CompositionPrincipes actifs
Brexpiprazole.
Excipients
Noyau du comprimé:
0,5 mg: lactose monohydraté 47,9 mg, amidon de maïs, cellulose microcristalline, hydroxypropylcellulose, hydroxypropylcellulose à faible substitution, stéarate de magnésium.
1 mg: lactose monohydraté 47,4 mg, amidon de maïs, cellulose microcristalline, hydroxypropylcellulose, hydroxypropylcellulose à faible substitution, stéarate de magnésium.
2 mg: lactose monohydraté 46,4 mg, amidon de maïs, cellulose microcristalline, hydroxypropylcellulose, hydroxypropylcellulose à faible substitution, stéarate de magnésium.
3 mg: lactose monohydraté 45,5 mg, amidon de maïs, cellulose microcristalline, hydroxypropylcellulose, hydroxypropylcellulose à faible substitution, stéarate de magnésium.
4 mg: lactose monohydraté 44,4 mg, amidon de maïs, cellulose microcristalline, hydroxypropylcellulose, hydroxypropylcellulose à faible substitution, stéarate de magnésium.
Pelliculage du comprimé:
0.5 mg: hypromellose, talc, colorant: dioxyde de titane (E171), oxyde de fer jaune et rouge (E172).
1 mg: hypromellose, talc, colorant: dioxyde de titane (E171), oxyde de fer jaune (E172).
2 mg: hypromellose, talc, colorant: dioxyde de titane (E171), oxyde de fer jaune et noir (E172).
3 mg: hypromellose, talc, colorant: dioxyde de titane (E171), oxyde de fer rouge et noir (E172).
4 mg: hypromellose, talc, colorant: dioxyde de titane (E171).
Forme pharmaceutique et quantité de principe actif par unité0,5 mg comprimé pelliculé: 0,5 mg brexpiprazole.
1 mg comprimé pelliculé: 1 mg brexpiprazole.
2 mg comprimé pelliculé: 2 mg brexpiprazole.
3 mg comprimé pelliculé: 3 mg brexpiprazole.
4 mg comprimé pelliculé: 4 mg brexpiprazole.
Comment se présentent les comprimés pelliculés
Tous les comprimés sont ronds, légèrement convexes et à bords biseautés.
0.5 mg: orange pâle, gravé «BRX» et «0.5» d'un côté.
1 mg: jaune pâle, gravé «BRX» et «1» d'un côté.
2 mg: vert pâle, gravé «BRX» et «2» d'un côté.
3 mg: violet pâle, gravé «BRX» et «3» d'un côté.
4 mg: blanc, gravé «BRX» et «4» d'un côté.
Indications/Possibilités d’emploiREXULTI est indiqué pour le traitement de la schizophrénie chez l'adulte et les adolescents de 13 ans et plus.
REXULTI est indiqué pour le traitement de l'agitation dans la démence d'Alzheimer (ADA) chez le patient adulte ne répondant pas aux interventions non pharmacologiques.
Posologie/Mode d’emploiSchizophrénie
Adulte
La posologie initiale recommandée de REXULTI pour le traitement de patients atteints de schizophrénie est de 1 mg une fois par jour du 1er au 4e jour.
La fourchette posologique recommandée est de 2–4 mg une fois par jour.
Le jour 5, la prise du médicament est augmentée à 2 mg, puis à 4 mg le jour 8 selon la tolérance et la réponse clinique du patient. La dose quotidienne maximale recommandée est de 4 mg.
Adolescents de 13 ans et plus
La dose initiale recommandée de brexpiprazole est de 0,5 mg une fois par jour du jour 1 au jour 4.
La dose de brexpiprazole doit être augmentée progressivement jusqu'à 1 mg une fois par jour du jour 5 au jour 7, puis jusqu'à 2 mg le jour 8. Des augmentations hebdomadaires de 1 mg peuvent être effectuées en fonction de la réponse clinique et de la tolérance.
La posologie cible recommandée est de 2 mg à 4 mg une fois par jour. La dose quotidienne maximale recommandée est de 4 mg.
Agitation dans la démence d'Alzheimer
La posologie initiale recommandée de REXULTI pour traiter l'agitation dans la démence d'Alzheimer (ADA) est de 0,5 mg une fois par jour du 1er au 7ème jour. La posologie est augmentée à 1 mg du 8ème au 14ème jour, puis à 2 mg à partir du 15ème jour.
Avant et pendant le traitement par REXULTI, les patients atteints de la démence d'Alzheimer doivent être évalués afin de détecter les facteurs réversibles pouvant provoquer une agitation (par exemple douleurs, infections, polypharmacie, délire aigu) et, le cas échéant, être traités de manière appropriée. Les interventions non pharmacologiques doivent s'être révélées inefficaces avant le début du traitement (voir Efficacité clinique).
REXULTI ne doit pas être utilisé comme traitement « si besoin » de la ADA.
Traitement d'entretien
Schizophrénie
La fourchette posologique recommandée est de 2-4 mg/jour. La nécessité du traitement d'entretien et la posologie appropriée doivent être contrôlées régulièrement.
Agitation dans la démence d'Alzheimer
La fourchette posologique recommandée est de 2 à 3 mg une fois par jour. Après au moins 4 semaines de traitement à raison de 2 mg une fois par jour, la dose peut être augmentée jusqu'à la dose journalière maximale recommandée de 3 mg si cela semble cliniquement nécessaire. L'efficacité doit être vérifiée au-delà de 12 semaines, puis périodiquement afin de déterminer la nécessité de poursuivre le traitement et la posologie appropriée de REXULTI. Le traitement par REXULTI doit être arrêté en cas de réponse insuffisante.
Mode d'administration
REXULTI se prend par voie orale pendant ou en dehors des repas. Les comprimés de REXULTI doivent être avalés entiers.
Instructions posologiques particulières
Patients âgés
La sécurité et l'efficacité de REXULTI pour le traitement de la schizophrénie chez le sujet âgé (plus de 65 ans) n'ont pas été établies. L'exposition au brexpiprazole observée était similaire lors de l'administration de REXULTI à des sujets âgés en bonne santé (plus de 65 ans) et à des sujets adultes (18-45 ans) (voir la rubrique Pharmacocinétique). La posologie doit être définie avec prudence et à l'extrémité inférieure de la fourchette posologique pour les patients âgés afin de tenir compte des atteintes plus fréquentes des fonctions hépatiques, rénales et cardiaques, ainsi que des comorbidités et des autres traitements médicamenteux.
Patients présentant des troubles de la fonction hépatique
Chez les patients atteints de schizophrénie qui présentent une insuffisance hépatique modérée à sévère (score de Child-Pugh ≥7), la dose maximale recommandée est de 3 mg, une fois par jour, et de 2 mg une fois par jour pour les patients atteints de ADA.
Patients présentant des troubles de la fonction rénale
Chez les patients atteints de schizophrénie qui présentent une insuffisance rénale modérée, sévère ou terminale (clairance de la créatinine CLcr < 60 ml/min), la dose maximale recommandée est de 3 mg, une fois par jour, et de 2 mg une fois par jour pour les patients atteints de ADA.
Autres groupes de patients particuliers
Aucun ajustement de la dose de REXULTI n'est nécessaire en raison du sexe, de l'origine ethnique ou du tabagisme (voir la rubrique Pharmacocinétique).
Tableau 1: Ajustements posologiques chez les métaboliseurs lents du CYP2D6 et lors de l'utilisation concomitante d'inhibiteurs ou d'inducteurs du CYP
|
|
Posologie ajustée
| |
Métaboliseurs lents du CYP2D6
| |
Métaboliseurs lents du CYP2D6 connus
|
Administration de la moitié de la dose habituelle
| |
Métaboliseurs lents du CYP2D6 connus, qui prennent un inhibiteur modéré/puissant du CYP3A4
|
Administration d'un quart de la dose habituelle
| |
Patients prenant un inhibiteur du CYP2D6 et/ou un inhibiteur du CYP3A4
| |
Inhibiteur puissant du CYP2D6
|
Administration de la moitié de la dose habituelle
| |
Inhibiteur puissant du CYP3A4
| |
Inhibiteur puissant/modéré du CYP2D6 avec inhibiteurs puissants/modérés du CYP3A4
|
Administration d'un quart de la dose habituelle
| |
Patients prenant des inducteurs du CYP3A4
| |
Inducteurs puissants du CYP3A4*
|
Doublement de la dose habituelle pendant 1 à 2 semaines
|
* Si l'inducteur du CYP3A4 pris simultanément est arrêté, la posologie doit être réduite au niveau initial pendant 1-2 semaines.
Enfants de moins de 13 ans
Rexulti n'est pas approuvé pour une utilisation chez les enfants de moins de 13 ans. Aucune recommandation posologique ne peut être donnée.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients mentionnés sous Composition.
Mises en garde et précautionsMortalité accrue chez les patients âgés atteints de psychose démentielle
Effet de classe: les patients âgés atteints de psychose démentielle sous traitement antipsychotique présentent un risque de décès plus élevé que ceux recevant un placebo.
Les analyses de 17 études contrôlées par placebo sur les psychoses démentielles (durée modale de 10 semaines), portant principalement sur des patients sous antipsychotiques atypiques, ont démontré que les patients traités par médicaments présentaient un risque de mortalité 1.6 à 1.7 fois plus élevé que les patients sous placebo. Au cours d'une étude contrôlée type sur 10 semaines, le taux de mortalité des patients traités par médicaments a été de 4.5% environ contre 2.6% chez les patients sous placebo. Chez les patients atteints de la démence d'Alzheimer, REXULTI n'est pas autorisé pour le traitement d'une psychose, mais uniquement pour le traitement de l'agitation dans le cadre de la maladie sous-jacente.
Risque de suicide
Effet de classe: les maladies psychotiques augmentent le risque de tentatives de suicide. Les patients à haut risque doivent être étroitement surveillés et accompagnés cliniquement pendant le traitement médicamenteux.
Les patients atteints de ADA présentant des signes de risque grave de suicide n'ont pas été évalués dans le cadre du programme de développement clinique REXULTI.
Affections cérébrovasculaires
Effet de classe: dans des études contrôlées par placebo portant sur la rispéridone, l'aripiprazole et l'olanzapine administrés à des patients âgés atteints de psychose démentielle, on a observé une incidence plus élevée d'événements indésirables cérébrovasculaires (accidents vasculaires cérébraux et troubles circulatoires transitoires), y compris des cas de décès, comparativement au placebo.
Les patients atteints de ADA présentant une pathologie préexistante du SNC, y compris des affections cérébrovasculaires et des formes de démence mixte, ont été exclus du programme de développement clinique REXULTI.
Affections cardiovasculaires
Les patients atteints de ADA présentant une affection cardiovasculaire préexistante cliniquement significative (y compris fibrillation auriculaire non contrôlée, insuffisance cardiaque ou cardiopathie ischémique) n'ont pas été évalués dans le cadre du programme de développement clinique REXULTI.
Allongement de l'intervalle QT
Les patients atteints de ADA présentant la préexistence d'un intervalle QTcF ≥450 ms pour les hommes et ≥470 ms pour les femmes et/ou une co-médication pouvant induire un allongement de l'intervalle QT n'ont pas été étudiés dans le cadre du programme de développement clinique REXULTI.
