CompositionPrincipes actifs
Rosuvastatinum ut Rosuvastatinum calcium
Excipients
Noyau du comprimé:
5 mg/10 mg/20 mg Cellulosum microcristallinum, Lactosum monohydricum, Crospovidonum (type A), Natrii carbonas monohydricus, Magnesii stearas, Natrii laurilsulfas
Pellicule:
Comprimés pelliculés 5 mg: Poly(alcohol vinylicus), Titanii dioxidum, Macrogol 3350, Talcum, Ferrum oxydatum flavum
Comprimés pelliculés 10 mg et 20 mg: Poly(alcohol vinylicus), Titanii dioxidum, Macrogol 3350, Talcum, Ferrum oxydatum rubrum, Ferrum oxydatum flavum, rouge allura AC (E 129), Indigotinum I
1 comprimé pelliculé de Rosuvastax 5 mg contient 2,84 mg de sodium et 42,55 mg de lactose.
1 comprimé pelliculé de Rosuvastax 10 mg contient 2,84 mg de sodium, 37,35 mg de lactose et 0,01 mg de rouge allura AC (E 129).
1 comprimé pelliculé de Rosuvastax 20 mg contient 5,68 mg de sodium, 74,70 mg de lactose et 0,02 mg de rouge allura AC (E 129).
Indications/Possibilités d’emploiAdultes
Traitement de l’hypercholestérolémie
Hypercholestérolémie primaire (type IIa incluant les hypercholestérolémies familiales hétérozygotes) ou dyslipidémies mixtes (type IIb), en complément d'un régime lorsque la réponse au régime et aux autres traitements non pharmacologiques (par ex. exercice, perte de poids) n'est pas suffisante.
En cas d’hypercholestérolémie familiale homozygote, en complément d'un régime et d'autres mesures à but hypolipémiant (par ex. l'aphérèse des LDL) ou lorsque ces mesures ne sont pas appropriées.
Prévention des complications cardio-vasculaires
Rosuvastax est utilisé pour réduire le risque d’événements cardio-vasculaires sévères chez les patients adultes présentant un taux normal de LDL-cholestérol, mais dont le risque de maladies cardio-vasculaires athéroscléreuses est accru en raison de l’âge (hommes ≥50 ans, femmes ≥60 ans), d’un taux accru de hsCRP (≥2,0 mg/l) et au moins d'un autre facteur de risque cardio-vasculaire tel qu’hypertension, tabagisme, faible taux de HDL-C ou antécédent familial de cardiopathie coronarienne prématurée.
Chez les patients hypercholestérolémiques, il est nécessaire de surveiller les valeurs lipidiques et de respecter les recommandations posologiques ci-dessous. Il est en outre nécessaire de traiter les autres causes connues de morbi-mortalité cardio-vasculaire conformément aux directives applicables.
Enfants et adolescents de 10 à 17 ans
Garçons et filles (au moins un an après la ménarche) souffrant d’hypercholestérolémie familiale hétérozygote, en complément d’un régime alimentaire, lorsque la réponse au régime et aux autres mesures non pharmacologiques (p. ex. exercice physique, perte de poids) reste insuffisante.
Posologie/Mode d’emploiPosologie usuelle
Traitement de l’hypercholestérolémie
Avant de débuter le traitement, le patient doit suivre un régime hypocholestérolémiant standard qu'il devra continuer pendant toute la durée du traitement. La posologie sera adaptée individuellement selon l'objectif thérapeutique et la réponse du patient.
La dose recommandée se situe entre 5 et 20 mg une fois/jour.
La plus grande partie des patients peuvent poursuivre le traitement avec la dose initiale. Une adaptation posologique peut toutefois se faire après 4 semaines si besoin (voir «Propriétés/Effets»). L’administration d’une dose de 40 mg ne sera envisagée que chez les patients présentant une hypercholestérolémie sévère et un risque cardiovasculaire élevé (en particulier ceux présentant une hypercholestérolémie familiale) n'ayant pas atteint l'objectif thérapeutique fixé avec la dose de 20 mg et qui feront l'objet d'un suivi régulier. L’administration de la posologie de 40 mg sera contrôlée par un spécialiste. Chez les patients avec des facteurs prédisposant au développement d’une myopathie, la dose initiale est de 5 mg (voir «Mises en garde et précautions»).
Rosuvastax peut être administré à tout moment de la journée, indépendamment des repas ou avec un repas.
Hypercholestérolémie primaire (y compris hypercholestérolémie familiale hétérozygote), dyslipidémie mixte
La posologie usuelle en début de traitement est de 5–10 mg une fois par jour aussi bien pour les patients nouvellement traités que pour ceux qui passent d’une autre statine à Rosuvastax. Le choix de la dose d’attaque se fera en tenant compte du taux de cholestérol individuel du patient, de ses risques cardiovasculaires et de ses risques de développer des effets indésirables.
Chez les patients présentant une hypercholestérolémie sévère (y compris une hypercholestérolémie familiale hétérozygote), une dose initiale de 10 mg peut être envisagée.
Hypercholestérolémie familiale homozygote
Chez les patients présentant une hypercholestérolémie homozygote familiale, la dose initiale recommandée est de 10 mg une fois/jour.
Prévention des complications cardio-vasculaires
Par analogie avec la dose utilisée dans l’étude sur la réduction du risque de complications cardio-vasculaires, la dose recommandée est de 20 mg une fois par jour.
Patients pédiatriques
Enfants et adolescents de 10 à 17 ans
Chez les enfants et adolescents souffrant d’hypercholestérolémie familiale hétérozygote, la posologie usuelle est de 5 à 20 mg une fois par jour. La dose doit être correctement augmentée de façon progressive au début du traitement jusqu’à ce que l’objectif thérapeutique soit atteint. La sécurité et l’efficacité de doses dépassant 20 mg par jour n’ont pas été testées chez ce groupe d’âge.
Enfants de moins de 10 ans
L'expérience chez les enfants de moins de 10 ans est limitée à un petit nombre d'enfants (âgés de 8 ans ou plus) présentant une hypercholestérolémie familiale homozygote. En conséquence, Rosuvastax n'est pas recommandé chez l'enfant de moins de 10 ans.
Patients âgés
Une dose initiale de 5 mg est recommandée chez les patients âgés (>70 ans).
Patients présentant des troubles de la fonction rénale
En cas d'insuffisance rénale légère à modérée, la dose initiale recommandée est de 5 mg. La dose de 40 mg est contre-indiquée chez les patients présentant une insuffisance rénale modérée. La prise de Rosuvastax est contre-indiquée chez les patients présentant une insuffisance rénale sévère (voir «Contreindications»).
Patients présentant des troubles de la fonction hépatique
Chez les patients présentant une insuffisance hépatique légère ou modérée, aucune adaptation de la dose n’est nécessaire. Chez les patients présentant une limitation sévère de la fonction hépatique (Child-Pugh >9), le traitement débutera par Rosuvastax 5 mg. Chez ces patients, une augmentation de l’exposition systémique à la rosuvastatine a été observée. En conséquence, l’administration de dosages supérieurs à Rosuvastax 5 mg sera soigneusement évaluée (voir «Pharmacocinétique»).
