|
-48
|
-42
|
4
| |
20 mg
|
17
|
-55
|
-40
|
8
|
-23
|
-51
|
-46
|
5
| |
40 mg
|
18
|
-63
|
-46
|
10
|
-28
|
-60
|
-54
|
0
|
Tableau 3: Effets dose-dépendants chez les patients avec hypertriglycéridémie primaire, types IIb et IV (moyenne en % par rapport à la valeur initiale)
|
Dose
|
N
|
TG
|
LDL-C
|
C total
|
HDL-C
|
Non HDL-C
|
VLDL-C
|
VLDL-TG
| |
Placebo
|
26
|
1
|
5
|
1
|
-3
|
2
|
2
|
6
| |
5 mg
|
25
|
-21
|
-28
|
-24
|
3
|
-29
|
-25
|
-24
| |
10 mg
|
23
|
-37
|
-45
|
-40
|
8
|
-49
|
-48
|
-39
| |
20 mg
|
27
|
-37
|
-31
|
-34
|
22
|
-43
|
-49
|
-40
| |
40 mg
|
25
|
-43
|
-43
|
-40
|
17
|
-51
|
-56
|
-48
|
Les données indiquées dans les tableaux 2 et 3 ont été confirmées lors d'un vaste programme clinique incluant plus de 5300 patients traités par rosuvastatine.
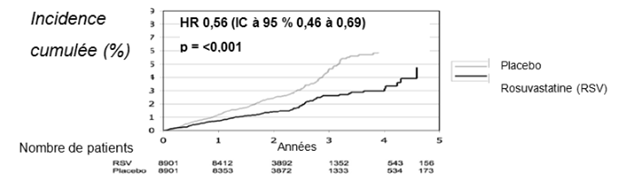
Dans l'étude JUPITER (Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin), les effets de la rosuvastatine sur l'incidence d'événements athéroscléreux majeurs de maladies cardio-vasculaires ont été examinés chez 17'802 hommes (≥50 ans) et femmes (≥60 ans) sans maladie cardio-vasculaire connue. Les autres critères d'inclusion ont englobé un taux de LDL-C <3,3 mmol/l (130 mg/dl), un taux de hsCRP- ≥2 mg/l et un taux de triglycérides <5,6 mmol/l (500 mg/dl). La population de l'étude présentait initialement un risque de cardiopathies coronariennes estimé à 11,3% selon les critères de risque de Framingham et comprenait un pourcentage élevé de patients ayant une hypertension (57%), un faible taux de HDL-C (23%), l'habitude de fumer (16%) ou un antécédent familial de cardiopathie coronarienne prématurée (12%). Les participants ont été assignés par randomisation à un traitement par placebo (n= 8901) ou par 20 mg de rosuvastatine une fois par jour (n= 8901) et suivis pendant une durée médiane de 2 ans.
Le critère primaire était composite et défini comme la durée jusqu'à la survenue d'un des événements cardio-vasculaires suivants: décès cardio-vasculaire, infarctus non mortel du myocarde, AVC non mortel, angor instable ou revascularisation artérielle.
La rosuvastatine a permis une réduction significative du risque d'événements cardio-vasculaires (252 événements sous placebo par rapport à 142 événements sous rosuvastatine), avec une réduction statistiquement significative (p <0,001) du risque relatif de 44% (voir la figure 1). Le bénéfice était constatable en l'espace de 6 mois après le début du traitement. La réduction du risque a été systématique dans diverses sous-populations prédéfinies. Les sous-populations analysées étaient définies en fonction de l'âge, du sexe, de l'origine ethnique, du tabagisme, de l'indice de masse corporelle et des taux initiaux de LDL-C, de HDL-C et de hsCRP. On a observé une réduction statistiquement significative de 48% pour le critère composite englobant les décès cardio-vasculaires, les AVC et les infarctus du myocarde (HR: 0,52, IC à 95%: 0,40 à 0,68, p <0,001). Les infarctus mortels ou non du myocarde étaient réduits de 54 % (HR: 0,46, IC à 95%: 0,30 à 0,70) et les AVC mortels ou non étaient réduits de 48 % (HR: 0,52, IC à 95%: 0,34 à 0,79, p= 0,002). La mortalité totale était réduite de 20% dans le groupe sous rosuvastatine (HR: 0,80, IC à 95%: 0,67 à 0,97, p= 0,02). Les taux de LDL-C étaient réduits de 45% sous rosuvastatine par rapport au placebo (p <0,001).