Hypotension orthostatique et syncope
L'hypotension orthostatique peut être associée à des effets indésirables tels que des vertiges, des étourdissements et une tachycardie. Le risque est généralement maximal en début de traitement et pendant une phase d'augmentation de la dose. Les patients déshydratés, hypovolémiques, sous traitement antihypertenseur, ayant des antécédents de maladies cardiovasculaires (par ex. insuffisance cardiaque, infarctus du myocarde, ischémie ou troubles de la conduction) ou cérébrovasculaires, ainsi que les patients prenant un traitement antipsychotique pour la première fois, présentent un risque accru de subir ces effets indésirables ou des complications dues à l'hypotension. Une dose initiale moins forte et une augmentation progressive plus lente doivent être envisagées chez ces patients, dont les paramètres vitaux orthostatiques doivent être surveillés.
Schizophrénie
Dans les études cliniques à court terme contrôlées par placebo avec REXULTI administré à des patients atteints de schizophrénie, l'incidence des événements indésirables liés à l'hypotension orthostatique observée sous REXULTI par rapport au placebo a été la suivante: vertiges (2,3 % contre 1,4 %), hypotonie orthostatique (0,4 % contre 0,2 %) et syncope (0,1 % contre 0 %).
Agitation dans la démence d'Alzheimer
Les patients atteints de ADA ayant des antécédents d'hypotension orthostatique ont été exclus du programme de développement clinique REXULTI.
Dans les études cliniques contrôlées par placebo sur 12 semaines portant sur REXULTI, avec dose fixe ou flexible à des patients atteints de ADA (âgés de 51 à 90 ans), l'incidence des événements indésirables liés à l'hypotension orthostatique était comparable chez les patients traités par REXULTI et chez les patients traités par placebo, notamment pour la sensation vertigineuse (3,2 % contre 3,4 %), l'hypotension orthostatique (0,5 % contre 0,5 %) et la syncope (0,2 % contre 0,8 %).
Thromboembolie veineuse
Effet de classe: des cas de thromboembolie veineuse (TEV) ont été rapportés sous antipsychotiques. Les patients traités par antipsychotiques présentent souvent des facteurs de risque acquis de TEV. Les facteurs de risque de TEV possibles doivent donc être testés scrupuleusement avant et pendant le traitement par REXULTI et des précautions doivent être prises.
Les patients atteints de la démence d'Alzheimer ayant des antécédents de TEV n'ont pas été étudiés dans le cadre du programme de développement clinique REXULTI.
Syndrome neuroleptique malin (SNM)
Le SMN est un ensemble potentiellement mortel de symptômes qui a été observé avec l'emploi d'antipsychotiques, y compris le brexiprazole. Les manifestations cliniques d'un SMN sont: hyperthermie sévère, raideur musculaire, altération de la conscience et signes d'instabilité du système nerveux autonome (pouls ou tension artérielle irréguliers, tachycardie, diaphorèse, troubles du rythme cardiaque). Les autres signes peuvent être: taux élevé de créatine phosphokinase, myoglobinurie (rhabdomyolyse) et insuffisance rénale aiguë. Si un patient développe des signes et symptômes faisant penser à un SMN ou présente une forte fièvre inexpliquée sans autres manifestations cliniques du SMN, tous les antipsychotiques, y compris REXULTI, doivent être arrêtés. Si un patient a besoin d'un traitement antipsychotique après avoir guéri d'un SMN, la reprise éventuelle d'un traitement médicamenteux doit être évaluée soigneusement. Le paient doit être surveillé étroitement, car des récidives de SMN ont été rapportées.
Les patients atteints de la démence d'Alzheimer présentant des antécédents de SMN n'ont pas été étudiés dans le cadre du programme de développement clinique REXULTI.
Convulsions
Effet de classe: comme les autres antipsychotiques, REXULTI doit être utilisé avec prudence chez les patients ayant des antécédents de convulsions ou dont le tableau clinique indique des seuils de déclenchement potentiellement réduits. Les patients atteints de ADA avec des antécédents d'épilepsie et/ou sous médicaments antiépileptiques n'ont pas été évalués dans le cadre du programme de développement clinique REXULTI.
Dyskinésies tardives
Effet de classe: les patients sous traitement antipsychotique peuvent développer un syndrome se manifestant par des mouvements dyskinétiques involontaires, et qui peut être potentiellement irréversible. La prévalence maximale de ce syndrome semble concerner les patients âgés, plus particulièrement les femmes âgées. Malgré les estimations de prévalence, il n'est pas possible de prévoir en début de traitement antipsychotique quels patients risquent de présenter ce syndrome.
Si des signes et symptômes de dyskinésie tardive surviennent pendant le traitement par REXULTI, une réduction de la dose ou l'arrêt du médicament doivent être envisagés. Les symptômes peuvent s'aggraver temporairement ou même apparaître après l'arrêt du traitement.
Dystonie
Effet de classe: pendant les premiers jours du traitement, les personnes sensibles peuvent ressentir des symptômes de dystonie, c'est-à-dire des contractions anormales et constantes de certains groupes musculaires. Les symptômes de la dystonie se manifestent notamment par des spasmes de la musculature du cou susceptibles de provoquer une sensation d'oppression au niveau de la gorge, des troubles de la déglutition, un essoufflement et/ou une protrusion de la langue. Bien que ces symptômes soient susceptibles d'apparaître à des faibles doses, ils se manifestent plus souvent et plus intensément avec les médicaments antipsychotiques de première génération qui sont plus puissants et plus forts. Un risque accru de dystonie aiguë a été observé chez les patients jeunes et de sexe masculin.
Troubles du contrôle des impulsions
Chez les patients sous brexpiprazole, des cas de troubles du contrôle des impulsions, y compris une addiction aux jeux, ont été rapportés. Les patients ayant des antécédents de troubles du contrôle des impulsions sont susceptibles de présenter un risque accru et doivent être étroitement suivis. Dans toutes les indications, il est à noter que les symptômes d'un trouble du contrôle des impulsions peuvent être liés à la maladie sous-jacente.
Maladies du SNC autres que la maladie d'Alzheimer (AD)
Les patients atteints de ADA présentant un délire aigu ou un délire dans les 30 jours, une démence ou d'autres troubles de la mémoire non imputables à la démence d'Alzheimer, ont été exclus du programme de développement clinique REXULTI. REXULTI ne doit pas être utilisé pour traiter la confusion aiguë ou pour traiter l'agitation associée à des affections autres que la démence d'Alzheimer (par exemple, délire aigu, psychose, autres types de démence).
Hyperglycémie et diabète sucré
Effet de classe: une hyperglycémie parfois très marquée, accompagnée d'une acidocétose ou d'un coma hyperosmolaire, voire un décès, a été rapportée chez les patients traités par antipsychotiques.
Chez ces patients, les signes et symptômes de l'hyperglycémie (par ex. polydipsie, polyurie, polyphagie et faiblesse) doivent être surveillés. Les patients diabétiques ou présentant des facteurs de risque de diabète (par ex. obésité ou antécédents familiaux de diabète) doivent être contrôlés régulièrement pour détecter toute détérioration du métabolisme du glucose. Chez les patients présentant une hyperglycémie liée au traitement significative, l'arrêt de REXULTI doit être envisagé.
Les patients atteints de ADA qui présentent un diabète sucré instable ou non contrôlé n'ont pas été étudiés dans le cadre du programme de développement clinique REXULTI.
Schizophrénie
Dans les études cliniques avec REXULTI chez des patients atteints de schizophrénie (études 331-10-231 et 331-10-230 (voir Efficacité clinique), les fluctuations de la glycémie à jeun étaient comparables sous REXULTI et sous placebo.
Dans l'étude sur le traitement d'entretien à long terme (étude 331-10-232 [voir Efficacité clinique]), les écarts moyens des taux de glucose sérique entre la visite de référence et la dernière visite étaient faibles, aussi bien sous REXULTI (2,11 mg/dl) que sous placebo (−1,62 mg/dl), et n'ont pas été considérées comme cliniquement significatives.
Dans les études en ouvert à long terme, l'écart moyen des valeurs de glucose sérique à jeun entre la visite de référence et à la dernière visite était de 2,31 mg/dl (N = 1120).
Agitation dans la démence d'Alzheimer
Dans les études contrôlées par placebo sur 12 semaines menées auprès de patients (âgés de 51 à 90 ans) atteints de ADA, la proportion de patients ayant vu leur glycémie à jeun passer de normale (< 100 mg/dL) ou altérée (≥100 et < 126 mg/dL) à trop élevée (≥126 mg/dL) était comparable chez les patients traités par REXULTI (14 %) et les patients traités par placebo (14 %).
Parmi les patients (âgés de 55 à 90 ans) de l'étude clinique contrôlée par placebo sur 12 semaines 331-14-213 transférés dans l'étude d'extension sur 12 semaines 331-201-00182 avec traitement actif, 18 % des patients dont la glycémie à jeun de référénce était normale ou altérée sont passés à une glycémie à jeun élevée (> 126 mg/dL) .
Prise de poids et dyslipidémie
Effet de classe: des modifications du métabolisme, comme une prise de poids et une dyslipidémie, se sont produites sous traitement antipsychotique. Les antipsychotiques atypiques peuvent être à l'origine de modifications indésirables de la lipidémie. Avant ou peu de temps après l'instauration du traitement antipsychotique, il est recommandé d'établir un profil lipidique à jeun au début et de le surveiller régulièrement pendant le traitement.
Une augmentation de la fréquence de prise de poids a été observée en cas de traitement prolongé par brexpiprazole. Une surveillance clinique du poids est recommandée en début et en cours de traitement (pour la prise de poids, voir la description des effets indésirables spécifiques et les informations supplémentaires).
Dyslipidémie
Schizophrénie
Chez les personnes sous traitement par antipsychotiques atypiques, des modifications indésirables de la lipidémie peuvent survenir.
Dans les études 331-10-231 et 331-10-230, les modifications du cholestérol total à jeun, du cholestérol LDL et du cholestérol HDL étaient comparables chez les patients sous REXULTI et sous placebo. Le tableau 2 montre les pourcentages de patients présentant des modifications des triglycérides à jeun.
Tableau 2: Modification des triglycérides à jeun dans les études 331-10-231 et 331-10-230
|
Proportion de sujets présentant des écarts entre les valeurs de référence et les valeurs suivantes
| |
|
Placebo
|
1 mg/jour
|
2 mg/jour
|
4 mg/jour
| |
Triglycérides
|
|
|
|
| |
Valeur normale à élevée
(< 150 à ≥200 mg/dl et < 500 mg/dl)
|
6% (15/253)*
|
10% (7/72)*
|
8% (19/232)*
|
10% (22/226)*
| |
Valeur normale/de la limite de la normale à très élevée
(< 200 à ≥500 mg/dl)
|
0 % (0/303)*
|
0 % (0/94)*
|
0 % (0/283)*
|
0,4 % (1/283)*
|
* Désigne n/N, où N = nombre total de patients avec une mesure de référence et au moins une mesure prise après la visite de référence; n = nombre de patients présentant un écart.
Dans l'étude 331-10-232, aucune différence cliniquement significative n'a été observée entre les groupes de traitement en termes de fréquence de valeurs métaboliques potentiellement pertinentes sur le plan clinique.
Dans les études ouvertes à long terme, les triglycérides à jeun entre la visite de référence et la dernière visite présentaient un écart moyen de -2,14 mg/dl (N = 1 123).