Origine ethnique
Une augmentation des taux plasmatiques de rosuvastatine a été observée chez les sujets asiatiques (voir «Mises en garde et précautions» et «Pharmacocinétique»). Une dose initiale de 5 mg est recommandée chez les patients ayant des origines asiatiques et la dose de 40 mg est contre-indiquée.
Génotype/polymorphismes génétiques
On connaît des types spécifiques de polymorphismes génétiques pouvant entraîner une augmentation de l'exposition à la rosuvastatine (voir «Pharmacocinétique»).
Il a été démontré que les génotypes SLCO1B1 (OATP1B1) c.521CC et ABCG2 (BCRP) c.421AA sont associés à une augmentation de l'exposition à la rosuvastatine (AUC) par rapport à SLCO1B1 c.521TT et ABCG2 c.421CC. Pour les patients présentant un génotype c.521CC ou c.421AA connu, la dose journalière maximale recommandée est de 20 mg de Rosuvastax. (voir aussi les rubriques Mises en garde et précautions, Interactions et Pharmacocinétique).
Traitements concomitants
La rosuvastatine est un substrat de différentes protéines de transport (p.ex. OATP1B1 et BCRP). Différents médicaments capables d'interagir avec ces protéines de transport (par ex. ciclosporine, ticagrelor, inhibiteurs de protéase, y compris les combinaisons de ritonavir avec l'atazanavir, le lopinavir et/ou le tipranavir), peuvent causer une exposition accrue à la rosuvastatine et augmenter ainsi le risque de divers effets indésirables (p.ex. myopathie, y compris rhabdomyolyse) (voir «Interactions», Tableau 1).
Si Rosuvastax doit nécessairement être utilisé en même temps que d'autres médicaments susceptibles d'accroître l'exposition à la rosuvastatine (voir «Interactions», Tableau 1), la dose de Rosuvastax doit être vérifiée et éventuellement ajustée en conséquence.
Rosuvastax est contre-indiqué chez les patients prenant de la ciclosporine (voir «Contreindications»).
Contre-indicationsRosuvastax est contreindiqué:
·chez les patients ayant une hypersensibilité avérée à la rosuvastatine ou à l'un des excipients,
·chez les patients présentant une affection hépatique évolutive, y compris élévations inexpliquées et prolongées des transaminases sériques et toute augmentation des transaminases sériques au-delà de 3 fois la limite supérieure de la norme (LSN),
·chez les patients présentant une insuffisance rénale sévère (clairance de la créatinine <30 ml/min),
·chez les patients présentant une myopathie,
·chez les patients sous traitement simultané par ciclosporine,
·pendant la grossesse, l’allaitement et chez les femmes en âge de procréer n'utilisant pas de moyens contraceptifs appropriés.
La dose de 40 mg est contre-indiquée chez les patients asiatiques (voir «Posologie/Mode d’emploi») et chez les patients présentant des facteurs prédisposant au développement d’une myopathie/rhabdomyolyse. Ces facteurs incluent:
·insuffisance rénale modérée (clairance de la créatinine <60 ml/min),
·hypothyroïdie,
·antécédents personnels ou familiaux de maladies musculaires génétiques,
·antécédents personnels d'atteinte toxico-musculaire en rapport avec l’administration d’un autre inhibiteur de l'HMG-CoA réductase ou d'un fibrate,
·consommation excessive d'alcool,
·situations favorisant une élévation des taux plasmatiques de rosuvastatine,
·association aux fibrates.
Mises en garde et précautionsEffets sur les muscles squelettiques
Chez les patients traités avec Rosuvastax, pour tous les dosages, des effets sur les muscles squelettiques ont été observés, par exemple, une myalgie, une myopathie et dans de rares cas, une rhabdomyolyse. Ceci vaut en particulier pour les dosages supérieurs à 20 mg de rosuvastatine. Après utilisation d'ézétémib associé à des inhibiteurs de l'HMG-CoA réductase, de rares cas de rhabdomyolyse ont été observés. Une interaction pharmacodynamique ne peut pas être exclue (voir paragraphe »Interactions»). La prudence est donc de rigueur lors d'une co-administration.
Dans quelques cas, il a été rapporté qu'une statine déclenche une myasthénia gravis ou une aggravation d'une myasthénia gravis existante ou d'une myasthénie oculaire (voir «Effets indésirables»).
La rosuvastatine doit être interrompue en cas d’aggravation des symptômes. Des récidives ont été rapportées en cas de (nouvelle) administration de la même statine ou d’une autre.
Dosage de la créatine-phosphokinase
Un dosage de la créatine-phosphokinase (CPK) ne sera pas pratiqué après des efforts physiques intenses ou si une autre cause est susceptible de provoquer une élévation du taux de CPK, car l’interprétation des données s’en trouverait compliquée. Si, au début du traitement, les taux de CPK sont nettement élevés (plus de 5 fois la valeur supérieure de la norme), il faudra procéder, à des fins de vérification, à un nouveau dosage en l’espace de 5 à 7 jours. Si la répétition du dosage confirme le taux initial de CPK >5 fois la valeur supérieure de la norme, le traitement ne sera pas instauré.
Examen avant le début du traitement
Les statines devraient être prescrites avec prudence aux patients présentant des facteurs prédisposant à l’apparition d’une rhabdomyolyse. Des mesures de créatine-phosphokinase devraient être effectuées avant un traitement par statines uniquement dans les cas suivants:
·altération de la fonction rénale;
·hypothyroïdie;
·antécédents personnels ou familiaux de maladie musculaire génétique;
·antécédents personnels d'atteinte toxico-musculaire en rapport avec l’administration d’une statine ou d’un fibrate;
·consommation excessive d'alcool;
·patients âgés (>70 ans). Chez ces patients, la nécessité d'un tel dosage sera prise en considération si d’autres facteurs prédisposant à la survenue d'une myopathie/rhabdomyolyse sont présents.
Dans ces situations, une analyse soigneuse du rapport bénéfices/risques est nécessaire et le patient sera surveillé au plan clinique. Si le taux de CPK est significativement élevé avant le début du traitement (supérieur à 5 fois la valeur supérieure de la norme), le traitement sera interrompu.
Surveillance pendant le traitement
Le dosage des CPK sera pratiqué si, durant un traitement par des statines, un patient souffre de douleurs musculaires, de faiblesse musculaire ou de crampes musculaires. Si le taux sanguin est nettement élevé (plus de 5 fois la valeur supérieure de la norme), le traitement sera interrompu.
L’arrêt du traitement devrait être envisagé lorsque les symptômes musculaires se révèlent importants et qu'ils constituent une gêne permanente, même si les taux de CPK sont inférieurs à 5 fois la valeur supérieure de la norme.
La reprise du traitement avec la même statine ou une statine alternative peut être envisagée avec un dosage minimal et une surveillance étroite lorsque les symptômes disparaissent et que le taux de CPK s’est normalisé.