Figure 1. Temps jusqu'à la survenue d'événements cardio-vasculaires majeurs dans l'étude JUPITER
Le profil de sécurité a été généralement comparable chez les sujets sous rosuvastatine 20 mg et les sujets sous placebo. 1,6% des sujets sous rosuvastatine et 1,8 % des sujets sous placebo ont quitté l'étude en raison d'un événement indésirable, indépendamment d'un rapport causal avec le traitement. Les événements indésirables ayant conduit le plus souvent à un arrêt du traitement ont englobé des myalgies (0,3% sous rosuvastatine, 0,2% sous placebo), des douleurs abdominales (0,03% sous rosuvastatine, 0,02% sous placebo) et des éruptions cutanées (0,02% sous rosuvastatine, 0,03% sous placebo). Les événements indésirables rapportés chez ≥5% des patients et au moins aussi souvent ou plus souvent sous placebo ont englobé des infections urinaires (8,7% sous rosuvastatine, 8,6% sous placebo), des rhinopharyngites (7,6% sous rosuvastatine, 7,2% sous placebo), des douleurs dorsales (7,6% sous rosuvastatine, 6,9% sous placebo), des myalgies (7,6% sous rosuvastatine, 6,6% sous placebo), des bronchites (7,2% sous rosuvastatine, 7,1% sous placebo), des arthrites (5,8% sous rosuvastatine, 5,6% sous placebo), de la toux (5,3% sous rosuvastatine, 5,3% sous placebo) et des diarrhées (4,7% sous rosuvastatine, 4,6% sous placebo).
Dans l'étude JUPITER, une augmentation statistiquement significative du diabète sucré a été rapportée, avec une incidence de 2,8% chez les patients sous rosuvastatine et de 2,3% chez les patients sous placebo (HR: 1,27, IC à 95%: 1,05-1,53, p=0,015). La différence des variations moyennes de la valeur de l'HbA1c par rapport à la valeur initiale au début de l'étude dans les deux bras de traitement (rosuvastatine versus placebo) était d'environ 0,1%. L'analyse suggère qu'un nouveau diagnostic de diabète s'est révélé nécessaire essentiellement chez les patients qui présentaient déjà un risque élevé de développement d'un diabète. Dans l'ensemble de la population de l'étude (tout comme chez les patients qui avaient déjà une prédisposition au diabète), le bénéfice en termes cardiovasculaires et en termes de mortalité conféré par le traitement à la rosuvastatine l'a emporté sur le risque de diagnostic d'un diabète nouvellement apparu (voir «Mises en garde et précautions», «Effets indésirables»).
Enfants et adolescents de 10 à 17 ans
Une étude multicentrique, randomisée, réalisée en double aveugle contrôlée par placebo, a inclus au total 176 patients (97 de sexe masculin, 79 de sexe féminin, tous âgés de 10 à 17 ans, aux stades de Tanner II à V, patientes au moins 1 an après la ménarche) souffrant d'hypercholestérolémie hétérozygote. Ces patients ont reçu pendant 12 semaines 5 mg, 10 mg ou 20 mg de rosuvastatine par jour ou un placebo. Après cette phase, tous les 173 patients (96 de sexe masculin, 77 de sexe féminin) ont reçu de la rosuvastatine chaque jour pendant 40 semaines. Au début de l'étude, 30% des patients environ étaient âgés de 10 à 13 ans. Parmi les patients, 17%, 18%, 40% et 25% présentaient respectivement un stade de Tanner de II, III, IV et V.
Au bout de 12 semaines, la rosuvastatine avait baissé le taux de LDL-cholestérol (critère primaire), le taux de cholestérol total et le taux d'ApoB (voir le tableau 4).
Tableau 4: Effet hypolipémiant de la rosuvastatine chez des enfants et adolescents souffrant d'hypercholestérolémie hétérozygote familiale (moyenne des moindres carrés à 12 semaines par rapport aux valeurs initiales, en %)
|
Dose
|
N
|
LDL-C
|
C total
|
HDL-C
|
TG
|
Non HDL-C
|
ApoB
|
ApoA-l
| |
Placebo
|
46
|
-0,7
|
-0,0
|
6,9
|
5,1
|
-0,9
|
-1,7
|
2,8
| |
5 mg
|
42
|
-38,3
|
-29,9
|
4,2
|
0,3
|
-36,1
|
-31,7
|
1,8
| |
10 mg
|
44
|
-44,6
|
-34,2
|
11,2
|
-13,6
|
-43,0
|
-38,1
|
5,4
| |
20 mg
|
44
|
-50,0
|
-38,7
|
8,9
|
-8,1
|
-47,5
|
-40,7
|
4,0
|
À la fin des 40 semaines (phase ouverte avec adaptation de la dose en fonction de la cible, doses allant jusqu'à 20 mg par jour au maximum), 70 (40,5%) des 173 patients avaient atteint le taux cible de cholestérol inférieur à 2,8 mmol/l.
L'analyse de la croissance linéaire (taille), du poids, de l'IMC (indice de masse corporelle) et des caractères sexuels secondaires reflétant le degré de maturité sexuelle (stades de Tanner) chez les patients pédiatriques (âgés de 10 à 17 ans) traités à la rosuvastatine est limitée à une période d'un an. Au bout de 52 semaines de traitement dans le cadre de l'étude, aucun impact sur la croissance, le poids, l'IMC ou la maturité sexuelle n'a été constaté.
L'impact du traitement par rosuvastatine sur la morbi-mortalité cardio-vasculaire n'a pas été examiné chez les enfants et les adolescents.
|