Agitation dans la démence d'Alzheimer
Dans les études cliniques contrôlées par placebo sur 12 semaines menées auprès de patients atteints de ADA (âgés de 51 à 90 ans), la fréquence des modifications des taux de cholestérol total (de normal < 200 mg/dL à élevé ≥240 mg/dL), de cholestérol LDL (de normal < 100 mg/dL à élevé ≥160 mg/dL) et de cholestérol HDL (de normal ≥40 mg/dL à faible < 40 mg/dL) était comparable entre les patients traités par REXULTI (6,6 %, 6,1 %, 17,1 %) et ceux sous placebo (9,4 %, 5,9 %, 13,7 %). La fréquence de la variation des taux de triglycérides à jeun, de l'intervalle normal/limite (< 200 mg/dL) à l'intervalle fortement augmenté (≥500 mg/dL) était comparable entre les patients atteints de ADA traités par REXULTI (0,4 %) et ceux recevant le placebo (0,3 %).
Chez les patients (âgés de 55 à 90 ans) qui sont passés de l'étude clinique 331-14-213 sur 12 semaines contrôlée par placebo à l'étude d'extension 331-201-00182 sur 12 semaines avec traitement actif, 10 % des patients recevant REXULTI ont vu leur cholestérol total (à jeun) passer de l'intervalle normal (< 200 mg/dL à l'examen de référence) à l'intervalle élevé (≥240 mg/dL) et chez 14 % des patients recevant REXULTI, le cholestérol HDL est passé de l'intervalle normal (≥40 mg/dL à l'examen de référence) à l'intervalle faible (< 40 mg/dL). De même, 12 % des patients ont vu leur taux de triglycérides de départ passer de normal à la visite de référence (< 150 mg/dL) à élevé (200 à < 500 mg/dL).
Leucopénie, neutropénie et agranulocytose
Effet de classe: une leucopénie/neutropénie a été rapportée sous traitement par antipsychotiques. Pour les autres principes actifs de cette classe, des agranulocytoses (allant jusqu'au décès) ont été observées.
Les facteurs de risque possibles de la leucopénie/neutropénie sont, entre autres, la préexistence d'une numération leucocytaire basse, ainsi que des antécédents de leucopénie/neutropénie induite par les médicaments. Chez les patients présentant ces facteurs de risque, un hémogramme complet doit être effectué fréquemment au cours des premiers mois de traitement. En l'absence d'autres facteurs déclencheurs, REXULTI doit être arrêté dès les premiers signes de chute des valeurs leucocytaires.
Chez les patients présentant une neutropénie, toute fièvre ou tout autre signe ou symptôme d'infection doivent être surveillés attentivement et, en cas de manifestation de ces symptômes, un traitement doit être instauré immédiatement. Chez les patients présentant une neutropénie sévère (numération absolue des neutrophiles < 1000/mm3), REXULTI doit être arrêté et la numération des leucocytes doit être contrôlée jusqu'à la guérison.
Thermorégulation
Effet de classe: les antipsychotiques peuvent compromettre la capacité du corps à abaisser la température interne. Chez les patients présentant un risque d'élévation de la température interne, par ex. en raison d'une activité physique intense, d'une chaleur extrême, d'une déshydratation ou d'un traitement concomitant par médicaments anticholinergiques, REXULTI doit être prescrit avec la diligence appropriée.
Dysphagie
Effet de classe: des troubles de la motilité de l'œsophage et de l'aspiration ont été observés sous traitement antipsychotique. REXULTI et les autres antipsychotiques doivent être utilisés avec prudence chez les patients présentant un risque de pneumonie d'aspiration. REXULTI n'a pas été étudié chez les patients présentant une dysphagie, une absorption orale limitée et/ou nourris par voie entérale.
Prolactine
Le brexpiprazole peut augmenter les taux de prolactine. Les augmentations associées au traitement par brexpiprazole sont généralement légères et peuvent s'atténuer au cours du traitement. Cependant, dans de rares cas, cet effet peut persister (voir la rubrique « Effets indésirables »).
Personnes âgées
Schizophrénie
Des études cliniques avec REXULTI ont été menées sur un nombre limité de sujets âgés de 65 ans ou plus pour déterminer la possibilité qu'ils réagissent différemment des sujets plus jeunes. Les sujets âgés (> 65 ans) ont présenté une exposition systémique au brexpiprazole comparable à celle des sujets adultes (18-45 ans) (voir les rubriques Pharmacocinétique et Posologie/Mode d'emploi). Les patients âgés atteints de psychose démentielle sous traitement antipsychotique présentent un risque de décès plus élevé que sous placebo (voir ci-dessus: Mortalité accrue chez les patients âgés atteints de psychose démentielle).
Agitation dans la démence d'Alzheimer
Un nombre total de 556 patients âgés de 65 ans et plus (216 patients âgés de 65 à 74 ans, 273 patients âgés de 75 à 84 ans et 67 patients âgés de 85 ans) ont été traités par REXULTI dans les études cliniques sur la démence d'Alzheimer sur 12 semaines contrôlées par placebo. Dans l'étude d'extension en ouvert 331-201-00182 (NCT03594123), 259 patients ont été traités, dont 100 patients âgés de 65 à 74 ans, 108 patients âgés de 75 à 84 ans et 22 patients âgés de 85 ans ou plus.
Dans les études cliniques contrôlés sur 12 semaines menées auprès de patients gériatriques (65 ans et plus) sur le traitement de ADA, l'incidence des chutes (2%) et des vertiges (3%) chez les patients traités par dose fixe ou flexible de REXULTI était similaire à celle des patients traités par placebo (chutes et vertiges, 3 % dans chaque cas).
Valeurs de laboratoire anormales
Les patients atteints de la maladie d'Alzheimer qui présentaient des valeurs de laboratoire anormales significatives sur le plan clinique, par ex. des valeurs élevées de créatine phosphokinase, de chimie sérique, de paramètres hépatiques et thyroïdiens, ont été exclus du programme de développement clinique REXULTI.
Troubles de la fonction hépatique
La posologie doit être ajustée chez les patients présentant des troubles de la fonction hépatique (voir Pharmacocinétique et posologie/administration).
Troubles de la fonction rénale
La posologie doit être ajustée chez les patients présentant des troubles de la fonction rénale (voir Pharmacocinétique et Posologie/Mode d'emploi).
Lactose
REXULTI comprimés pelliculés contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
InteractionsREXULTI est principalement métabolisé par les cytochromes CYP3A4 et CYP2D6. Sur la base des résultats d'études d'interaction du médicament chez les personnes prenant de puissants inhibiteurs du CYP2D6 et du CYP3A4, il est recommandé d'ajuster la dose d'entretien en la réduisant de moitié. D'après les estimations effectuées dans le cadre de l'analyse pharmacocinétique de population, il y a lieu de considérer que les métaboliseurs rapides du CYP2D6 qui reçoivent un inhibiteur du CYP3A4 et du CYP2D6 ou les métaboliseurs lents du CYP2D6 qui reçoivent un puissant inhibiteur du CYP3A4 présentent des concentrations en brexpiprazole 4 ou 5 fois supérieures. Dans ce cas, la prise de REXULTI doit donc être réduite à un quart de la dose recommandée (voir la rubrique Posologie/Mode d'emploi).
Si REXULTI est utilisé conjointement à un puissant inducteur du CYP3A4 (par ex. rifampicine), la dose doit être multipliée par 2, puis adaptée en fonction de la réponse clinique (voir la rubrique Posologie/Mode d'emploi).
Inhibiteurs enzymatiques
Inhibiteur puissant du CYP2D6
En cas d'utilisation concomitante de REXULTI 2 mg en dose unique par voie orale et de quinidine (324 mg/jour pendant 7 jours), un puissant inhibiteur du CYP2D6 (non autorisé en Suisse), l'aire sous la courbe (area under the curve [AUC]) du brexpiprazole a augmenté de 94%.
Kétoconazole et autres puissants inhibiteurs du CYP3A4
En cas d'utilisation concomitante de kétoconazole (200 mg deux fois par jour pendant 7 jours), un puissant inhibiteur du CYP3A4 et de REXULTI 2 mg en dose unique par voie orale, l'AUC du brexpiprazole a augmenté de 97%.
Inhibiteurs du CYP2B6
L'utilisation concomitante de REXULTI 2 mg en dose unique par voie orale et de ticlopidine (250 mg deux fois par jour pendant 7 jours), un inhibiteur du CYP2B6 (non autorisé en Suisse), n'a aucun effet sur la pharmacocinétique du brexpiprazole.
Inducteurs enzymatiques
Rifampicine et autres inducteurs du CYP3A4
L'utilisation concomitante de rifampicine (600 mg deux fois par jour pendant 12 jours), un inducteur puissant du CYP3A4, avec une dose orale unique de 4 mg de REXULTI, a réduit la Cmax du brexpiprazole d'environ 31 % et l'AUC d'environ 73 %.
Autres interactions
Modificateurs de pH de l'acide gastrique
L'utilisation concomitante d'oméprazole (40 mg une fois par jour pendant 5 jours), un inhibiteur de la pompe à protons (IPP) largement utilisé, et de REXULTI 4 mg en dose unique par voie orale n'a aucun effet sur l'absorption du brexpiprazole.
Effet de REXULTI sur d'autres médicaments
Les données in vitro ont montré une inhibition faible, voire nulle, de l'isoenzyme CYP450 par le brexpiprazole. L'inhibition potentielle in vitro des transporteurs de MDR1 (P-gp), OAT1, OAT3, OCT2, d'extrusion de multiples médicaments et toxines (MATE1), MATE2-K, OATP1B1, OATP1B3 et OCT1 par le brexpiprazole a également été étudiée. Le brexpiprazole ou son métabolite principal ont seulement été identifiés comme des inhibiteurs potentiels du transporteur d'efflux-BCRP de BCRP, OATP1B1, MATE1 et MATE2-K, mais pas comme des inhibiteurs des autres transporteurs testés.
Des études cliniques montrent que REXULTI par voie orale (2 mg/jour pendant 5 jours) n'a aucun effet sur la pharmacocinétique du dextrométhorphane (substrat du CYP2D6), de la lovastatine (substrat du CYP3A4 non autorisé en Suisse), du bupropion (substrat du CYP2B6) ou de la fexofénadine (substrat du transporteur de P-gp). REXULTI (6 mg en dose unique) n'a eu aucun effet sur l'absorption de la rosuvastatine (substrat des transporteurs de BCRP et OATP).
Alcool
REXULTI n'a pas été étudié chez les patients ayant consommé de l'alcool de façon concomitante.
Grossesse, allaitementGrossesse
L'utilisation sûre de REXULTI avant la conception, pendant la grossesse ou l'allaitement n'a pas été démontrée. REXULTI ne doit être utilisé ni pendant la grossesse ni chez la mère allaitante, ni chez les patients en âge de procréer qui n'utilisent pas de contraception.
Les expérimentations animales sur la reproduction n'ont révélé aucune tératogénicité, mais une augmentation des cas de mortalité périnatale a été constatée (voir la rubrique Données précliniques).
Pour l'instant, aucune étude suffisamment contrôlée et adéquate n'a été menée sur la femme enceinte pour déterminer les risques liés à l'utilisation de REXULTI. Chez les nouveau-nés dont la mère a pris des antipsychotiques comme REXULTI au cours du troisième trimestre de grossesse, il existe un risque de syndrome extrapyramidal et/ou de manque après la naissance. Des cas d'agitation, de tonus musculaire anormalement élevé ou faible, de tremblements, de somnolence, de difficultés à respirer ou de problèmes pour s'alimenter ont été rapportés chez ces enfants. Ces complications présentent différents niveaux de gravité. Dans certains cas, les symptômes étaient auto-limitatifs, tandis que dans d'autres cas, un traitement en soins intensifs et une hospitalisation plus longue ont été nécessaires.
L'effet de REXULTI sur le travail et l'accouchement n'est pas connu chez l'humain.