Le risque de rhabdomyolyse est accru en cas d’utilisation concomitante du rosuvastatine et de certains médicaments:
Une augmentation de l’incidence des cas de myosites et de myopathies a été observée chez les patients sous traitement associant les inhibiteurs de la HMG-CoA réductase et les dérivés de l’acide fibrique, y compris gemfibrozil, ciclosporine, acide nicotinique, antifongiques azolés, inhibiteurs de la protéase et les antibiotiques macrolides. Le gemfibrozil augmente le risque de myopathie lorsqu’il est associé à certains inhibiteurs de la HMG-CoA réductase. En conséquence, l’association de Rosuvastax et du gemfibrozil n'est pas recommandée. Le bénéfice obtenu par l'association de Rosuvastax avec les fibrates ou la niacine sur les modifications des paramètres lipidiques sera soigneusement évalué par rapport au risque potentiel de telles associations. De très rares cas de rhabdomyolyse ont été observés après l’utilisation d’ézétimibe en association avec des inhibiteurs de la HMG-CoA réductase. Une interaction pharmacodynamique ne peut pas être exclue. La prudence est donc de rigueur lors d’une co-administration (voir «Interactions» et «Effets indésirables»).
Rosuvastax ne doit pas être administré aux patients présentant des symptômes graves, aigus suggérant une myopathie ou prédisposant au développement d'une insuffisance rénale secondaire à une rhabdomyolyse (par ex. septicémie, hypotension, intervention chirurgicale majeure, traumatisme, troubles métaboliques, endocriniens ou électrolytiques sévères ou épilepsie non contrôlée).
Les inhibiteurs de la HMG-CoA-réductase (statines), dont Rosuvastax, ne doivent pas être utilisés avec les préparations d'acide fusidique systémique. Des cas de rhabdomyolyse (dont certains décès) ont été signalés chez des patients qui recevaient des préparations d'acide fusidique systémique en association avec des statines (voir «Interactions»). Le traitement par Rosuvastax doit être suspendu pendant un traitement indispensable par acide fusidique systémique. Les patients doivent être informés de la nécessité de demander sans délai conseil à un médecin lorsqu'ils remarquent des signes de faiblesse musculaire, de douleurs musculaires ou de sensibilité musculaire.
Le traitement par statines peut être poursuivi 7 jours après la dernière dose d'acide fusidique.
Dans les situations exceptionnelles où un traitement systémique durable à l'acide fusidique est nécessaire, l'utilisation simultanée de Rosuvastax et de l'acide fusidique ne doit être envisagée qu'au cas par cas et sous étroite surveillance médicale.
Effets hépatiques
Comme les autres inhibiteurs de l'HMG-CoA réductase, Rosuvastax doit être utilisé avec précaution chez les patients consommant d'importantes quantités d'alcool et/ou présentant des antécédents de maladie hépatique. Il est recommandé de pratiquer des tests fonctionnels hépatiques avant le début du traitement par Rosuvastax et 3 mois après l’instauration du traitement. Une élévation des transaminases supérieure à 3 fois la limite supérieure de la norme doit conduire à l'arrêt du traitement par Rosuvastax ou à une diminution de la dose.
Chez les patients ayant une hypercholestérolémie secondaire à une hypothyroïdie ou à un syndrome néphrotique, la pathologie sous-jacente devra être traitée avant l’instauration d'un traitement par Rosuvastax.
Origine ethnique
Les études de pharmacocinétique montrent une augmentation de l'exposition chez les sujets asiatiques par rapport aux caucasiens (voir «Posologie/Mode d'emploi» et «Pharmacocinétique»).
Intolérance au lactose
Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne devraient pas prendre ce médicament.
Maladie pulmonaire interstitielle
De rares cas de maladie pulmonaire interstitielle (MPI) ont été signalés en rapport avec les statines, en particulier chez des patients recevant un traitement au long cours (voir sous «Effets indésirables»). Les caractéristiques d’une MPI peuvent englober des symptômes tels qu’une dyspnée, une toux non productive et une détérioration de l’état de santé général (fatigue, perte de poids et fièvre). Si une MPI est suspectée chez un patient, il est recommandé d’arrêter le traitement aux statines.
Diabète
Comme avec d'autres inhibiteurs de la HMG-CoA-réductase, des élévations de l'HbA1c et du glucose sérique ont été observées chez des patients sous rosuvastatine. Chez quelques patients, essentiellement des patients qui présentaient déjà un risque de développement d'un diabète, il a été nécessaire de poser un diagnostic de diabète nouvellement apparu (voir «Effets indésirables», «Efficacité clinique»).
Ce médicament contient moins de 1 mmol de sodium (23 mg) par comprimé pelliculé, c.-à-d. qu’il est essentiellement sans sodium.
Les patients présentant une intolérance héréditaire rare au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas utiliser ce médicament.
Peut provoquer des réactions allergiques.
InteractionsEffets d'autres médicaments administrés simultanément sur la rosuvastatine
Des examens in vitro et in vivo ont montré l’absence d’interactions cliniquement significatives de la rosuvastatine avec le cytochrome P450 (que ce soit en tant que substrat, inhibiteur ou inducteur). La rosuvastatine est un substrat de certaines protéines de transport, dont le transporteur hépatique d'influx OATP1B1 et le transporteur d'efflux BCRP. L'utilisation concomitante de Rosuvastax et de médicaments inhibiteurs de ces protéines de transport peut entraîner une concentration plasmatique accrue de rosuvastatine, et par là un risque accru de myopathies. S’il est prévu d’administrer de tels produits avec Rosuvastax, il est recommandé aux prescripteurs d’en consulter l’information professionnelle.
Interactions nécessitant un ajustement de la posologie de rosuvastatine (voir également Tableau 1):
S'il est nécessaire d'administrer Rosuvastax de façon concomitante avec d'autres médicamens connus pour augmenter l'exposition à la rosuvastatine, la posologie de Rosuvastax doit être ajustée. S'il est prévu d'administrer de tels produits avec Rosuvastax, il est recommandé aux prescripteurs d'en consulter l'information professionnelle.
S'il est établi que le médicament en question augmente l'AUC de la rosuvastatine d'un facteur d'environ 2 ou plus, la dose initiale de Rosuvastax doit être au maximum de 5 mg une fois par jour. La dose maximale journalière de Rosuvastax doit être ajustée de telle sorte que l'exposition à la rosuvastatine attendue n'excède pas l'exposition à une dose journalière de 40 mg de Rosuvastax prise en l'absence de médicament interagissant – p. ex. 5 mg de Rosuvastax avec de la ciclosporine (augmentation de l'exposition d'un facteur de 7,1), 10 mg de Rosuvastax avec l'association ritonavir/atazanavir (augmentation d'un facteur de 3,1) et 20 mg de Rosuvastax avec du gemfibrozil (augmentation d'un facteur de 1,9).
S'il est établi que le médicament en question augmente l'AUC de la rosuvastatine d'un facteur inférieur à 2, la dose intiale ne doit pas être diminuée. La prudence est en revanche de rigueur lorsque la dose de Rosuvastax est augmentée au-delà de 20 mg.