Allaitement
On ne sait pas si le brexpiprazole ou ses métabolites passent dans le lait maternel chez l'humain. Le brexpiprazole est excrété dans le lait de rats. Compte-tenu des effets indésirables potentiels graves sur le nourrisson, il convient de décider si l'allaitement doit être interrompu ou si le traitement par REXULTI doit être arrêté en tenant compte du risque lié à l'arrêt du traitement pour la mère.
Fertilité
L'effet du brexpiprazole sur la fertilité humaine n'a pas été étudié. Les études animales ont révélé une diminution de la fertilité féminine (voir la rubrique Données précliniques).
Effet sur l’aptitude à la conduite et l’utilisation de machinesComme avec les autres antipsychotiques susceptibles d'altérer le jugement et les capacités de réflexion et motrice, les patients doivent s'abstenir de conduire des véhicules ou d'utiliser des machines dangereuses par mesure de précaution jusqu'à ce qu'il soit sûr que le traitement par REXULTI n'affecte pas ces activités chez ces patients.
Dans les études à court terme contrôlées par placebo menées sur des patients schizophrènes, 5% des patients traités par REXULTI et 4% des patients sous placebo ont rapporté une somnolence (y compris sédation et besoin de sommeil accru). Dans les études ouvertes à long terme, l'incidence de la somnolence et des effets similaires était de 3%.
La démence affecte l'aptitude à conduire des véhicules et à utiliser des machines. Dans les études cliniques sur 12 semaines contrôlées par placebo menées auprès de patients atteints de ADA (âgés de 51 à 90 ans), une somnolence (y compris une sédation) a été rapportée chez 4 % des patients traités par REXULTI et, comparativement, chez 2 % des patients traités par placebo.
Effets indésirablesSchizophrénie
Les effets indésirables les plus fréquemment observés chez les adultes étaient akathisie (5,6%) et prise de poids (3,9%), et chez les adolescents nausées (6,4 %), somnolence (4,5 %) et akathisie (3,6 %). Les événements indésirables étaient pour la plupart légers à modérés et ont généralement conduit à une interruption de l'étude.
Résumé des effets indésirables
La fréquence des effets indésirables associés au traitement par brexpiprazole est indiquée ci-dessous. Ces effets indésirables ont été rapportés lors d'études de phase 2 et 3 à court terme, contrôlées par placebo, menées chez l'adulte aux doses thérapeutiques pertinentes (2 à 4 mg) et lors d'études de phase 3 à court terme, contrôlées par placebo, menées chez l'enfant et l'adolescent aux doses thérapeutiques pertinentes (1 à 4 mg). Tous les effets indésirables sont classés par classe de systèmes d'organes et par fréquence: très fréquents (≥1/10), fréquents (≥1/100 à <1/10), occasionnels (≥1/1 000 à <1/100), rares (≥1/10 000 à <1/1 000), très rares (<1/10 000), fréquence inconnue (ne peut être estimée sur la base des données disponibles). Au sein de chaque catégorie de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Troubles du système immunitaire:
Fréquent : Éruption cutanée.
Occasionnel : Angio-œdème, urticaire, gonflement du visage.
Troubles du métabolisme et de la nutrition:
Fréquent : prise de poids, augmentation de la créatine-phosphokinase.
Affections psychiatriques :
Fréquent : agitation*.
Occasionnel : tentatives de suicide, pensées suicidaires.
Fréquence indéterminée : dépendance au jeu, comportements impulsifs, crises de boulimie, achats compulsifs, comportements sexuels compulsifs.
* L'agitation peut être associée à l'effet pharmacologique du brexpiprazole. Elle est citée, bien que la différence par rapport au placebo soit inférieure à 0.5%.
Affections du système nerveux:
Fréquent : acathisie, tremblements, somnolence**, vertiges.
Occasionnel : parkinsonisme.
Fréquence indéterminée : crises d'épilepsie, syndrome neuroleptique malin#.
** Inclut la sédation et l'hypersomnie
# Effets indésirables signalés après commercialisation
Troubles cardiaques:
Fréquence indéterminée: allongement de l'intervalle QT à l'électrocardiogramme.
Troubles vasculaires:
Occasionnel : thromboembolie veineuse (y compris embolie pulmonaire et thrombose veineuse profonde), hypotension orthostatique.
Affections respiratoires, thoraciques et médiastinales:
Occasionnel : toux.
Affections gastro-intestinales:
Fréquent : diarrhée, nausée, sècheresse buccale, douleurs abdominales hautes.
Occasionnel : carie dentaire, aérophagie.
Affections de la peau et du tissu sous-cutané:
Fréquent : exanthème.
Affections musculo-squelettiques et systémiques:
Fréquent : maux de dos, douleurs dans les membres.
Occasionnel : myalgie.
Fréquence inconnue : Rhabdomyolyse.
Troubles généraux et anomalies au site d'administration:
Fréquent : douleurs.
Grossesse, période post-partum et troubles périnatals
Fréquence inconnue : Syndrome de sevrage néonatal.
Examens complémentaires
Très fréquent : Hyperprolactinémie.***
Occasionnel : Hypertension artérielle, hypertriglycéridémie, élévation des enzymes hépatiques.
*** La classification de l'hyperprolactinémie repose sur le critère des valeurs potentiellement cliniquement pertinentes (VPC) supérieures à 1 fois la limite supérieure de la normale (LSN).
Agitation dans la démence d'Alzheimer
La sécurité d'emploi de REXULTI a été évaluée auprès de 655 patients (âgés de 51 à 90 ans) ayant reçu un diagnostic présumé de ADA qui ont participé à trois études cliniques sur 12 semaines contrôlées par placebo dans lesquelles REXULTI était administré à des doses quotidiennes ≤1 mg (157 patients), de 2 mg (213 patients), de 3 mg (153 patients) ou à des doses flexibles entre 0,5 et 2 mg (132 patients). Les effets indésirables les plus fréquemment observés dans les études cliniques sur 12 semaines contrôlées par placebo étaient l'insomnie (3,7 %), la somnolence (3,4 %), les infections des voies urinaires (2,6 %) et la diarrhée (2,0 %). Tous les événements sont survenus plus fréquemment dans le groupe brexpiprazole que dans le groupe placebo.
De plus, les données de sécurité issues d'une étude d'extension en ouvert ont été prises en compte:
Les patients éligibles ont également pu participer à cette étude (331-201-00182) pendant 12 semaines supplémentaires sous traitement actif. Au total, 259 personnes ont été examinées, incluses et traitées dans cette étude. Dans l'étude principale 331-14-213 (NCT03548584), 163 personnes ont reçu du brexpiprazole et 96 ont reçu un placebo. Les patients randomisés à 2 mg/jour ou 3 mg/jour dans l'étude principale 331-14-213 ont également conservé cette dose dans l'étude d'extension 331-201-00182. Les effets indésirables les plus fréquemment observés sous traitement actif dans l'étude d'extension ont été les céphalées (3,5 %), les chutes (2,3 %), la somnolence (1,9 %), les vertiges (1,9 %), la rhinopharyngite (1,9 %) et l'agitation (1,5 %).
Résumé des effets indésirables
Les effets indésirables (EI) rapportés dans ces études cliniques menées sur le brexpiprazole dans la ADA sont classés rangés par classe de système d'organes et par fréquence selon la convention suivante: très fréquents (≥1/10), fréquents (≥1/100 à <1/10), occasionnels (≥1/1 000 à <1/100), rares (≥1/10 000 à <1/1000), très rares (<1/10 000), fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Infections et infestations:
Fréquent: infection des voies urinaires, pneumonie, rhinopharyngite.
Occasionnel: bronchite, cystite, infections virales des voies respiratoires.
Affections hématologiques et du système lymphatique:
Occasionnel: anémie.
Troubles du métabolisme et de la nutrition:
Fréquent: augmentation de l'appétit.
Affections psychiatriques:
Fréquent: insomnie, agitation.
Occasionnel: confusion, hallucinations, retard psychomoteur.
Affections du système nerveux:
Fréquent: somnolence, céphalées, vertiges, bradykinèse.
Occasionnel: acathisie, troubles extrapyramidaux.
Affections cardiaques
Occasionnel: allongement de l'intervalle QT à l'électrocardiogramme.
Affections vasculaires
Fréquent: augmentation de la pression artérielle.
Affections respiratoires, thoraciques et médiastinales:
Fréquent: difficultés respiratoires.
Occasionnel: épistaxis.
Affections gastro-intestinales:
Fréquent: diarrhée, bouche sèche, nausées.
Occasionnel: hypersécrétion salivaire.
Affections de la peau et du tissu sous-cutané:
Occasionnel: exanthème.
Affections musculosquelettiques et du tissu conjonctif:
Fréquent: augmentation de la créatine-phosphokinase dans le sang.
Occasionnel: spasmes musculaires.
Troubles généraux et anomalies au site d'administration:
Fréquent: asthénie, fatigue, prise de poids.
Occasionnel: fièvre.
Investigations :
Occasionnel: augmentation de la lactate déhydrogénase dans le sang.
Lésions, intoxications et complications d'interventions
Fréquent: chute.
Données de sécurité complémentaires
Le brexpiprazole a été étudié dans d'autres indications non autorisées en Suisse en tant que traitement d'appoint aux antidépresseurs pour traiter le trouble dépressif majeur (TDM) et en tant que traitement d'appoint du TDAH.
Dans ces études, les effets indésirables suivants, non décrits dans les indications approuvées de schizophrénie et d'ADA, ont été notés: la constipation, la dyspepsie, l'anxiété, l'hypersensibilité et l'augmentation du taux de prolactine.
Description d'effets indésirables spécifiques et informations complémentaires
Indication: Schizophrénie chez l'adulte
Autres symptômes extrapyramidaux, notamment l'akathisie
Dans les études 331-10-231 (NCT01396421) et 331-10-230 (NCT01393613), la fréquence des événements connexes au SEP (sans acathisie) a été de 5,1% chez les patients sous REXULTI contre 3,5% sous placebo. La fréquence des événements d'acathisie a été de 5,4% chez les patients sous REXULTI contre 4,9% sous placebo. Dans tous les groupes de traitement, les nouveaux cas d'acathisie ont été rapportés au cours des 3 premières semaines suivant le début du traitement et ont été légers à modérés.
Les échelles objectives SAS (Simpson Angus Scale) pour le syndrome extrapyramidal, BARS (Barnes Akathisia Rating Scale) pour l'acathisie et AIMS (Abnormal Involuntary Movement Scale) pour la dyskinésie ont été utilisées dans les études 331-10-231 et 331-10-230. Sur les échelles SAS, BARS et AIMS (-0,10; 0,02 et -0,08), les écarts moyens entre la valeur de départ et celle de la visite de clôture observés chez les patients sous REXULTI étaient comparables à ceux constatés avec le placebo (0,00; 0,01 et -0,07).
Dans l'étude 331-10-232 (NCT01668797) également, les écarts moyens entre la valeur de départ et celle de la visite de clôture étaient comparables entre les patients sous REXULTI et sous placebo sur les échelles SAS, BARS et AIMS.
Prise de poids
Dans les études à court terme 331-10-231 et 331-10-230 (voir la section « Efficacité clinique »), la proportion de patients présentant une prise de poids potentiellement cliniquement significative (≥7 % du poids corporel) était de 10,5 % dans le groupe REXULTI 2 mg/jour et de 10,2 % dans le groupe REXULTI 4 mg/jour, contre 4,1 % dans le groupe placebo. La prise de poids moyenne lors de la dernière visite était de 1,2 kg dans les deux groupes REXULTI et de 0,2 kg dans le groupe placebo.
Dans l'étude 331-10-232 (voir la section « Efficacité clinique »), la proportion de patients présentant une prise de poids ≥7 % lors de la dernière visite était de 3,1 % dans le groupe REXULTI, contre 1,0 % dans le groupe placebo.
Dans les études ouvertes à long terme sur la schizophrénie (331-10-237 (NCT01397786), 331-08-210 (NCT01649557), 14644B (NCT01810783)) (voir la section « Efficacité clinique »), la variation moyenne du poids corporel entre l'inclusion et la dernière visite était de 1,1 kg. Le pourcentage de patients présentant une prise de poids potentiellement cliniquement significative (≥7 %) était de 18 %, et 0,4 % des sujets ont interrompu le traitement en raison d'une prise de poids.
Indication: Schizophrénie chez les adolescents âgés de 13 ans et plus
La fréquence, le type et la gravité des effets indésirables chez les adolescents devraient être similaires à ceux observés chez les adultes.
Symptômes extrapyramidaux (SEP)
Dans les études à court terme, l'akathisie était l'effet indésirable lié aux SEP le plus fréquemment rapporté dans le groupe brexpiprazole (1 mg/jour à 4 mg/jour) (3,6 %) comparativement à 2,9 % dans le groupe placebo. Parmi les autres effets indésirables liés aux SEP rapportés dans les études contrôlées à court terme menées chez des patients pédiatriques, on note la rigidité musculaire (0,9 %), l'hypokinésie (0,9 %) et les tremblements (0,9 %).
Akathisie
Dans une étude randomisée, en double aveugle et à court terme, l'incidence de l'akathisie chez les enfants et les adolescents traités par brexpiprazole était de 3,6 %, contre 2,9 % sous placebo.
Dans l'étude ouverte à long terme en cours, l'incidence de l'akathisie était de 5,1 %. Voir la section « Efficacité clinique ».
Prise de poids
Dans une étude contrôlée à court terme, le pourcentage de participants présentant une prise de poids cliniquement significative (augmentation du poids corporel ≥7 % par rapport à la valeur initiale) était de 8,2 % dans le groupe traité par brexpiprazole, contre 4,9 % dans le groupe placebo. La prise de poids moyenne entre l'inclusion et l'examen final était de 0,8 kg chez les participants traités par brexpiprazole et de 0,0 kg chez ceux du groupe placebo. Afin de tenir compte de la croissance normale, des scores Z (exprimés en écarts-types [ET]) ont été calculés. Ces scores permettent de normaliser la croissance naturelle des enfants et des adolescents par comparaison avec les normes de la population, ajustées selon l'âge et le sexe. Une variation du score Z < 0,5 ET est considérée comme non cliniquement significative. Dans cette étude, aucune variation du score Z moyen n'a été observée entre l'inclusion et l'examen final, ni dans le groupe traité par brexpiprazole, ni dans le groupe placebo. Chez 4,5 % des patients traités par brexpiprazole et chez 3,9 % de ceux sous placebo, le score Z du poids corporel, ajusté pour l'âge et le sexe, a augmenté d'au moins 0,5 écart-type par rapport à la valeur initiale. Des événements indésirables transitoires (EITT) (prise de poids) ont été rapportés chez 1,7 % des patients du groupe brexpiprazole, contre 3,4 % dans le groupe placebo.
Dans cette étude ouverte à long terme, la proportion de patients présentant une prise de poids cliniquement significative (augmentation du poids corporel ≥7 % par rapport à la valeur initiale) à chaque visite était de 44,6 % dans le groupe brexpiprazole. La variation moyenne du score Z du poids corporel entre la valeur initiale et la dernière visite était de 0,10 écart-type, tandis que 20 % des patients ont présenté une augmentation du score Z du poids corporel, ajusté pour l'âge et le sexe, d'au moins 0,5 écart-type par rapport à la valeur initiale. Des événements indésirables liés à une prise de poids (EILP) ont été observés chez 11,5 % des sujets, tandis que d'autres EILP liés à une prise de poids, tels qu'une augmentation de l'IMC et du tour de taille, sont survenus chez un seul sujet chacun.
Prolactine
Dans une étude contrôlée à court terme, l'incidence d'une élévation du taux de prolactine sanguine était de 32,4 % dans le groupe brexpiprazole (2 à 4 mg) contre 10,2 % dans le groupe placebo. Cette élévation était plus fréquente chez les femmes que chez les hommes (26,8 % contre 24,5 %). L'incidence d'une élévation de la prolactine supérieure à 1 × LSN était de 26,8 % chez les femmes du groupe brexpiprazole (2 à 4 mg) contre 6,3 % dans le groupe placebo, et de 24,5 % chez les hommes contre 6 % dans le groupe placebo. Dans les études à long terme, 1,7 % des participants ont présenté une élévation du taux de prolactine sanguine et 0,7 % une hyperprolactinémie.
Somnilance, incluant sédation et hypersomnie
Dans les études à court terme, l'incidence des événements indésirables liés au traitement (EILT) de type somnolence (sédation, somnolence, hypersomnie) dans le groupe traité par brexpiprazole 2-4 mg était de 7,3 %, contre 6,7 % dans le groupe placebo. Dans une étude ouverte à long terme, l'incidence des EILT de type somnolence (sédation, somnolence, hypersomnie) était de 11,9 %. Ces EILT étaient d'intensité légère à modérée.
Comportement suicidaire
Dans une étude contrôlée à court terme, un événement indésirable lié au traitement (EILT) associé à un comportement suicidaire a été rapporté chez un sujet (0,9 %, événement non grave) du groupe brexpiprazole et chez aucun sujet du groupe placebo. Dans une étude ouverte à long terme, des EIG associés à un comportement suicidaire ont été rapportés chez 8 sujets (2,7 %) (voir Mises en garde et précautions).
Indication: Agitation dans la démence d'Alzheimer
L'incidence rapportée des effets indésirables connexes aux EPS était de 5,3 % chez les patients traités par REXULTI contre 3,1% chez les patients traités par placebo. L'incidence des événements d'acathisie chez les patients traités par REXULTI était de 1,8 %, contre 0,3 % chez les patients traités par placebo.
Des données objectives ont été collectées à l'aide de l'échelle de Simpson et Angus (SAS) pour les EPS, l'échelle de Barnes (Barnes Akathisia Rating Scale, BARS) pour l'acathisie et l'AIMS (Abnormal Involuntary Movement Scale) pour la dyskinésie dans les études cliniques contrôlées sur 12 semaines portant sur la ADA.
La variation moyenne entre les valeurs de référence et celles de la dernière visite étaient comparables entre les patients sous REXULTI et sous placebo sur les échelles SAS, BARS et AIMS.
Pour l'échelle SAS, le pourcentage de patients qui sont passés d'un état normal à un état anormal était plus élevé chez les patients traités par REXULTI que chez ceux sous placebo (6 % contre 2 %).
Prise de poids
Dans les études cliniques sur 12 semaines contrôlées par placebo menées auprès de patients (âgés de 51 à 90 ans) atteints de la ADA (étude 331-12-283 [NCT01862640] à dose fixe, étude 331-14-213 à dose fixe [voir Efficacité clinique] et étude à dose flexible 331-12-284 [NCT01922258]), la proportion de patients présentant une prise de poids en kg de ≥7 % à chaque visite était de 1,7% dans le groupe REXULTI, contre 0,8 % dans le groupe placebo.
Parmi les patients (âgés de 55 à 90 ans) qui sont passés de l'étude clinique 12 semaines à dose fixe contrôlée par placebo 331-14-213 à l'étude d'extension sur 12 semaines avec traitement actif 331-201-00182, 3 % des patients ont présenté une augmentation du poids corporel de ≥7 % et 3 % une diminution du poids corporel de ≥7 % entre la visite de référence et la dernière visite de l'étude.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageTraitement
Il n'existe pas d'informations spécifiques sur le traitement du surdosage par REXULTI. Un lavage gastrique et un traitement émétique peuvent être utiles juste après un surdosage. Un ECG doit être effectué et, en cas d'allongement de l'intervalle QT, une surveillance cardiovasculaire doit être mise en place. Globalement, le traitement d'un surdosage doit se concentrer sur des mesures de soutien, le dégagement des voies aériennes, l'apport d'oxygène, la respiration et le traitement symptomatique. Une étroite surveillance médicale et un contrôle jusqu'à la guérison sont nécessaires.
L'administration de charbon actif et de sorbitol par voie orale (50 g/240 ml) une heure après la prise de REXULTI a abaissé la valeur Cmax du brexpiprazole d'environ 5% à 23% et l'AUC de 31% à 46%. Les informations disponibles sur le potentiel thérapeutique du charbon actif pour le traitement d'un surdosage par REXULTI ne sont toutefois pas suffisantes. Bien que l'on ne dispose pas d'expériences concrètes sur l'effet d'une hémodialyse, celle méthode devrait être peu utile pour le traitement d'un surdosage par REXULTI compte-tenu de la liaison élevée du brexpiprazole aux protéines plasmatiques.
Propriétés/EffetsCode ATC
N05AX16
Mécanisme d'action
REXULTI est un nouveau principe actif antipsychotique atypique, dont l'activité pharmacologique repose sur la modulation de l'activité sérotonine-dopamine. Bien que le mécanisme d'action précis du brexpiprazole dans le traitement de la schizophrénie ou de l'agitation dans la démence d'Alzheimer ne soit pas totalement connu, on suppose que la pharmacologie du brexpiprazole repose principalement sur l'association d'une haute affinité de liaison et d'une activité fonctionnelle sur plusieurs récepteurs monoaminergiques. Son effet modulateur sur les systèmes sérotoninergique et dopaminergique repose sur l'association d'une activité partiellement agoniste sur les récepteurs sérotoninergiques 5-HT1A et les récepteurs dopaminergiques D2 et d'une activité antagoniste sur les récepteurs sérotoninergiques 5-HT2A, les affinités pour tous ces récepteurs étant similaires et élevées (Ki : 0,1–0,5 nM).
Le brexpiprazole affiche en outre une activité antagoniste sur les récepteurs noradrénergiques α1B/2C dans la même zone Ki sous-nanomolaire (Ki : 0,2–0,6 nM). L'activité partiellement agoniste sur le récepteur 5HT1A/D2 combinée à l'activité antagoniste sur les récepteurs 5-HT2A et α1B/2C est susceptible de contribuer à l'effet antipsychotique du brexpiprazole.
Pharmacodynamique
Le brexpiprazole a une affinité élevée (Ki < 5 nM) pour plusieurs récepteurs monoarminergiques, notamment les récepteurs sérotoninergiques 5-HT1A, 5-HT2A, 5-HT2B et 5-HT7, les récepteurs dopaminergiques D2 et D3, ainsi que les récepteurs noradrénergiques α1A, α1B, α1D et α2C. Le brexpiprazole est un agoniste partiel des récepteurs 5-HT1A, D2 et D3 et un antagoniste des récepteurs 5-HT2A, 5-HT2B, 5-HT7, α1A, α1B, α1D et α2C.
Le brexpiprazole possède une faible activité intrinsèque sur les récepteurs dopaminergiques D2. Le brexpiprazole a une affinité modérée pour les récepteurs histaminiques H1 (19 nM) et une affinité très faible pour les récepteurs muscariniques M1 (inhibition de 67% à 10 µM). Le rapport dose/efficacité et le ratio d'exposition cerveau/plasma ont été déterminés dans des études précliniques in vivo ou ex vivo sur les récepteurs D2/D3, 5-HT2A, 5-HT1A, 5-HT6 et 5-HT7 ainsi que sur le transporteur de 5-HT. Les résultats concordent avec les affinités de liaison relatives in vivo et démontrent l'activité du brexpiprazole sur plusieurs points d'ancrage du système nerveux central en cas d'exposition plasmatique significative.