Inhibiteurs de la protéase
En cas d'utilisation concomitante de rosuvastatine et de certains inhibiteurs de la protéase ou d'une association d'inhibiteurs de la protéase, l'exposition à la rosuvastatine (AUC) peut augmenter d'un facteur allant jusqu'à 7 (voir Tableau 1). Selon le degré d'impact sur l'exposition à la rosuvastatine, un ajustement de la posologie est nécessaire (voir «Posologie/Mode d'emploi» et «Mises en garde et précautions»).
|
Tableau 1: Influence de médicaments administrés de façon concomitante sur l'exposition à la rosuvastatine (AUC; Cmax par ordre décroissant de la taille de l'effet) d'après les études cliniques publiées
| |
|
Augmentation de l'AUC de la rosuvastatine d'un facteur de 2 ou plus
| |
Régime médicamenteux interagissant
|
Régime de rosuvastatine
|
Modification de l'AUC de la rosuvastatine
|
Modification de la Cmax de la rosuvastatine
| |
Sofosbuvir/velpatasvir/voxilaprévir (400 mg/100 mg/100 mg) + voxilaprévir (100 mg) 1x /j pendant 15 jours
|
10 mg, dose unitaire
|
7,39x ↑
|
18,88x ↑
| |
Ciclosporine 75 mg 2x/j à 200 mg 2x/j, 6 mois
|
10 mg 1x/j., 10 jours
|
7,1x ↑
|
11x ↑
| |
Darolutamide 600 mg 2x/j, 5 jours
|
5 mg, dose unitaire
|
5,2x ↑
|
~5x ↑
| |
Regorafénib 160 mg 1x/j, 14 jours
|
5 mg, dose unitaire
|
3,8x ↑
|
4,6x ↑
| |
Atazanavir 300 mg/ritonavir 100 mg 1x/j, 8 jours
|
10 mg, dose unitaire
|
3,1x ↑
|
7x ↑
| |
Roxadustat 200 mg 1x/jour
|
10 mg, dose unitaire
|
2,9x ↑
|
4,47x ↑
| |
Velpatasvir 100 mg 1x/j
|
10 mg, dose unitaire
|
2,69x ↑
|
2,61x ↑
| |
Momelotinib 200 mg 1x/jour, 6 jours
|
10 mg, dose unitaire
|
2,7x
|
| |
Ticagrelor 90 mg 2x/jour, 2 jours
|
20 mg, dose unitaire
|
2,6x
|
| |
Ombitasvir 25 mg/paritaprévir 150 mg/ritonavir 100 mg/dasabuvir 400 mg 2x/j
|
5 mg, dose unitaire
|
2,59x ↑
|
7,13x ↑
| |
Tériflunomide, Leflunomide
|
non disponible
|
2,51x ↑
|
2,65x ↑
| |
Énasidénib 100 mg 1x/j, 28 jours
|
10 mg, dose unitaire
|
2,4x ↑
|
3,66 ↑
| |
Grazoprévir 200 mg/elbasvir 50 mg 1x/j
|
10 mg, dose unitaire
|
2,26x ↑
|
5,49x ↑
| |
Glecaprévir 400 mg/pibrentasvir 120 mg 1x/j, 7 jours
|
5 mg, dose unitaire
|
2,2x ↑
|
5,62x ↑
| |
Lopinavir 400 mg/ritonavir 100 mg 2x/j, 17 jours
|
20 mg 1x/j, 7 jours
|
2,1x ↑
|
5x ↑
| |
Capmatinib 400 mg 2x/j
|
10 mg, dose unitaire
|
2,08x ↑
|
3.04x ↑
| |
Clopidogrel 300 mg comme «dose de charge», puis 75 mg après 24 heures
|
20 mg, dose unitaire
|
2x ↑
|
2x ↑
| |
Tafamidis 61 mg 2x/j les jours 1 et 2, puis 1x/j les jours 3 à 9
|
10 mg, dose unitaire
|
1,97x ↑
|
1,86x ↑
| |
Fostamatinib 100 mg 2x/j
|
20 mg, dose unitaire
|
1,96x ↑
|
1,88x ↑
| |
Febuxostat 120 mg 1x/j
|
10 mg, dose unitaire
|
1,9x ↑
|
2,1x ↑
| |
Gemfibrozil 600 mg 2x/j, 7 jours
|
80 mg, dose unitaire
|
1,9x ↑
|
2,2x ↑
| |
Augmentation de l'AUC de la rosuvastatine d'un facteur inférieur à 2
| |
Régime médicamenteux interagissant
|
Régime de rosuvastatine
|
Modification de l'AUC de la rosuvastatine
|
Modification de la Cmax de la rosuvastatine
| |
Eltrombopag 75 mg 1x/j, 5 jours
|
10 mg, dose unitaire
|
1,6x ↑
|
2x ↑
| |
Darunavir 600 mg/ ritonavir 100 mg 2x/j, 7 jours
|
10 mg 1xj, 7 jours
|
1,5x ↑
|
2,4x ↑
| |
Tipranavir 500 mg/ritonavir 200 mg 2x/j, 11 jours
|
10 mg, dose unitaire
|
1,4x ↑
|
2,2x ↑
| |
Dronédarone 400 mg 2x/j
|
sans données
|
1,4x ↑
|
sans données
| |
Itraconazole 200 mg 1x/j, 5 jours
|
10 mg ou 80 mg, dose unitaire
|
1,4x ↑
|
1,4x ↑
| |
Ézétimibe 10 mg 1x/j, 14 jours
|
10 mg 1x/j, 14 jours
|
1,2x ↑
|
1,2x ↑
| |
Diminution de l'AUC de la rosuvastatine
| |
Régime médicamenteux interagissant
|
Régime de rosuvastatine
|
Modification de l'AUC de la rosuvastatine
|
Modification de la Cmax de la rosuvastatine
| |
Érythromycine 500 mg 4x/j, 7 jours
|
80 mg, dose unitaire
|
20% ↓
|
31% ↓
| |
Baicaline 50 mg 3x/j, 14 jours
|
20 mg, dose unitaire
|
47% ↓
|
19% ↓
|
* Les données indiquées sous la forme d'une variation d'un facteur x représentent un ratio simple entre l'administration concomitante de rosuvastatine et l'administration de rosuvastatine seule.
Les données qui sont indiquées sous la forme d'une variation en pourcentage représentent la différence en pourcentage comparativement à l'administration de rosuvastatine seule.
Une augmentation est symbolisée par «↑», et une diminution par «↓».
AUC = aire sous la courbe (Area Under the Curve); 1x/2jours = une fois par deux jours, 1x/j = une fois par jour; 2x/j = deux fois par jour; 3x/j = trois fois par jour; 4x/j = quatre fois par jour
Les médicaments suivants/associations suivantes n'ont eu aucun effet cliniquement significatif sur l'AUC de la rosuvastatine en cas d'administration concomitante: aléglitazar 0,3 mg, 7 jours; fénofibrate 67 mg 3x/j, 7 jours; fluconazole 200 mg 1x/j, 11 jours; fosamprénavir 700 mg//ritonavir 100 mg 2x/j, 8 jours; kétoconazole 200 mg 2x/j, 7 jours; rifampicine 450 mg, 7 jours; silymarine 140 mg 3x/j, 5 jours.
Effet d'autres médicaments sur Rosuvastax
Antiacides
L'administration concomitante de rosuvastatine et d'une suspension antiacide à base d'hydroxyde d'aluminium ou d'hydroxyde de magnésium a provoqué une réduction d'environ 50% du taux plasmatique de rosuvastatine. Cet effet a toutefois pu être affaibli (à 20% environ) en administrant l'antiacide deux heures après la dose de rosuvastatine.
Préparations systémiques de l'acide fusidique
L'association de statines, dont Rosuvastax, avec l'acide fusidique peut entraîner des rhabdomyolyses à évolution éventuellement fatale. Des cas de rhabdomyolyse (dont certains décès) ont été signalés chez des patients qui recevaient des préparations d'acide fusidique systémique en association avec des statines (voir «Mises en garde et précautions»). Le mécanisme de cette interaction n'est pas connu.
Le traitement par Rosuvastax doit être suspendu pendant un traitement indispensable par acide fusidique systémique.
Le traitement par Rosuvastax peut être repris sept jours après la dernière dose d'acide fusidique.
Ticagrelor
Le ticagrelor inhibe le transporteur BCRP, ce qui entraîne une augmentation de 2,6 fois de l'AUC de la rosuvastatine et peut conduire à un risque accru de myopathie. Les avantages de la prévention des événements cardiovasculaires indésirables par l'utilisation de la rosuvastatine et les risques d'une concentration plasmatique accrue doivent être pris en compte.
Effet de Rosuvastax sur d'autres médicaments
Antivitamines K
Comme avec les autres inhibiteurs de l'HMG-CoA réductase, l'administration simultanée de Rosuvastax et de la warfarine comparée à celle de la warfarine seule peut entraîner une augmentation de l'INR. Chez les patients prenant des antivitamines K, une surveillance de l'INR est recommandée aussi bien au début du traitement par Rosuvastax que lors de son arrêt, de même qu'après une adaptation posologique.
Fénofibrate, autres fibrates
D'après les données des études spécifiques sur les interactions, on n'attend aucune interaction d'importance pharmacocinétique avec le fénofibrate, mais la survenue d'une interaction pharmacodynamique reste possible. Le gemfibrozil, le fénofibrate, d'autres fibrates et des doses hypolipémiantes (≥1 g/jour) de niacine (acide nicotinique) accroissent le risque de myopathie lors d'une utilisation en association avec un inhibiteur de la HMG-CoAréductase. L'association de Rosuvastax avec le gemfibrozil exige en outre un ajustement de la dose de Rosuvastax (voir le Tableau 1). La prudence est donc de rigueur lors d'une utilisation de ces médicaments en association avec Rosuvastax.
Ciclosporine
L'administration concomitante de ciclosporine et de rosuvastatine ne modifie pas les concentrations plasmatiques de la ciclosporine.
Contraception orale/traitement hormonal substitutif (THS)
L'utilisation concomitante de rosuvastatine et d'une contraception orale entraîne une augmentation de 26% de l'AUC de l'éthinylestradiol et une augmentation de 34% de celle du norgestrel. Ces augmentations des taux plasmatiques doivent être prises en compte lors du choix de la dose du contraceptif oral. Bien qu'il n'existe pas de données pharmacocinétiques disponibles pour des patients traités simultanément par rosuvastatine et un THS, un effet similaire ne peut être exclu. Toutefois, cette association a été largement utilisée chez des femmes durant les essais cliniques et elle a été bien tolérée.
Autres médicaments
Aucune interaction cliniquement significative avec la digoxine, l’ézétimibe ou le fénofibrate n'est attendue.
Grossesse, allaitementGrossesse
Rosuvastax est contre-indiqué pendant la grossesse et l'allaitement.
Les femmes en âge de procréer doivent prendre des mesures de contraception adéquates. Comme le cheolestérol et d'autres produits issus de la synthèse du cholestérol sont nécessaires pour le bon développement du fœtus, le risque potentiel lié à l'inhibition de l'HMG-CoA réductase l'emporte sur les bénéfices du traitement pendant la grossesse. Les données issues des études d'expérimentation animale ont apporté des signes limités d'une toxicité sur la reproduction (préclinique). Le traitement sera immédiatement interrompu si une patiente débute une grossesse durant la prise de Rosuvastax.
Allaitement
L'allaitement est contre-indiqué pendant le traitement par Rosuvastax. Des données limitées issues de publications indiquent que Rosuvastax passe dans le lait maternel. En raison du mécanisme d'action de Rosuvastax, il existe un risque potentiel d'effets indésirables chez le nourrisson allaité. On ne dispose d'aucune donnée sur les effets du médicament sur le nourrisson allaité ni sur les effets du médicament sur la lactation.
Effet sur l’aptitude à la conduite et l’utilisation de machinesRosuvastax peut avoir une légère influence sur l'aptitude à la conduite ou l’utilisation de machines. Aucune étude n'a été réalisée. Toutefois, en raison des propriétés pharmacodynamiques de rosuvastatine, il est peu probable que Rosuvastax altèrent ces aptitudes. Lors d’une participation active au trafic routier ou de l’utilisation de machine, prendre en compte la survenue possible de vertiges.
Effets indésirablesDans les études cliniques contrôlées, moins de 4% des patients traités par rosuvastatine ont arrêté prématurément le traitement en raison d'effets indésirables.
Dans une étude clinique de 52 semaines menée auprès d'enfants et d'adolescents, des augmentations des valeurs de CPK de plus de 10 fois la limite supérieure de la normale ainsi que l'apparition de symptômes musculaires après la pratique du sport et une augmentation de l'activité physique ont été observées plus fréquemment que chez les patients adultes. Pour le reste, le profil de sécurité de la rosuvastatine chez l'enfant et l'adolescent a été comparable à celui observé chez l'adulte.
Les mêmes mises en garde et précautions que pour l’adulte s’appliquent néanmoins également aux enfants et adolescents (voir «Mises en garde et précautions»).
Définitions adoptées pour la fréquence des effets indésirables: très fréquents (≥1/10), fréquents (≥1/100 à <1/10), occasionnels (≥1/1000 à <1/100), rares (≥1/10 000 à <1/1000), très rares (<1/10 000); fréquence inconnue sur la base des données disponibles.
Affections hématologiques et du système lymphatique
Fréquence inconnue: thrombocytopénie.
Affections du système immunitaire
Rares: réactions d'hypersensibilité, y compris angio-œdème.
Affections endocriniennes
Fréquents: diabète (dans l’étude JUPITER: 2,8% sous rosuvastatine vs 2,3% sous placebo). Cet effet indésirable a été rapporté surtout chez des patients présentant déjà un risque élevé de développement d'un diabète.
Affections du système nerveux
Fréquents: céphalées, vertiges.
Très rares: polynévrite, perte de mémoire.
Fréquence inconnue: neuropathie périphérique, myasthenia gravis.
Affections oculaires
Fréquence inconnue: myasthenia gravis oculaire.
Affections respiratoires, thoraciques et médiastinales
Fréquence inconnue: toux, dyspnée.
Affections gastro-intestinales
Fréquents: douleurs abdominales, constipation, nausées.
Rares: pancréatite.
Fréquence inconnue: diarrhée.
Affections hépatobiliaires
Rares: élévation des transaminases hépatiques.
Très rares: ictère, hépatite.