Dans une étude complète sur l'intervalle QTc menée auprès de patients atteints de schizophrénie ou d'un trouble schizo-affectif, REXULTI n'a pas provoqué d'allongement de l'intervalle QTcF après 12 jours à une dose thérapeutique (4 mg /jour) ou extra-thérapeutique (12 mg /jour). Aucune corrélation n'a été observée entre les concentrations en brexpiprazole et l'allongement de l'intervalle QTcF.
Efficacité clinique
Schizophrénie
Adulte:
L'efficacité de REXULTI dans le traitement de patients adultes atteints de schizophrénie selon les critères DSM-IV-TR a été démontrée dans deux études randomisées en double aveugle contrôlées par placebo sur 6 semaines à dose fixe (études 331-10-231 et 331-10-230) et dans une étude à long terme sur le traitement d'entretien (étude 331-10-232).
Pour les patients concernés, une hospitalisation ou un allongement de la durée d'hospitalisation pour le traitement d'une rechute aiguë de la schizophrénie s'est avérée bénéfique. Ils ont dû au préalable recevoir un traitement ambulatoire approprié par antipsychotiques et ont bien répondu à ce traitement (sauf à la clozapine) au cours des 12 derniers mois ayant précédé le début de l'étude.
Dans les études 331-10-231 und 331-10-230, les patients ont été randomisés aux doses journalières de REXULTI 2 mg ou 4 mg ou au placebo. Les patients des groupes REXULTI ont commencé le traitement à 1 mg une fois par jour le jour 1. Le jour 5, la dose de REXULTI a été augmentée à 2 mg une fois par jour. Le jour 8, la dose a été soit maintenue à 2 mg pendant les 5 semaines restantes de l'étude, soit augmentée à 4 mg une fois par jour selon le groupe de traitement.
Les études 331-10-231 und 331-10-230 comprenaient également deux groupes de traitement recevant une dose de REXULTI plus faible de 0,25 mg/jour et 1 mg/jour. Celles-ci n'ont toutefois pas été reprises dans l'analyse principale.
Le principal critère d'évaluation de l'efficacité des deux études était la modification du score global sur l'échelle PANSS (Positive and Negative Syndrome Scale) entre le départ et la semaine 6. L'échelle PANSS comporte 30 items et mesure les symptômes positifs (7 items) et négatifs (7 items) de la schizophrénie, ainsi que la physiopathologie générale (16 items). Chaque item est évalué sur une échelle de 1 (inexistant) à 7 (extrême). Le score global sur l'échelle PANSS s'étend de 30 (meilleur résultat) à 210 (pire résultat). Le principal critère d'évaluation secondaire des études était la modification de la valeur entre le départ et la semaine 6 sur l'échelle CGI-S (Clinical Global Impression – Severity of Illness Scale). CGI-S est une échelle validée, évaluée par le médecin, qui détermine l'état clinique actuel du patient en termes de gravité des symptômes. Les autres critères d'évaluation secondaires étaient entre autres l'échelle PSP (Personal and Social Performance Scale) validée et évaluée par le médecin, qui mesure les compétences personnelle et sociale.
Dans l'étude 331-10-231, REXULTI a été étudié aux deux posologies (2 mg/jour et 4 mg/jour) par rapport au placebo à l'aide du score global sur l'échelle PANSS (tableau 3). Dans l'étude 331-10-230, REXULTI a été étudié à la posologie de 4 mg/jour par rapport au placebo à l'aide du score global sur l'échelle PANSS (tableau 3). Pour la posologie de 2 mg/jour, seule l'étude 331-10-231 a démontré une efficacité statistiquement significative. L'analyse des groupes de population par âge, sexe et origine ethnique n'a pas démontré de différences de sensibilité de la réponse.
Le tableau 3 récapitule les résultats concernant le critère d'évaluation principal de l'efficacité pour les études 331-10-231 et 331-10-230.
Tableau 3: Principaux résultats d'efficacité pour les études sur 6 semaines concernant la schizophrénie (études 331-10-231 und 331-10-230)
|
|
Paramètre d'efficacité principal: PANSS
| |
Etude
|
Groupe de traitement
|
N
|
Valeur de départ moyenne (SD)
|
Ecart moyen LS par rapport à la valeur de départ (SE)
|
Différence LS moyennea (IC à 95%)
|
Valeur P
| |
331-10-231
|
REXULTI (2 mg/jour)*
|
180
|
95,85 (13,75)
|
-20,73 (1,55)
|
-8,7
(-13,1; -4,4)
|
<0,0001
| |
REXULTI (4 mg/jour)*
|
178
|
94,70 (1,06)
|
-19,65 (1,54)
|
-7,6
(-12,0; -3,1)
|
-0,0006
| |
Placebo
|
178
|
95,69 (11,46)
|
-12,01 (1,60)
|
--
|
-
| |
331-10-230
|
REXULTI
(2 mg/jour)
|
179
|
96,30 (12,91)
|
-16,61 (1,49)
|
-3.1
(-7,2; 1,1)
|
0,1448
| |
REXULTI (4 mg/jour)*
|
181
|
94,99 (12,38)
|
-20,00 (1,48)
|
-6.5
(-10,6; -2,4)
|
-0,0022
| |
Placebo
|
180
|
94,63 (12,84)
|
-13,53 (1,52)
|
--
|
-
|
SD: écart type; SE: erreur standard; LS: moindres carrés; IC: intervalle de confiance non ajusté.
* Supériorité statistiquement significative de REXULTI par rapport au placebo
a Différence (REXULTI moins placebo) de modification moyenne des moindres carrés entre la valeur de départ et à la semaine 6
REXULTI a été supérieur au placebo aux posologies de 2 mg/jour et 4 mg/jour concernant les critères d'évaluation secondaires (score CGI-S et score global sur l'échelle PSP). Les résultats des critères d'évaluation secondaires (scores CGI-S et PSP) confirment donc la supériorité de REXULTI par rapport au placebo et son effet cliniquement significatif.
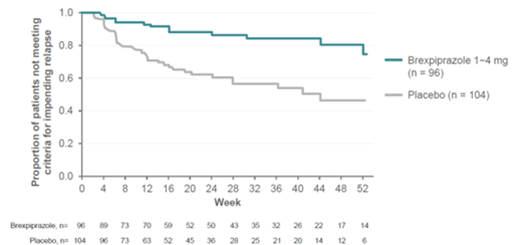
Une analyse intermédiaire établie précédemment a démontré que la durée avant rechute imminente était statistiquement significativement plus longue chez les patients randomisés dans le groupe REXULTI (1-4 mg/jour) que chez patients randomisés dans le groupe placebo (p = 0,0008, test Log-Rank). L'analyse finale a confirmé la durée statistiquement significativement plus longue avant rechute imminente pour les patients du groupe REXULTI par rapport au groupe placebo (p = 0,0001, Log-Rank Test). Les courbes de Kaplan-Meier du nombre cumulé de patients du dernier groupe d'analyse, ayant subi une rechute durant la phase de traitement en double aveugle dans les groupes REXULTI et placebo sont représentés dans l'illustration 1. Lors de l'analyse finale, le rapport de risque déduit du modèle de risque proportionnel de Cox était de 3,42 pour la comparaison entre le placebo et REXULTI (IC à 95%: 1,82; 6,41, taux de rechute 13,54% contre 38,46%). Les patients du groupe placebo présentaient donc un risque 3.42 fois plus élevé de rechute imminente que ceux du groupe REXULTI.
Illustration 1: Courbes de Kaplan-Meier concernant le temps avant rechute imminente (phase d'entretien en double aveugle - échantillon aléatoire d'efficacité) – analyse finale, étude 3
Adolescents âgés de 13 ans et plus
L'efficacité et l'innocuité du brexpiprazole dans le traitement des patients pédiatriques atteints de schizophrénie ont été évaluées dans le cadre d'une étude randomisée, en double aveugle, contrôlée par placebo, d'une durée de 6 semaines (étude 6), et dans le cadre d'une étude ouverte à long terme, d'une durée de 24 mois, actuellement en cours. L'étude à court terme a inclus 110 patients randomisés pour recevoir du brexpiprazole, 101 patients randomisés pour recevoir de l'aripiprazole en fonction de la sensibilité du test, et 104 patients randomisés pour recevoir un placebo. L'âge moyen était de 15 ans.
Dans l'étude à court terme (étude 6), les patients du groupe brexpiprazole ont débuté le traitement par 0,5 mg une fois par jour du jour 1 au jour 4, puis par 1 mg par jour du jour 5 au jour 7. La dose de brexpiprazole a été augmentée à 2 mg du jour 8 au jour 14. Elle a ensuite été maintenue à 2 mg ou augmentée à 3 mg une fois par jour du jour 15 au jour 21. Après la phase de titration, les patients ont soit maintenu la dose d'entretien, soit l'ont augmentée ou diminuée de 1 mg, jusqu'à un maximum de 4 mg de brexpiprazole par jour.
Le critère d'évaluation principal de l'efficacité était la variation moyenne du score total de l'échelle PANSS (Positive and Negative Syndrome Scale) entre l'inclusion et la semaine 6.
Le brexpiprazole à des doses de 2 à 4 mg/jour a montré une amélioration statistiquement significative de la variation moyenne du score total PANSS par rapport à l'inclusion, comparativement au placebo.
Tableau 4: Résultats d'efficacité primaires des études de 6 semaines sur la schizophrénie chez les patients pédiatriques
|
Étude
|
Groupe de traitement
|
N
|
Paramètre d'efficacité principal: PANSS
| |
|
|
|
Valeur de référence moyenne (écart-type)
|
Écart moyen des moindres carrés par rapport à la valeur de référence (erreur standard)
|
Différence moyenne des moindres carrésa (IC à 95 %)
|
Valeur p
| |
6
|
Brexpiprazole (2 mg/jour à 4 mg/jour)*
|
110
|
101.06
(14.87)
|
-22.75
(1.49)
|
-5.33
(-9.55, -1.10)
|
0.0136
| |
|
Aripiprazole (10 mg/jour à 20 mg/jour)
|
101
|
101.03
(13.08)
|
-23.95
(1.57)
|
-6.53
(-10.8, -2.21)
|
0.0032
| |
|
Placebo
|
103b
|
102.17
(16.30)
|
-17.42
(1.58)
|
--
|
--
|
ET: Écart type
ES: Erreur standard
MMC: Moyenne des moindres carrés
IC: Intervalle de confiance non ajusté
* Traitement statistiquement significativement supérieur au placebo
a Différence (brexpiprazole moins placebo) des moindres carrés de la variation moyenne par rapport à la valeur initiale, à la semaine 6
b L'échantillon d'efficacité comprend les sujets traités ayant une évaluation initiale et au moins une évaluation d'efficacité post-inclusion pour le score total PANSS.
Étude à long terme chez des adolescents de 13 ans et plus atteints de schizophrénie
Les analyses intermédiaires des études à long terme avec des doses flexibles de brexpiprazole de 1 à 4 mg/jour ont montré une amélioration durable des symptômes entre l'inclusion et le 24e mois, mesurée par le score total PANSS.
Agitation dans la démence d'Alzheimer
L'efficacité de REXULTI dans le traitement de l'agitation dans la démence d'Alzheimer (ADA) a été démontrée dans deux études randomisées en double aveugle contrôlées par placebo sur 12 semaines à dose fixe (études 331-12-283 et 331-14-213). Les patients de l'étude devaient satisfaire à un diagnostic de maladie d'Alzheimer probable selon les critères du National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA) et à un score de ≥5 à ≤22 sur l'échelle Mini-mental Examination (MMSE).
Lors de l'inclusion dans l'étude, les patients devaient présenter un score total de ≥4 pour l'item agitation/agressivité du NPI/NPI-NH (Neuropsychiatric Inventory-Nursing Home Version). Les patients devaient présenter au moment de l'inclusion dans l'étude un tel degré d'agitation qu'une pharmacothérapie était justifiée après exclusion d'autres causes.
Les patients atteints de la ADA qui n'avaient pas répondu à deux ou plusieurs antipsychotiques par le passé étaient exclus du programme de développement clinique REXULTI.
L'étude 331-12-283 incluait 433 patients âgés en moyenne de 74 ans (intervalle: 51 à 90 ans). L'étude 331-14-213 incluait 345 patients âgés en moyenne de 74 ans (intervalle: 56 à 90 ans).
Dans l'étude 331-12-283, 78 (18 %) des patients traités par REXULTI et 36 (8%) des patients traités par placebo présentaient des symptômes psychotiques avant le début de l'étude. Dans l'étude 331-14-213, 44 (19 %) des patients traités par REXULTI et 21 (18 %) des patients traités par placebo ont rapporté des antécédents de symptômes psychotiques.
Le principal critère d'évaluation de l'efficacité dans l'ensemble des études était la modification des scores sur l'échelle d'agitation de Cohen-Mansfield (Cohen-Mansfield Agitation Inventory, CMAI) entre l'inclusion et la semaine 12. Le CMAI est un questionnaire évalué par les médecins composé de 29 items qui évalue la fréquence des manifestations d'agitation chez les patients âgés en fonction des commentaires des soignants.
Les patients de l'étude 331-14-213 devaient remplir les critères du 1er facteur du CMAI (comportement agressif) en plus des critères généraux d'inclusion au début de l'étude. Pour cela, les patients doivent présenter l'une des caractéristiques suivantes: ≥1 type de comportement agressif plusieurs fois par semaine, ou ≥2 types de comportement agressif une ou deux fois par semaine, ou ≥3 types de comportement agressif moins d'une fois par semaine. Le terme de comportement agressif comprend les actions suivantes: donner des coups (y compris à soi-même), donner des coups de pied, griffer, agripper, bousculer, se blesser soi-même ou autrui, lancer des objets, jurer ou agresser verbalement, cracher, briser des objets ou détruire des biens, crier et mordre. De plus, dans l'étude 331-14-213, les patients devaient répondre au critère d'agitation chez les patients présentant des troubles cognitifs selon la définition consensuelle préliminaire de l'International Psychogeriatric Association (IPA) (2014).
Les patients de l'étude 331-12-283 ont été randomisés pour recevoir une dose fixe de REXULTI 1 mg une fois par jour, 2 mg une fois par jour ou un placebo. Dans cette étude, les patients recevant REXULTI 2 mg une fois par jour ont présenté une amélioration des scores CMAI à la semaine 12 par rapport aux patients recevant le placebo. Les patients de l'étude 331-14-213 ont été randomisés pour recevoir une dose fixe de REXULTI 2 mg ou 3 mg une fois par jour (bras de traitement combiné) ou un placebo. Dans cette étude, les patients recevant REXULTI 2 mg ou 3 mg une fois par jour ont présenté une amélioration des scores CMAI à la semaine 12 par rapport aux patients recevant le placebo.
Comme le montre le Tableau 5, la variation moyenne du score CMAI à la semaine 12 était significativement plus importante chez les patients traités par REXULTI 2 mg/jour ou 3 mg/jour que chez les patients sous placebo. À la dose de 1 mg une fois par jour, aucune variation significative du score CMAI par rapport à la valeur de référence n'a été observée dans cette population de patients.
Tableau 5: Modification du score global sur l'échelle CMAI à la semaine 12 par rapport au score de référence chez les patients agités atteints de la démence d'Alzheimer dans les études 331-12-283 et 331-14-213
|
Étude
|
Groupe de traitement
|
N
|
Valeur moyenne de référence
(SD)
|
Variation de la moyenne des moindres carrés (SE)
|
Différence entre les traitements†
(IC à 95 %)
| |
331-12-283
|
REXULTI 1 mg/jour
|
134
|
70,5 (16,0)
|
-17,6 (1,3)
|
0,2 (-3,4; 3,9)
| |
REXULTI 2 mg/jour‡
|
138
|
71,0 (16,6)
|
-21,6 (1,3)
|
-3,8 (-7,4; -0,2)
| |
Placebo
|
131
|
72,2 (17,9)
|
-17,8 (1,3)
|
—
| |
331-14-213*
|
REXULTI 2 mg/jour ou 3 mg/jour‡
|
225
|
80,6 (16,6)
|
-22,6 (1,1)
|
-5,3 (-8,8; -1,9)
| |
Placebo
|
116
|
79,2 (17,5)
|
-17,3 (1,4)
|
—
|
SD: écart-type, SE: erreur standard; IC: intervalle de confiance non ajusté
† Différence (médicament moins placebo) entre les variations des moyennes ajustées depuis la valeur de référence
‡ Supériorité statistiquement significative des posologies par rapport au placebo
* Échantillon d'efficacité enrichi (sujets atteints de la ADA avec un comportement agressif minimum défini comme critère CMAI facteur 1).
L'étude du critère d'évaluation principal dans les sous-groupes de population (selon l'âge, l'origine ethnique ou le sexe) n'a révélé aucune différence de réponse entre ces groupes.
Pour l'étude 331-14-213, une analyse de réaction post-hoc a été réalisée dans laquelle une variation cliniquement significative (Meaningful Within Patient Change, MWPC) sous brexpiprazole a été définie comme une variation du score global sur l'échelle CMAI à la semaine 12 d'au moins 20 points par rapport à la valeur de référence. La proportion de patients présentant un MWPC était de 57 % chez les patients traités par REXULTI et de 37 % chez les patients traités par placebo.
Données à long terme
Schizophrénie
L'étude à long terme (étude 331-10-232) était une étude randomisée en double aveugle contrôlée par placebo de mesure de l'efficacité, la sécurité et la tolérabilité de REXULTI 1-4 mg/jour comme traitement d'entretien chez l'adulte atteint de schizophrénie. Dans cette étude, la posologie du brexpiprazole a pu être ajustée à la discrétion de l'investigateur. La dose modale la plus fréquente pour les patients stabilisés randomisés dans le groupe du brexpiprazole était de 4 mg/jour (64 patients, 66%), suivie de 3 mg/jour (25 patients, 25.8%), 2 mg/jour (7 patients, 7.2%) et 1 patient (1%) a reçu une dose modale de 1 mg/jour.
Agitation dans la démence d'Alzheimer
Une étude d'extension en ouvert sur 12 semaines (étude 331-201-00182) évaluant la sécurité et la tolérance du brexpiprazole chez des patients souffrant d'agitation associée à une démence de type Alzheimer a mis en évidence des valeurs globales moyennes sur l'échelle CMAI similaires après 12 semaines de traitement chez des patients précédemment traités par brexpiprazole ou placebo dans l'étude contrôlée précédente. Pour différentes valeurs CMAI moyennes à l'inclusion (groupe brexpiprazole précédent 57,3 [SD 17,1] versus groupe placebo précédent 62,9 [18,1], la variation moyenne (SD) par rapport à l'inclusion était, comme prévu, plus importante chez les 86 sujets du groupe placebo précédent (-12,5 [SD 16,6]) que chez les 140 sujets du groupe brexpiprazole précédent (−7,1 [SD 12,3]).
PharmacocinétiqueAbsorption
Suite à l'administration de doses uniques de REXULTI en comprimés, le brexpiprazole est bien absorbé, les concentrations plasmatiques maximales étant atteintes sous 4,0 à 8 heures. La biodisponibilité orale absolue de la formulation en comprimés est de 95,1%. La concentration de brexpiprazole à l'état d'équilibre est atteinte 10 à 12 jours après l'administration et est environ 4 fois supérieure à celle de la première dose. REXULTI peut se prendre pendant ou en-dehors des repas. La prise d'un comprimé de REXULTI 4 mg pendant un repas ordinaire riche en lipides n'a pas eu d'effet significatif sur la valeur Cmax ni sur l'AUC du brexpiprazole. Après l'administration de doses uniques ou multiples atteignant 8 mg une fois par jour, l'exposition au brexpiprazole (Cmax et AUC) a augmenté proportionnellement à la dose administrée. Les études in vitro n'ont pas démontré que le brexpiprazole était un substrat pour les transporteurs d'efflux comme MDR1 (P-gp) et BCRP.
Distribution
Le volume de distribution du brexpiprazole après administration par voie intraveineuse est élevé (1,56 ± 0,418 l/kg), ce qui suggère une distribution extravasculaire. Dans le plasma, le brexpiprazole affiche une forte liaison protéique à la sérum-albumine (supérieure à 99%) et à l'α1 glycoprotéine acide. Sa liaison protéique n'est pas influencée par l'insuffisance rénale ou hépatique. Les résultats des études in vitro ne démontrent aucune influence de la warfarine, du diazépam ou de la digoxine sur la liaison protéique du brexpiprazole.
Métabolisme
Les études in vitro du métabolisme du brexpiprazole utilisant le cytochrome recombinant humain P450 (CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 et 3A4) démontrent que le brexpiprazole est principalement métabolisé par le CYP3A4 et le CYP2D6.
In vivo, le brexpiprazole est principalement métabolisé par les enzymes CYP3A4 et CYP2D6. Après une ou plusieurs utilisations, le brexpiprazole et un métabolite principal, DM-3411, sont les principaux constituants du médicament présents dans le circuit systémique. A l'état stable, DM-3411 représente entre 23,1 et 47,7% de l'exposition plasmatique (AUC) au brexpiprazole (AUC). Toutefois, des études précliniques in vivo démontrent qu'en cas d'expositions plasmatiques cliniquement significatives du brexpiprazole, l'exposition du DM-3411 dans le cerveau est inférieure à la limite de détection. DM-3411 ne contribue donc vraisemblablement pas à l'effet thérapeutique du brexpiprazole.
Élimination
Après une dose unique par voie orale de brexpiprazole marqué au [14C], environ 24,6% de la radioactivité administrée a été excrétée dans l'urine et 46%, dans les fèces. Moins de 1% du brexpiprazole inchangé a été excrété dans l'urine et environ 14% de la dose orale ont été excrétés sous une forme inchangée dans les fèces. La clairance orale apparente du brexpiprazole après administration d'un comprimé une fois par jour était de 19,8 (±11,4) ml/h/kg. Après l'administration répétée de REXULTI une fois par jour, la demi-vie d'élimination terminale du brexpiprazole est de 91,4 heures et de 85,7 heures pour le métabolite principal, DM-3411.
Cinétique pour certains groupes de patients
Âge / Sexe
Après administration d'une dose unique de REXULTI (2 mg), les sujets âgés (plus de 65 ans) en bonne santé ont présenté une exposition systémique au brexpiprazole similaire (Cmax et AUC) à celle des sujets adultes (18–45 ans). Les sujets de sexe féminin ont affiché une exposition systémique au brexpiprazole environ 40-50% supérieure (Cmax et AUC) à celle des sujets de sexe masculin. Lors de l'analyse de la pharmacocinétique au sein de la population, l'âge et le sexe féminin ont été définis comme des co-variables statistiquement significatives ayant un effet sur la pharmacocinétique du brexpiprazole, mais non cliniquement significatives.