Comme les autres inhibiteurs de l'HMG-CoA réductase, une élévation dose-dépendante des transaminases hépatiques et des valeurs des CPK a été observée chez un petit nombre de patients traités par rosuvastatine.
Affections de la peau et du tissu sous-cutané
Occasionnels: prurit, éruption cutanée, urticaire.
Fréquence inconnue: syndrome de Stevens-Johnson, réaction médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), exanthème médicamenteux lichénoïde.
Affections musculosquelettiques et du tissu conjonctif
Fréquents: myalgie.
Rares: myopathie (y compris myosite), rhabdomyolyse, arthralgie.
Fréquence inconnue: myopathie nécrosante à médiation immunitaire.
Des effets sur les muscles squelettiques comme par ex. des myalgies, des myopathies et rarement des rhabdomyolyses, ont été observés chez des patients traités par rosuvastatine à toutes les doses.
Une augmentation dose-dépendante des CPK a été observée chez les patients prenant de la rosuvastatine. La majorité des cas étaient bénins, asymptomatiques et transitoires. Si les taux de CPK sont élevés (>5 fois la valeur supérieure de la norme), le traitement doit être interrompu (voir «Mises en garde et précautions»).
Fréquence inconnue: troubles tendineux, rarement avec rupture tendineuse.
Affections du rein et des voies urinaires
Très rares: hématurie.
Une protéinurie, détectée par bandelette urinaire et principalement d'origine tubulaire, a été observée chez des patients traités par rosuvastatine. Les modifications des protéines urinaires passant de zéro ou traces à ++ ou plus, ont été observées chez moins de 1% des patients traités par 10 mg et 20 mg, et chez approximativement 3% des patients traités par 40 mg. Une augmentation mineure des modifications passant de zéro ou traces à +, a été observée avec la dose de 20 mg. Dans la plupart des cas, la protéinurie diminue ou disparaît spontanément avec la poursuite du traitement. Cette protéinurie ne s’est pas avérée être un facteur prédictif d'une affection rénale aiguë ou évolutive.
Affections des organes de reproduction et du sein
Fréquence inconnue: gynécomastie.
Troubles généraux
Fréquents: asthénie.
Fréquence inconnue: œdèmes.
Autres effets
Une étude clinique contrôlée et menée à long terme a démontré que rosuvastatine n’a aucun effet délétère sur le cristallin.
Aucune altération de la fonction corticosurrénale n’a été observée chez les patients traités par rosuvastatine.
Les effets indésirables suivants ont été observés en rapport avec les statines: dépressions, troubles du sommeil (insomnies, cauchemars), troubles de la fonction sexuelle, rares cas de maladie pulmonaire interstitielle (surtout dans le cadre de traitements au long cours).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageII n'existe pas de traitement spécifique en cas de surdosage. La prise en charge du patient en cas de surdosage sera symptomatique et d'autres mesures de soutien éventuellement nécessaires seront mises en œuvre. La fonction hépatique et le taux de CPK doivent être surveillés. L'hémodialyse n'est probablement pas utile.
Propriétés/EffetsCode ATC: C10AA07
Mécanisme d'action
La rosuvastatine est un inhibiteur sélectif et compétitif de l'HMG-CoA réductase, l’enzyme limitant la vitesse de transformation de la 3-hydroxy-3-méthylglutaryl coenzyme A en mévalonate, précurseur du cholestérol.
L’effet de la rosuvastatine sur le profil lipidique a lieu de deux manières: d'une part, la rosuvastatine augmente le nombre des récepteurs des LDL à la surface des hépatocytes, augmentant ainsi la captation et le catabolisme des LDL, et d’autre part elle inhibe la synthèse hépatique des VLDL, réduisant ainsi le nombre total de particules de VLDL et de LDL.
Pharmacodynamique
Voir aussi sous «Mécanisme d'action».
Efficacité clinique
Rosuvastatine réduit les taux élevés de LDL-cholestérol, de cholestérol total et de triglycérides, et augmente le taux de HDL-cholestérol. Il réduit également les taux des ApoB, du non HDL-C, du VLDL-C, du VLDL-TG et augmente le taux d'ApoA-I (voir tableaux 2 et 3).
Rosuvastatine réduit également les ratios LDL-C/HDL-C, cholestérol total/HDL-C, non HDL-C/HDL-C et ApoB/ApoA-I.
Un effet thérapeutique de rosuvastatine est obtenu durant la première semaine de traitement et 90% de la réponse maximale sont normalement observés après 2 semaines. La réponse maximale est habituellement atteinte en 4 semaines et persiste par la suite.
Tableau 2: Effets dose-dépendants chez les patients avec hypercholestérolémie primaire, types IIa et IIb (moyenne ajustée en % par rapport à la valeur initiale)
|
Dose
|
N
|
LDL-C
|
C total
|
HDL-C
|
TG
|
Non HDL-C
|
ApoB
|
ApoA-I
| |
Placebo
|
13
|
–7
|
–5
|
3
|
–3
|
–7
|
–3
|
0
| |
5 mg
|
17
|
–45
|
–33
|
13
|
–35
|
–44
|
–38
|
4
| |
10 mg
|
17
|
–52
|
–36
|
14
|
–10
|
–48
|
–42
|
4
| |
20 mg
|
17
|
–55
|
–40
|
8
|
–23
|
–51
|
–46
|
5
| |
40 mg
|
18
|
–63
|
–46
|
10
|
–28
|
–60
|
–54
|
0
|
Tableau 3: Effets dose-dépendants chez les patients avec hypertriglycéridémie primaire, types IIb et IV (moyenne en % par rapport à la valeur initiale)
|
Dose
|
N
|
TG
|
LDL-C
|
C total
|
HDL-C
|
Non HDL-C
|
VLDL-C
|
VLDL-TG
| |
Placebo
|
26
|
1
|
5
|
1
|
–3
|
2
|
2
|
6
| |
5 mg
|
25
|
–21
|
–28
|
–24
|
3
|
–29
|
–25
|
–24
| |
10 mg
|
23
|
–37
|
–45
|
–40
|
8
|
–49
|
–48
|
–39
| |
20 mg
|
27
|
–37
|
–31
|
–34
|
22
|
–43
|
–49
|
–40
| |
40 mg
|
25
|
–43
|
–43
|
–40
|
17
|
–51
|
–56
|
–48
|
Les données indiquées dans les tableaux 2 et 3 ont été confirmées lors d’un vaste programme clinique incluant plus de 5300 patients traités par rosuvastatine.
Dans l’étude JUPITER (Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin), les effets de la rosuvastatine sur l’incidence d'événements athéroscléreux majeurs de maladies cardio-vasculaires ont été examinés chez 17'802 hommes (≥50 ans) et femmes (≥60 ans) sans maladie cardio-vasculaire connue. Les autres critères d’inclusion ont englobé un taux de LDL-C <3,3 mmol/l (130 mg/dl), un taux de hsCRP- ≥2 mg/l et un taux de triglycérides <5,6 mmol/l (500 mg/dl). La population de l’étude présentait initialement un risque de cardiopathies coronariennes estimé à 11,3% selon les critères de risque de Framingham et comprenait un pourcentage élevé de patients ayant une hypertension (57%), un faible taux de HDL-C (23%), l’habitude de fumer (16%) ou un antécédent familial de cardiopathie coronarienne prématurée (12%). Les participants ont été assignés par randomisation à un traitement par placebo (n= 8901) ou par 20 mg de rosuvastatine une fois par jour (n= 8901) et suivis pendant une durée médiane de 2 ans.