Enfants et adolescents
Une étude pharmacocinétique à doses multiples (0,5, 1, 2, 3 ou 4 mg/jour) a été menée chez 43 patients pédiatriques âgés de 13 à 17 ans. Une analyse pharmacocinétique populationnelle a montré que la biodisponibilité systémique (Cmax et AUC) du brexpiprazole chez les patients pédiatriques (13 à 17 ans) était comparable à celle observée chez les patients adultes sur l'ensemble de la gamme de doses de 0,5 à 4 mg.
Origine ethnique
Bien qu'aucune étude pharmacocinétique spéciale n'ait été menée concernant l'effet de l'origine ethnique sur la disposition du brexpiprazole, l'évaluation pharmacocinétique au sein de la population n'a pas démontré de différences cliniquement significatives liées à l'origine ethnique dans la pharmacocinétique du brexpiprazole.
Métaboliseurs lents du CYP2D6
Environ 8% des personnes caucasiennes et 3-8% des personnes de couleur/afro-américaines n'ont pas la capacité de métaboliser le substrat du CYP2D6. Elles sont classées dans la catégorie des métaboliseurs lents (PM), par opposition aux métaboliseurs rapides (EM). L'exposition au brexpiprazole est environ 1.8 fois plus élevée chez les PM du CYP2D6 que chez les EM (voir la rubrique Posologie/Mode d'emploi).
Tabagisme
Des études in vitro menées sur des enzymes hépatiques humaines démontrent que le brexpiprazole n'est pas un substrat du CYP1A2. Le tabagisme ne devrait donc pas avoir d'effet sur la pharmacocinétique du brexpiprazole.
Troubles de la fonction hépatique
Les patients atteints d'insuffisance hépatique modérée à sévère (classification de Child-Pugh ≥7) sont en général davantage exposés au brexpiprazole que les patients dont la fonction hépatique est normale. La dose maximale recommandée doit donc être réduite. Chez les sujets atteints d'insuffisance hépatique de divers grades (classes Child-Pugh A, B et C), l'AUC du brexpiprazole par voie orale (2 mg en dose unique) a augmenté d'environ 24% en cas d'insuffisance hépatique légère et de 60% en cas d'insuffisance hépatique modérée par rapport aux sujets sains, tandis qu'en cas d'insuffisance hépatique sévère, elle est restée inchangée.
Troubles de la fonction rénale
Les patients atteints d'insuffisance rénale (CLcr < 60 ml/min) ont été plus exposés au brexpiprazole que les patients dont la fonction rénale était normale. La dose maximale recommandée doit donc être réduite. Chez les sujets atteints d'insuffisance rénale sévère (CLcr < 30 ml/min), l'AUC du brexpiprazole par voie orale (2 mg en dose unique) a augmenté de 69% par rapport aux sujets sains et la valeur Cmax est restée inchangée.
Données précliniquesPharmacologie de sécurité
Chez le rat, le brexpiprazole à hautes doses (ED50 = 20 mg/kg) a induit une catalepsie (modèle animal pour le risque de syndrome extrapyramidal [SEP]). Cela démontre que le brexpiprazole présente un faible risque de déclencher un SEP dans des conditions cliniques.
Chez le chien et le singe, une baisse de la tension artérielle et un allongement des intervalles QT et QTC ont été constatés. L'effet sur la tension artérielle a été imputé à un blocage des récepteurs adrénergiques α1 dans les vaisseaux périphériques, ce qui concorde avec le profil pharmacologique du brexpiprazole. L'allongement des intervalles QT et QTc est susceptible de dépendre de la diminution de la température interne induite pharmacologiquement, mais le brexpiprazole a également inhibé le transport des ions par le canal hERG (avec une valeur IC50 de 0,117 µM).
Les principaux signes cliniques permettant de définir la dose sans effet observable (NOAEL) dans les études de toxicité en cas d'administration répétée à des rats et des singes étaient liés à la diminution induite pharmacologiquement de l'activité du système nerveux central (réduction des mouvements, tremblements, somnolence, yeux fermés, hypothermie). Certains signes cliniques, comme par exemple les convulsions cloniques, l'hyperactivité et l'agressivité, observés chez le rat ont été de courte durée seulement.
Comme les autres antipsychotiques, l'index thérapeutique, mesuré à l'aide de l'AUC chez les rats et les singes, ainsi que de l'exposition de l'humain à la dose maximale recommandée de 4 mg/jour, est relativement faible (hommes/femmes: 0,1/0,8 et 0,4/0,4 chez les rats et les singes).
Globalement, les effets observés chez les ratons et les chiots après administration de brexpiprazole ont également été constatés chez les animaux adultes; les jeunes animaux ne sont donc pas les seuls à avoir eu une réaction sensible au brexpiprazole.
Mutagénicité
Le potentiel mutagène du brexpiprazole a été analysé dans le cadre de tests in vitro de rétro-mutation sur des bactéries, de tests in vitro de mutation génétique sur les ganglions lymphatiques de la souris, de tests in vitro d'aberration chromosomique sur les cellules ovariennes du hamster de Chine (CHO), de tests micronucléiques in vivo sur le rat, ainsi que de tests de la synthèse de l'ADN imprévue sur le rat. Dans les tests in vitro sur les cellules de mammifères, le brexpiprazole était mutagène et clastogène, mais seulement aux doses induisant une cytotoxicité. Dans d'autres études, aucune mutagénicité et aucune génotoxicité n'ont été observées. Sur la base des éléments factuels, le brexpiprazole n'est pas considéré comme présentant un risque génotoxique pour l'humain.
Effet sur la prolactine sérique
Suite à l'administration chronique d'antipsychotiques à des rongeurs, des modifications prolifératives et/ou néoplasiques des glandes mammaires et de l'hypophyse vraisemblablement provoquées par la prolactine ont été observées. Le brexpiprazole a démontré un potentiel d'augmentation du taux de prolactine sérique, aussi bien chez la souris que chez le rat. La pertinence de ces données concernant les tumeurs endocrines médiées par la prolactine chez les rongeurs n'est pas connue en termes de risque chez l'humain.
Carcinogénicité
Le potentiel carcinogène tout au long de la vie du brexpiprazole a été analysé dans une étude de deux ans sur des souris ICR et des rats Sprague-Dawley. Le brexpiprazole a été administré aux souris pendant deux ans par voie orale (sonde œsophagienne) à une posologie de 0,75; 2 et 5 mg/kg/jour (AUC plasmatique 0,3-3,1 fois supérieure (mâles) et 0,2-1,1 fois supérieure (femelles) à l'exposition clinique de 4 mg par voie orale, soit la dose maximale recommandée chez l'homme [MRHD]). Chez les animaux mâles, la fréquence des tumeurs n'a augmenté dans aucun groupe de dose. Chez les souris femelles, la fréquence des adénocarcinomes mammaires, des carcinomes adénosquameux et des adénomes dans la partie distale de l'hypophyse a augmenté. Le brexpiprazole a été administré aux rats pendant deux ans par voie orale (sonde œsophagienne) à une posologie de 1, 3 et 10 mg/kg/jour pour les rats et de 3, 10 et 30 mg/kg/jour pour les rates (AUC plasmatique d'exposition jusqu'à 1,0 fois supérieure (mâles) et 4,4 fois supérieure (femelles) à l'exposition à la MRHD de 4 mg/jour). Chez le rat, le brexpiprazole n'a pas été carcinogène.
Troubles de la fertilité
Les effets du brexpiprazole sur la fertilité ont été analysés sur des rates et des rats. Le brexpiprazole a été administré aux rates avant l'accouplement et aux rats après la conception et l'implantation des ovules une fois par jour par voie orale par sonde œsophagienne à des doses de 0; 0,3; 3 ou 30 mg/kg/jour avec une exposition (AUC plasmatique) jusqu'à 4,1 fois supérieure à l'exposition clinique à la MRHD). À 3 et 30 mg/kg/jour, un allongement de l'œstrus et une diminution de la fertilité ont été observés, l'exposition (plasma) étant ≥0.2 fois supérieure à l'exposition clinique à la MRHD. À 30 mg/kg/jour (AUC d'exposition 4.1 fois supérieure à l'exposition clinique à la MHRD), un léger allongement de la phase d'accouplement, ainsi qu'une hausse significative des pertes avant implantation des ovules ont été observés. La dose sans effet observable du brexpiprazole était de 0,3 mg/kg/jour (0,7 fois la MRHD par voie orale de 4 mg pour un patient de 60 kg, calculée à l'aide d'une surface corporelle comparative).
Le brexpiprazole a été administré aux rats une fois par jour par voie orale par sonde œsophagienne à des posologies de 0, 3 et 10 ou 100 mg/kg/jour, l'exposition (AUC plasmatique) étant jusqu'à 11,6 fois supérieure à l'exposition clinique à la MRHD. Après 63 jours d'administration, les mâles traités pendant 14 jours maximum et les femelles non traitées ont été réunis. Il n'y a pas eu de différence détectable avec les groupes traités par brexpiprazole concernant la durée d'accouplement ou les indices de fertilité.
Toxicité au stade du développement et lactation
Dans les études de toxicité au stade du développement, le brexpiprazole n'a pas été tératogène et n'a pas eu d'effet indésirable sur le développement. Dans ces études, les rates et les lapines en gestation ont reçu des doses de brexpiprazole allant jusqu'à 30 mg/kg/jour pendant la phase d'organogenèse, l'exposition (AUC plasmatique) étant 4,1 et 5,9 fois supérieure à l'exposition clinique à la MRHD.
Dans les études sur le développement embryon-fœtal menées sur le lapin, il a été observé qu'à une dose orale entraînant une toxicité maternelle (150 mg/kg/jour, AUC d'exposition 16,5 fois supérieure à l'exposition clinique), les fœtus avaient un poids corporel plus faible, une formation osseuse tardive et plus souvent des anomalies des viscères et du squelette.
Lorsque le brexpiprazole a été administré à des rates en gestation de la période d'organogenèse jusqu'à la lactation, le nombre de décès périnataux des portées a augmenté à 30 mg/kg/jour (AUC plasmatique 4,1 fois supérieure à l'exposition clinique à la MRHD de 4 mg); la dose sans effet observable était de 10 mg/kg/jour (exposition 0.8 fois supérieure à l'exposition clinique).
Potentiel addictif
Le brexpiprazole n'a pas démontré de potentiel à induire une dépendance physique chez le rat, ni d'effet de renforcement chez le singe rhésus. Dans une étude sur le potentiel addictif du médicament chez le rat, aucun syndrome de manque suggérant une dépendance physique n'a été démontré. Le brexpiprazole n'a pas le potentiel d'induire une dépendance physique.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Tenir hors de portée des enfants.
Conserver dans l'emballage d'origine.
Ne pas conserver au-dessus de 30°C.
Numéro d’autorisation66475 (Swissmedic)
PrésentationREXULTI (brexpiprazole) comprimés pelliculés est disponible en:
Comprimés pelliculés 0.5 mg: 7 [B]
Comprimés pelliculés 1 mg: 10, 28 [B]
Comprimés pelliculés 2 mg: 28 [B]
Comprimés pelliculés 3 mg: 28 [B]
Comprimés pelliculés 4 mg: 28 [B]
Titulaire de l’autorisationLundbeck (Schweiz) AG, Opfikon
Mise à jour de l’informationSeptembre 2025
09122025FI
|