Le critère primaire était composite et défini comme la durée jusqu’à la survenue d’un des événements cardio-vasculaires suivants: décès cardio-vasculaire, infarctus non mortel du myocarde, AVC non mortel, angor instable ou revascularisation artérielle.
La rosuvastatine a permis une réduction significative du risque d’événements cardio-vasculaires (252 événements sous placebo par rapport à 142 événements sous rosuvastatine), avec une réduction statistiquement significative (p <0,001) du risque relatif de 44% (voir la figure 1). Le bénéfice était constatable en l’espace de 6 mois après le début du traitement. La réduction du risque a été systématique dans diverses sous-populations prédéfinies. Les sous-populations analysées étaient définies en fonction de l’âge, du sexe, de l'origine ethnique, du tabagisme, de l’indice de masse corporelle et des taux initiaux de LDL-C, de HDL-C et de hsCRP. On a observé une réduction statistiquement significative de 48% pour le critère composite englobant les décès cardio-vasculaires, les AVC et les infarctus du myocarde (HR: 0,52, IC à 95%: 0,40 à 0,68, p <0,001). Les infarctus mortels ou non du myocarde étaient réduits de 54 % (HR: 0,46, IC à 95%: 0,30 à 0,70) et les AVC mortels ou non étaient réduits de 48 % (HR: 0,52, IC à 95%: 0,34 à 0,79, p= 0,002). La mortalité totale était réduite de 20% dans le groupe sous rosuvastatine (HR: 0,80, IC à 95%: 0,67 à 0,97, p= 0,02). Les taux de LDL-C étaient réduits de 45% sous rosuvastatine par rapport au placebo (p <0,001).
Figure 1. Temps jusqu’à la survenue d’événements cardio-vasculaires majeurs dans l’étude JUPITER
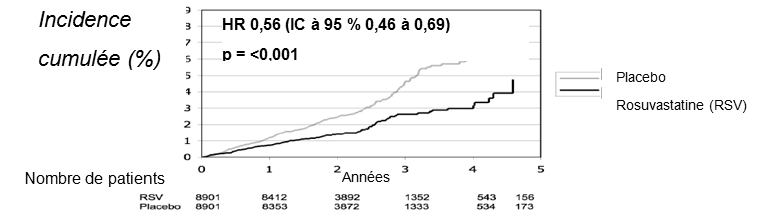
Le profil de sécurité a été généralement comparable chez les sujets sous rosuvastatine 20 mg et les sujets sous placebo. 1,6% des sujets sous rosuvastatine et 1,8 % des sujets sous placebo ont quitté l’étude en raison d’un événement indésirable, indépendamment d’un rapport causal avec le traitement. Les événements indésirables ayant conduit le plus souvent à un arrêt du traitement ont englobé des myalgies (0,3% sous rosuvastatine, 0,2% sous placebo), des douleurs abdominales (0,03% sous rosuvastatine, 0,02% sous placebo) et des éruptions cutanées (0,02% sous rosuvastatine, 0,03% sous placebo). Les événements indésirables rapportés chez ≥5% des patients et au moins aussi souvent ou plus souvent sous placebo ont englobé des infections urinaires (8,7% sous rosuvastatine, 8,6% sous placebo), des rhinopharyngites (7,6% sous rosuvastatine, 7,2% sous placebo), des douleurs dorsales (7,6% sous rosuvastatine, 6,9% sous placebo), des myalgies (7,6% sous rosuvastatine, 6,6% sous placebo), des bronchites (7,2% sous rosuvastatine, 7,1% sous placebo), des arthrites (5,8% sous rosuvastatine, 5,6% sous placebo), de la toux (5,3% sous rosuvastatine, 5,3% sous placebo) et des diarrhées (4,7% sous rosuvastatine, 4,6% sous placebo).
Dans l'étude JUPITER, une augmentation statistiquement significative du diabète sucré a été rapportée, avec une incidence de 2,8% chez les patients sous rosuvastatine et de 2,3% chez les patients sous placebo (HR: 1,27, IC à 95%: 1,05-1,53, p=0,015). La différence des variations moyennes de la valeur de l'HbA1c par rapport à la valeur initiale au début de l'étude dans les deux bras de traitement (rosuvastatine versus placebo) était d'environ 0,1%. L'analyse suggère qu'un nouveau diagnostic de diabète s'est révélé nécessaire essentiellement chez les patients qui présentaient déjà un risque élevé de développement d'un diabète. Dans l'ensemble de la population de l'étude (tout comme chez les patients qui avaient déjà une prédisposition au diabète), le bénéfice en termes cardiovasculaires et en termes de mortalité conféré par le traitement à la rosuvastatine l'a emporté sur le risque de diagnostic d'un diabète nouvellement apparu (voir «Mises en garde et précautions», «Effets indésirables»).
Enfants et adolescents de 10 à 17 ans
Une étude multicentrique, randomisée, réalisée en double aveugle contrôlée par placebo, a inclus au total 176 patients (97 de sexe masculin, 79 de sexe féminin, tous âgés de 10 à 17 ans, aux stades de Tanner II à V, patientes au moins 1 an après la ménarche) souffrant d’hypercholestérolémie hétérozygote. Ces patients ont reçu pendant 12 semaines 5 mg, 10 mg ou 20 mg de rosuvastatine par jour ou un placebo. Après cette phase, tous les 173 patients (96 de sexe masculin, 77 de sexe féminin) ont reçu de la rosuvastatine chaque jour pendant 40 semaines. Au début de l'étude, 30% des patients environ étaient âgés de 10 à 13 ans. Parmi les patients, 17%, 18%, 40% et 25% présentaient respectivement un stade de Tanner de II, III, IV et V.
Au bout de 12 semaines, la rosuvastatine avait baissé le taux de LDL-cholestérol (critère primaire), le taux de cholestérol total et le taux d’ApoB (voir le tableau 4).
Tableau 4: Effet hypolipémiant de la rosuvastatine chez des enfants et adolescents souffrant d’hypercholestérolémie hétérozygote familiale (moyenne des moindres carrés à 12 semaines par rapport aux valeurs initiales, en %)
|
Dose
|
N
|
LDL-C
|
C total
|
HDL-C
|
TG
|
Non HDL-C
|
ApoB
|
ApoA-I
| |
Placebo
|
46
|
–0,7
|
–0,0
|
6,9
|
5,1
|
–0,9
|
–1,7
|
2,8
| |
5 mg
|
42
|
–38,3
|
–29,9
|
4,2
|
0,3
|
–36,1
|
–31,7
|
1,8
| |
10 mg
|
44
|
–44,6
|
–34,2
|
11,2
|
–13,6
|
–43,0
|
–38,1
|
5,4
| |
20 mg
|
44
|
–50,0
|
–38,7
|
8,9
|
–8,1
|
–47,5
|
–40,7
|
4,0
|
À la fin des 40 semaines (phase ouverte avec adaptation de la dose en fonction de la cible, doses allant jusqu’à 20 mg par jour au maximum), 70 (40,5%) des 173 patients avaient atteint le taux cible de cholestérol inférieur à 2,8 mmol/l.
L’analyse de la croissance linéaire (taille), du poids, de l'IMC (indice de masse corporelle) et des caractères sexuels secondaires reflétant le degré de maturité sexuelle (stades de Tanner) chez les patients pédiatriques (âgés de 10 à 17 ans) traités à la rosuvastatine est limitée à une période d’un an. Au bout de 52 semaines de traitement dans le cadre de l’étude, aucun impact sur la croissance, le poids, l'IMC ou la maturité sexuelle n’a été constaté.
L'impact du traitement par rosuvastatine sur la morbi-mortalité cardio-vasculaire n’a pas été examiné chez les enfants et les adolescents.
PharmacocinétiqueAbsorption
Rosuvastatine est administré sous forme active par voie orale. Les concentrations plasmatiques maximales sont atteintes 5 heures après la prise. L’absorption augmente de manière linéaire avec la dose dans l'intervalle posologique. Le métabolisme de la rosuvastatine a lieu dans le foie, l'organe principal de la synthèse du cholestérol et de la clairance des LDL-C. La biodisponibilité absolue est de 20%. Une accumulation minime s’observe après administration répétée de la dose unitaire quotidienne.
Distribution
Approximativement 90% de rosuvastatine sont liés aux protéines plasmatiques et principalement à l'albumine.
Métabolisme
La rosuvastatine subit un métabolisme limité de 10% environ, principalement en dérivé Ndesméthylé. La molécule mère est responsable de plus de 90% de l'activité de la forme circulante active inhibitrice sur l'HMG-CoA réductase.
Élimination
La demi-vie d'élimination de la rosuvastatine s’élève à 19 heures et n'augmente pas avec l’augmentation des doses. Environ 90% sont excrétés sous forme inchangée dans les selles, le reste étant excrété dans les urines.
Cinétique pour certains groupes de patients
Âge et sexe des patients
Aucun effet cliniquement significatif de l’âge ou du sexe sur la pharmacocinétique de la rosuvastatine n’a été mis en évidence chez les adultes. La pharmacocinétique de Rosuvastax chez les enfants et les adolescents présentant une hypercholestérolémie familiale hétérozygote a été comparable à celle observée chez des volontaires sains adultes (voir ci-dessous).
Appartenance ethnique
Les études de pharmacocinétique menées en Asie montrent une multiplication par 2 environ de l'AUC moyenne chez les asiatiques d'Asie par rapport aux caucasiens vivant en Asie ou en Europe. L'influence des facteurs environnementaux et génétiques sur ces différences observées n'a pas été étudiée. Une analyse de pharmacocinétique n'a mis en évidence aucune différence cliniquement significative entre les populations caucasiennes, hispaniques et noires ou les personnes d’origine afro-caribéenne.
Enfants et adolescents
Les paramètres pharmacocinétiques n’ont pas été complètement étudiés chez les patients pédiatriques de 10 à 17 ans souffrant d’hypercholestérolémie familiale hétérozygote. Dans une étude pharmacocinétique, 18 patients pédiatriques (âgés de 10 à 17 ans) souffrant d’hypercholestérolémie familiale hétérozygote ont reçu de la rosuvastatine sous forme de comprimé. Après administration d’une dose unique croissante de rosuvastatine (10 mg, puis 40 mg et finalement 80 mg), une augmentation de l’exposition systémique à la rosuvastatine a été constatée. Les résultats suggèrent que la pharmacocinétique de la rosuvastatine est linéaire dans ce domaine de dosage chez les patients pédiatriques et que les paramètres pharmacocinétiques observés sont comparables à ceux observés chez des volontaires sains adultes.
Troubles de la fonction rénale
Dans une étude incluant des volontaires avec différents degrés d'insuffisance rénale, une altération légère à modérée de la fonction rénale a marginalement influencé les concentrations plasmatiques de rosuvastatine. Cependant, une insuffisance rénale sévère (clairance de la créatinine <30 ml/min) entraîne une multiplication par 3 des concentrations plasmatiques par rapport aux valeurs observées chez les volontaires sains (voir «Contreindications»). En conséquence, l’utilisation de la rosuvastatine est contre-indiquée dans ce groupe de patients.
Troubles de la fonction hépatique
Dans une étude incluant des volontaires avec différents degrés d'insuffisance hépatique, aucun indice en faveur d'une augmentation de l'exposition à la rosuvastatine n’a été mis en évidence, sauf pour 2 personnes présentant une affection hépatique très sévère (scores de Child-Pugh de 8 et 9). Chez ces patients, une augmentation de l’exposition systémique au moins double de celle observée chez des volontaires avec des scores de Child-Pugh inférieurs a été notée.
Polymorphismes génétiques
La disponibilité des inhibiteurs de la HMG-CoA-réductase tels que la rosuvastatine dépend entre autres des protéines de transport OATP1B1 et BCRP. Les patients présentant une variation particulière de la séquence du gène SLCO1B1 (OATP1B1) et/ou du gène ABCG2 (BCRP) présentent un risque d’exposition supérieure à la rosuvastatine. Les variantes de génotypes SLCO1B1 c.521CC ou ABCG2 c.421AA sont associées à des expositions 1,7 fois supérieures (AUC) ou 2,4 fois supérieures à la rosuvastatine que les variantes SLCO1B1 c.521TT et ABCG2 c.421CC.
Données précliniquesSur la base des études conventionnelles de pharmacologie de sécurité, de génotoxicité et de cancérogénicité, les données précliniques ne révèlent aucun risque particulier pour l'être humain. Des études spécifiques sur l'influence de hERG n'ont pas été évaluées. Les effets indésirables ci-après n'ont pas été observés dans les études cliniques mais sont apparus sur les animaux après exposition dans la fourchette thérapeutique humaine: dans les études de toxicité à doses multiples, des modifications histopathologiques du foie, probablement dues à l'action pharmacologique de la rosuvastatine, ont été observées chez la souris, le rat et, dans une moindre mesure, avec des effets dans la vésicule biliaire chez le chien, mais pas chez le singe. En outre, une toxicité testiculaire a été observée chez les singes et les chiens à des doses plus élevées. Chez le rat, la toxicité pour la reproduction a été démontrée par une réduction de la taille et du poids des portées et par une diminution du taux de survie des jeunes animaux. Ces effets ont été observés à des doses toxiques pour la mère, où l'exposition systémique était plusieurs fois supérieure au niveau d'exposition thérapeutique.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Conserver hors de portée des enfants et à une température inférieure à 30°C dans l’emballage d’origine.
Numéro d’autorisation66447 (Swissmedic)
PrésentationRosuvastax 5 mg: 30 et 100 comprimés pelliculés. [B]
Rosuvastax 10 mg: 30 et 100 comprimés pelliculés. [B]
Rosuvastax 20 mg: 30 et 100 comprimés pelliculés. [B]
Titulaire de l’autorisationDrossapharm SA, 4002 Bâle
Mise à jour de l’informationJanvier 2025
|